

Abstract
Due to the high-level of metastatic and relapsed rates, rhabdomyosarcoma (RD) patients have a poor prognosis, and novel treatment strategies are required. Thereby, the present study evaluated the efficacy of PFK15, a PFKFB3 inhibitor, in RD cells to explore its potential underlying mechanism on the regulation of autophagy and proliferation in these cells. The effects of PFK15 on cell viability loss and cell death in different treatment groups, were evaluated by MTS assay, colony growth assay and immunoblotting, respectively. In addition, the autophagy levels were detected by electron microscopy, fluorescence microscopy and immunoblotting following PFK15 treatment, and the autophagic flux was analyzed with the addition of chloroquine diphosphate salt or by monitoring the level of p62. PFK15 was observed to evidently decrease the viability of RD cells, inhibit the colony growth and cause abnormal nuclear morphology. Furthermore, PFK15 inhibited the autophagic flux and cell proliferation, as well as induced apoptotic cell death in RD cells through downregulation of the adenosine monophosphate-activated protein kinase (AMPK) signaling pathway. An AMPK agonist rescued the inhibited cell proliferation and autophagy induced by PFK15. In conclusion, PFK15 inhibits autophagy and cell proliferation via downregulating the AMPK signaling pathway in RD cells.
Keywords: rhabdomyosarcoma, PFK15, autophagy, proliferation, adenosine monophosphate-activated protein kinase
Introduction
Rhabdomyosarcoma (RD) is a type of soft tissue sarcoma that commonly occurs in children and adolescents (1,2). RD is originated from mesenchymal progenitors of myogenic lineages (3) and is characterized by undifferentiated myoblast-like cells (4). Although various therapeutic strategies have been applied, including intense chemotherapy, irradiation and surgery, patients with RD exhibit poor prognosis (5). Thus, effective drugs with minimal toxicity are urgently needed for RD treatment.
Energy consumption from metabolic activities of cancer cells depends on glycolysis, even under oxygen-rich conditions. Phosphofructokinase-1 (PFK-1) serves an important role in glycolysis, which is activated by fructose 2,6-bisphosphate (F2,6BP) (6). F2,6BP has been reported to increase the glucose uptake and allosterically activate PFK-1 (7,8). In cancer cells, F2,6BP has been observed to be significantly increased compared with that in normal tissue cells, indicating that upregulated F2,6BP may contribute to cell proliferation in cancer tissues (9,10).
PFKFB, also known as 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase, has been regarded as a key glycolysis regulator in modulating F2,6BP, lactate production and glucose uptake (11). PFKFB3, a member of the PFKFB family, has a significantly high kinase activity, enhances the F2,6BP level and glycolysis effect (12). The protein levels of PFKFB3 are commonly elevated in various types of human cancer, including breast (13), gastric (14) and bladder cancer (15). In addition, previous studies have demonstrated that the inhibition of PFKFB3 was able to suppress the glucose metabolism and cell proliferation in cancer cells (16,17). Therefore, strategies of downregulating PFKFB3 activity may be a promising target for anticancer therapy.
Targeting PFKFB3 suppression inhibited head and neck squamous cell carcinoma growth and metastasis (18). In addition, the selective antagonist of PFKFB3 suppressed the proliferation of the transplanted tumor in several cells (19,20). PFK15, also known as 1-(4-pyridinyl)-3-(2-quinolinyl)-2-propen-1-one, is a small molecular PFKFB3 inhibitor that has displayed a powerful activity against PFKFB3 and anti-tumor effects (21). However, the functions of PFK15 and the adenosine monophosphate-activated protein kinase (AMPK) signaling pathway in RD cells remain largely unknown.
In the present study, the effects of PFK15 on the viability, apoptosis, autophagy and proliferation of RD cells were investigated. Furthermore, the study explored the possible roles of the AMPK signaling pathway during these processes.
Materials and methods
Reagents and chemicals
PFK15 (cat. no. SML1009), 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO; cat. no. 407325), 3-bromopyruvate (3BP), 3-methyladenine (3MA), chloroquine diphosphate salt (CQ; cat. no. C6628) and a polyclonal antibody against light chain 3 (LC3; cat. no. L7543) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). An antibody against t-ERK1/2 (cat.no. 9102), antibody against poly (ADP-ribose) polymerase 1 (PARP-1; cat. no. 9542), antibody against pAMPK (cat. no. 2535; Thr 172), antibody against mTOR (cat. no. 2971; Ser2448), antibody against P-AKT (cat.no. 9721; Ser 473), antibody against t-Akt (cat. no. T308) and an antibody against phosphorylated acetyl-CoA carboxylase [pACC (cat. no. 3661; Ser79)] were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). An antibody against p62 (cat. no. sc-28359) was obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA), while an antibody against β-actin (cat. no. TA-09) was purchased from OriGene Technologies, Inc. (Beijing, China). MTS [also known as 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; cat. no. G1111] was acquired from Promega Corporation (Madison, WI, USA). The Alexa Fluor 594 goat anti-rabbit IgG (H+L) antibody (cat. no. R37117), fetal bovine serum (FBS; Gibco) and enhanced chemiluminescence reagent (Pierce) were obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR; Sigma-Aldrich; Merck KGaA; cat. no. A9978), an activator of AMPK. DAPI (VECTASHIELD® with DAPI; cat. no. H-1200) was purchased from Fushen Biotechnology Co., Ltd. (Shanghai, China). 3PO was provided by Dr. Y.Q. Huo (Georgia Regents University, Augusta, GA, USA). The siRNA specific for Autophagy Related 5 (Atg5; cat. no. sc-41445), Autophagy Related 7 (Atg7; cat. no. sc-41447) was purchased from Santa Cruz Biotechnology, Inc. together with the control (mock) siRNA (cat. no. sc-37007).
Cell culture and treatments
Human RD (ATCC® CCL-136™) cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Cell cultures were maintained in Dulbecco's modified Eagle's medium (DMEM) (4.5 g/l glucose,) containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.) and 1% Penicillin-Streptomycin (100 IU/ml) and incubated in an atmosphere with 95% humidified air and 5% CO2 at 37°C. Prior to treatment, the cells were grown to 70–80% confluence. Subsequently, PFK15 was diluted in dimethyl sulfoxide (DMSO) at different concentrations (2, 4 and 6 mM) and then added to the cells in growth medium. Cells in the control group were only treated with DMSO. The concentration of DMSO in DMEM was 1:2,000. 3PO with concentration of 10 μM was also used for comparison.
Immunoblotting
Cells were homogenized with lysis buffer containing Triton X-100/glycerol buffer (1% Triton X-100, 10% glycerol), 50 mM Tris-HCl (pH 7.4), 1 mM dithiothreitol, 2 mM EGTA and 4 mM EDTA, supplemented with 1% SDS, and 1% protease inhibitor cocktail (cat. no. 04693159001; Roche Diagnostics, Basel, Switzerland). Next, the cells were subjected to 13, 10 or 8% SDS-PAGE and transferred onto a polyvinylidene difluoride membrane. The membrane was blocked in 5% milk for 1 h at the room temperature. The appropriate primary antibodies (1:1,000) was incubated for 12 h at 4°C and horseradish peroxidase-conjugated suitable secondary antibodies (1:5,000) were used for immunoblotting. The secondary antibodies were incubated for 1 h at room temperature. Subsequently, the blots were detected by enhanced chemiluminescence. Several X-ray films were analyzed to verify the linear range of the chemiluminescence signals, and the densitometry was performed using ImageJ software version 1.46 (National Institutes of Health, Bethesda, MD, USA).
Cell viability
For viability analysis, cells in the PFK15-treated and control groups were seeded into a 96-well plate at a density of 5,000–10,000 cells per well and allowed to adhere by culturing at 37°C overnight. For dose-dependent experiments, PFK15 was added in increasing concentrations (2, 4 and 6 μM) and incubated for 24 h. For time-dependent experiments, PFK15 (6 μM) or 3BP (25 μM) was added and incubated for 12, 24 or 48 h. For synergistic effect analysis, PFK15 (4 μM) combined with CQ (10 μM) or 3MA (1 mM) were added to the cells and incubated for 48 h. Subsequent to these different treatments, 0.8 mg/ml MTS was added to the samples and incubated for 3 h. The absorbance of samples were then read at 492nm using a microtiter plate spectrophotometer (BioTek Instruments, Inc., Winooski, VT, USA).
Colony growth assay
For colony growth assay, cells were seeded in a 6 cm dish at a concentration of 300 cells/ml, PFK15-treated and cultured for 2 weeks. To examine the association of AMPK activity with autophagy and cell death caused by PFK15 in RD cells, the AMPK agonist AICAR (100 μM) was used in subsequent experiments. Next, cells were fixed in paraformaldehyde (4%) and stained by trypan blue. The numbers of colonies in each sample were counted by ImageJ software version 1.46 (National Institutes of Health, Bethesda, MD, USA).
Fluorescence microscopy
Cells were fixed with paraformaldehyde (4%) for 12 min at room temperature and then examined by fluorescence microscopy. Briefly, cells were blocked by PBS containing 3% bovine serum albumin and 0.2% Triton X-100 for 1 h. Following washing three times with PBS, the cells were incubated with the LC3 antibody (1:200) for 2 h at room temperature, washed with PBS for three times, and then incubated with Alexa Fluor 594 goat anti-rabbit IgG (H+L) antibody for 2 h at room temperature. The nuclei were subsequently counterstained with DAPI/Antifade and observed using a fluorescence microscope system. The number of the punctate LC3 structures in each cell was counted, and at least 30 cells were included for each group in the immunofluorescent assay.
Transmission electron microscopy
Electron microscopy was conducted as described previously (22). Briefly, cells were initially treated with DMSO (control group) or PFK15 6 μM (treatment group). Samples were then washed, trypsinized and collected by centrifugation at 1,500 × g, at 4°C for 5 min. Next, cells were fixed with 4% paraformaldehyde overnight and post-fixed with 1% OsO4 in cacodylate buffer at room temperature for 1 h, followed by stepwise dehydration with ethanol. The dehydrated pellets were subsequently washed with propylene oxide for 30 min at room temperature and embedded in Spurr resin for sectioning. A transmission electron microscope (JEM1230; JEOL, Ltd., Tokyo, Japan) was used for observation of the thin sections. Morphometric analysis of the area fractions between autophagosomes and whole cells were calculated using ImageJ software, and the data were presented in a histogram graph.
Lactate assay
Cells were seeded in 6-well plates, after PFK15-treated, the suspension was collected and subjected to lactate assay. Levels of the secreted lactate were detected using the lactate assay kit (K-DATE, Megazyme, Bray, Co. Wicklow, Ireland). Data represent three independent experiments.
Statistical analysis
GraphPad Prism version 5 software (GraphPad Software, Inc., La Jolla, CA, USA) was used for statistical analyses. All data are expressed as the mean ± standard deviation. Student's t-test was used to evaluate the statistical significant differences between two groups. Multigroup comparisons of the means were conducted by one-way analysis of variance with a post hoc Student-Newman-Keuls test. A statistically significant difference was regarded when the P-value was <0.05. Experiments were repeated for a minimum of three times for each condition.
Results
PFK15 suppresses the viability of RD cells
As a potent small molecule antagonist of PFKFB3, PFK15 has been verified to inhibit the proliferation and viability of various cancer cells at low concentrations (14,21). In the present study, it was observed that PFK15 significantly suppressed the glycolysis in RD cells by monitoring the final product lactate, and the inhibitory effect of PFK15 was dose-dependent. Treatment with 4 and 6 μM PFK15 significantly attenuated the lactate production by 20 and 27%, respectively, as compared with the control group (4 μM vs. control: Mean difference (MD)=−0.2, q=6.928, P<0.05; 6 μM vs. control: MD=−0.3, q=10.39, P<0.01; Fig. 1A). Thus, the concentration of PFK15 was selected at 6 μM for the following experiments.
Figure 1.

Embryonal RD cells are sensitive to PFK15. (A) RD cells were treated with different PFK15 concentrations (0–6 μM) for 24 h, and the supernatant lactate level was analyzed by a lactate assay. The viability of RD cells treated with (B) 6 μM PFK15 and (C) 25 μM 3BP for 12, 24 or 48 h was analyzed by an MTS assay. (D) RD cells were treated with PFK15 (6 μM) combined with CQ (10 μM) or 3MA (1 mM) for 48 h, and the cell viability was analyzed by an MTS assay. *P<0.05 and **P<0.01 vs. corresponding Ctrl group. RD, rhabdomyosarcoma; 3BP, 3-bromopyruvate; 3MA, 3-methyladenine; CQ, chloroquine diphosphate salt; Ctrl, control.
The cell viability was then detected by an MTS assay in RD cells following PFK15 treatment for different time periods. At the 12-h time point, PFK15 evidently decreased the viability of RD cells compared with the control group (P<0.01; Fig. 1B). With longer treatment time, the survival rate of RD cells decreased from ~31% at 24 h to 19% at 48 h compared with that in the control group (P<0.01; Fig. 1B).
3BP is an inhibitor of key enzyme hexokinase 2 in glycolysis and is also an inhibitor of phosphatidylinositol 3-kinase, which is widely used as an inhibitor of autophagy (23). 3BP inhibits the activity of PI3K, which is a known negative regulator of autophagy. Theoretically 3BP could promotes autophagy, however, our un-shown data indicated that 3BP inhibited autophagy. In addition, CQ is a known inhibitor of autolysosome degradation that is widely used in monitoring the autophagic flux. In the current study, after 12 h of treatment with 3BP (25 μM), the cell viability was significantly reduced as compared with the control group (Fig. 1C). Furthermore, when treated with PFK15 and 3MA (1 mM) but not CQ, the cell viability was further decreased as a result of a synergistic effect (Fig. 1D). One single drug treatment can induce various types of cell death, thus PFK15 may cause apoptotic and autophagic cell death, as well as other types of cell death. However, as the autophagy initiation inhibitor 3MA further increased the cell viability loss, it is suggested that autophagy may serve a protective role upon PFK15 treatment. As inhibition of autophagy exacerbated the cell viability loss upon PFK15 treatment, it was hypothesized that autophagy may in part ameliorate the PFK15-induced cell viability loss. Furthermore, although widely used as an inhibitor of autophagic flux, CQ functions during the autolysosome fusion and degradation processes, which may be reason for the distinct phenotype between CQ and 3MA on PFK15-induced cell viability loss in Fig. 1D. These results indicated that PFK15 was able to decrease RD cell autophagy.
PFK15 inhibits clone formation and causes abnormal nuclear morphology
A colony formation assay was performed to further analyze the effect of PFK15 on the cell proliferation. As a result, the number of clones were significantly reduced in the 2 and 6 μM PFK15 treatment groups as compared with the controls (6 μM vs. control: MD=−13, q=22.52, P<0.01; 2 μM vs. control: MD=−9, q=15.59, P<0.01; Fig. 2A). Furthermore, the nuclear component was distinct from the primary nuclei in RD cells treated with 6 μM PFK15 for 2 h, as observed by immunofluorescence microscopy (Fig. 2B) and transmission electron microscopy (Fig. 2C). Vehicle control cells exhibited a wavy plasma membrane edge, which is a characteristic of normal morphology. By contrast, the integrity of the nuclear membrane of PFK15-treated cells disappeared, while protuberance was detected in the nuclear membrane. The nucleus condensed and the chromosomes gathered to assemble chromatin. Gradually, the protuberance was separated from nucleus and formed micronuclei, which subsisted in the cytoplasm. Compared with the control group, an evident accumulation of micronuclei was observed in PFK15-treated RD cells (P<0.01; Fig. 2B). Similar results were observed by transmission electron microscopy (Fig. 2C). The cells were spindle-shaped and well-attached to the culture plate in the control group, while the PFK15-treated cells were loosely attached, rounded and randomly oriented. Following PFK15 treatment for 24 h, the cells exhibited reduction in size, karyorrhexis and even death (Fig. 2D).
Figure 2.

PFK15 induces an abnormal nuclear morphology. (A) Colony growth assays were performed in RD cells treated with PFK15 (2 and 6 μM). Representative microphotographs of RD cells in adherent conditions after 24 h of treatment in the absence or presence of PFK15 are shown (bar=100 μm). (B) RD cells were cultured for 12 h with PFK15 (6 μM), and immunofluorescence analysis using DAPI was performed to stain nuclei (top panels, magnification ×400; bottom panels, magnification ×1,000). (C) Transmission electron microscopy images of nuclear structures in RD cells are displayed (arrows indicate the micronuclei). (D) RD cells were treated with PFK15 (6 μM) for 24 h, and the morphology of cells were observed by bright field microscopy (magnification, ×40). *P<0.05 vs. control and **P<0.01 vs. control. RD, rhabdomyosarcoma; Ctrl, control.
PFK15 inhibits autophagy in RD cells
Transmission electron microscopy, considered as one of the most convincing approaches to detect autophagosome formation (24,25), was used in the current study to determine the effect of PFK15 on autophagy. Compared with the DMSO control, PFK15 markedly attenuated the accumulation of membrane vacuoles (Fig. 3A). When autophagy occurs, LC3-I can be converted into its phosphatidylethanolamine-conjugated form, LC3-II, which is associated with autophagosomes. As observed in the immunoblotting assay, PFK15 increased the level of LC3-II and p62 after 2 h of treatment in RD cells (Fig. 3B). After 12 h of treatment, PFK15 further increased the LC3-II level, whereas it decreased the p62 level (Fig. 3C). However, combined incubation with CQ, a known inhibitor of autolysosome degradation that is widely used in monitoring autophagic flux, failed to further increase the level of LC3-II and block p62 degradation in the PFK15-treated cells, suggesting that PFK15 inhibited the autophagic flux in RD cells. An immunofluorescence assay was also performed to monitor the aggregation of LC3 in RD cells. Consistently, the punctate of LC3 was significantly decreased when cells were treated with PFK15 (Fig. 3D).
Figure 3.

PFK15 suppresses autophagy in RD cells. (A) Transmission electron microscopy examination of vacuolar structures in RD cells (arrows represent the autophagosome). Morphometric analysis of the area fractions between autophagosomes and cytoplasm were calculated using ImageJ software; area ratio data were not normally distributed and are presented as the mean of at least 20 cells counted for each group. (B) RD cells treated for 2 h or (C) 12 h with PFK15 (6 μM) in the presence or absence of CQ (10 μM) were examined by an immunoblotting assay. Densitometry was performed for quantification, and the adjusted ratios of LC3-II and p62 to actin are presented under the blots. (D) RD cells were cultured for 2 h with PFK15 (6 μM), and an immunofluorescence assay using LC3 antibody was performed, using DAPI as a nuclear stain. Data represent three independent experiments. *P<0.05 vs. control and **P<0.01 vs. control. RD, rhabdomyosarcoma; CQ, chloroquine diphosphate salt; LC3, light chain 3; Ctrl, control.
PFK15 induces the cleavage of PARP-1
PARP-1 has been reported to be involved in several cellular processes, including cell autophagy and apoptosis, and the cleavage of PARP-1 has been used as a marker of caspase-dependent apoptosis (26). In the current study, immunoblotting analysis was performed to evaluate the effect of PFK15 on PARP-1, and the results revealed that PFK15 induced the cleavage of PARP-1 in a dose-dependent manner (Fig. 4A and B), indicating that PFK15 activated the apoptotic pathway. However, addition of CQ did not significantly induce cleavage of PARP-1 in RD cells (Fig. 4B). The autophagy-associated proteins Atg5 and Atg7 serve important roles in the maturation of autophagosomes by promoting the elongation of phagophores (27). In order to further analyze the association between cell autophagy and apoptosis caused by PFK15, an autophagy inhibition model was constructed by silencing the Atg5 and Atg7 genes. The results demonstrated that the cleavage of PARP-1 was decreased when Atg5 and Atg7 were knocked down, indicating that the cell apoptosis caused by PFK15 was suppressed by the inhibition of earlier autophagy (Fig. 4C).
Figure 4.
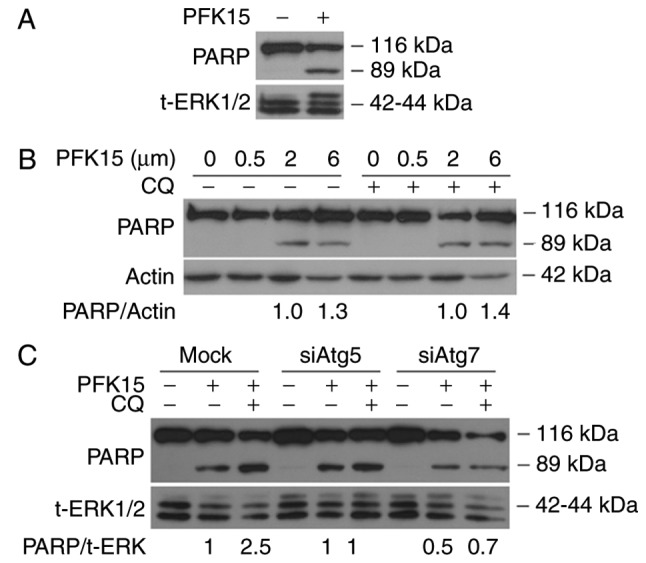
PFK15 induces the cleavage of PARP-1. (A) Western blot analysis of RD cells, demonstrating that PFK15 promotes apoptosis and induces cleavage of PARP-1. RD cells were cultured for 12 h with or without 6 μM PFK15, and then collected for immunoblotting analysis. (B) RD cells were cultured for 12 h with different concentrations of PFK15 (0–6 μM) in the presence or absence of CQ (10 μM) and then collected for immunoblotting analysis. (C) Effects of Atg5 and Atg7 knockdown on the cleavage of PARP-1 in PFK15-treated RD cells. RD, rhabdomyosarcoma; CQ, chloroquine diphosphate salt; PARP-1, poly (ADP-ribose) polymerase 1.
PFK15 downregulates the activity of AMPK, pAKT and mammalian target of rapamycin complex 1 (mTORC1)
mTORC1 is a protein complexes, in which mTOR is the core component. The p-mTOR (S2448) could reflect the activity of mTORC1. mTORC1 is usually known as a negative regulator of autophagy. AMPK, a sensor for intracellular adenosine nucleotides, responds to alterations in energy status, and coordinates the cell growth, autophagy and metabolism (28). Since it is generally phosphorylation of PFKFB3 on Ser461, the present study attempted to clarify whether AMPK serves a regulatory role in the PFK15-mediated effects in RD cells (29). The results demonstrated that, after 2-h treatment, PFK15 suppressed the level of phosphorylated AMPK (pAMPK) and its substrate pACC by 44 and 20%, respectively. Similar trends were also identified at the 4 and 12 h time points (Fig. 5A). At the 12 h time point, PFK15 had a significant inhibitory effect on pAMPK and pACC, reducing their levels by 88 and 61%, respectively. Similar results were also obtained upon treatment with 3PO, another inhibitor of PFKFB3. Inhibition of PFKFB3 using 3PO was successful in reducing cancer growth and increasing apoptosis, but only at certain time points within the circadian cycle (30). Considering that the decreased glycolysis caused by PFK15 failed to activated the AMPK activity in a feedback loop, whereas it decreased the levels of pAMPK and pACC, it is suggested that PFKFB3 may function upstream of AMPK under certain circumstance. The activity of AKT, which is another negative regulator of autophagy, was also investigated in the present study. It was observed that PFK15 evidently upregulated the pAKT (Ser473) level, which inhibits autophagy by activating mTORC1 (Fig. 5B). As glycolysis is closely associated with the energy sensor AMPK, the present study only focused on the AMPK related data; other data will be reported in future studies.
Figure 5.

PFK15 regulates AMPK proteins expression. (A) RD cells were treated for 2, 4 and 12 h with 6 μM PFK15, and western blot analysis was conducted to examine the levels of t-AMPK, p-AMPK and p-ACC proteins. Total ERK was used as a loading control. (B) Immunoblotting assay was performed to determine the p-mTOR, p-AKT and t-AKT levels in cells treated with PFK15 (6 μM). RD, rhabdomyosarcoma; AMPK, adenosine monophosphate-activated protein kinase; AKT, protein kinase B; ACC, acetyl-CoA carboxylase; ERK, extracellular signal-regulated kinase; mTOR, mammalian target of rapamycin; p-, phosphorylated; t-, total; Ctrl, control.
AICAR rescues the downregulation of PFK15 on autophagy and activity of AMPK
It was observed that, compared with PFK15 treatment alone, the combination of AICAR and PFK15 decreased LC3-II expression with CQ treatment and decreased p62 degradation at the 2 and 12 h time points. Furthermore, CQ addition further increased the LC3-II level, as well as blocked the p62 degradation, indicating a complete autophagic flux (Fig. 6A). Thus, it is likely that AICAR rescued the PFK15-dependent inhibition of autophagic flux in RD cells. Subsequently, the AMPK activity was detected following the indicated treatment in RD cells. Addition of AICAR inhibited the downregulation of AMPK phosphorylation at the 2 and 12 h time points (Fig. 6B). This consistency of AMPK activity and autophagy supported that PFK15 inhibited autophagy by decreasing AMPK signaling. The current study also examined the effect of AICAR on PFK15-dependent cell death, and the results demonstrated that pre-treatment with AICAR provided protection against PFK15-induced cell death at the 24 and 48 h time points (Fig. 6C). These findings indicated that PFK15 inhibited the cell growth, while AICAR partly recovered the cell growth (PFK15 vs. DMSO: Mean diff.= −13, q=14.16, P<0.01; PFK15+AICAR vs. DMSO: Mean diff.= −6.667, q=7.263, P<0.01; Fig. 6D).
Figure 6.

AICAR attenuates PFK15-induced suppression of autophagy and cell viability loss, partly blocking the decrease of p-AMPK. (A) Western blots analyzing the expression levels of p62 and LC3 in RD cells treated with AICAR and PFK15 in the presence or absence of CQ (10 μM). (B) Effect of AICAR (0.5 mM) treatment for 2 or 12 h on AMPK activity in the absence and presence of PFK15 (6 μM), as reflected by the phosphorylation of AMPK. (C) RD cells were treated with PFK15 and AICAR for 24 and 48 h, and cell viability was analyzed by an MTS assay. (D) Colony growth assays were performed in RD cells treated with PFK15 (6 μM) in the absence and presence of AICAR. RD, rhabdomyosarcoma; AMPK, adenosine monophosphate-activated protein kinase; *P<0.05 vs. control and **P<0.01 vs. control. AICAR, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside; CQ, chloroquine diphosphate salt; LC3, light chain 3; p-, phosphorylated; t-, total; Ctrl, control.
Discussion
Currently, the deregulation of cellular energetics by anti-glycolysis is a novel strategy in the treatment of cancer cells, which has attracted much attention (31). As an antagonist of PFKFB3, PFK15 has been suggested to suppress the tumor cell viability by inhibiting the glycolysis (21). However, the underlying pharmacological mechanism is not yet clearly understood. In the present study, the appearance of cleaved PARP-1 and micronucleus protuberance was different in PFK15-treated cells compared with control group, indicating that PFK15 inhibited RD cell viability by inducing apoptosis.
Autophagy, the 'self-eating' system (32), is known as one of the most important systems in solid tumor. In cancer, autophagy-mediated recycling can be used to maintain energy homeostasis and mitochondrial function, for providing elevated metabolic demand of growth and proliferation (33). At the beginning of autophagy, the isolation membrane is developed to package the organelles and other materials in the cytoplasm to form autophagosomes. During this process, LC3 is cleaved into a soluble form known as LC3-I, which is then modified into a membrane-bound form, termed LC3-II (34). At the second step, the packaged cell organelles are digested by lysosomal enzymes, while LC3 and the other autophagosomes are digested. In the present study, treatment with PFK15 increased the LC3-II level in the RD cells. However, the autophagy inhibitor CQ did not further increase the PFK15-induced LC3-II level and failed to block the degradation of p62, suggesting that PFK15 inhibited the second step of autophagy, in which the accumulated LC3-II was not digested by lysosomal enzymes.
PARP-1 is a nuclear enzyme that catalyzes the transfer of ADP-ribose polymers onto itself and other proteins in response to DNA strand breaks (35). It is one of the main cleavage targets of caspase-3 in vivo (36), and cleavage of PARP-1 serves as a marker of cells undergoing apoptosis (37). During apoptosis, PARP-1 breaks into two fragments (89 and 24 kDa), which is a useful hallmark in cell death (38). In the present study, it was observed that PFK15 induced the cleavage of PARP-1 in RD cells, indicating that PFK15 activated the apoptotic pathway. There is a close connection between autophagy and apoptosis, since autophagy is able to promote, suppress and accompany apoptosis. The current study observed that inhibition of autophagy by silencing Atg5 and Atg7 attenuated the PFK15-induced caspase-dependent apoptosis, particularly by silencing Atg7. Along with the promoting effect of 3MA on PFK15-induced cell viability loss, these results indicated that PFK15 induced multi-type cell death other than caspase-dependent apoptosis. In addition, the distinct effect of CQ on PFK15-induced PARP-1 cleavage between siRNA knockdown mock group and PFK15 treated group (Fig. 4B) may be due to siRNA transfection altering the cell state via an uncertain mechanism. As demonstrated in the present study, there was crosstalk between autophagy and apoptosis. Xi et al (39) reported that, in RD cells, inhibition of autophagosomes at the stage of autophagosome and lysosome fusion promoted apoptosis. Notably, another previous study confirmed that induction of autophagy was a useful therapeutic approach for overcoming drug resistance to certain therapeutic agents, particularly those that typically induce an apoptotic response (40). Consistent with the results of the present study, PARP serves an important function in the crosstalk between autophagy and apoptosis. Furthermore, PFK15-induced apoptosis was suppressed by autophagy inhibition.
In addition, the present results indicated that PFK15-induced cell death was mediated by AMPK; however, AICAR was able to attenuate this effect. As an evolutionary conserved fuel-sensing enzyme, AMPK is activated in shortage of energy and suppressed by its surfeit (41). Previous studies have demonstrated that AMPK serves a dual role in cancer (42). Under some conditions, activation of AMPK signaling inhibited cancer cell growth and tumorigenesis (43,44). Convincing evidence has accumulated indicating that AMPK signaling is a conditional tumor suppressor pathway (45,46). Hadad et al (47) have also reported that reduced pAMPK and pACC signals were inversely correlated with the histological grade, as well as axillary node metastasis in breast cancer. However, in certain cancer cells, AMPK downregulation is beneficial to therapy, while with the administration of pharmacological activators of AMPK the antineoplastic effect disappears or is decreased (42,48). In the present study, it was observed that PFK15 suppressed the levels of pAMPK and pACC at different treated times, and upon treatment with AICAR, an agonist of AMPK, the RD cell activity and autophagic flux were partially recovered. Taken together, these findings suggested that PFK15 was able to inhibit autophagy and cell viability through the AMPK signaling pathway.
There are also certain limitations to the present study. Due to the limitation of experimental conditions, experiments involving an in vivo xenograft model were not performed In the future, PFK15-induced inhibition of autophagy and proliferation in an in vivo xenograft model will be researched.
In conclusion, the present study provided novel insights into the antitumor activity of PFK15 in RD cells. PFK15 inhibited autophagy and cell viability through AMPK signaling, and AMPK functioned downstream of PFKFB3. These findings may provide a theoretical basis for the use of PFKFB3 as a target for the clinical treatment of RD.
Acknowledgments
Not applicable.
Funding
This study was supported by a grant from the National Natural Science Foundation of China (grant no. H0605).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
CW and JQ designed the present study, SY performed the research, QG analyzed the data, and SH and DZ wrote the paper.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interests exist.
References
- 1.Huh WW, Skapek SX. Childhood rhabdomyosarcoma: New insight on biology and treatment. Curr Oncol Rep. 2010;12:402–410. doi: 10.1007/s11912-010-0130-3. [DOI] [PubMed] [Google Scholar]
- 2.Leaphart C, Rodeberg D. Pediatric surgical oncology: Management of rhabdomyosarcoma. Surg Oncol. 2007;16:173–185. doi: 10.1016/j.suronc.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Ther. 2002;1:97–104. doi: 10.4161/cbt.51. [DOI] [PubMed] [Google Scholar]
- 4.Keller C, Guttridge DC. Mechanisms of impaired differentiation in rhabdomyosarcoma. FEBS J. 2013;280:4323–4334. doi: 10.1111/febs.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malempati S, Hawkins DS. Rhabdomyosarcoma: Review of the Children's Oncology Group (COG) soft-tissue sarcoma committee experience and rationale for current COG studies. Pediatr Blood Cancer. 2012;59:5–10. doi: 10.1002/pbc.24118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Schaftingen E, Jett MF, Hue L, Hers HG. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA. 1981;78:3483–3486. doi: 10.1073/pnas.78.6.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, Makita Z, Mitchell R, Bucala R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002;62:5881–5887. [PubMed] [Google Scholar]
- 8.Van Schaftingen E, Hue L, Hers HG. Fructose 2,6-bisphosphate, the probably structure of the glucose- and glucagon-sensitive stimulator of phosphofructokinase. Biochem J. 1980;192:897–901. doi: 10.1042/bj1920897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miralpeix M, Azcon-Bieto J, Bartrons R, Argiles JM. The impairment of respiration by glycolysis in the Lewis lung carcinoma. Cancer Lett. 1990;50:173–178. doi: 10.1016/0304-3835(90)90261-U. [DOI] [PubMed] [Google Scholar]
- 10.Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, Yoshioka N, Bucala R, Koike T. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res. 2005;11:5784–5792. doi: 10.1158/1078-0432.CCR-05-0149. [DOI] [PubMed] [Google Scholar]
- 11.Ros S, Schulze A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013;1:8. doi: 10.1186/2049-3002-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatases in cancer. Exp Mol Pathol. 2009;86:174–179. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Ge X, Lyu P, Gu Y, Li L, Li J, Wang Y, Zhang L, Fu C, Cao Z. Sonic hedgehog stimulates glycolysis and proliferation of breast cancer cells: Modulation of PFKFB3 activation. Biochem Biophys Res Commun. 2015;464:862–868. doi: 10.1016/j.bbrc.2015.07.052. [DOI] [PubMed] [Google Scholar]
- 14.Zhu W, Ye L, Zhang J, Yu P, Wang H, Ye Z, Tian J. PFK15, a small molecule inhibitor of PFKFB3 induces cell cycle arrest, apoptosis and inhibits invasion in gastric cancer. PLoS One. 2016;11:e0163768. doi: 10.1371/journal.pone.0163768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu KY, Wang de G, Liu PF, Cao YW, Wang YH, Yang XC, Hu CX, Sun LJ, Niu HT. Targeting of MCT1 and PFKFB3 influences cell proliferation and apoptosis in bladder cancer by altering the tumor microenvironment. Oncol Rep. 2016;36:945–951. doi: 10.3892/or.2016.4884. [DOI] [PubMed] [Google Scholar]
- 16.O'Neal J, Clem A, Reynolds L, Dougherty S, Imbert-Fernandez Y, Telang S, Chesney J, Clem BF. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res Treat. 2016;160:29–40. doi: 10.1007/s10549-016-3968-8. [DOI] [PubMed] [Google Scholar]
- 17.Feng Y, Wu L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem Biophys Res Commun. 2017;483:897–903. doi: 10.1016/j.bbrc.2017.01.031. [DOI] [PubMed] [Google Scholar]
- 18.Li HM, Yang JG, Liu ZJ, Wang WM, Yu ZL, Ren JG, Chen G, Zhang W, Jia J. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. 2017;36:7. doi: 10.1186/s13046-016-0481-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calvo MN, Bartrons R, Castaño E, Perales J, Navarro-Sabaté A, Manzano A. PFKFB3 gene silencing decreases glycolysis, induces cell-cycle delay and inhibits anchorage-independent growth in HeLa cells. FEBS Lett. 2006;580:3308–3314. doi: 10.1016/j.febslet.2006.04.093. [DOI] [PubMed] [Google Scholar]
- 20.Lea MA, Guzman Y, Desbordes C. Inhibition of growth by combined treatment with inhibitors of lactate dehydrogenase and either phenformin or inhibitors of 6-phosphofructo-2-kinase/fructose-2,6-bis phosphatase 3. Anticancer Res. 2016;36:1479–1488. [PubMed] [Google Scholar]
- 21.Clem BF, O'Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA, II, Klarer AC, Redman R, Miller DM, Trent JO, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. 2013;12:1461–1470. doi: 10.1158/1535-7163.MCT-13-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scagliusi SM, Jorge V, Hugo K. Cytopathology of callus cells infected with grapevine leafroll-associated virus 3. Trop Plant Pathol. 2002;27:384–388. [Google Scholar]
- 23.Liu D, Yang Y, Liu Q, Wang J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med Oncol. 2011;28:105–111. doi: 10.1007/s12032-009-9397-3. [DOI] [PubMed] [Google Scholar]
- 24.Lucocq JM, Hacker C. Cutting a fine figure: On the use of thin sections in electron microscopy to quantify autophagy. Autophagy. 2013;9:1443–1448. doi: 10.4161/auto.25570. [DOI] [PubMed] [Google Scholar]
- 25.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muñozgámez JA, Rodríguezvargas JM, Quilespérez R, Aguilar-Quesada R, Martín-Oliva D, de Murcia G, Menissier de Murcia J, Almendros A, Ruiz de Almodóvar M, Oliver FJ. PARP-1 is involved in autophagy induced by DNA damage. Autophagy. 2009;5:61–74. doi: 10.4161/auto.5.1.7272. [DOI] [PubMed] [Google Scholar]
- 27.Pham DL, Kim SH, Losol P, Yang EM, Shin YS, Ye YM, Park HS. Association of autophagy related gene polymorphisms with neutrophilic airway inflammation in adult asthma. Korean J Intern Med. 2016;31:375–385. doi: 10.3904/kjim.2014.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carling D. The AMP-activated protein kinase cascade-a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Novellasdemunt L, Obach M, Millán-Ariño L, Manzano A, Ventura F, Rosa JL, Jordan A, Navarro-Sabate A, Bartrons R. Progestins activate 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) in breast cancer cells. Biochem J. 2012;442:345–356. doi: 10.1042/BJ20111418. [DOI] [PubMed] [Google Scholar]
- 30.Telang S, Clem BF, Klarer AC, Clem AL, Trent JO, Bucala R, Chesney J. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. J Transl Med. 2012;10:95. doi: 10.1186/1479-5876-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 33.White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015;21:5037–5046. doi: 10.1158/1078-0432.CCR-15-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim DK, Yang JS, Maiti K, Hwang JI, Kim K, Seen D, Ahn Y, Lee C, Kang BC, Kwon HB, et al. A gonadotropin-releasing hormone-II antagonist induces autophagy of prostate cancer cells. Cancer Res. 2009;69:923–931. doi: 10.1158/0008-5472.CAN-08-2115. [DOI] [PubMed] [Google Scholar]
- 35.D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342:249–268. doi: 10.1042/bj3420249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amé JC, Spenlehauer C, de Murcia G. The PARP super-family. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 38.Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53:3976–3985. [PubMed] [Google Scholar]
- 39.Xi X, Zhang X, Wang B, Wang T, Wang J, Huang H, Wang J, Jin Q, Zhao Z. The interplays between autophagy and apoptosis induced by enterovirus 71. PLoS One. 2013;8:e56966. doi: 10.1371/journal.pone.0056966. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Yokoyama T, Kondo Y, Bögler O, Kondo S. The role of autophagy and apoptosis in the drug resistance of cancer. Springer US, Drug Resistance in Cancer Cells. 2009:53–71. doi: 10.1007/978-0-387-89445-4_3. [DOI] [Google Scholar]
- 41.Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol. 2010;6:457–470. doi: 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 43.Jose C, Hébert-Chatelain E, Bellance N, Larendra A, Su M, Nouette-Gaulain K, Rossignol R. AICAR inhibits cancer cell growth and triggers cell-type distinct effects on OXPHOS biogenesis, oxidative stress and Akt activation. Biochim Biophys Acta. 2011;1807:707–718. doi: 10.1016/j.bbabio.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 44.Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med. 2014;2:57. doi: 10.3978/j.issn.2305-5839.2014.06.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2015;356:165–170. doi: 10.1016/j.canlet.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 46.Zadra G, Batista JL, Loda M. Dissecting the dual role of AMPK in cancer: From experimental to human studies. Mol Cancer Res. 2015;13:1059–1072. doi: 10.1158/1541-7786.MCR-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG, et al. Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer. 2009;9:307. doi: 10.1186/1471-2407-9-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.