

Abstract
Neuronal α3‐containing nicotinic acetylcholine receptors (nAChRs) in the peripheral nervous system (PNS) and non‐neuronal tissues are implicated in a number of severe disease conditions ranging from cancer to cardiovascular diseases and chronic pain. However, despite the physiological characterization of mouse models and cell lines, the precise pathophysiology of nAChRs outside the CNS remains not well understood, in part because there is a lack of subtype‐selective antagonists. α‐Conotoxins isolated from cone snail venom exhibit characteristic individual selectivity profiles for nAChRs and, therefore, are excellent tools to study the determinants for nAChR‐antagonist interactions. Given that human α3β4 subtype selective α‐conotoxins are scarce and this is a major nAChR subtype in the PNS, the design of new peptides targeting this nAChR subtype is desirable. Recent studies using α‐conotoxins RegIIA and AuIB, in combination with nAChR site‐directed mutagenesis and computational modelling, have shed light onto specific nAChR residues, which determine the selectivity of the α‐conotoxins for the human α3β2 and α3β4 subtypes. Publications describing the selectivity profile and binding sites of other α‐conotoxins confirm that subtype‐selective nAChR antagonists often work through common mechanisms by interacting with the same structural components and sites on the receptor.
Linked Articles
This article is part of a themed section on Nicotinic Acetylcholine Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.11/issuetoc
Abbreviations
- AChBP
ACh‐binding protein
- DRG
dorsal root ganglion
- MD
molecular dynamics
- nAChRs
nicotinic ACh receptors
- PNS
peripheral nervous system
Structure and physiology of neuronal nAChRs
The cation‐selective nicotinic ACh receptors (http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=76) are pentameric ligand‐gated ion channels of the Cys‐loop receptor superfamily (Figure 1), which also includes GABA type A (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=72)‐, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=73‐ and serotonin (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=68) receptors (Changeux, 2012). Channel opening is triggered by binding of the endogenous ligand http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=294 or exogenous ligands such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2585 to the binding site at the interface of two adjacent subunits (Itier and Bertrand, 2001; Jensen et al., 2005; Albuquerque et al., 2009; Cecchini and Changeux, 2015).
Figure 1.
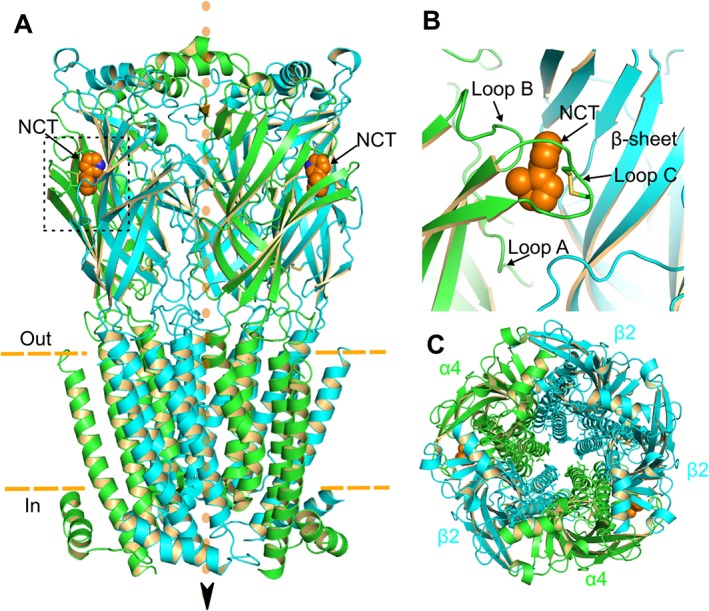
Crystal structure of human α4β2 nAChR with bound nicotine molecules (PDB ID: 5KXI) (Morales‐Perez et al., 2016). (A) Side view. Two nicotine molecules (NCT, orange) are bound at the extracellular domain of the receptor. Arrow and orange dots indicate the direction of cation movement. Dashed orange line indicates the plasma membrane. The α4 and β2 subunits are coloured green and cyan respectively. (B) Magnification of the nicotine binding site (dotted box from A), formed at the interface of α4 and β2 subunits. ACh, the endogenous nAChR agonist, occupies the same site. The binding pocket consists of loops A, B and C of the α4 subunit, indicated by the arrows, and loops D, E and F of the β2 subunit (not shown). (C) Top view of the α4β2 nAChR pentamer. Note the central ion‐conducting pore.
Seventeen nAChR subunits have been identified in vertebrate species (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=462–http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=470, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=471–http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=474, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=475, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=476 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=477). All of these subunits, except α8, which has been identified only in avian species, are expressed in humans and in other mammalian species (McGehee and Role, 1995; Lukas et al., 1999; Millar and Gotti, 2009). Nine α (2–10) and three β (2–4) subunits are generally classified as neuronal nAChRs (Albuquerque et al., 2009; Millar and Gotti, 2009). http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=468 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=469 nAChR subunits form functional homopentameric receptors, but most native nAChR subtypes consist of different subunits assembling to heteropentameric receptors (Figure 1A, C).
Neuronal nAChRs are present in a variety of CNS neurons and also in autonomic ganglion neurons, vagal afferent neurons and adrenal chromaffin cells of the peripheral nervous system (PNS) (Sargent, 1993; Cooper, 2001; Campanucci et al., 2010; Rudchenko et al., 2014). Most neuronal nAChRs are localized presynaptically where they modulate neurotransmitter release either through the depolarization of the presynaptic bouton (Dani and Bertrand, 2007) and/or an increase of intracellular Ca2+ concentration (Albuquerque et al., 2009). Neuronal nAChR subtypes are also located postsynaptically, where nAChR activation initiates depolarization and action potential firing.
Numerous nAChR subunit configurations are expressed in the CNS (Dani and Bertrand, 2007; Gotti et al., 2007; Zoli et al., 2015) where they have been implicated in neurological disorders such as Parkinson's disease, Alzheimer's disease, schizophrenia, Tourette's syndrome and epilepsy, as well as opiate and nicotine addiction (Lindstrom, 1997; Ripoll et al., 2004; Dani and Bertrand, 2007; Changeux, 2010; Hurst et al., 2013; Muldoon et al., 2014). Furthermore, a variety of nAChR subunits are expressed in the PNS and non‐neuronal cells, resulting in a vast array of subunit combinations. This diversity leads to the customized pharmacology and distinct function of non‐CNS nAChRs, including http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=464‐containing nAChRs. The focus of this review is on α3 subunit‐containing nAChRs, as their physiological functions are not well understood due to the lack of subtype‐selective antagonists.
α3β4 and α3β2 nAChR subtypes expressed in the PNS and non‐neuronal cells
In the PNS, the α3 subunit is widely expressed in autonomic ganglia (Jensen et al., 2005; Millar and Gotti, 2009; Zoli et al., 2015), adrenal chromaffin cells and enteric nervous system (Tachikawa et al., 2001; Zhou et al., 2002), preferentially forming heteromeric nAChRs with the β4 subunit (Table 1). Studies using α3 knockout mice indicate that this subunit is an essential component of the nAChRs mediating normal function of the autonomic nervous system. Phenotypic abnormalities of the mice include impaired growth and increased mortality after weaning, dysfunctionally enlarged bladder, urinary stones and widely dilated ocular pupils (Xu et al., 1999a). Detailed histological examination of the α3‐null mice revealed no significant abnormalities in the brain or peripheral tissues, whereas inflammation was prominently observed in the urinary bladder. Electrophysiological recordings showed reduced open probability of ACh‐activated single channel currents in neurons of the superior cervical ganglia (Xu et al., 1999a). Furthermore, postganglionic sympathetic neurons of α3‐null mice are devoid of excitatory synaptic potentials, confirming that postsynaptic α3‐containing receptors are a crucial component for synaptic transmission in autonomic ganglia (Rassadi et al., 2005). Interestingly, a http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=472–β4 double knockout mouse showed phenotypic abnormalities and physiological impairments similar to the α3‐null mouse, despite both β2 and β4 single‐knockout mice appearing superficially normal (Xu et al., 1999b). This finding suggests an apparent redundancy between β2 and β4 in forming physiologically intact receptors. Indeed, no overt physiological abnormalities were identified in β2‐null mice (Wang et al., 2005). In contrast, ion channel function in the superior cervical ganglion and bladder contractility were functionally impaired in the β4‐null mouse (Xu et al., 1999b), indicating that α3 and β4 subunits are the major components of ganglionic nAChRs.
Table 1.
Tissue distribution of α3, α5, α6, β2 and β4 nAChR subunits in the PNS and non‐neuronal cells determined by protein and/or functional expression of α3‐containing nAChRs
| Tissue | nAChR subunit | Reference |
|---|---|---|
| PNS | ||
| Autonomic ganglia | α3, α5, β2, β4 | Xu et al., 1999a; Xu et al., 1999b; Bibevski et al., 2000; Del Signore et al., 2004; Purnyn et al., 2004; Rassadi et al., 2005; Wang et al., 2005; Park et al., 2006; Campanucci et al., 2010; Rudchenko et al., 2014 |
| Adrenal chromaffin cells | α3, α5, β2, β4 | Campos‐Caro et al., 1997; Tachikawa et al., 2001; Sala et al., 2008 |
| Dorsal root ganglia | α3, α6, β2, β4 | Genzen et al., 2001; Khan et al., 2003; Fucile et al., 2005; Spies et al., 2006; Hone et al., 2012; Smith et al., 2013; Zhang et al., 2015 |
| Enteric ganglia | α3, α5, β2, β4 | Zhou et al., 2002; Galligan and North, 2004; Glushakov et al., 2004; Foong et al., 2015 |
| Nodose ganglia | α3, α5, β2, β4 | Mao et al., 2006 |
| Non‐neuronal cells | ||
| Keratinocytes | α3, α5, β2, β4 | Grando et al., 1995; Zia et al., 2000; Chernyavsky et al., 2004; Kurzen et al., 2004 |
| Lung epithelial cells | α3, α5, β2, β4 | Zia et al., 1997; Maus et al., 1998; Conti‐Fine et al., 2000; Proskocil et al., 2004; Sekhon et al., 2005 |
| Endothelial cells | α3, α5, β2, β4 | Macklin et al., 1998; Heeschen et al., 2002; Bruggmann et al., 2003; Moccia et al., 2004; Hawkins et al., 2005 |
| Astrocytes | α3, α5, β2, β4 | Graham et al., 2003 |
| Lymphocytes | α3, α5, β2, β4 | Hiemke et al., 1996; Skok et al., 2007 |
| Granulocytes | α3, β2, β4 | Benhammou et al., 2000; Blanchet et al., 2007 |
| Mast cells | α3, α5, β2, β4 | Kindt et al., 2008 |
| Macrophages | α3, β2, β4 | Yang et al., 2016 |
| Fibroblasts | α3, α5, β2, β4 | Arredondo et al., 2003 |
ACh release in the adrenal medulla, triggered by stimulation of the splanchnic nerve from the sympathetic nervous system, activates nAChRs in chromaffin cells. Opening of the nAChRs causes membrane depolarization, triggering catecholamine secretion (Sala et al., 2008). Expression analysis by in situ hybridization indicated that all chromaffin cells co‐express α3, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=466 and β4 transcripts (Campos‐Caro et al., 1997), and as these subunits can form functional nAChRs, it is postulated that they are responsible for the release of catecholamines (Sala et al., 2008).
The α3β4 nAChR has been identified as the major subtype expressed in both sympathetic and parasympathetic major pelvic ganglion neurons of the male rat, as ACh‐induced currents were inhibited by the α3β4 nAChR antagonists, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3990 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3973 (Park et al., 2006). Rat superior cervical ganglia of the sympathetic nervous system also reportedly express α3‐containing receptors, which are associated with β2 and/or β4 subunits (Del Signore et al., 2004).
Multiple distinct subtypes of nAChRs are expressed and pharmacologically active in cultured dorsal root ganglion (DRG) neurons (Genzen et al., 2001; Dube et al., 2005; Hone et al., 2012) Table 1. Rodent DRG neurons can be divided into four different subclasses based on their nAChR expression, with one of them predominantly expressing β4‐containing nAChRs with α3 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=467 (Smith et al., 2013). Given that α3‐containing nAChRs, including the α3β4 subtype, are predominantly expressed in nociceptive DRG neurons (Fucile et al., 2005; Spies et al., 2006), it suggests that this subtype may be involved in neuronal signalling of nociception and inflammation. A recent electrophysiological study on cutaneous rat DRG neurons showing that inflammation induces an increase in current density of slow nicotine‐evoked currents is consistent with the involvement of α3/β4‐containing nAChRs (Zhang et al., 2015). Furthermore, in cultured mouse sympathetic neurons and adrenal chromaffin cells, α3‐containing nAChRs were identified as key components for the hyperglycaemia‐induced inactivation of nAChR‐mediated currents. This finding is relevant in the context of diabetes, as impaired sympathetic neuronal signalling of the autonomic nervous system is commonly observed among diabetics (Campanucci et al., 2010; Rudchenko et al., 2014).
In addition to their presence in many cells of the PNS, α3β4 and α3β2 nAChRs have also been detected in a large variety of non‐neuronal cells such as human vascular endothelial cells, epithelial cells, lymphocytes, macrophages, mast cells, granulocytes, keratinocytes, fibroblasts, microglia and astrocytes (Sharma and Vijayaraghavan, 2002; Graham et al., 2003; Gotti and Clementi, 2004; Wessler and Kirkpatrick, 2008; Arias et al., 2009) (Table 1). For example, α3‐containing nAChRs play a pivotal role in regulating the inflammatory responses in endothelial cells and macrophages (Yang et al., 2016). It can be expected that these subtypes are expressed in other tissues, yet to be analysed (Sharma and Vijayaraghavan, 2002; Egleton et al., 2008).
To add to the complexity of non‐neuronal distribution of nAChRs, their expression pattern not only varies according to phenotypic cell function but also changes in response to internal and external environmental conditions. For example, it has been shown that during the maturation of keratinocytes, steroid hormones trigger changes in the expression pattern of nicotinic and http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=2, resulting in the formation of completely different subtypes (Wessler and Kirkpatrick, 2008).
Given that Ca2+ appears to be a primary mediator of nAChR signalling in non‐neuronal cells, the downstream effects are likely to be diverse as well and dependent on the cell type. Activation of α3‐containing nAChRs in these cells can control a number of general cellular processes like cell migration, survival, differentiation, proliferation, regulation of gene expression and other cell type‐specific roles in an autocrine and paracrine manner (Sharma and Vijayaraghavan, 2002; Egleton et al., 2008; Wessler and Kirkpatrick, 2008). Not surprisingly, non‐neuronal signalling of these nAChRs has considerable implications for pathophysiological conditions such as cancer and cardiovascular disease. Due to their involvement in cell proliferation, non‐neuronal nAChRs are linked to nicotine‐induced cancer. Although nicotine is believed to stimulate cell proliferation via signalling pathways mainly involving the α7 subtype, the anti‐apoptotic activity of nicotine in bronchial epithelial cells is mediated by α3‐ and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=465‐containing nAChRs (West et al., 2003; Egleton et al., 2008). Furthermore, epidemiologic data suggests the role of α3‐containing nAChRs in tobacco‐related cancers in smokers (Hallden et al., 2016).
From physiology to pharmacology
Molecular techniques such as in situ hybridization, immunoassays as well as studies on transgenic mice provided significant insight into the expression and distribution of nAChR subunits and their physiological role. However, these studies fail to provide detailed knowledge about the tissue‐specific physiological role of individual nAChR subtypes. To this end, pharmacological studies can contribute crucial information in filling this gap. For example, at presynaptic terminals, the α3β4 nAChR subtype has been shown to induce http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=484 release, whereas β2‐containing receptors with α3 and α6 subunits induce http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=940 release (Soliakov and Wonnacott, 1996; Kulak et al., 2001). Subtype‐selective α‐conotoxins such as AuIB and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3970 (described in more detail below) can provide direct evidence of the physiological functions of nAChRs, for example, inhibition of noradrenaline and dopamine release, respectively (Luo et al., 1998), or the specific role of α3β4 in small cell lung carcinomas (Improgo et al., 2013). Furthermore, some α‐conotoxins that specifically target nAChR subtypes have been shown to be effective at reducing mechanical allodynia in neuropathic pain models. In vitro patch clamp electrophysiology of primary afferent synaptic transmission showed that the mode of action of these conotoxins presumably involves the inhibition of nAChRs containing α3 and α7 subunits (Napier et al., 2012). These findings provided valuable information about receptor targets for the effective treatment of neuropathic pain.
Conotoxins from cone snail venom, a rich source of pharmacological compounds – a short overview
Conopeptides are peptides isolated from the venom of predatory marine snails of the genus Conus, commonly called cone snails. More than 800 species of cone snails are known (Puillandre et al., 2015), and the venom of each species is a complex and unique mixture of at least 200 distinct peptides (Olivera, 2006; Lavergne et al., 2015). Cone snails inject their venom via a hollow harpoon‐like tooth to immobilize their prey; mostly worms, molluscs and other invertebrates. Approximately 50 species of cone snail prey on fish, and the venom of one species (Conus geographus) is even dangerous to humans (Gray et al., 1988; Livett et al., 2004; Kohn, 2016) with a human lethal dose estimated to be between 0.0038 and 0.029 mg·kg−1 (Dutertre et al., 2014). Of the estimated >100 000 peptides present in Conus venoms, to date, only a relatively small portion (<0.1%) have been structurally and pharmacologically characterized (Lewis et al., 2012; Schroeder and Craik, 2012; Akondi et al., 2014). Thus, cone snail venoms are still a vast source of potential pharmacologically active substances. The short disulfide‐rich conotoxins, which specifically target different voltage‐ and ligand‐gated ion channels, are the best characterized biologically active cone snail venom peptides. For an in‐depth overview of the different classes of conotoxins and their pharmacological profiles, see the following comprehensive reviews (Becker and Terlau, 2008; Lewis et al., 2012; Akondi et al., 2014; Robinson and Norton, 2014).
α‐Conotoxins, a class of conotoxins active at nAChRs
Peptides with pharmacological activity at nAChRs have been identified in several conotoxin superfamilies (Akondi et al., 2014), but the largest and best known subgroup of conotoxins inhibiting nAChRs are the α‐conotoxins. α‐Conotoxins are 12–20 amino acid disulfide‐bonded peptides and represent the largest known group of nAChR antagonists. Some α‐conotoxins exhibit high selectivity towards specific nAChR subtypes. Consequently, they are excellent tools to pharmacologically study the distribution and functional role of nAChR subtypes in specific tissues and the nervous system.
The structure of α‐conotoxins is defined by the two disulfide bonds, contributed by four cysteine residues (CI–IV) (Figure 2). The characteristic framework for α‐conotoxins is CICIIXmCIIIXnCIV, where Xm and Xn refer to the number of non‐cysteine residues, which define loops 1 and 2 of the peptide. Loop 1 comprises four residues (m = 4) and loop 2 has between three and seven residues (n = 3, 6 or 7) (Millard et al., 2004). Due to the shuffling of disulfide bonds, α‐conotoxins can fold into three different isomers with completely altered three‐dimensional structure. Most native and pharmacologically active α‐conotoxins adopt the globular isomer with a CI–CIII and CII–CIV connectivity (Figure 2). The two other possible isomers are the ribbon (CI–CIV and CII–CIII) and bead (CI–CII and CIII–CIV) (Millard et al., 2004; Muttenthaler et al., 2011). Comprehensive reviews of α‐conotoxins and their pharmacological activities at muscle and neuronal nAChRs subtypes have been published (Azam and McIntosh, 2009; Muttenthaler et al., 2011; Lebbe et al., 2014; Lin et al., 2016a).
Figure 2.
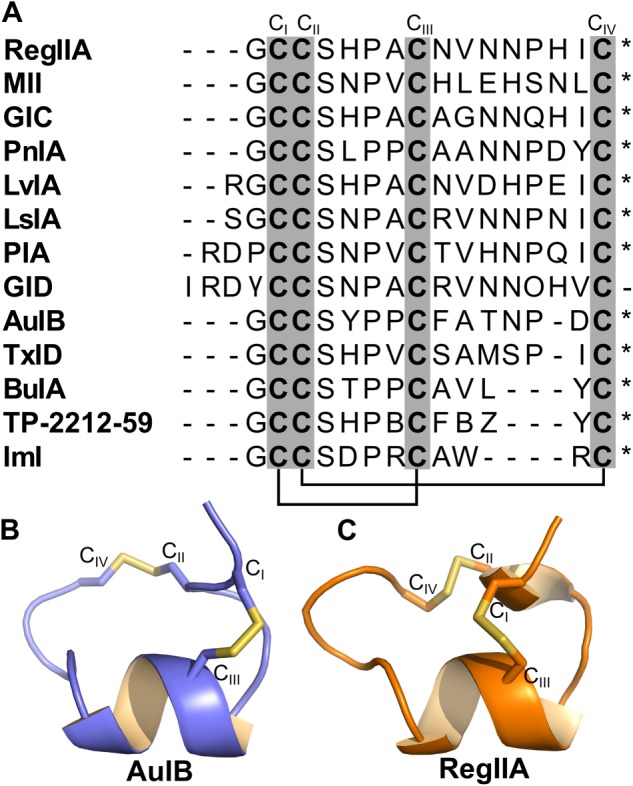
Sequence and structure of α3β2 and α3β4 nAChR‐targeting α‐conotoxins. (A) Sequence alignment of 13 α‐conotoxins that antagonize α3β2 and α3β4 nAChRs. Cysteine residues CI–CIV (grey columns) form the disulfide bridges between CI–CIII and CII–CIV (black lines) in native α‐conotoxins. * indicates C‐terminal amidation. γ in GID sequence refers to γ‐carboxyglutamate residue. B and Z in the TP‐2212‐59 sequence refer to 2‐aminobutyric acid and norvaline, respectively. (B and C) Structures of α‐conotoxins AuIB and RegIIA respectively.
Pharmacologically relevant α‐conotoxins active at α3β2 nAChRs
The only known α‐conotoxin with a 4/3 disulfide framework active at α3β2 nAChR is http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3965, isolated from the venom of the worm‐hunting cone snail Conus imperialis. It was reported to be a selective antagonist for neuronal over muscle nAChRs at the mouse neuromuscular junction (McIntosh et al., 1994). It most potently blocked the human α3β2 subtype with an IC50 of 40.8 nM while also being active at human α7 (IC50 595 nM), and weakly active at α3β4 nAChRs (IC50 3.39 μM) (Ellison et al., 2004) (Table 2). BuIA, from the venom of the fish‐eating cone snail Conus bullatus , with an uncommon 4/4 spacing of disulfide loops, potently inhibits several rat nAChR subtypes, including α3β2 with an IC50 of 5.7 nM (Azam et al., 2005) (Table 2).
Table 2.
Selected α‐conotoxins that exhibit inhibitory activity at α3β2 and α3β4 nAChRs expressed in Xenopus oocytes
|
α‐Conotoxin Conus species |
Framework | aIC50 (nM) at recombinant nAChRs | bReference | |
|---|---|---|---|---|
| α3β2 | α3β4 | |||
|
ImI Conus imperialis |
4/3 |
40.8 (h) (Ellison et al., 2004) |
3390 (h) (Ellison et al., 2004) |
McIntosh et al., 1994 |
|
BuIA Conus bullatus |
4/4 | 5.72 | 27.7 | Azam et al., 2005 |
| TP‐2212‐59 (synthetic) | 4/4 | >10 000 | 2.3 | Chang et al., 2014 |
|
AuIB Conus aulicus |
4/6 |
>10 000 (Luo et al., 1998) >30 000 (h) (Cuny et al., 2016) |
750 (Luo et al., 1998) >30 000 (h) (Cuny et al., 2016) |
Luo et al., 1998 |
|
TxID Conus textile |
4/6 | >10 000 | 12.5 | Luo et al., 2013 |
|
MII Conus magus |
4/7 |
1.7 (Dowell et al., 2003) |
>200 | Cartier et al., 1996 |
|
PIA Conus purpurascens |
4/7 | 74.2 | 518 | Dowell et al., 2003 |
|
RegIIA Conus regius |
4/7 |
33 (Franco et al., 2012) 132.4 (h) (Cuny et al., 2016) |
97 (Franco et al., 2012) 45.6 (h) (Cuny et al., 2016) |
Franco et al., 2012 |
|
GIC Conus geographus |
4/7 | 1.1 (h) | 755 (h) | McIntosh et al., 2002 |
|
GID C. geographus |
4/7 | 3.1 | >10 000 | Nicke et al., 2003 |
|
PnIA (synthetic)c |
4/7 |
9.56 (Luo et al., 1999) |
>1000 (Everhart et al., 2003) |
Fainzilber et al., 1994 |
|
LsIA Conus limpusi |
4/7 | 10 | >1000 | Inserra et al., 2013 |
|
LvIA Conus lividus |
4/7 |
8.67 17.5 (h) |
148 | Luo et al., 2014 |
Most α‐conotoxins listed were tested on a range of nAChR subtypes; however, only IC50 values for subtypes covered in this review are shown. For full details see the cited references.
If not indicated otherwise, IC50 values were obtained at rat nAChRs. References to individual values indicate the publication in which the data were reported. h, human.
Reference in which the α‐conotoxin was first described.
IC50 values were obtained with the synthetic analogue PnIA[sTy15Y], which is commonly referred to as PnIA, albeit amino acid 15 differs from native PnIA of Conus pennaceus.
A large number of α‐conotoxins acting on α3β2 are from the 4/7 cysteine framework group. One of the earliest and well‐characterized 4/7 α‐conotoxins, MII from Conus magus, was shown to have a remarkable selectivity for α3β2 with an IC50 in the low nanomolar range, whereas other nAChR subtypes are >200‐fold less sensitive (Cartier et al., 1996; Harvey et al., 1997; Dowell et al., 2003). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4009 isolated from the venom of C. geographus selectively and potently inhibits human α3β2 with an IC50 of 1.1 nM, whereas potency at human α3β4 is 755 nM (Table 2). Furthermore, it has a remarkable 100 000‐fold selectivity for α3β2 over muscle nAChRs (McIntosh et al., 2002; Lin et al., 2016b). Another 4/7 α‐conotoxin with nanomolar affinity for α3β2 nAChRs is http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3985 from Conus purpurascens. Interestingly, PIA displayed a 75‐fold higher affinity for rat α6‐containing nAChRs (specifically the α6/α3β2http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=473 subtype) than for rat α3β2 (Dowell et al., 2003).
Luo et al. (2014) recently reported the discovery of 4/7 α‐conotoxin LvIA as a potent α3β2‐selective conotoxin, which exhibited low nanomolar potency at rat α3β2 (IC50 8.7 nM) whereas its IC50 values were >100 nM at α6/α3β2β3, α6/α3β4 and α3β4 nAChRs. At the homologous human nAChR subtypes, an even clearer preference for α3β2 over α6/α3β2β3 (IC50 17.5 nM compared with 5342 nM) was observed.
α‐Conotoxins pharmacologically active at α3β4 nAChRs and their molecular determinants for selectivity
To date, only few pharmacological compounds have been discovered that selectively block the α3β4 nAChR subtype. As this is the predominant subtype in the PNS and its involvement in several important pathophysiological processes has been shown, there remains a need for pharmacological compounds with high affinity and selectivity for this subtype.
The first α3β4‐selective α‐conotoxin described was the 4/6 toxin AuIB from Conus aulicus (Luo et al., 1998) (Figure 2B). AuIB inhibits rat α3β4 with >100‐fold higher potency than other nAChR subtypes, even though the potency at α3β4 itself is relatively low (IC50 = 0.75 μM) (Table 2). Closer analysis of α3β4‐mediated ACh‐evoked current inhibition by AuIB revealed properties unique to this peptide. First, the non‐native ribbon isomer of AuIB exhibits a lower IC50 at inhibiting α3β4 compared with the native globular isomer. This is in contrast to the general assumption that conotoxins lose activity when not folded in the native form. However, analysis of concentration‐dependent inhibition of ACh‐evoked currents mediated by α3β4 revealed that the maximal block (efficacy) of the ribbon isomer is only ~50%, whereas complete block was achieved by sufficiently high concentrations of the globular isomer (Grishin et al., 2010). Furthermore, ribbon AuIB behaves as a competitive antagonist, and its inhibition is dependent on the receptor subunit stoichiometry. In contrast, inhibition by the globular isomer is independent of receptor stoichiometry, and surprisingly, globular AuIB inhibits α3β4 by a non‐competitive antagonism, which is unusual for α‐conotoxins. Molecular docking simulation suggested that ribbon AuIB binds to the ACh‐binding pocket of the α3(3)β4(2) nAChR stoichiometry (Grishin et al., 2010), whereas globular AuIB is likely to bind to both α3(3)β4(2) and α3(2)β4(3) stoichiometries at a location outside the ACh‐binding pocket.
In a subsequent study, the mechanism and structural determinants of AuIB binding to α3β4 were analysed in more detail (Grishin et al., 2013). Alanine scan mutagenesis revealed that two AuIB analogues, AuIB[P6A] and AuIB[F9A], did not inhibit α3β4 nAChR. The AuIB[P6A] analogue lost its secondary structure and consequently the ability to inhibit α3β4, but the AuIB[F9A] analogue retained its globular structure, and the loss in activity was due to disruption of the specific peptide‐receptor pairwise contacts. Molecular docking and receptor mutagenesis revealed that the main determinants of AuIB activity at α3β4 lie on the β4 subunit. Residues W59 and K61 form a binding pocket for F9 of AuIB in which its aromatic ring is inserted between the two aforementioned β4 residues and stabilized by them. Alanine substitution of K61 reduced inhibition, whereas substitution of W59 completely abolished inhibition, suggesting W59 is the main determinant of peptide activity. Interestingly, W59 is common to all neuronal nAChRs and hence cannot determine AuIB selectivity by itself. K61, however, is unique to the β4 subunit and likely determines AuIB selectivity for α3β4 (Grishin et al., 2013).
Luo et al. (2013) reported the discovery of conotoxin TxID, the most potent α‐conotoxin for α3β4 nAChRs described to date. TxID inhibited rat α3β4 nAChRs with an IC50 of 12.5 nM and the closely related α6/α3β4 nAChRs with sevenfold less potency (IC50 94 nM), but exhibited minimal activity at other nAChR subtypes. Interestingly, TxID is the only α‐conotoxin sharing the 4/6 disulfide framework with AuIB, but is 60‐fold more potent than AuIB at α3β4 nAChRs.
Other α‐conotoxins inhibiting ACh‐evoked currents mediated by α3β4 with low nanomolar IC50 have been reported, but none are selective for this nAChR subtype. Therefore, effort has been focused on understanding the selectivity profile of α‐conotoxins and, more specifically, the molecular determinants for specificity towards α3β4. An example is the 4/7 α‐conotoxin RegIIA, isolated from Conus regius (Figure 2C). RegIIA exhibited low IC50 values at rat α3β4 (97 nM) and α3β2 (33 nM), and human α7 (103 nM) nAChRs (Franco et al., 2012). Therefore, despite the potent activity at α3β4, RegIIA is far from being a selective pharmacological probe. Alanine‐scanning mutagenesis revealed [N11A] and [N12A]RegIIA analogues were threefold more selective for α3β4 compared with α3β2, and the double‐alanine analogue RegIIA[N11A,N12A] was even more selective for α3β4 with an IC50 of 370 nM (Kompella et al., 2015b). Although less potent at α3β4 compared with TxID, RegIIA[N11A,N12A] exhibits a >14‐fold selectivity for α3β4 over the α6/α3β4 subtype (IC50 ~ 5 μM) (Kompella et al., 2015b).
Species‐specific activity differences of α‐conotoxins help elucidate key residues for selective binding to α3‐containing nAChR subtypes
An impediment to the effective development of nAChR subtype‐selective α‐conotoxins as pharmacological probes with therapeutic potential is the fact that most α‐conotoxins have only been characterized in heterologous expression systems using cloned nAChR subunits of one species, usually rat. However, there is growing evidence that α‐conotoxins can exhibit considerably different potencies on homologous nAChR subtypes of different species (Azam and McIntosh, 2012; Yu et al., 2013; Azam et al., 2015).
Interestingly, α‐conotoxin RegIIA was significantly less potent at the human α3β2 nAChR subtype compared with rat, whereas at α3β4, no species‐specific differences in sensitivity were observed (Kompella et al., 2015a). As the homologous receptor subtypes of rat and human are overall relatively conserved, it is believed that the binding site of RegIIA at α3β2 and α3β4 nAChRs overlaps with the agonist binding site (Kompella et al., 2015a). Therefore, only the few residues in the ACh binding domains that differ between the species were predicted to play a role in determining selectivity. It was predicted that the determinants for the species difference would reside on the β subunit, because at α3β2, no species difference in activity was observed. The extracellular domains of the β4 subunit differ in only three residues, and surprisingly, when any of these residues in the rat β4 subunit was mutated to the human counterpart, only a modest loss in activity was observed. Conversely, exchange of a single residue in loop F of the α3 subunit (Q198 to P) was sufficient to reduce the affinity of RegIIA to a value similar to that observed for the human nAChR. Mutagenesis experiments and molecular dynamic (MD) simulations showed that the residue exchange causes structural changes at the ACh binding site by reducing the flexibility of nearby receptor residues and preventing them from effective interaction with RegIIA residues due to steric hindrance (Kompella et al., 2015a). A similar effect has been reported for α4β2, whereby the reverse mutation at the homologous position in rat α4, P195Q, enhanced α‐conotoxin TxIA potency at α4β2 by increasing the number of contacts between toxin and receptor (Beissner et al., 2012).
As mentioned above, AuIB was the first rat α3β4 selective α‐conotoxin discovered, and it has represented a prime example of a specific antagonist of this receptor subtype. However, AuIB was largely inactive with no activity observed at 1 μM when tested on human α3β4 (compared with an IC50 of 0.75 μM at rat α3β4), and at 30 μM, only ~20% of ACh‐evoked current amplitude mediated by human α3β4 was inhibited, indicating that the IC50 is >40‐fold higher for rat α3β4. At the human α3β2 subtype, AuIB exhibited a similar low inhibitory potency (Cuny et al., 2016).
Molecular determinants in the agonist binding loops of human α3β2 and α3β4 nAChRs that define subtype selectivity of α‐conotoxins
As the extracellular domains of human β2 and β4 subunits are reasonably conserved, mutational studies on non‐conserved amino acids have identified critical residues for the differences in nAChR subtype response to α‐conotoxins. With molecular modelling and MD simulations, the lack of activity of AuIB at human α3β4 nAChR could be interpreted on a molecular level. At the rat α3β4 nAChR, residue F9 of AuIB closely interacts with residues W59 and K61 of the β4 subunit WLK (Trp‐Leu‐Lys) pocket, hypothetically via π–π and cation–π interactions. In contrast, at the human α3β4 nAChR, residue F9 of AuIB only binds outside the WLK pocket (Figure 3A), where although interaction is still possible with β4 residues K59 and L119, the crucial π–π interaction with W57 is absent (Cuny et al., 2016). At the rat homologue, the β4 W59 residue was indispensable for AuIB inhibition and K61 appeared to play an auxiliary role (Grishin et al., 2013). Hence, the lack of interaction between AuIB F9 and human β4 W57 could explain the loss of AuIB activity at the human α3β4 subtype.
Figure 3.
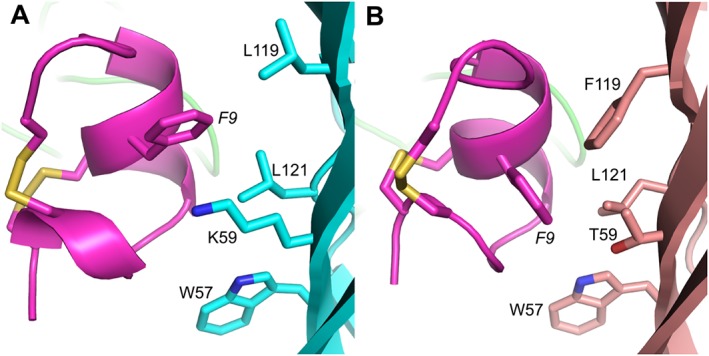
MD simulation models of AuIB (magenta) binding at human α3β4 and α3β2 nAChRs. (A) Residue F9 of AuIB is oriented outside the WLK pocket (residues W57, L119 and K59) of human β4 (cyan), thereby preventing direct interaction. (B) AuIB F9 is oriented in close proximity to W57 at human β2 (pink) and can form the crucial π–π interaction, but the short and hydrophilic side chain of β2 T59 is ineffective at stabilizing AuIB F9 and forming a binding pocket.
At human α3β2, molecular modelling revealed contacts between AuIB and β2 W57, T59 and other residues. Although AuIB F9 can form the crucial π–π interaction with W57 in loop D, unlike β4 K59, the hydrophilic side chain of β2 residue T59 does not form an effective binding pocket for AuIB F9 because its interaction with the aromatic phenyl ring of AuIB F9 is energetically unfavourable (Figure 3B) (Cuny et al., 2016). In summary, AuIB at hα3β2 and hα3β4 nAChRs is stabilized ineffectively due to the lack of key interactions between AuIB F9 and β subunit agonist binding loop residues.
The key residues for receptor binding, which consequently dictate the selectivity profile, are not precisely the same for all 4/7 α‐conotoxins despite overall structural similarity among these peptides. For α‐conotoxins MII, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3982 and GID, which all exhibit high potency at α3β2, residues β2‐F119 and β2‐V111 have a major effect on peptide binding. When the bulky side chains of those residues in β2 were exchanged to Ala or Gly, the peptides exhibited increased inhibitory activity for the mutant α3β2 nAChRs, indicating that their affinity to the receptor was enhanced. Investigation of PnIA analogues indicated that high affinity binding to aforementioned mutated β2 residues was conferred by amino acids at position 10 with long side chains (Dutertre et al., 2005).
For α‐conotoxin RegIIA, the human β2 and β4 subunit selectivity was dependent on two key residues located at position 59 in loop D and position 113 in loop E respectively. Exchange of β2 T59 to K, increased RegIIA potency at the mutant human α3β2 similar to that of α3β4, whereas the opposite exchange of β4 K59 to T resulted in ~60‐fold decrease in potency compared with wild‐type human α3β4 (Cuny et al., 2016). Swapping loop E residues S113 of β2 subunit with R113 of β4 subunit considerably improved the sensitivity of the receptor to RegIIA (twice as sensitive as wild‐type α3β4). Conversely, β4 R113S mutation reduced the sensitivity to RegIIA by ~20‐fold compared with wild‐type human α3β4 (Cuny et al., 2016). To our knowledge, the finding that β4 residue R113 plays an important role in nAChR subtype selectivity for RegIIA (Cuny et al., 2016) is novel in identifying β4 R113 as a key determinant for antagonist binding at human nAChRs.
The sensitivity changes of the mutant receptors compared with their wild‐type counterparts suggested that the affinity was affected, as RegIIA is competing with ACh for the agonist binding site. In agreement with this notion, the RegIIA wash‐off kinetics was also determined by the aforementioned residues. Recovery from block by RegIIA differed significantly between human α3β2 and α3β4 nAChRs (1 min compared with 13–14 min), and the substitution of β2–β4 residues 59 and 113 switched the rate of recovery towards the opposite subtype (Cuny et al., 2016). Interestingly, residue 59 of the rat β subunits has also been identified as a determinant for α‐conotoxin LvIA potency and wash‐off kinetics (Luo et al., 2014). LvIA exhibited ~20‐fold higher selectivity for rat α3β2 over α3β4. The rat β2[T59K] mutant further increased the peptide activity by approximately 10‐fold and considerably slowed the recovery from block by LvIA compared with wild‐type α3β2 (Luo et al., 2014).
MD simulation studies provided further details on the interaction between human β2 and β4 residues 59 and 113 with RegIIA. RegIIA N9/β4 K59 contact (via hydrogen bond) is proposed to be critical for RegIIA activity at human α3β4 (Figure 4A, E). In contrast, such contacts are not possible between RegIIA and human α3β2, as the side chain of the β2 corresponding residue, T59, is not long enough to interact with loop 2 of RegIIA (Figure 4B, F). The increased potency and slower wash‐off caused by the β2 T59K point mutation could then be explained by the gain of these additional hydrogen bonds, resulting in tighter binding of the peptide to the mutant nAChR. Computational modelling also revealed a crucial role of the long and positively charged side‐chain residue R113 in β4 for forming an H‐bond with RegIIA (Figure 4A, C). When the β4 R113 is introduced into the β2 backbone, it forms H‐bonds with the peptide similar to hα3β4 thereby stabilizing nAChR‐RegIIA binding, improving its potency and significantly slowing its wash‐off rate (Cuny et al., 2016). Other hα3β4 mutations, primarily within loop F, also decreased RegIIA sensitivity and increased ACh affinity, albeit less pronounced than the aforementioned residues 59 and 113. Therefore, it was concluded that other residues within the agonist binding loops probably play an auxiliary role for the subtype selectivity of RegIIA by stabilizing the main interacting residues and shaping the topology of the agonist binding pocket (Cuny et al., 2016).
Figure 4.
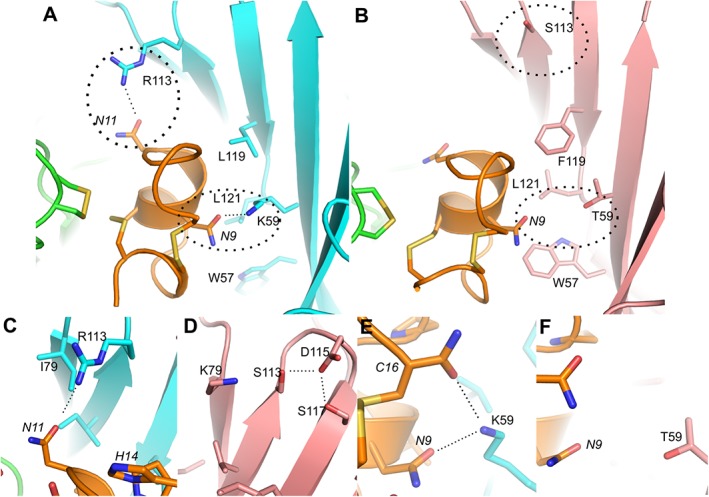
Molecular docking models illustrating the binding of RegIIA to human α3β4 (A) and α3β2 (B) nAChRs respectively. Several H‐bonds (dotted lines) with threshold distance 3.2 Å are formed between pairwise interacting residues of different loops or β‐sheets, thereby affecting their local conformation or dynamics, which in turn affects the binding of RegIIA. The α3(+) interface is shown in green, β2(−) in pink, β4(−) in cyan and RegIIA in orange. Residues nearby the agonist binding site that are essential for the interaction with RegIIA are shown as liquorice models. Residues from the receptor and RegIIA are labelled using normal and italic fonts respectively. Non‐conserved residues forming H‐bonds are highlighted with circles. (C–F) Magnification of the interaction sites in α3β4 (C, E) and α3β2 (D, F). The neutral and short β2 subunit side chain residues T59 and S113 cannot interact with backbone atoms of N9 and N11 of RegIIA. In contrast, the long positively charged side chains of K59 and R113 of β4 form H‐bonds with RegIIA N9 and C16, and N11 residues respectively.
α‐Conotoxin BuIA was also investigated for nAChR subtype selectivity and wash‐off kinetics (Azam et al., 2005; Shiembob et al., 2006). In different species, including human, a slow recovery from BuIA block at α3β4 compared with significantly faster recovery at α3β2 was observed (Azam et al., 2005). Residues 59, 111 and 119 of rat β2 and β4 subunits were identified as being critical for the off‐rate differences. Similar to findings with RegIIA, the off‐rate of BuIA was slower at the β2[T59K] mutant compared with wild‐type α3β2 (Shiembob et al., 2006).
Additional studies support the conclusion that residue 59 on the β subunit plays an important role for agonist and antagonist binding. Mutation β2 T59K has been shown to be critical for http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4006 and neuronal http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3964 (antagonists) sensitivity of α3β2 nAChR (Harvey and Luetje, 1996) as well as affecting the affinity of ACh and nicotine (agonists) to http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=463β2 nAChR (Parker et al., 2001). The high selectivity of α‐conotoxin MII for the α3β2 nAChR subtype (>200‐fold more active over other nAChR subtypes) was mapped to three sequence segments using β2–β4 chimeras. Within the segments, β2 residue T59 was identified as one factor determining the higher sensitivity of β2 to MII compared with β4 (Harvey et al., 1997).
ACh‐binding protein (AChBP) isolated from neurons of the giant sea slug Aplysia californica (Ac‐AChBP) is homologous to the ligand‐binding domains of nAChRs and also pharmacologically similar. Since the time when its crystal structure was determined, it has proved to be a valuable tool to study conotoxin‐nAChR interactions. Co‐crystal structures of α‐conotoxins in complex with Ac‐AChBP have been reported for PnIA[A10L,D14K] (PDB code 2BR8), TxIA[A10L] (PDB code 2UZ6), ImI (PDB code 2C9T), BuIA (PDB code 4EZ1) and GIC (PDB code 5CO5) (Lin et al., 2016a).
Lin et al. (2016b) compared their Ac‐AChBP/GIC crystal structure with MD simulation models of GIC docking to α3β2 and α3β4, respectively, to obtain accurate toxin‐receptor binding models and identify the key amino acid residues on the complementary receptor interface that appear to be responsible for selectivity of GIC for α3β2 over α3β4. The authors concluded that H5 and Q3 of GIC are the important residues interacting with the receptors. GIC H5 appeared to mainly interact with the principal binding interface of human α3, whereas on the complementary β2 interface residues E61, V111, S113, S117 and F119 form a binding pocket for GIC Q13. In contrast, the non‐conserved residues in human β4 and most importantly R113 (labelled R115 in this publication) lead to steric hindrance and even steric clashes with GIC Q13, resulting in overall strongly diminished binding affinity (Lin et al., 2016b). Note that β4 R113 was shown to be a major determinant for the selectivity of RegIIA towards α3β4 (as discussed above). This example shows again that common structural features of the receptors can be responsible for markedly different selectivity profiles of α‐conotoxins.
Designing α‐conotoxin analogues to improve nAChR subtype selectivity
Scientific advances such as MD simulation, co‐crystal structures of α‐conotoxins with AChBP and functional studies with mutated receptors significantly improved our understanding of how these peptides can confer their remarkably unique selectivity profiles. The next step is to custom‐design peptide analogues with improved affinity towards a specific receptor subtype while decreasing affinity at other subtypes to reduce off‐target effects. Some studies provided the proof of concept that this is indeed possible. For example, as mentioned above, RegIIA gained enhanced selectivity for α3β4 when its residues N11 and N12 are exchanged for alanine residues (Kompella et al., 2015b). Similarly, single amino acid changes in PnIA yielded improved potency and selectivity. Hogg et al. (1999) showed that PnIA[A10L] was an order of magnitude more potent than native PnIA at inhibiting ACh‐evoked currents in rat parasympathetic neurons, albeit a maximal inhibition of only 45% of the peak current amplitude was observed, compared with almost complete block by the native toxin. It was proposed that position 10 of PnIA influences potency and determines selectivity among α7 and other nAChR subtypes, including α3β2 and α3β4 (Hogg et al., 1999). Subsequent studies aimed at revealing the role of PnIA position 10 on potency and selectivity for the α7 and α3β2 nAChR subtypes (Kasheverov et al., 2011; Hopping et al., 2014). Electrophysiological evaluation of various PnIA[A10L] analogues revealed that hydrophobic residues in position 10 maintained potency at both subtypes whereas charged or polar residues abolished α7 binding thereby shifting selectivity towards α3β2 (Hopping et al., 2014). In a recent study, an α‐conotoxin LsIA[R10F,N12L] double mutant was designed to specifically inhibit the α3β2 versus α7 nAChR subtype (Abraham et al., 2017). The wild‐type LsIA is about 150 times more potent at α7 than α3β2, whereas LsIA[R10F,N12L] possesses >250‐fold selectivity for α3β4 over α7 nAChR (Abraham et al., 2017).
In addition to these targeted approaches to improve α‐conotoxin selectivity based on known structural data, large‐scale synthetic combinatorial screens are also a powerful tool to elucidate peptide analogues with improved pharmacological properties. A synthetic combinatorial library derived from α‐conotoxin BuIA sequence revealed 11 analogues with inhibitory activity at the α3β4 nAChR. One of these analogues, termed TP‐2212‐59, is one of the most potent and selective α3β4 antagonists known to date, with a calculated IC50 of 2.3 nM at α3β4 and more than 1000‐fold less activity at α3β2 and α7 subtypes (Chang et al., 2014).
Conclusions
Recent findings with RegIIA, AuIB and other α‐conotoxins confirm that subtype‐selective nAChR antagonists often work through a common mechanism, by interacting with the same structural components and sites on the receptor. In the future, with increasing knowledge about the interactions necessary for selective and potent antagonist binding, new pharmacological probes can be custom‐designed to target specific nAChR subtypes. These probes will not only help to elucidate the physiological roles of non‐CNS nAChRs but also the discovery of novel drug candidates to treat diseases in which a particular nAChR subtype is the underlying cause.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/2016 (Alexander et al., 2015).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the Australian Research Council (Discovery Project Grant DP150103990 to D.J.A.).
Cuny, H. , Yu, R. , Tae, H.‐S. , Kompella, S. N. , and Adams, D. J. (2018) α‐Conotoxins active at α3‐containing nicotinic acetylcholine receptors and their molecular determinants for selective inhibition. British Journal of Pharmacology, 175: 1855–1868. doi: 10.1111/bph.13852.
References
- Abraham N, Healy M, Ragnarsson L, Brust A, Alewood PF, Lewis RJ (2017). Structural mechanisms for α‐conotoxin activity at the human α3β4 nicotinic acetylcholine receptor. Sci Rep 7: 45466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akondi KB, Muttenthaler M, Dutertre S, Kaas Q, Craik DJ, Lewis RJ et al. (2014). Discovery, synthesis, and structure‐activity relationships of conotoxins. Chem Rev 114: 5815–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89: 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The concise guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias HR, Richards VE, Ng D, Ghafoori ME, Le V, Mousa SA (2009). Role of non‐neuronal nicotinic acetylcholine receptors in angiogenesis. Int J Biochem Cell Biol 41: 1441–1451. [DOI] [PubMed] [Google Scholar]
- Arredondo J, Hall LL, Ndoye A, Nguyen VT, Chernyavsky AI, Bercovich D et al. (2003). Central role of fibroblast α3 nicotinic acetylcholine receptor in mediating cutaneous effects of nicotine. Lab Invest 83: 207–225. [DOI] [PubMed] [Google Scholar]
- Azam L, Dowell C, Watkins M, Stitzel JA, Olivera BM, McIntosh JM (2005). α‐Conotoxin BuIA, a novel peptide from Conus bullatus, distinguishes among neuronal nicotinic acetylcholine receptors. J Biol Chem 280: 80–87. [DOI] [PubMed] [Google Scholar]
- Azam L, McIntosh JM (2009). α‐Conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol Sin 30: 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, McIntosh JM (2012). Molecular basis for the differential sensitivity of rat and human α9α10 nAChRs to α‐conotoxin RgIA. J Neurochem 122: 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, Papakyriakou A, Zouridakis M, Giastas P, Tzartos SJ, McIntosh JM (2015). Molecular interaction of α‐conotoxin RgIA with the rat α9α10 nicotinic acetylcholine receptor. Mol Pharmacol 87: 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Terlau H (2008). Toxins from cone snails: properties, applications and biotechnological production. Appl Microbiol Biotechnol 79: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beissner M, Dutertre S, Schemm R, Danker T, Sporning A, Grubmuller H et al. (2012). Efficient binding of 4/7 α‐conotoxins to nicotinic α4β2 receptors is prevented by Arg185 and Pro195 in the α4 subunit. Mol Pharmacol 82: 711–718. [DOI] [PubMed] [Google Scholar]
- Benhammou K, Lee M, Strook M, Sullivan B, Logel J, Raschen K et al. (2000). [3H]Nicotine binding in peripheral blood cells of smokers is correlated with the number of cigarettes smoked per day. Neuropharmacology 39: 2818–2829. [DOI] [PubMed] [Google Scholar]
- Bibevski S, Zhou Y, McIntosh JM, Zigmond RE, Dunlap ME (2000). Functional nicotinic acetylcholine receptors that mediate ganglionic transmission in cardiac parasympathetic neurons. J Neurosci 20: 5076–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet MR, Langlois A, Israel‐Assayag E, Beaulieu MJ, Ferland C, Laviolette M et al. (2007). Modulation of eosinophil activation in vitro by a nicotinic receptor agonist. J Leukoc Biol 81: 1245–1251. [DOI] [PubMed] [Google Scholar]
- Bruggmann D, Lips KS, Pfeil U, Haberberger RV, Kummer W (2003). Rat arteries contain multiple nicotinic acetylcholine receptor alpha‐subunits. Life Sci 72: 2095–2099. [DOI] [PubMed] [Google Scholar]
- Campanucci V, Krishnaswamy A, Cooper E (2010). Diabetes depresses synaptic transmission in sympathetic ganglia by inactivating nAChRs through a conserved intracellular cysteine residue. Neuron 66: 827–834. [DOI] [PubMed] [Google Scholar]
- Campos‐Caro A, Smillie FI, Dominguez del Toro E, Rovira JC, Vicente‐Agullo F, Chapuli J et al. (1997). Neuronal nicotinic acetylcholine receptors on bovine chromaffin cells: cloning, expression, and genomic organization of receptor subunits. J Neurochem 68: 488–497. [DOI] [PubMed] [Google Scholar]
- Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM (1996). A new α‐conotoxin which targets α3β2 nicotinic acetylcholine receptors. J Biol Chem 271: 7522–7528. [DOI] [PubMed] [Google Scholar]
- Cecchini M, Changeux JP (2015). The nicotinic acetylcholine receptor and its prokaryotic homologues: structure, conformational transitions & allosteric modulation. Neuropharmacology 96: 137–149. [DOI] [PubMed] [Google Scholar]
- Chang YP, Banerjee J, Dowell C, Wu J, Gyanda R, Houghten RA et al. (2014). Discovery of a potent and selective α3β4 nicotinic acetylcholine receptor antagonist from an α‐conotoxin synthetic combinatorial library. J Med Chem 57: 3511–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP (2010). Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci 11: 389–401. [DOI] [PubMed] [Google Scholar]
- Changeux JP (2012). The nicotinic acetylcholine receptor: the founding father of the pentameric ligand‐gated ion channel superfamily. J Biol Chem 287: 40207–40215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyavsky AI, Arredondo J, Marubio LM, Grando SA (2004). Differential regulation of keratinocyte chemokinesis and chemotaxis through distinct nicotinic receptor subtypes. J Cell Sci 117: 5665–5679. [DOI] [PubMed] [Google Scholar]
- Conti‐Fine BM, Navaneetham D, Lei S, Maus AD (2000). Neuronal nicotinic receptors in non‐neuronal cells: new mediators of tobacco toxicity? Eur J Pharmacol 393: 279–294. [DOI] [PubMed] [Google Scholar]
- Cooper E (2001). Nicotinic acetylcholine receptors on vagal afferent neurons. Ann N Y Acad Sci 940: 110–118. [DOI] [PubMed] [Google Scholar]
- Cuny H, Kompella SN, Tae HS, Yu R, Adams DJ (2016). Key structural determinants in the agonist binding loops of human β2 and β4 nicotinic acetylcholine receptor subunits contribute to α3β4 subtype selectivity of α‐conotoxins. J Biol Chem 291: 23779–23792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Bertrand D (2007). Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol 47: 699–729. [DOI] [PubMed] [Google Scholar]
- Del Signore A, Gotti C, Rizzo A, Moretti M, Paggi P (2004). Nicotinic acetylcholine receptor subtypes in the rat sympathetic ganglion: pharmacological characterization, subcellular distribution and effect of pre‐ and postganglionic nerve crush. J Neuropathol Exp Neurol 63: 138–150. [DOI] [PubMed] [Google Scholar]
- Dowell C, Olivera BM, Garrett JE, Staheli ST, Watkins M, Kuryatov A et al. (2003). α‐Conotoxin PIA is selective for α6 subunit‐containing nicotinic acetylcholine receptors. J Neurosci 23: 8445–8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube GR, Kohlhaas KL, Rueter LE, Surowy CS, Meyer MD, Briggs CA (2005). Loss of functional neuronal nicotinic receptors in dorsal root ganglion neurons in a rat model of neuropathic pain. Neurosci Lett 376: 29–34. [DOI] [PubMed] [Google Scholar]
- Dutertre S, Jin AH, Alewood PF, Lewis RJ (2014). Intraspecific variations in Conus geographus defence‐evoked venom and estimation of the human lethal dose. Toxicon 91: 135–144. [DOI] [PubMed] [Google Scholar]
- Dutertre S, Nicke A, Lewis RJ (2005). β2 Subunit contribution to 4/7 α‐conotoxin binding to the nicotinic acetylcholine receptor. J Biol Chem 280: 30460–30468. [DOI] [PubMed] [Google Scholar]
- Egleton RD, Brown KC, Dasgupta P (2008). Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol Sci 29: 151–158. [DOI] [PubMed] [Google Scholar]
- Ellison M, Gao F, Wang HL, Sine SM, McIntosh JM, Olivera BM (2004). α‐Conotoxins ImI and ImII target distinct regions of the human α7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry 43: 16019–16026. [DOI] [PubMed] [Google Scholar]
- Everhart D, Reiller E, Mirzoian A, McIntosh JM, Malhotra A, Luetje CW (2003). Identification of residues that confer α‐conotoxin‐PnIA sensitivity on the α3 subunit of neuronal nicotinic acetylcholine receptors. J Pharmacol Exp Ther 306: 664–670. [DOI] [PubMed] [Google Scholar]
- Fainzilber M, Hasson A, Oren R, Burlingame AL, Gordon D, Spira ME et al. (1994). New mollusc‐specific α‐conotoxins block Aplysia neuronal acetylcholine receptors. Biochemistry 33: 9523–9529. [DOI] [PubMed] [Google Scholar]
- Foong JP, Hirst CS, Hao MM, McKeown SJ, Boesmans W, Young HM et al. (2015). Changes in nicotinic neurotransmission during enteric nervous system development. J Neurosci 35: 7106–7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco A, Kompella SN, Akondi KB, Melaun C, Daly NL, Luetje CW et al. (2012). RegIIA: an α4/7‐conotoxin from the venom of Conus regius that potently blocks α3β4 nAChRs. Biochem Pharmacol 83: 419–426. [DOI] [PubMed] [Google Scholar]
- Fucile S, Sucapane A, Eusebi F (2005). Ca2+ permeability of nicotinic acetylcholine receptors from rat dorsal root ganglion neurones. J Physiol 565: 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan JJ, North RA (2004). Pharmacology and function of nicotinic acetylcholine and P2X receptors in the enteric nervous system. Neurogastroenterol Motil 16 (Suppl 1): 64–70. [DOI] [PubMed] [Google Scholar]
- Genzen JR, Van Cleve W, McGehee DS (2001). Dorsal root ganglion neurons express multiple nicotinic acetylcholine receptor subtypes. J Neurophysiol 86: 1773–1782. [DOI] [PubMed] [Google Scholar]
- Glushakov AV, Voytenko LP, Skok MV, Skok V (2004). Distribution of neuronal nicotinic acetylcholine receptors containing different alpha‐subunits in the submucosal plexus of the guinea‐pig. Auton Neurosci 110: 19–26. [DOI] [PubMed] [Google Scholar]
- Gotti C, Clementi F (2004). Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol 74: 363–396. [DOI] [PubMed] [Google Scholar]
- Gotti C, Moretti M, Gaimarri A, Zanardi A, Clementi F, Zoli M (2007). Heterogeneity and complexity of native brain nicotinic receptors. Biochem Pharmacol 74: 1102–1111. [DOI] [PubMed] [Google Scholar]
- Graham AJ, Ray MA, Perry EK, Jaros E, Perry RH, Volsen SG et al. (2003). Differential nicotinic acetylcholine receptor subunit expression in the human hippocampus. J Chem Neuroanat 25: 97–113. [DOI] [PubMed] [Google Scholar]
- Grando SA, Horton RM, Pereira EF, Diethelm‐Okita BM, George PM, Albuquerque EX et al. (1995). A nicotinic acetylcholine receptor regulating cell adhesion and motility is expressed in human keratinocytes. J Invest Dermatol 105: 774–781. [DOI] [PubMed] [Google Scholar]
- Gray WR, Olivera BM, Cruz LJ (1988). Peptide toxins from venomous Conus snails. Annu Rev Biochem 57: 665–700. [DOI] [PubMed] [Google Scholar]
- Grishin AA, Cuny H, Hung A, Clark RJ, Brust A, Akondi K et al. (2013). Identifying key amino acid residues that affect α‐conotoxin AuIB inhibition of α3β4 nicotinic acetylcholine receptors. J Biol Chem 288: 34428–34442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin AA, Wang CI, Muttenthaler M, Alewood PF, Lewis RJ, Adams DJ (2010). α‐Conotoxin AuIB isomers exhibit distinct inhibitory mechanisms and differential sensitivity to stoichiometry of α3β4 nicotinic acetylcholine receptors. J Biol Chem 285: 22254–22263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallden S, Sjogren M, Hedblad B, Engstrom G, Hamrefors V, Manjer J et al. (2016). Gene variance in the nicotinic receptor cluster (CHRNA5‐CHRNA3‐CHRNB4) predicts death from cardiopulmonary disease and cancer in smokers. J Intern Med 279: 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Luetje CW (1996). Determinants of competitive antagonist sensitivity on neuronal nicotinic receptor β subunits. J Neurosci 16: 3798–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, McIntosh JM, Cartier GE, Maddox FN, Luetje CW (1997). Determinants of specificity for α‐conotoxin MII on α3β2 neuronal nicotinic receptors. Mol Pharmacol 51: 336–342. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Egleton RD, Davis TP (2005). Modulation of cerebral microvascular permeability by endothelial nicotinic acetylcholine receptors. Am J Physiol Heart Circ Physiol 289: H212–H219. [DOI] [PubMed] [Google Scholar]
- Heeschen C, Weis M, Aicher A, Dimmeler S, Cooke JP (2002). A novel angiogenic pathway mediated by non‐neuronal nicotinic acetylcholine receptors. J Clin Invest 110: 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemke C, Stolp M, Reuss S, Wevers A, Reinhardt S, Maelicke A et al. (1996). Expression of alpha subunit genes of nicotinic acetylcholine receptors in human lymphocytes. Neurosci Lett 214: 171–174. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Miranda LP, Craik DJ, Lewis RJ, Alewood PF, Adams DJ (1999). Single amino acid substitutions in α‐conotoxin PnIA shift selectivity for subtypes of the mammalian neuronal nicotinic acetylcholine receptor. J Biol Chem 274: 36559–36564. [DOI] [PubMed] [Google Scholar]
- Hone AJ, Meyer EL, McIntyre M, McIntosh JM (2012). Nicotinic acetylcholine receptors in dorsal root ganglion neurons include the α6β4* subtype. FASEB J 26: 917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopping G, Wang CI, Hogg RC, Nevin ST, Lewis RJ, Adams DJ et al. (2014). Hydrophobic residues at position 10 of α‐conotoxin PnIA influence subtype selectivity between α7 and α3β2 neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 91: 534–542. [DOI] [PubMed] [Google Scholar]
- Hurst R, Rollema H, Bertrand D (2013). Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther 137: 22–54. [DOI] [PubMed] [Google Scholar]
- Improgo MR, Soll LG, Tapper AR, Gardner PD (2013). Nicotinic acetylcholine receptors mediate lung cancer growth. Front Physiol 4: 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inserra MC, Kompella SN, Vetter I, Brust A, Daly NL, Cuny H et al. (2013). Isolation and characterization of α‐conotoxin LsIA with potent activity at nicotinic acetylcholine receptors. Biochem Pharmacol 86: 791–799. [DOI] [PubMed] [Google Scholar]
- Itier V, Bertrand D (2001). Neuronal nicotinic receptors: from protein structure to function. FEBS Lett 504: 118–125. [DOI] [PubMed] [Google Scholar]
- Jensen AA, Frolund B, Liljefors T, Krogsgaard‐Larsen P (2005). Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem 48: 4705–4745. [DOI] [PubMed] [Google Scholar]
- Kasheverov IE, Zhmak MN, Khruschov AY, Tsetlin VI (2011). Design of new α‐conotoxins: from computer modeling to synthesis of potent cholinergic compounds. Mar Drugs 9: 1698–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I, Osaka H, Stanislaus S, Calvo RM, Deerinck T, Yaksh TL et al. (2003). Nicotinic acetylcholine receptor distribution in relation to spinal neurotransmission pathways. J Comp Neurol 467: 44–59. [DOI] [PubMed] [Google Scholar]
- Kindt F, Wiegand S, Niemeier V, Kupfer J, Loser C, Nilles M et al. (2008). Reduced expression of nicotinic α subunits 3, 7, 9 and 10 in lesional and nonlesional atopic dermatitis skin but enhanced expression of α subunits 3 and 5 in mast cells. Br J Dermatol 159: 847–857. [DOI] [PubMed] [Google Scholar]
- Kohn AJ (2016). Human injuries and fatalities due to venomous marine snails of the family Conidae. Int J Clin Pharmacol Ther 54: 524–538. [DOI] [PubMed] [Google Scholar]
- Kompella SN, Cuny H, Hung A, Adams DJ (2015a). Molecular basis for differential sensitivity of α‐conotoxin RegIIA at rat and human neuronal nicotinic acetylcholine receptors. Mol Pharmacol 88: 993–1001. [DOI] [PubMed] [Google Scholar]
- Kompella SN, Hung A, Clark RJ, Mari F, Adams DJ (2015b). Alanine scan of α‐conotoxin RegIIA reveals a selective α3β4 nicotinic acetylcholine receptor antagonist. J Biol Chem 290: 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak JM, McIntosh JM, Yoshikami D, Olivera BM (2001). Nicotine‐evoked transmitter release from synaptosomes: functional association of specific presynaptic acetylcholine receptors and voltage‐gated calcium channels. J Neurochem 77: 1581–1589. [DOI] [PubMed] [Google Scholar]
- Kurzen H, Berger H, Jager C, Hartschuh W, Naher H, Gratchev A et al. (2004). Phenotypical and molecular profiling of the extraneuronal cholinergic system of the skin. J Invest Dermatol 123: 937–949. [DOI] [PubMed] [Google Scholar]
- Lavergne V, Harliwong I, Jones A, Miller D, Taft RJ, Alewood PF (2015). Optimized deep‐targeted proteotranscriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc Natl Acad Sci U S A 112: E3782–E3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebbe EK, Peigneur S, Wijesekara I, Tytgat J (2014). Conotoxins targeting nicotinic acetylcholine receptors: an overview. Mar Drugs 12: 2970–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RJ, Dutertre S, Vetter I, Christie MJ (2012). Conus venom peptide pharmacology. Pharmacol Rev 64: 259–298. [DOI] [PubMed] [Google Scholar]
- Lin B, Xiang S, Li M (2016a). Residues responsible for the selectivity of α‐conotoxins for Ac‐AChBP or nAChRs. Mar Drugs 14: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Xu M, Zhu X, Wu Y, Liu X, Zhangsun D et al. (2016b). From crystal structure of α‐conotoxin GIC in complex with Ac‐AChBP to molecular determinants of its high selectivity for α3β2 nAChR. Sci Rep 6: 22349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom J (1997). Nicotinic acetylcholine receptors in health and disease. Mol Neurobiol 15: 193–222. [DOI] [PubMed] [Google Scholar]
- Livett BG, Gayler KR, Khalil Z (2004). Drugs from the sea: conopeptides as potential therapeutics. Curr Med Chem 11: 1715–1723. [DOI] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK et al. (1999). International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 51: 397–401. [PubMed] [Google Scholar]
- Luo S, Kulak JM, Cartier GE, Jacobsen RB, Yoshikami D, Olivera BM et al. (1998). α‐Conotoxin AuIB selectively blocks α3β4 nicotinic acetylcholine receptors and nicotine‐evoked norepinephrine release. J Neurosci 18: 8571–8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Nguyen TA, Cartier GE, Olivera BM, Yoshikami D, McIntosh JM (1999). Single‐residue alteration in α‐conotoxin PnIA switches its nAChR subtype selectivity. Biochemistry 38: 14542–14548. [DOI] [PubMed] [Google Scholar]
- Luo S, Zhangsun D, Schroeder CI, Zhu X, Hu Y, Wu Y et al. (2014). A novel α4/7‐conotoxin LvIA from Conus lividus that selectively blocks α3β2 vs. α6/α3β2β3 nicotinic acetylcholine receptors. FASEB J 28: 1842–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Zhangsun D, Zhu X, Wu Y, Hu Y, Christensen S et al. (2013). Characterization of a novel α‐conotoxin TxID from Conus textile that potently blocks rat α3β4 nicotinic acetylcholine receptors. J Med Chem 56: 9655–9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklin KD, Maus AD, Pereira EF, Albuquerque EX, Conti‐Fine BM (1998). Human vascular endothelial cells express functional nicotinic acetylcholine receptors. J Pharmacol Exp Ther 287: 435–439. [PubMed] [Google Scholar]
- Mao D, Yasuda RP, Fan H, Wolfe BB, Kellar KJ (2006). Heterogeneity of nicotinic cholinergic receptors in rat superior cervical and nodose ganglia. Mol Pharmacol 70: 1693–1699. [DOI] [PubMed] [Google Scholar]
- Maus AD, Pereira EF, Karachunski PI, Horton RM, Navaneetham D, Macklin K et al. (1998). Human and rodent bronchial epithelial cells express functional nicotinic acetylcholine receptors. Mol Pharmacol 54: 779–788. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Role LW (1995). Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol 57: 521–546. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Dowell C, Watkins M, Garrett JE, Yoshikami D, Olivera BM (2002). α‐Conotoxin GIC from Conus geographus, a novel peptide antagonist of nicotinic acetylcholine receptors. J Biol Chem 277: 33610–33615. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Yoshikami D, Mahe E, Nielsen DB, Rivier JE, Gray WR et al. (1994). A nicotinic acetylcholine receptor ligand of unique specificity, α‐conotoxin ImI. J Biol Chem 269: 16733–16739. [PubMed] [Google Scholar]
- Millar NS, Gotti C (2009). Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 56: 237–246. [DOI] [PubMed] [Google Scholar]
- Millard EL, Daly NL, Craik DJ (2004). Structure‐activity relationships of α‐conotoxins targeting neuronal nicotinic acetylcholine receptors. Eur J Biochem 271: 2320–2326. [DOI] [PubMed] [Google Scholar]
- Moccia F, Frost C, Berra‐Romani R, Tanzi F, Adams DJ (2004). Expression and function of neuronal nicotinic ACh receptors in rat microvascular endothelial cells. Am J Physiol Heart Circ Physiol 286: H486–H491. [DOI] [PubMed] [Google Scholar]
- Morales‐Perez CL, Noviello CM, Hibbs RE (2016). X‐ray structure of the human α4β2 nicotinic receptor. Nature 538: 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muldoon PP, Jackson KJ, Perez E, Harenza JL, Molas S, Rais B et al. (2014). The α3β4* nicotinic ACh receptor subtype mediates physical dependence to morphine: mouse and human studies. Br J Pharmacol 171: 3845–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muttenthaler M, Akondi KB, Alewood PF (2011). Structure‐activity studies on α‐conotoxins. Curr Pharm Des 17: 4226–4241. [DOI] [PubMed] [Google Scholar]
- Napier IA, Klimis H, Rycroft BK, Jin AH, Alewood PF, Motin L et al. (2012). Intrathecal α‐conotoxins Vc1.1, AuIB and MII acting on distinct nicotinic receptor subtypes reverse signs of neuropathic pain. Neuropharmacology 62: 2202–2207. [DOI] [PubMed] [Google Scholar]
- Nicke A, Loughnan ML, Millard EL, Alewood PF, Adams DJ, Daly NL et al. (2003). Isolation, structure, and activity of GID, a novel α 4/7‐conotoxin with an extended N‐terminal sequence. J Biol Chem 278: 3137–3144. [DOI] [PubMed] [Google Scholar]
- Olivera BM (2006). Conus peptides: biodiversity‐based discovery and exogenomics. J Biol Chem 281: 31173–31177. [DOI] [PubMed] [Google Scholar]
- Park KS, Cha SK, Kim MJ, Kim DR, Jeong SW, Lee JW et al. (2006). An α3β4 subunit combination acts as a major functional nicotinic acetylcholine receptor in male rat pelvic ganglion neurons. Pflugers Arch 452: 775–783. [DOI] [PubMed] [Google Scholar]
- Parker MJ, Harvey SC, Luetje CW (2001). Determinants of agonist binding affinity on neuronal nicotinic receptor β subunits. J Pharmacol Exp Ther 299: 385–391. [PubMed] [Google Scholar]
- Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J et al. (2004). Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology 145: 2498–2506. [DOI] [PubMed] [Google Scholar]
- Puillandre N, Duda TF, Meyer C, Olivera BM, Bouchet P (2015). One, four or 100 genera? A new classification of the cone snails. J Moll Stud 81: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purnyn HE, Rikhalsky OV, Skok MV, Skok VI (2004). Functional nicotinic acetylcholine receptors in the neurons of rat intracardiac ganglia. Fiziol Zh 50: 79–84. [PubMed] [Google Scholar]
- Rassadi S, Krishnaswamy A, Pie B, McConnell R, Jacob MH, Cooper E (2005). A null mutation for the α3 nicotinic acetylcholine (ACh) receptor gene abolishes fast synaptic activity in sympathetic ganglia and reveals that ACh output from developing preganglionic terminals is regulated in an activity‐dependent retrograde manner. J Neurosci 25: 8555–8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll N, Bronnec M, Bourin M (2004). Nicotinic receptors and schizophrenia. Curr Med Res Opin 20: 1057–1074. [DOI] [PubMed] [Google Scholar]
- Robinson SD, Norton RS (2014). Conotoxin gene superfamilies. Mar Drugs 12: 6058–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudchenko A, Akude E, Cooper E (2014). Synapses on sympathetic neurons and parasympathetic neurons differ in their vulnerability to diabetes. J Neurosci 34: 8865–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala F, Nistri A, Criado M (2008). Nicotinic acetylcholine receptors of adrenal chromaffin cells. Acta Physiol (Oxf) 192: 203–212. [DOI] [PubMed] [Google Scholar]
- Sargent PB (1993). The diversity of neuronal nicotinic acetylcholine receptors. Annu Rev Neurosci 16: 403–443. [DOI] [PubMed] [Google Scholar]
- Schroeder CI, Craik DJ (2012). Therapeutic potential of conopeptides. Future Med Chem 4: 1243–1255. [DOI] [PubMed] [Google Scholar]
- Sekhon HS, Song P, Jia Y, Lindstrom J, Spindel ER (2005). Expression of lynx1 in developing lung and its modulation by prenatal nicotine exposure. Cell Tissue Res 320: 287–297. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S (2002). Nicotinic receptor signaling in nonexcitable cells. J Neurobiol 53: 524–534. [DOI] [PubMed] [Google Scholar]
- Shiembob DL, Roberts RL, Luetje CW, McIntosh JM (2006). Determinants of α‐conotoxin BuIA selectivity on the nicotinic acetylcholine receptor β subunit. Biochemistry 45: 11200–11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skok MV, Grailhe R, Agenes F, Changeux JP (2007). The role of nicotinic receptors in B‐lymphocyte development and activation. Life Sci 80: 2334–2336. [DOI] [PubMed] [Google Scholar]
- Smith NJ, Hone AJ, Memon T, Bossi S, Smith TE, McIntosh JM et al. (2013). Comparative functional expression of nAChR subtypes in rodent DRG neurons. Front Cell Neurosci 7: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliakov L, Wonnacott S (1996). Voltage‐sensitive Ca2+ channels involved in nicotinic receptor‐mediated [3H]dopamine release from rat striatal synaptosomes. J Neurochem 67: 163–170. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies M, Lips KS, Kurzen H, Kummer W, Haberberger RV (2006). Nicotinic acetylcholine receptors containing subunits α3 and α5 in rat nociceptive dorsal root ganglion neurons. J Mol Neurosci 30: 55–56. [DOI] [PubMed] [Google Scholar]
- Tachikawa E, Mizuma K, Kudo K, Kashimoto T, Yamato S, Ohta S (2001). Characterization of the functional subunit combination of nicotinic acetylcholine receptors in bovine adrenal chromaffin cells. Neurosci Lett 312: 161–164. [DOI] [PubMed] [Google Scholar]
- Wang N, Orr‐Urtreger A, Chapman J, Ergun Y, Rabinowitz R, Korczyn AD (2005). Hidden function of neuronal nicotinic acetylcholine receptor β2 subunits in ganglionic transmission: comparison to α5 and β4 subunits. J Neurol Sci 228: 167–177. [DOI] [PubMed] [Google Scholar]
- Wessler I, Kirkpatrick CJ (2008). Acetylcholine beyond neurons: the non‐neuronal cholinergic system in humans. Br J Pharmacol 154: 1558–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West KA, Brognard J, Clark AS, Linnoila IR, Yang X, Swain SM et al. (2003). Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest 111: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Gelber S, Orr‐Urtreger A, Armstrong D, Lewis RA, Ou CN et al. (1999a). Megacystis, mydriasis, and ion channel defect in mice lacking the α3 neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A 96: 5746–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Orr‐Urtreger A, Nigro F, Gelber S, Sutcliffe CB, Armstrong D et al. (1999b). Multiorgan autonomic dysfunction in mice lacking the β2 and the β4 subunits of neuronal nicotinic acetylcholine receptors. J Neurosci 19: 9298–9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Li Z, Yan S, He Y, Dai R, Leung GP et al. (2016). Role of the nicotinic acetylcholine receptor α3 subtype in vascular inflammation. Br J Pharmacol 173: 3235–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Kompella SN, Adams DJ, Craik DJ, Kaas Q (2013). Determination of the α‐conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J Med Chem 56: 3557–3567. [DOI] [PubMed] [Google Scholar]
- Zhang XL, Albers KM, Gold MS (2015). Inflammation‐induced increase in nicotinic acetylcholine receptor current in cutaneous nociceptive DRG neurons from the adult rat. Neuroscience 284: 483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Ren J, Brown E, Schneider D, Caraballo‐Lopez Y, Galligan JJ (2002). Pharmacological properties of nicotinic acetylcholine receptors expressed by guinea pig small intestinal myenteric neurons. J Pharmacol Exp Ther 302: 889–897. [DOI] [PubMed] [Google Scholar]
- Zia S, Ndoye A, Lee TX, Webber RJ, Grando SA (2000). Receptor‐mediated inhibition of keratinocyte migration by nicotine involves modulations of calcium influx and intracellular concentration. J Pharmacol Exp Ther 293: 973–981. [PubMed] [Google Scholar]
- Zia S, Ndoye A, Nguyen VT, Grando SA (1997). Nicotine enhances expression of the α3, α4, α5, and α7 nicotinic receptors modulating calcium metabolism and regulating adhesion and motility of respiratory epithelial cells. Res Commun Mol Pathol Pharmacol 97: 243–262. [PubMed] [Google Scholar]
- Zoli M, Pistillo F, Gotti C (2015). Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96: 302–311. [DOI] [PubMed] [Google Scholar]