

Abstract
Background and Purpose
GAT107 ((3aR,4S,9bS)‐4‐(4‐bromo‐phenyl)‐3a,4,5,9b‐tetrahydro‐3H‐cyclopenta‐[c]quinoline‐8‐sulfonamide) is a positive allosteric modulator (PAM) and agonist of α7 nicotinic acetylcholine receptors (nAChRs)that can cause a prolonged period of primed potentiation of acetylcholine responses after drug washout. NS6740 is a silent agonist of α7 nAChRs that has little or no efficacy for activating the ion channel but induces stable desensitization states, some of which can be converted into channel‐active states by PAMs. Although GAT107 and NS6740 appear to stably induce different non‐conducting states, both agents are effective treatment for inflammation and inflammatory pain models. We sought to better understand how both of these drugs that have opposite effects on channel activation could regulate signal transduction.
Experimental Approach
Voltage‐clamp experiments were conducted with α7 nAChRs expressed in Xenopus oocytes.
Key Results
Long‐lived sensitivity to a PAM or to an agonist was produced by NS6740 or GAT107 respectively. With sequential applications, these two drugs induced varying levels of persistent activation, which is a unique condition for a receptor that is known for rapid desensitization. The non‐conducting states induced by NS6740 or GAT107 differ in their sensitivity to an α7 nAChR‐selective antagonist and in how effectively they promote current.
Conclusions & Implications
Our data suggest that the persistent currents represent a dynamic interconversion between different stable desensitized states and the PAM‐inducible conducting states. However, the similarity of NS6740 and GAT107 effects on inflammation and pain suggests that the different stable non‐conducting states have common activity on signal transduction.
Linked Articles
This article is part of a themed section on Nicotinic Acetylcholine Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.11/issuetoc
Abbreviations
- PNU‐120596
N‐(5‐chloro‐2,4‐dimethoxyphenyl)‐N′‐(5‐methyl‐3‐isoxazolyl)‐urea
- GAT107
(3aR,4S,9bS)‐4‐(4‐bromo‐phenyl)‐3a,4,5,9b‐tetrahydro‐3H‐cyclopenta‐[c]quinoline‐8‐sulfonamide
- NS6740
1,4‐diazabicyclo‐[3.2.2]nonan‐4‐yl)(5‐(3‐(trifluoromethyl)‐phenyl)furan‐2‐yl) methanone
- tkP3BzPB
1,2,4,5‐tetra‐{5‐[1‐(3‐benzyl)‐pyridinium]pent‐1‐yl}benzene tetrabromide
Introduction
Homomeric http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=468 have a very low probability of opening, even under the most optimized conditions, a limitation that can be overcome by mutations (Bertrand et al., 1997) or positive allosteric modulators (PAMs) (Williams et al., 2012). PAMs work through different mechanisms with varying efficacy to promote channel activation in the presence of orthosteric agonists such as ACh. It has also been reported that GAT107 ((3aR,4S,9bS)‐4‐(4‐bromo‐phenyl)‐3a,4,5,9b‐tetrahydro‐3H‐cyclopenta‐[c]quinoline‐8‐sulfonamide) (Thakur et al., 2013) acts both as a PAM and an allosteric agonist, identifying it as a PAM and agonist (ago‐PAM). This activity involves binding to two distinct sites on the receptor, shown in Figure 1; the ‘P’ site in the transmembrane domains that is also bound by conventional PAMs and the ‘D’ site (Figure 1) in the extracellular domain of the receptor (Papke et al., 2014a; Horenstein et al., 2016).
Figure 1.
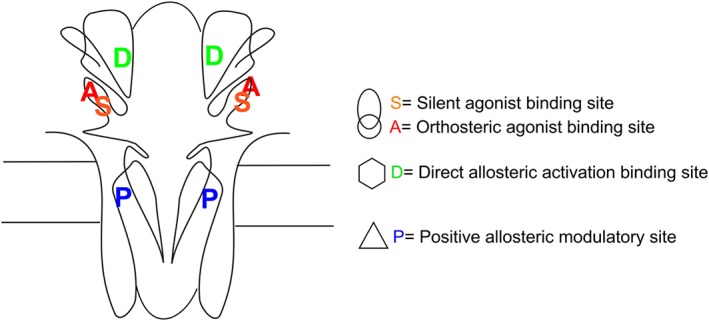
Putative binding sites in α7 nAChRs for most of the ligands used in these experiments (two of the five subunits are illustrated). The unique sites for the binding of ago‐PAMs to produce direct allosteric activation (D) have been hypothesized to be near the channel vestibule (Horenstein et al., 2016). The sites for orthosteric agonists (A) and silent agonists (S) are hypothesized to be distinct but overlapping (Chojnacka et al., 2013; Papke et al., 2014b). The binding sites for α7 PAMs are hypothesized to be within the transmembrane domains (Young et al., 2008).
Silent agonists (Clark et al., 2014; Papke et al., 2014b; Quadri et al., 2016) are a novel class of nicotinic drugs that produce little channel activation on their own, but their ability to manipulate the conformation equilibrium of the α7 receptor can be confirmed when they are used in conjunction with a PAM, which will destabilize the receptors' desensitized states and activate conducting states. The first silent agonist to be introduced was NS6740 (1,4‐diazabicyclo‐[3.2.2]nonan‐4‐yl)(5‐(3‐(trifluoromethyl)‐phenyl)furan‐2‐yl) methanone) (Briggs et al., 2009).
The binding sites for agonists and silent agonists (‘A’ and ‘S’ in Figure 1 respectively) are proximal to one another, such that agents binding to one of these sites compete with agents binding to the other (Papke et al., 2015). Data suggest that the S site is, in fact, an extension of the A site since silent agonists of simple structure are closely related to agonists and may differ by as little as a single methyl group in the core cationic element, which puts them over a critical size limitation for efficacious agonism (Papke et al., 2014b). Therefore, we may consider them jointly as the A/S site.
Just as efficacious agonists vary greatly in their extended structures (Horenstein et al., 2008), some silent agonists too must make multiple point‐to‐point contacts in an extended binding pocket (Wang et al., 2012; Papke et al., 2014b; Quadri et al., 2016), which precisely regulate their properties. The structure of NS6740 apparently exploits the features of this site to achieve long‐lasting effects on the receptors' conformational equilibrium (Papke et al., 2015).
The goal of the present study is to evaluate the dynamic interactions of the silent agonist NS6740 and the ago‐PAM GAT107 on the activation and desensitization of α7 nAChRs. It is interesting to note that, while these two ligands applied alone each have very different effects on receptor activation, it has recently been reported that they have a similar profile of anti‐inflammatory and anti‐allodynic activities with in vivo models of chronic neuropathic and inflammatory pain (Papke et al., 2015; Bagdas et al., 2016).
Methods
Animals
All animal care and experimental procedures were approved by the University of Florida Institutional Animal Care and Use Committee (https://iacuc.ufl.edu), protocol # 201502669. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Pigmented female Xenopus laevis >9 cm were purchased from Nasco (Ft. Atkinson, WI, USA). The frogs are maintained in the Animal Care Service facility of the University of Florida. Newly purchased frogs are held in quarantine for at least 30 days to ensure that they would not transmit any disease to the established colony. The light cycle is 12 h light/12 h dark. The water temperature is kept at 63–64F (17–18°C) utilizing a chiller. Tap water is conditioned with Amquel Water Detoxifier (Kordon, Hayword, CA, USA), StressCoat (MARS Fishcare North America, Chalfont, PA, USA), Cichlid Lake Salt (SeaChem Laboratories, Madison, GA, USA) and sodium bicarbonate (Aquatic Ecosystems, Apopka, FL, USA) in a separate 50 gallon drum for at least 1 h before being used. A 15–20% water change is performed daily. Frogs are checked daily by the husbandry staff and are fed sinking frog pellets (Xenopus Express, Brooksville, FL, USA) twice per week. The water quality parameters that are monitored biweekly include pH (6.5–8.5), total ammonia nitrogen (<0.5 mg·L−1), nitrite (<0.3 mg·L−1) and conductivity (800–1200 μS). The frogs are maintained in a recirculating water system from Pentair Aquatic Habitats (Apopka, FL). Water is filtered via a mechanical filtration, chemical filtration (using activated charcoal), biological filtration and UV light before it recirculates back to the housing tanks. Water exchange in the tanks is between 5–10 tank changes an hour. The frogs are group‐housed in 30 gallon polypropylene opaque tanks with a viewing window on the front. The tanks are each furnished with two plastic tunnels for enrichment.
Xenopus oocytes are internationally accepted as a valuable system of choice for the study of artificially expressed mammalian brain receptors. The use of Xenopus oocytes to study human or other mammalian brain receptors is a method of using the lowest phylogenetic species available and the recommended alternative to the use of mammalian tissue. Xenopus oocytes are used routinely to characterize numerous ion channels. Thus, our results can be directly compared with hundreds of other studies. Note that oocytes are the experimental units, not frogs.
Over the course of this project, a total of 26 surgeries to remove oocytes were performed on nine frogs. We may perform up to five survival surgeries on each animal, and a 6th surgery is terminal. The frog recovers at least 2 months between surgeries, and the surgeries are done on alternate sides of the belly.
The frog is fully anaesthetized after 20 min in 1.5 L frog tank water containing 1 g of 3‐aminobenzoate methanesulfonate (MS‐222) buffered with sodium bicarbonate to pH 7.2. The anaesthetic is absorbed through the skin. The basin is held in ice. The frog is then disinfected before the surgery. There are Pseudomonas/Aeromonas bacteria normally present in frog water and on their skin. These bacteria are highly mobile, very small and swift, and they could easily traverse the moist skin of the frog from the back to the belly during the surgery if we only disinfected the belly. Opthalmic ointment is placed on the frog's eyes to protect them from the disinfectant. The frog is placed in an autoclaved beaker containing 1% Nolvasan (chlorhexidine diacetate) solution, keeping the eyes and nose out of the solution. After no more than 3 min in the disinfectant, the frog is rinsed thoroughly with e‐pure water, the skin of the belly wiped gently with a sterile gauze pad to remove any loose debris and rinsed again with autoclaved water. The frog is then laid on the drape over the ice in the surgical area.
Clean surgical instruments (blunt forceps, tissue forceps, hemostats, fine scissors and blunt scissors) are autoclaved together with a section of surgical drape and a packet of sterile gauze all in a surgical autoclave pack. The surgical area, surrounding counter tops and the surgical tray are washed first with soap and water, rinsed, then wiped with 70% ethanol. A large autoclaved drape is draped over equipment in the back of the hood area where surgeries are performed. A germicidal light is turned on for 20 min.
The surgeon puts on sterile gloves and observes the sterile field. A window is cut in the autoclaved surgical drape and laid over the frog belly. The skin is cut with a #15 scalpel blade, holding the belly taut, making an incision approximately 1.5–2 cm long. Tissue forceps are used to grasp the muscle layer, which is then cut in an upward direction with a #11 scalpel blade. Fine pointed scissors are used to lengthen the incision to approximately 1–1.5 cm long. The incision in the muscle layer will be centred under the incision in the skin layer and will be a little shorter. Oocytes are usually presenting, in ovarian lobes, and can be easily and gently pulled out with forceps. The desired quantity is cut off with scissors and placed in a petri dish containing filter‐sterilized calcium‐free Barth's solution. There is no need to tie off the remaining ovarian tissue, which is allowed to slip back into the belly cavity. Care is taken that no oocytes are left between the muscle layer and the skin.
For a survival surgery, the muscle layer and the skin are sutured separately with 4–0 synthetic absorbable monofilament suture. Three knots are made for each stitch. Care is taken to include the membrane over the muscle layer. The first two knots are loose, just butting the sides of the incision together. The third knot is pulled tight. There are usually 2–3 stitches made in the muscle layer and 3–4 stitches in the skin. The incision is sealed with VetBond tissue adhesive. Gentamicin solution (2–4 mg·kg−1) is injected into muscle of a hind leg as a prophylactic measure. At the first surgery, coloured beads are attached to a hind toe using 4–0 nylon monofilament suture in order to mark the frog for identification. The surgery takes approximately 30 min.
The single use of MS‐222 provides post‐operative analgesia as well as pre‐operative anaesthesia (Lalonde‐Robert et al., 2012; Ramlochansingh et al., 2014). No signs of any pain, distress or discomfort are seen in the frogs before, during or after surgeries. For recovery, the frog is laid on her back on clean, wet paper towels, and the frog skin is kept continuously wetted with filtered distilled water. The frog is monitored closely until she flips herself over, and then she is placed in a basin containing an inch or two of frog tank water for transport back to the ACS facility. She is placed directly in the main tank with the other frogs, as we have found that it is a less stressful environment than an isolated tank. The behaviour of the frogs after recovery from the surgery is indistinguishable from their behaviour before a surgery. The frogs are typically motionless at the bottom of the tank, will swim when startled, and occasionally, they will swim to the surface for air. She is monitored daily for 3 days by research staff as well as by the Animal Care Services routine care.
Frogs are killed after the sixth surgery. If for some reason the frog did not recover after surgery (extremely rare), the frog would be killed instead of being brought back to Animal Care Services. A frog may also be killed if it has no more oocytes, if no oocytes are found, or if it consistently produced poor quality oocytes. Euthanasia is accomplished by first over‐anaesthetizing the frog with 5 g·L−1 MS‐222 buffered with sodium bicarbonate absorbed through skin for 1–2 h, followed by physical destruction of the brain.
Heterologous expression of nAChRs in Xenopus laevis oocytes
The human α7 nAChR clone was obtained from Dr J. Lindstrom (University of Pennsylvania, Philadelphia, PA, USA). The human resistance‐to‐cholinesterase 3 clone was co‐injected with α7 to improve the level and speed of α7 receptor expression without affecting the pharmacological properties of the receptors (Halevi et al., 2003). Subsequent to linearization and purification of the plasmid cDNAs, cRNAs were prepared using the mMessage mMachine in vitro RNA transcription kit (Ambion, Austin, TX, USA). Mutants were made as previously described (Papke et al., 2011).
The harvested oocytes are treated with 1.4 mg·mL−1 Type 1 collagenase (Worthington Biochemicals, Freehold, NJ, USA) for 2–4 h at room temperature in calcium‐free Barth's solution (88 mM NaCl, 1 mM KCl, 2.38 mM NaHCO3, 0.82 mM MgSO4, 15 mM HEPES and 12 mg·L−1 tetracycline, pH 7.6) to remove the ovarian tissue and the follicular layer from each oocyte. Stage V oocytes are subsequently isolated and injected with 50 nL of 5–20 ng nAChR subunit cRNA. Oocytes are maintained in Barth's solution with calcium (additional 0.32 mM Ca(NO3)2 and 0.41 mM CaCl2), and recordings were carried out 1–14 days after injection.
Two‐electrode voltage clamp electrophysiology
Experiments were conducted using OpusXpress 6000A (Molecular Devices, Union City, CA, USA) (Papke and Stokes, 2010). Both the voltage and current electrodes were filled with 3 M KCl. Oocytes were voltage‐clamped at −60 mV. The oocytes were bath‐perfused with Ringer's solution (115 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 10 mM HEPES and 1 μM http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=320, pH 7.2) at 2 mL·min−1. To evaluate the effects of experimental compounds compared with ACh‐evoked responses of various nAChR subtypes expressed in oocytes, control responses were defined as the average of two initial applications of ACh made before test applications. The solutions were applied from a 96‐well plate via disposable tips. Drug applications were 12 s in duration followed by a 181 s washout period. A typical recording for each set of oocytes constituted two initial control applications of ACh, one or more experimental compound applications and then a follow‐up control application(s) of ACh. The control ACh concentration was 60 μM. The responses were calculated as both peak current amplitudes and net charge, as previously described (Papke and Papke, 2002), and the average of the peak current amplitudes of the two initial controls was used for normalization purposes. Statistical comparisons were based on t‐tests of the normalized data where indicated.
Data were collected at 50 Hz, filtered at 20 Hz and analysed by Clampfit 9.2 or 10.0 (Molecular Devices) and Excel (Microsoft, Redmond, WA, USA). Data were expressed as means ± SEM from at least five oocytes for each experiment, unless otherwise stated, and plotted by Kaleidagraph 4.5.2 (Abelbeck Software, Reading, PA, USA). Multi‐cell averages were calculated for comparisons of complex responses. Averages of the normalized data were calculated for each of the 10 322 points in each of the 206.44 s traces (acquired at 50 Hz), as well as the standard errors for those averages.
Data and statistical analysis
The data and statistical analyses in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Comparisons of results were made using t‐tests between the groups of experimental values. A value of P < 0.05 was used to constitute the level of significance. The statistics were calculated using an Excel template provided in Microsoft Office. Note that some experiments (Figure 4C, D) were unusually long and the current amplitudes extremely high, at the margin of what the equipment could voltage clamp. These experiments began with groups of eight cells; however, due to the size of the responses, the voltage clamp failed in four of the original eight cells, while four remained well clamped. Due to the low n values, no statistical comparisons were calculated, although the data plotted as the point‐by‐point averages (±SEM) permitting qualitative evaluation of the data by visual inspection.
Figure 4.
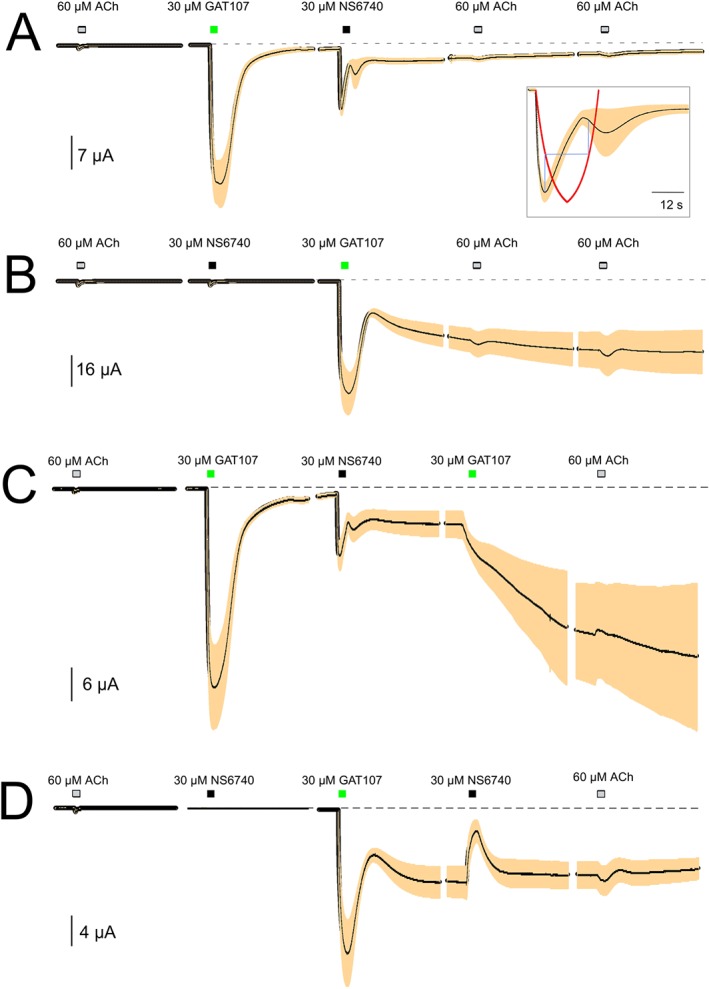
Interactions between GAT107 and NS6740 with sequential applications. (A) The averaged responses (normalized to average amplitude of two initial ACh controls, shown to provide scale) of four cells treated first with 30 μM GAT107 and then with 30 μM NS6740, followed by two further 60 μM ACh applications. The steady‐state current at the end of the upper set of traces was 1.5‐fold larger than the initial ACh controls. (B) The responses in a similar experiment (n = 5) when NS6740 was given prior to GAT107. The steady‐state current at the end of the lower set of traces was 15‐fold larger than the initial ACh controls. (C) Cells (n = 4) were treated with 30 μM GAT107, 30 μM NS6740 and then 30 μM GAT107, followed by a final 60 μM ACh application. The final steady‐state current was 27‐fold larger than the initial ACh controls. (D) Cells (n = 4) were treated with 30 μM NS6740, 30 μM GAT107 and then 30 μM NS6740, followed by a final 60 μM ACh application. The final steady‐state current was 14‐fold larger than the initial ACh controls. For all sets of cells, the second of the two initial ACh controls is shown to provide scale. Each trace segment is 206.44 s long, and the interval between each trace is 16.56 s. The dashed lines represent the original baselines. The inset in (A) enlarges a 30 s section of the data. The red line is an estimation of solution exchange with a 12 s drug application and a 5 s time constant of solution exchange (Papke and Papke, 2002). The average ACh controls were 915 ± 115 nA for (A), 2.2 ± 0.8 μA for (B), 750 ± 120 nA for (C) and 660 ± 165 nA for (D). Vertical scale bars are based on averaged ACh controls.
Materials
ACh chloride, atropine and other chemicals were purchased from Sigma‐Aldrich Chemical Company (St. Louis, MO, USA). NS6740 and GAT107 were prepared as described (Peters et al., 2004; Kulkarni and Thakur, 2013; Thakur et al., 2013; Papke et al., 2015) respectively. tkP3BzPB (1,2,4,5‐tetra‐{5‐[1‐(3‐benzyl)‐pyridinium]pent‐1‐yl}benzene tetrabromide) was provided by Peter Crooks (University of Arkansas). Fresh ACh stock solutions were made in Ringer's solution each day of experimentation. Stock solutions of tkP3BzPB were made in Ringer's solution and kept at 4°C for up to 1 week. Concentrated stock solutions of other test drugs were prepared in DMSO and held at −20°C. Working solutions were prepared freshly at the desired concentration from the stored stock.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Prolonged and reciprocal activities of GAT107 and NS6740 on α7 nAChRs
We noted that potentiating effects of GAT107 were present after washout of the drug from the bath (Figure 2A top trace), and after currents allosterically activated by the application of GAT107 alone had returned to baseline. Responses to ACh 6 min after washing out GAT107 were significantly higher than responses to the same dose of ACh prior to GAT107 application. In contrast to the effects of GAT107, the silent agonist NS6740 produced almost no channel activation but induced desensitization (Papke et al., 2015) and thereby blocked ACh activation even after washout (Figure 2B, top trace) (Papke et al., 2015). NS6740 does, however, prime the receptors for activation by the PAM http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3991 (N‐(5‐chloro‐2,4‐dimethoxyphenyl)‐N′‐(5‐methyl‐3‐isoxazolyl)‐urea). When the PAM, which does not usually produce ion currents on its own, was applied alone after NS6740 (Figure 2B, bottom trace), there was an effect that persisted after a prolonged washout of N6740 from the bath. As expected, an application of PNU‐120596 evoked no current after a priming application of GAT107 (Figure 2A bottom trace), as both of these agents target the P site.
Figure 2.
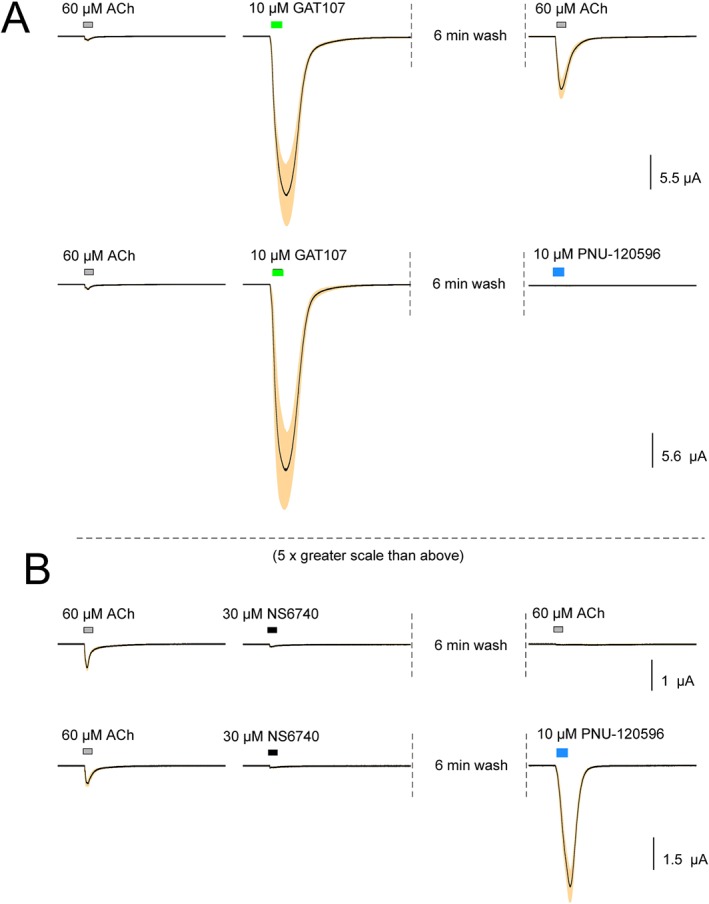
Averaged data showing the responses to GAT107 and NS6740 and the modulation of subsequent responses to ACh or a PAM. (A) Data from voltage‐clamped oocytes showing initial ACh control responses and then the direct allosteric activation by GAT107. Data for each cell were normalized to the average of the peak current amplitude of two initial ACh controls, which are shown to provide scale. The solid lines are the averages (n = 7), and the shaded areas are the standard errors of the mean for each of the 10 322 points (see Methods). Each trace segment is 206.44 s long, and the interval between traces is 16.56 s. After the acquisition of the GAT107 response data, cells were washed for an additional 6 min before the application of either 60 μM ACh or 10 μM PNU‐120596. Control ACh responses were 894 ± 129 and 779 ± 98 nA for the upper and lower traces respectively. (B) The data from oocytes, processed as described above, illustrating responses to 30 μM NS6740 and, following a 6 min wash, the application of either 60 μM ACh or 10 μM PNU‐120596. Control ACh responses were 804 ± 98 and 854 ± 212 nA for the upper (n = 7) and lower trace (n = 8) respectively. The amplitudes of the ACh controls were similar for other experiments. Note the difference in scale of the presentation between panels A and B. Vertical scale bars are based on averaged ACh controls.
In order to determine the duration of the GAT107 and NS6740 effects, we made single applications of the drugs followed by washes of varying duration. A single 12 s application of 30 μM GAT107 followed by washout left cells primed to generate large responses to NS6740 for more than 45 min (Figure 3A), at which time the potentiation appeared relatively stable. (An explanation for the unusual waveform of these currents is given below.) Likewise, even after an 87 min washout period, cells exposed to a single application of NS6740 gave robust responses to an application of 30 μM PNU‐120596 (Figure 3B), which on its own or following GAT107 application produced no currents (Figure 2A bottom).
Figure 3.
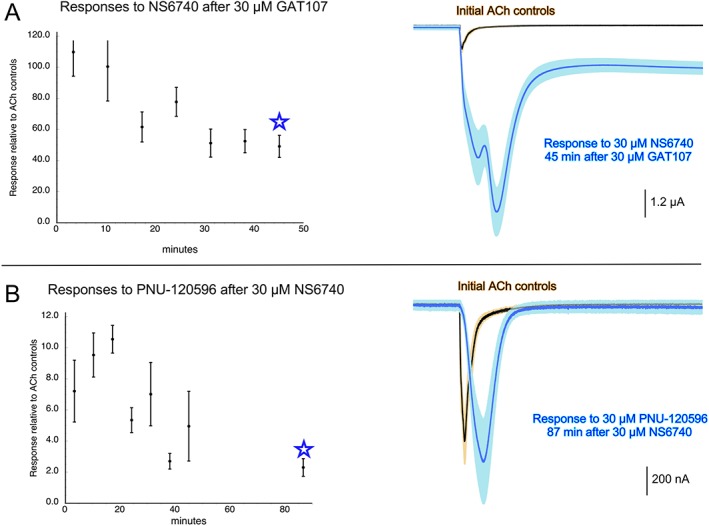
Duration of GAT107 and NS6740 effects on α7 nAChR responses. Groups of cells expressing human α7 AChRs were given two control applications of 60 μM ACh prior to single 12 s applications of either 30 μM GAT107 (A) or 30 μM NS6740 (B). Cells were continuously perfused with Ringers for varying periods of time and then probed with single applications of 30 μM NS6740 (A) or 30 μM PNU‐120596 (B). Peak current responses evoked by the probe applications were normalized to the initial responses to 60 μM ACh, which in these experiments averaged 930 ± 70 nA. For all points in the plots, n ≥ 5 cells. Traces displayed on the right are the averaged raw data for the eight cells in (A) and the seven cells in (B) that were obtained at the long wash times tested for each condition (indicated by the stars). They are compared with the averaged ACh responses obtained prior to the initial applications of GAT107 (A) or NS6740 (B). Each trace segment is 206.44 s long. Vertical scale bars are based on averaged ACh controls.
Dynamic long‐lasting interactions of GAT107 and NS6740
GAT107 produced a long‐lived state primed for potentiated activation, and we determined that NS6740, which produces little activation on its own, could exploit this potentiated state to produce both a transient and relatively persistent current (see Figure 3A right‐hand panel). The application of NS6740 to the receptors primed with GAT107 produced a large response but with peculiar kinetic features (Figures 3A right and 4A). Following a brief period of rapid synchronized activation, the response relaxed, rebounded and then showed a slowing decaying period of low‐level persistent current that was 1.5‐fold the amplitude of the initial ACh controls. The unusual waveform was a consistent feature of NS6740 responses with this experimental protocol and was probably related to the narrow inverted‐U concentration–response function previously reported for PAM‐dependent responses to this compound (Papke et al., 2015). Responses reached a peak on the rising edge of the 30 μM applications and then rebounded as concentrations fell (Figure 4A inset, red line indicates changes in the drug's concentration).
When receptors were primed with an application of 30 μM NS6740 shortly before an application of 30 μM GAT107, the results were even more striking than those shown in Figure 3B for PNU‐120596, with a large transient response being followed by a persistent current that after 10 min was 15 ± 4.5‐fold higher than the initial ACh‐evoked responses (measured at the end of the traces in Figure 4B). The pattern of activation of a large initial transient followed by an approach to a large persistent level of activation is similar to that observed when there is a constant bath perfusion of ACh and PNU‐120596 (Williams et al., 2011b). As with the currents produced by an NS6740 application followed by GAT107, the currents primed by applications in the reverse order were largely insensitive to further applications of ACh.
The dynamic character of the interactions between NS6740 and GAT107 is further illustrated with protocols utilizing additional applications. When the application order was GAT107‐NS6740‐GAT107 (Figure 4C), the activation after the NS6740 application was essentially the same as shown in Figure 4A, and the further application of GAT107 resulted in the stimulation of a current similar to the late phase of NS6740‐primed GAT107 response in Figure 4B. However, the response to the initial application of GAT107, prior to the application of NS6740, appeared to suppress any additional transient phase with the second application of GAT107. The application order in Figure 4D was NS6740‐GAT107‐NS6740. As expected, up to the point of the second NS6740 application, the response was similar to that of Figure 4B. However, under the conditions when the receptors were entering a phase of increasing current, the second NS6740 application produced a transient decrease rather than an increase in current and seemed to curtail the progressive rise in currents.
The waveforms associated with applications of these long‐acting ligands are consistent with a complex landscape for the energy associated with the various conformational states of the receptor and the non‐stationary processes that shift the receptors gradually through a series of states (Williams et al., 2011b; Papke et al., 2014a,b; Horenstein et al., 2016). Initially, as ligands bind, receptors will tend to move over the lowest energy barriers connecting to alternative conformational states, initially including the activated channel states. However, over time, receptors shift to more stable, non‐conducting states. This dynamic process is reflected in the currents shown on an expanded time scale in Figure 5A. Once the free GAT107 was washed out of solution, releasing the D site, the persistent current began a slow decay until the receptors were primarily in the non‐conducting PAM‐insensitive desensitized (Di) state (Williams et al., 2011b). The time constant for this process was approximately 10 min (Figure 5B). However, even after an hour, a small persistent current could still be detected, as illustrated by the effect of a 100 μM application of the channel blocker http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3990 (Figure 5C). This is consistent with the long duration of the single agent effects shown in Figure 3 and supports the hypothesis that the persistent current is the product of an equilibration between conducting (open) and non‐conducting (desensitized) states.
Figure 5.
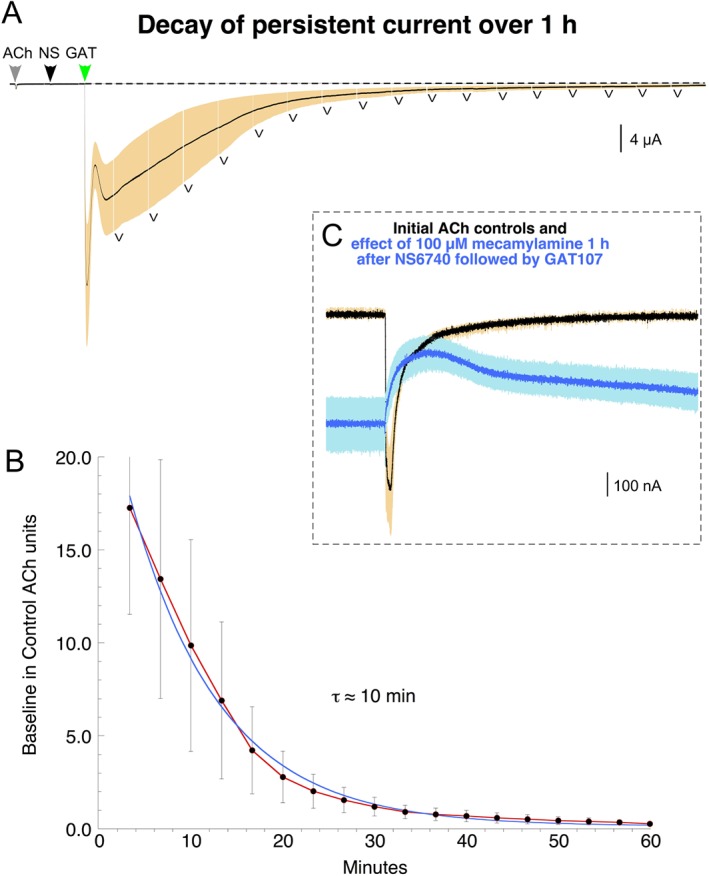
Decay of persistent current activated by GAT107 after NS6740. The upper trace shows the averaged response of four cells to a control 60 μM ACh application followed by 30 μM NS6740 and μM GAT104 (see Methods). During the subsequent periods of washout following the peak of the GAT107 activated current, data were sampled for 30 s at the beginning of every episode (223 s intervals), as indicated by the ‘v’ symbols below the trace and the mean values calculated and normalized to the respective ACh controls. These are plotted (red line ± S.E.M) and fit (blue line) with an exponential function that had a time constant of 10 ± 0.5 min and a correlation coefficient of 0.99716. The inset shows the effect of an application of 100 μM mecamylamine after 1 h of wash compared with the ACh controls and original baseline. Vertical scale bars are based on averaged ACh controls.
Modelling the conformational states of α7 nAChRs regulated by NS6740 and GAT107
Binding of ligands (Figure 6) to the allosteric or orthosteric sites changes the probability of the receptor making transitions among its multiple conformational forms. Minimal models for these conformational landscapes, based on previous studies (Williams et al., 2011b; Papke et al., 2014a,b; Horenstein et al., 2016) are presented in Figure 6.
Figure 6.
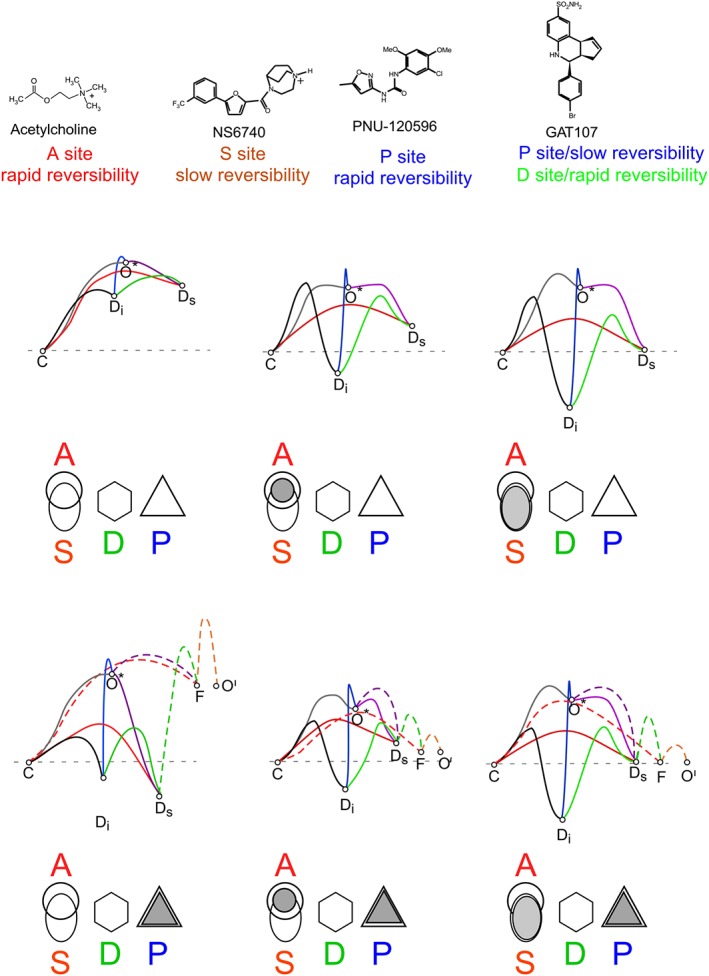
Ligands and hypothetical energy landscapes. Shown at the top are the orthosteric and allosteric ligands used in these experiments. Shown below are hypothetical energy landscapes for conformational state transitions based on occupancy of A, S and P sites, as indicated by shading are shown below. These schemes are based on previously published models (Williams et al., 2011b; Papke et al., 2014a,b; Horenstein et al., 2016) and include the resting closed state (C) the normal brief open state (O*), PAM‐insensitive (Di) and PAM‐sensitive (Ds) desensitized states, the PAM‐dependent open state (O') and the intraburst closed ‘flip’ state F (Lape et al., 2008). Note that since GAT107 allosteric activation does not seem to be important for the generation of persistent current, we omit showing models for receptors with ligand bound to the direct allosteric activation (D) site, as these have previously been presented (Horenstein et al., 2016). The selection of the colours for the lines connecting the states was arbitrary. The dotted lines in the lower landscapes represent the interstate conversion that can only occur when the P site is occupied by a PAM.
Two different classes of desensitized states have been identified that are stabilized by the binding of ligands to the A/S sites. One class (Ds) is able to be connected to a novel open state (O′) via the flip state (F) (Lape et al., 2008) when the P site is occupied (Papke et al., 2009), while the other (Di) is insensitive to the effects of PAM binding (Williams et al., 2011b). Specific agonists and silent agonists vary in whether they favour the induction of Ds or Di (Papke et al., 2014b). Binding to the P site alone appears to also be able to permit induction of Ds.
It should be noted that the conformational transitions of α7 receptors are regulated not only by which sites are bound but also by the level of occupancy of those sites within the pentameric receptor. We have previously shown that opening of the receptor in the absence of a PAM most likely occurs when only one or two A sites are bound (Williams et al., 2011a). The occupancy of more of the A/S sites also appears to shift the distribution of receptors between the Ds and Di sites, favouring Di when more of the sites are bound. This is certainly the case for silent agonists, since for NS6740 (Papke et al., 2015) and several of the small tetra‐ethyl‐ammonium silent agonists (Papke et al., 2014b), the concentration–response function for activation with PNU‐120596 is an inverted U, as explained for Figure 4A. The effectiveness and dynamics of PAMs for inducing currents is also sensitive to the PAM concentration and likely to the level of PAM site occupancy (Williams et al., 2011b; Papke et al., 2014a,b). Therefore, the energy landscapes shown are intended to represent the condition of relatively low fractional site occupancy (one to three of the five possible sites of each type), the best condition for sustained channel activation (Williams et al., 2011b).
In the lower part of Figure 6 are schematic models for the effects of binding Type II PAMs (Gronlien et al., 2007). We have previously proposed models with the Ds state directly connected to the O' state, which would have implied that residence in the Ds state was very short lived and represented the brief intraburst closures previously observed. However, NS6740 and GAT107 induce long‐lived non‐conducting states that are qualitatively different in how they react to the binding of other ligands, consistent with differences in the preferential induction of Ds or Di. This suggests that Ds itself is not a state within the PAM‐promoted bursts but rather a gateway state to the bursting states. Therefore, in the revised models presented in the lower tier of Figure 6, we have included a specific intraburst closed state (F) corresponding to the ‘flip’ state proposed to precede channel activation (Lape et al., 2008). While it is unlikely for channel opening to occur with binding to the P site alone in wild‐type receptors, certain mutations, such W55A, which faces the A/S site across the subunit interface (Gay et al., 2008; Williams et al., 2009), increase this probability (Papke et al., 2014a,b). However, while binding to P alone introduces the two new states to the conformational landscape, in wild‐type receptors, additional binding to A/S is required to stabilize these states.
Figure 7 illustrates how these landscapes can change with differences in the levels of ligand binding at the S site by NS6740 or the P site by GAT107 in order to explain the behaviour observed in the lower two traces of Figure 4. The centre landscape represents the condition during a persistent current when partial occupancy of both the S and P sites was achieved by the previous applications of NS6740 and GAT107, and receptors are variously in the C, Di, Ds, F or O' states (relative occupancy represented by the size of the encircled dots). When there is an additional application of GAT107 (moving from the centre to the right), the barrier is reduced between the Di and Ds states, favouring an increase in the persistent current (see Figure 4C). Conversely, when there is an additional application of NS6740 (moving from the centre to the left), the barrier is reduced between the Ds and Di states, favouring a transient reduction in current (see Figure 4D).
Figure 7.
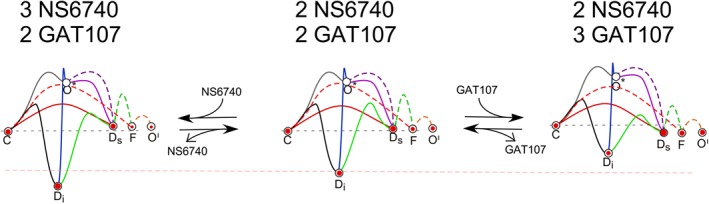
Hypothetical changes in the energy landscapes for α7 state transitions at differing levels of NS6740 and GAT107 binding. Shown in the centre is the landscape for occupancy of the S site and the P site from Figure 6, which assumes an intermediate level of binding at both sites. The left‐ and right‐most models hypothesize what might be changing with additional applications of either NS6740 or GAT107 respectively. These models are discussed in the text regarding the dynamic changes in currents shown in Figure 4C, D. The pink dashed line is provided to reference the starting energy for the Di state in the centre landscape for comparison to the other landscapes. Also shown in the centre landscape is hypothetical occupancy of the states relative to each other for a population of receptors during the phase of persistent current prior to another drug application, indicated by the size of the enclosed red dots. As shown, the occupancy of the states by the receptors in the population is assumed to shift following a drug application, favouring transitions among the states where the energy barriers have been reduced.
Effects of antagonists on persistent current in α7 nAChRs
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4005 is generally thought to be an α7‐selective competitive antagonist. However, observations suggest that it may function more as an inverse agonist when there is constitutive activation of the receptor. For example, we have previously shown that removing ACh from a bursting PNU‐potentiated receptor did not stop the bursting, but applying MLA to a channel in the midst of a prolonged burst did rapidly terminate the bursting (Williams et al., 2011b). As shown in Figure 8A, the application of MLA to receptors that were made constitutively active with sequential applications of NS6740 and GAT107 decreased the persistent currents. In the case of 10 μM MLA, the reduction in current was almost down to the original baseline. However, the MLA effects were relatively short lived, and although there was no agonist in solution, the constitutive currents regenerated. One hypothesis to explain these results is that NS6740 stayed bound to at least some of the silent agonist (A/S) sites, while GAT107 was bound to the PAM (P) sites, and the coupling of these bound ligands promoted channel activation. Alternatively, the ligands may dissociate from one or both sites but with persistent effects on channel conformation. The persistent currents most likely represent protracted bursts arising from single receptors transitioning from the PAM‐insensitive desensitized state (Di) through the Ds state, which is coupled via the intraburst ‘flip state’ (F state) (Lape et al., 2008) to the PAM‐dependent open state (O' state) (Figure 6; Williams et al., 2011b). We hypothesize that this form of activity occurs when some of the orthosteric (A/S) sites on a single receptor are still bound by agonist (Williams et al., 2011a), or in this case by the silent agonist NS6740, permitting MLA to bind to other unoccupied A/S sites and, by doing so, reduce activation. Consistent with continued binding of the drugs are our previously reported results showing that constitutive activation of L119C mutant α7 receptors can be achieved if PNU‐120596 is bath‐applied after the cationic sulfhydryl reagent http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6272 (2‐aminoethyl methanethiosulfonate) has been reacted with the receptor to produce a covalently tethered agonist at the A/S site (Wang et al., 2010). These currents were also sensitive to MLA in a similar manner (Figure 8B).
Figure 8.
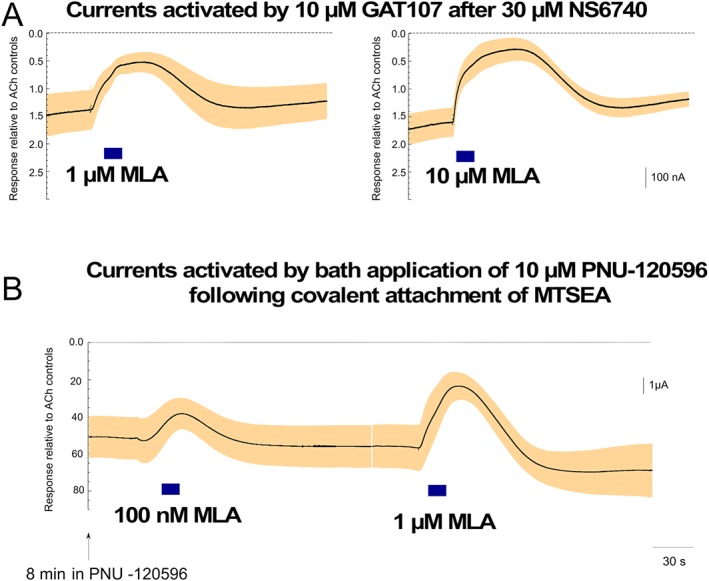
Transient inhibition of constitutive α7 nAChR currents by MLA. (A) Applications of 10 μM GAT107 were made to α7‐expressing cells following the application of 30 μM NS6740 (not shown) in order to establish steady‐state currents. Shown are the effects of 10 or 1 μM of the α7‐selective inverse agonist MLA. The dashed lines represent the original baselines. Data in the traces are averaged from six cells for 1 μM and seven cells for 10 μM, each normalized to the initial responses to 60 μM ACh obtained from the same cells (average ACh responses were 290 ± 90 nA). Each trace is 206.44 s long. (B) As previously described (Wang et al., 2010), L119C mutant α7 receptors were reacted with MTSEA, a sulfhydryl reagent that contains a charged amine, to produce a population of receptors with the amine functioning as an agonist tethered in the orthosteric binding site. Following a transient period of activation during the reaction process, these receptors are insensitive to further applications of ACh (not shown, see Wang et al., 2010). Constitutive activation was then achieved with the bath application of 10 μM PNU‐120596. Shown are the effects of 12 s applications of MLA at the concentrations indicated on the constitutive currents recorded 8.5 and 12.5 min after the beginning of the PNU‐120596 bath applications. The data are the averaged responses of six cells, normalized to the amplitude of their initial control responses to ACh. Note that for these data, the average amplitude of the ACh control responses was only 124 ± 22 nA since cells were used about 24 h after injection. Due to the large steady‐state current evoked, cells with larger baseline control responses would have been impossible to keep voltage clamped. Vertical scale bars are based on averaged ACh controls.
In experiments described by Williams et al. (2011b), steady‐state currents could be generated by continuous bath application of a PAM plus agonist. The amplitude and kinetics of the currents were dependent on both the PAM and agonist concentrations, such that with higher concentration of either, there was more transient current and less delayed current. Under those conditions, high concentrations of MLA produced transient inhibition of the delayed steady‐state current, while low concentrations of MLA perturbed the equilibrium in such a way as to cause a transient increase in current. In Figure 8, we illustrate the effects of relatively high MLA concentrations on the persistent currents stimulated by sequential application of NS6740 and GAT107. These transient decreases might be consistent with channel block or inverse agonism. Therefore, in order to determine whether at lower concentrations MLA showed the anomalous effects previously attributed to inverse agonism, we tested a broader range of MLA concentrations applied following the induction of the low‐amplitude persistent current by GAT107 and NS6740 (in that order, as in Figure 4A). While 100 nM MLA decreased current, lower concentrations produced transient increases in the current, and the positive effects of 1 nM MLA were independent of timing of the application (Figure 9).
Figure 9.
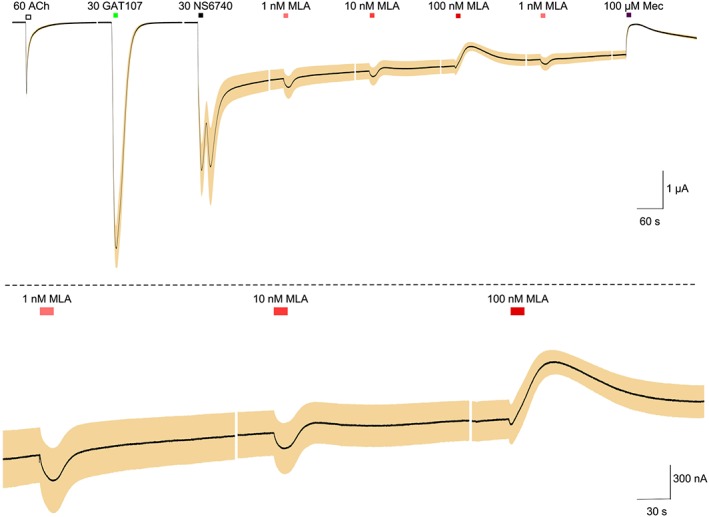
Concentration‐dependent modulation of persistent current by MLA. A lower level of persistent current was induced with the protocol illustrated in Figure 4A (30 μM NS6740 application following a 30 μM GAT107 application). MLA was then applied at progressively higher concentrations as indicated. Following the applications of MLA at 1, 10 and 100 nM, an additional application of 1 nM MLA was made in order to determine the reproducibility of the low concentration response. Following the sequence of four MLA applications, 100 μM mecamylamine (Mec) was applied in order to illustrate the channel blocking effect on the persistent current. Data in the traces are averaged from 7 cells, each normalized to the initial responses to 60 μM ACh obtained from the same cells (average ACh responses were 1.8 ± 0.3 μA). Vertical scale bars are based on averaged ACh controls. The upper trace shows the entire experiment. The lower trace shows just responses to the first three MLA applications at a scale threefold increased both vertically and horizontally. The dashed line represents the original baseline.
The tetrakis‐azaaromatic quaternary ammonium compound tkP3BzPB is a very potent and highly selective non‐competitive antagonist of α7 nAChRs (Lopez‐Hernandez et al., 2009; Peng et al., 2013) which, once bound, produces ery long‐lived inhibition. A 12 s application of just 1 μM tkP3BzPB profoundly inhibited the persistent current induced by the synergistic effects of NS6740 and GAT107 (Figure 10A upper trace). However, when tkP3BzPB was applied prior to induction of the persistent current, it had no significant effect (lower trace of Figure 8A), suggesting that tkP3BzPB may be acting as a use‐dependent open channel blocker, as reported for the non‐selective neuronal nAChR antagonist BTMPS (Francis et al., 1998). It was previously reported that at a concentration of 10 μM, the tkP3BzPB inhibition of α7 receptors activated by the endogenous orthosteric agonist ACh (Lopez‐Hernandez et al., 2009) was not use‐dependent. However, it has also been reported that currents potentiated by the PAM PNU‐120596 differ from those activated by ACh alone in their antagonist sensitivity (Peng et al., 2013). We therefore further investigated the state dependence of 1 μM tkP3BzPB inhibition of the persistent currents in α7 nAChRs.
Figure 10.
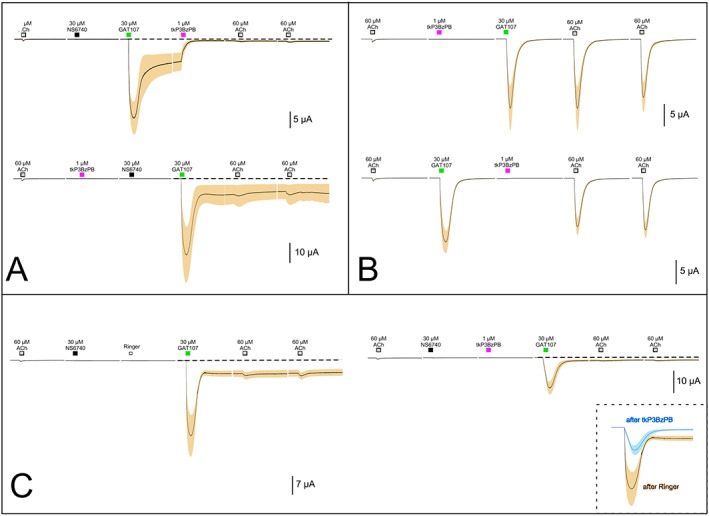
Antagonism of persistent currents in α7 nAChRs with the α7‐selective slowly reversible antagonist tkP3BzPB. (A) Upper trace: sequential application of 30 μM NS6740 and μM GAT107 followed by the application of 1 μM tkP3BzPB (n = 6, average ACh control responses 800 ± 325 nA). Lower trace: 1 μM tkP3BzPB application applied prior to 30 μM NS6740 and μM GAT107 (n = 6, average ACh control response 1.3 ± 0.3 μA). (B) Upper trace: the pre‐application of 1 μM tkP3BzPB prior to the allosteric activation of α7 receptors by 30 μM GAT107 (n = 6, average ACh control responses 750 ± 125 nA). Lower trace: the application of tkP3BzPB during the period of primed potentiation by GAT107 (n = 6, average ACh control responses 700 ± 67 nA). (C) Effects of tkP3BzPB on receptors desensitized by 30 μM NS6740. In the traces on the left, a 12 s application of control Ringer's solution was made following the application of NS6740 (n = 5 average ACh control responses 500 ± 100 nA), and in the traces on the right, a 12 s application of 1 μM tkP3BzPB was made following the NS6740, both prior to the initiation of persistent current by 30 μM GAT107 (n = 8, average ACh responses 600 ± 125 nA). The application of tkP3BzPB significantly inhibited both the GAT107 peak responses and the persistent current measured before the subsequent ACh applications. The inset shows an overlay of the two GAT107 responses.
The application of tkP3BzPB prior to GAT107, in the absence of priming by NS6740, decreased neither the direct allosteric activation produced by GAT107 nor the primed potentiation of subsequent ACh‐evoked responses (Figure 10B upper trace). Likewise, when tkP3BzPB was applied following direct allosteric activation of the receptors by GAT107, there was no significant effect on the subsequent ACh‐evoked responses potentiated by the GAT107 priming effect (Figure 10B lower trace). However, when receptors were primed for the generation of GAT107 persistent currents by an application of NS6740, they became sensitive to tkP3BzPB (Figure 10C). In the paired experiments (same batch of cells, same day and same drug solutions), when tkP3BzPB was delivered (right set of traces) instead of control Ringer's solution (left set of traces), both the transient peaks and persistent currents induced by GAT107 were significantly reduced (see inset). Specifically, the transient peaks induced by GAT107 were significantly reduced from 46 ± 12 times the ACh controls to 17.1 ± 3.6 times the ACh controls following the tkP3BzPB treatment, and persistent currents were significantly reduced from 8.1 ± 1.8 times the ACh control amplitudes to 1.5 ± 0.5 times the ACh control. These results indicate that tkP3BzPB is a state‐dependent antagonist, but not strictly a use‐dependent open channel blocker. Rather tkP3BzPB binds to yet unknown sites that are accessible in the Di state and stabilize that and/or other non‐conducting states.
Discussion
Previous work has shown that both NS6740 and GAT107 lead to long‐lasting non‐conducting states of α7 nAChRs that can promote activation by other drugs (Papke et al., 2014a,b; Papke et al., 2015). Our aim in the present study was to better understand these states and determine the degree to which they were independent or could interact. Novel results in this paper show unusual and unexpected channel activation properties following sequential application of the drugs. While we have previously reported that the effects of NS6740 at the A/S site (Papke et al., 2015) and GAT107 at the P site (Papke et al., 2014a,b) appear to be particularly long‐lasting, in the present study, we report that NS6740 or GAT107 applied alone lead to different distributions of non‐conducting states that are biased in how they respond to other ligands. Because this biasing or priming arises from activity at different sites (the S or P sites for NS6740 or GAT107 respectively), the combined effects of these drugs synergize to produce, depending on the specific sequence of drug applications and on the concentrations of each drug, varying amounts of persistent activation and insensitivity to other orthosteric ligands such as ACh or other allosteric ligands such as PNU‐120596, which need to bind to the same or overlapping sites.
One hypothesis is that the persistence of the NS6740 and GAT107 effects reflects persistent binding to their respective sites. It should also be noted that both of these compounds are relatively lipophilic (logP values for NS6740 and GAT107 are estimated to be 2.3 and 3.54 respectively), so it is possible that they may partition into the membrane, which could then act as a depot for the drugs. An alternative hypothesis is that the persistence of potentially interacting non‐conducting states following their induction does not rely on the continued binding of the ligands at their respective sites but rather on the stable induction of the downstream states. However, GTS‐21, a desensitizing weak partial agonist with relatively reversible desensitization (Papke et al., 2009), fails to produce large persistent currents when paired with GAT107 (not shown). Similarly, GAT904 and PNU‐120596, which are relatively reversible at the P site, will produce significant persistent current only for as long as they are present in the bath (Williams et al., 2011b; Horenstein et al., 2016). These observations suggest that the induction of the specific states alone is insufficient to generate persistent currents and that the specific ligands are required.
The antagonism of α7 receptor currents by MLA (Figure 8) is believed to be based on competitive binding to A/S sites (Lopez‐Hernandez et al., 2009), which invites the question of how MLA is able to antagonize constitutive currents that do not rely on agonists that are free in solution. We have shown that receptor activation arises most readily when only a fraction of the multiple binding sites are occupied (Williams et al., 2011a) and that, in fact, high levels of agonist binding site occupancy favour the preferential induction of Di (Williams et al., 2011b). Therefore, it is likely that the channels that are bursting during the periods of persistent current are ones that have one or more A/S binding sites unoccupied by ligand and are thus available for MLA to bind. The results would then be consistent with MLA not just passively blocking the accessibility of the A/S binding sites but rather acting as an inverse agonist, terminating the paroxysms of channel activation as was observed on the single channel level (Williams et al., 2011b), perhaps by favouring the induction of Di.
We see that the inhibition of persistent current by tkP3BzPB (Figure 10) is clearly state‐dependent and not restricted to the open states of the receptor since it was effective when delivered to receptors in the non‐conducting states induced by NS6740. The inhibition of ACh‐evoked currents by tkP3BzPB previously reported (Lopez‐Hernandez et al., 2009; Peng et al., 2013) therefore also likely involves binding of the agent to the ACh‐induced desensitized states because single applications of tkP3BzPB were almost entirely effective, and with any single episode of ACh activation, it is likely that only a small percent of the available channels were activated (Williams et al., 2012). Therefore, even though both NS6740 and GAT107 induce stable non‐conducting states, those states are conformationally distinct and differ in tkP3BzPB sensitivity and in their connection to the flip (F) state (Lape et al., 2008), the PAM‐dependent interburst state (Figure 6) that functions like an activated complex. In the case of the α7 PAM‐facilitated currents, dwells in the F state are observed as the unstable, intraburst closed state (Williams et al., 2011b; Horenstein et al., 2016). As proposed in the models presented in Figure 6, NS6740 may most effectively induce Di in preference to Ds, and, without occupancy at the PAM site, neither of these states leads to channel activation. With binding of GAT107, the balance between Ds and Di is shifted toward Ds, which can connect to the flip state. As suggested by the data in Figure 4, the balance between these states and the resultant amount of persistent current can be dynamically regulated by repeated applications of NS6740 or GAT107, but not by more reversible agents like ACh or PNU‐120596. The details of bursting behaviour will, of course, vary with recording temperature (Sitzia et al., 2011; Williams et al., 2012; Andersen et al., 2016), the specific agonist and PAM used and the concentrations of these agents (Williams et al., 2011b; Andersen et al., 2016), as well as, potentially, the recording configuration (Kalappa and Uteshev, 2013). The Xenopus oocyte expression system has proven to be a good predictor of α7 α7 nAChR pharmacology for mammalian cells that produce receptors that are competent for ion channel function, such as transfected HEK cells (Williams et al., 2012) and hippocampal interneurons (Alkondon et al., 1997; Frazier et al., 2003). Although our results clearly show that NS6740 and GAT107 have very different ion channel‐related activity profiles in the oocyte expression system, they have very similar activity profiles with animal models of inflammatory and neuropathic pain (Papke et al., 2015; Bagdas et al., 2016). It is well documented that α7 nAChRs can regulate calcium homeostasis (Koukouli and Maskos, 2015) and metabotropic functions (de Jonge et al., 2005; de Jonge and Ulloa, 2007; Egea et al., 2015). The conventional assumption is that calcium flux through the receptor's ion channel is a necessary initiator of these effects (Koukouli and Maskos, 2015). This is an untenable hypothesis in the case of immune cell data, where aside from astrocytes (Shen and Yakel, 2012), the nAChR detected appear to be incapable of ion channel activation (Villiger et al., 2002; Peng et al., 2004; Skok, 2009). It is also unlikely to be the complete story for the in vivo data and the in vitro immune cell effects, especially in the case of NS6740, which as a single agent has the primary effect of decreasing channel function in our assay system. Thus, effects of these drugs on inflammatory processes are likely not to depend on channel activity, rather the α7 receptor has been shown to possess a large intracellular interactome (Paulo et al., 2009; King et al., 2015), enabling it to function metabotropically.
Our data with GAT107 and NS6740 indicate that they each have long‐lasting effects on α7 receptor conformation. As single agents, they each favour the induction of states that are stable and non‐conducting but pharmacologically distinct. In concert GAT107 and NS6740 orchestrate a dynamic ballet of conformational transitions that can produce a unique mode of persistent channel activation, many orders of magnitude greater than could ever be produced by the endogenous agonists (ACh and choline) alone. Although, as single agents, GAT107 and NS6740 have disparate effects on channel activation and induce different distributions of stable non‐conducting states, they have similar analgesic activity, suggesting that convergent signal transduction may rely on induction of divergent stable non‐conducting states. Development of therapeutics targeting α7 nAChRs challenge us to think differently about this unique receptor and accept data that may blur the canonical distinction between ionotropic and metabotropic receptor functions.
Author contributions
R.L.P. and N.A.H. designed experiments. C.S. and K.M. performed electrophysiology experiments. A.R.K. and G.A.T. performed synthetic chemistry. C.S. and R.L.P. analysed electrophysiology experiments. R.L.P., N.A.H. and M.T. wrote the manuscript. M.I.D., D.B., C.S. and G.A.T. read, reviewed and offered revisions to the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by an NIH grant (GM57481) and an award from United States‐Israel Binational Science Foundation 2013055. tkP3BzPB was generously provided by Dr Peter Crooks.
Papke, R. L. , Stokes, C. , Damaj, M. I. , Thakur, G. A. , Manther, K. , Treinin, M. , Bagdas, D. , Kulkarni, A. R. , and Horenstein, N. A. (2018) Persistent activation of α7 nicotinic ACh receptors associated with stable induction of different desensitized states. British Journal of Pharmacology, 175: 1838–1854. doi: 10.1111/bph.13851.
References
- Alexander SP, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX (1997). Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci 9: 2734–2742. [DOI] [PubMed] [Google Scholar]
- Andersen ND, Nielsen BE, Corradi J, Tolosa MF, Feuerbach D, Arias HR et al. (2016). Exploring the positive allosteric modulation of human alpha7 nicotinic receptors from a single‐channel perspective. Neuropharmacology 107: 189–200. [DOI] [PubMed] [Google Scholar]
- Bagdas DA, Wilkerson JL, Kulkarni A, Toma W, AlSharari S, Gul Z et al. (2016). The alpha7 nicotinic receptor dual allosteric agonist and positive allosteric modulator GAT107 reverses nociception in mouse models of inflammatory and neuropathic pain. Br J Pharmacol 173: 2506–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand S, Devillers‐Thiery A, Palma E, Buisson B, Edelstein SJ, Corringer PJ et al. (1997). Paradoxical allosteric effects of competitive inhibitors on neuronal alpha7 nicotinic receptor mutants. Neuroreport 8: 3591–3596. [DOI] [PubMed] [Google Scholar]
- Briggs CA, Gronlien JH, Curzon P, Timmermann DB, Ween H, Thorin‐Hagene K et al. (2009). Role of channel activation in cognitive enhancement mediated by alpha7 nicotinic acetylcholine receptors. Br J Pharmacol 158: 1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnacka K, Papke RL, Horenstein NA (2013). Synthesis and evaluation of a conditionally‐silent agonist for the alpha7 nicotinic acetylcholine receptor. Bioorg Med Chem Lett 23: 4145–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RB, Lamppu D, Libertine L, McDonough A, Kumar A, LaRosa G et al. (2014). Discovery of novel 2‐((Pyridin‐3‐yloxy)methyl)piperazines as alpha7 nicotinic acetylcholine receptor modulators for the treatment of inflammatory disorders. J Med Chem 57: 3966–3983. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea J, Buendia I, Parada E, Navarro E, Leon R, Lopez MG (2015). Anti‐inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol 97: 463–472. [DOI] [PubMed] [Google Scholar]
- Francis MM, Choi KI, Horenstein BA, Papke RL (1998). Sensitivity to voltage‐independent inhibition determined by pore‐lining region of ACh receptor. Biophys J 74: 2306–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier CJ, Strowbridge BW, Papke RL (2003). Nicotinic acetylcholine receptors on local circuit neurons in the dentate gyrus: a potential role in the regulation of granule cell excitability. J Neurophysiol 89: 3018–3028. [DOI] [PubMed] [Google Scholar]
- Gay EA, Giniatullin R, Skorinkin A, Yakel JL (2008). Aromatic residues at position 55 of rat alpha7 nicotinic acetylcholine receptors are critical for maintaining rapid desensitization. J Physiol 586: 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronlien JH, Haakerud M, Ween H, Thorin‐Hagene K, Briggs CA, Gopalakrishnan M et al. (2007). Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol 72: 715–724. [DOI] [PubMed] [Google Scholar]
- Halevi S, Yassin L, Eshel M, Sala F, Sala S, Criado M et al. (2003). Conservation within the RIC‐3 gene family. Effectors of mammalian nicotinic acetylcholine receptor expression. J Biol Chem 278: 34411–34417. [DOI] [PubMed] [Google Scholar]
- Horenstein NA, Leonik FM, Papke RL (2008). Multiple pharmacophores for the selective activation of nicotinic alpha7‐type acetylcholine receptors. Mol Pharmacol 74: 1496–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horenstein NA, Papke RL, Kulkarni AR, Chaturbhuj GU, Stokes C, Manther K et al. (2016). Critical molecular determinants of alpha7 nicotinic acetylcholine receptor allosteric activation: separation of direct allosteric activation and positive allosteric modulation. J Biol Chem 291: 5049–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, Ulloa L (2007). The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151: 915–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, van der Zanden EP, The FO , Bijlsma MF, van Westerloo DJ, Bennink RJ et al. (2005). Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2‐STAT3 signaling pathway. Nat Immunol 6: 844–851. [DOI] [PubMed] [Google Scholar]
- Kalappa BI, Uteshev VV (2013). The dual effect of PNU‐120596 on alpha7 nicotinic acetylcholine receptor channels. Eur J Pharmacol 718: 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JR, Nordman JC, Bridges SP, Lin MK, Kabbani N (2015). Identification and characterization of a G protein‐binding cluster in alpha7 nicotinic acetylcholine receptors. J Biol Chem 290: 20060–20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koukouli F, Maskos U (2015). The multiple roles of the alpha7 nicotinic acetylcholine receptor in modulating glutamatergic systems in the normal and diseased nervous system. Biochem Pharmacol 97: 378–387. [DOI] [PubMed] [Google Scholar]
- Kulkarni AR, Thakur GA (2013). Microwave‐assisted expeditious and efficient synthesis of cyclopentene ring‐fused tetrahydroquinoline derivatives using three‐component Povarov reaction. Tetrahedron Lett 54: 6592–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde‐Robert V, Beaudry F, Vachon P (2012). Pharmacologic parameters of MS222 and physiologic changes in frogs (Xenopus laevis) after immersion at anesthetic doses. J Am Assoc Lab Anim Sci 51: 464–468. [PMC free article] [PubMed] [Google Scholar]
- Lape R, Colquhoun D, Sivilotti LG (2008). On the nature of partial agonism in the nicotinic receptor superfamily. Nature 454: 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Hernandez GY, Thinschmidt JS, Zheng G, Zhang Z, Crooks PA, Dwoskin LP et al. (2009). Selective inhibition of acetylcholine‐evoked responses of alpha7 neuronal nicotinic acetylcholine receptors by novel tris‐ and tetrakis‐azaaromatic quaternary ammonium antagonists. Mol Pharmacol 76: 652–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Papke JKP (2002). Comparative pharmacology of rat and human alpha7 nAChR conducted with net charge analysis. Br J Pharmacol 137: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Stokes C (2010). Working with OpusXpress: methods for high volume oocyte experiments. Methods 51: 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Kem WR, Soti F, López‐Hernández GY, Horenstein NA (2009). Activation and desensitization of nicotinic alpha7‐type acetylcholine receptors by benzylidene anabaseines and nicotine. J Pharmacol Exp Ther 329: 791–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Stokes C, Williams DK, Wang J, Horenstein NA (2011). Cysteine accessibility analysis of the human alpha7 nicotinic acetylcholine receptor ligand‐binding domain identifies L119 as a gatekeeper. Neuropharmacology 60: 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Horenstein NA, Kulkarni AR, Stokes C, Corrie LW, Maeng CY et al. (2014a). The activity of GAT107, an allosteric activator and positive modulator of alpha7 nicotinic acetylcholine receptors (nAChR), is regulated by aromatic amino acids that span the subunit interface. J Biol Chem 289: 4515–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Chojnacka K, Horenstein NA (2014b). The minimal pharmacophore for silent agonism of alpha7 nAChR. J Pharmacol Exp Therapeut 350: 665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Bagdas D, Kulkarni AR, Gould T, AlSharari S, Thakur GA et al. (2015). The analgesic‐like properties of the alpha7 nAChR silent agonist NS6740 is associated with nonconducting conformations of the receptor. Neuropharmacology 91: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo J, Brucker W, Hawrot E (2009). Proteomic analysis of an alpha7 nicotinic acetylcholine receptor interactome. J Proteome Res 8: 1849–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Ferris RL, Matthews T, Hiel H, Lopez‐Albaitero A, Lustig LR (2004). Characterization of the human nicotinic acetylcholine receptor subunit alpha (alpha) 9 (CHRNA9) and alpha (alpha) 10 (CHRNA10) in lymphocytes. Life Sci 76: 263–280. [DOI] [PubMed] [Google Scholar]
- Peng C, Kimbrell MR, Tian C, Pack TF, Crooks PA, Fifer EK et al. (2013). Multiple modes of alpha7 nAChR non‐competitive antagonism of control agonist‐evoked and allosterically enhanced currents. Mol Pharmacol 84: 459–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters D, Olsen G, Nielsen E, Jorgensen T, Ahring P (2004). Preparation of diazabicyclic aryl derivatives as cholinergic ligands at the nicotinic acetylcholine receptors. Neurosearch A/S.
- Quadri M, Papke RL, Horenstein NA (2016). Dissection of N,N‐diethyl‐N'‐phenylpiperazines as alpha7 nicotinic receptor silent agonists. Bioorg Med Chem 24: 286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlochansingh C, Branoner F, Chagnaud BP, Straka H (2014). Efficacy of tricaine methanesulfonate (MS‐222) as an anesthetic agent for blocking sensory‐motor responses in Xenopus laevis tadpoles. PLoS One 9: e101606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JX, Yakel JL (2012). Functional alpha7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J Mol Neurosci 48: 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitzia F, Brown JT, Randall AD, Dunlop J (2011). Voltage‐ and temperature‐dependent allosteric modulation of alpha7 nicotinic receptors by PNU120596. Front Pharmacol 2: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skok MV (2009). To channel or not to channel? Functioning of nicotinic acetylcholine receptors in leukocytes. J Leukoc Biol 86: 1–3. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur GA, Kulkarni AR, Deschamps JR, Papke RL (2013). Expeditious synthesis, enantiomeric resolution and enantiomer functional characterization of 4‐(4‐bromophenyl)‐3a,4,5,9b‐tetrahydro‐3H‐cyclopenta[c]quinoline‐8‐sulfonamide (4BP‐TQS) an allosteric agonist – positive allosteric modulator of alpha7 nAChR. J Med Chem 56: 8943–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villiger Y, Szanto I, Jaconi S, Blanchet C, Buisson B, Krause KH et al. (2002). Expression of an alpha7 duplicate nicotinic acetylcholine receptor‐related protein in human leukocytes. J Neuroimmunol 126: 86–98. [DOI] [PubMed] [Google Scholar]
- Wang J, Horenstein NA, Stokes C, Papke RL (2010). Tethered agonist analogs as site‐specific probes for domains of the human alpha7 nicotinic acetylcholine receptor that differentially regulate activation and desensitization. Mol Pharmacol 78: 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Papke RL, Stokes C, Horenstein NA (2012). Potential state‐selective hydrogen bond formation can modulate the activation and desensitization of the alpha7 nicotinic acetylcholine receptor. J Biol Chem 287: 21957–21969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DK, Stokes C, Horenstein NA, Papke RL (2009). Differential regulation of receptor activation and agonist selectivity by highly conserved tryptophans in the nicotinic acetylcholine receptor binding site. J Pharmacol Exp Ther 330: 40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DK, Stokes C, Horenstein NA, Papke RL (2011a). The effective opening of nicotinic acetylcholine receptors with single agonist binding sites. J Gen Physiol 137: 369–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DK, Wang J, Papke RL (2011b). Investigation of the molecular mechanism of the alpha7 nAChR positive allosteric modulator PNU‐120596 provides evidence for two distinct desensitized states. Mol Pharmacol 80: 1013–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DK, Peng C, Kimbrell MR, Papke RL (2012). The intrinsically low open probability of alpha7 nAChR can be overcome by positive allosteric modulation and serum factors leading to the generation of excitotoxic currents at physiological temperatures. Mol Pharmacol 82: 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GT, Zwart R, Walker AS, Sher E, Millar NS (2008). Potentiation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci U S A 105: 14686–14691. [DOI] [PMC free article] [PubMed] [Google Scholar]