

Abstract
The remarkably efficient suppression of amyloid fibril formation by the DNAJB6 chaperone is dependent on a set of conserved S/T-residues and an oligomeric structure, features unusual among DNAJ chaperones. We explored the structure of DNAJB6 using a combination of structural methods. Lysine-specific crosslinking mass spectrometry provided distance constraints to select a homology model of the DNAJB6 monomer, which was subsequently used in crosslink-assisted docking to generate a dimer model. A peptide-binding cleft lined with S/T-residues is formed at the monomer-monomer interface. Mixed isotope crosslinking showed that the oligomers are dynamic entities that exchange subunits. The purified protein is well folded, soluble and composed of oligomers with a varying number of subunits according to small-angle X-ray scattering (SAXS). Elongated particles (160 × 120 Å) were detected by electron microscopy and single particle reconstruction resulted in a density map of 20 Å resolution into which the DNAJB6 dimers fit. The structure of the oligomer and the S/T-rich region is of great importance for the understanding of the function of DNAJB6 and how it can bind aggregation-prone peptides and prevent amyloid diseases.
Subject terms: Peptides, Chaperones, Molecular neuroscience, Molecular modelling, SAXS
Introduction
The human molecular chaperone DNAJB6 is a member of the highly diverse family of DNAJ chaperones1,2 and a remarkably efficient suppressor of amyloid fibril formation, with a potential for disease targeting. The DNAJ-proteins, first recognized as co-chaperones in E. coli3, form the largest and functionally most diverse chaperone family4,5. The N-terminal domain (NTD) is a conserved so called J domain, folded into four α-helices. This domain is the defining domain for this class of chaperones and through a conserved HPD-motif it is essential for the functional interaction with Hsp706. There are 49 human DNAJ homologues divided into classes DNAJA, DNAJB and DNAJC, with large structural and functional diversities both within and between the classes. One of the most studied DNAJ-proteins is the heat-induced DNAJB1, the human homologue is known as Hdj17,8 and the yeast homologue as Sis19. DNAJB1 forms dimers through the peptide-binding C-terminal domain, which contains two β-barrel domains.
Human DNAJB6 was cloned in 1999, at that time named Mrj (mammalian relative to DNAJ), and found to be distributed in various tissues throughout the body10. It was also identified by gene trapping as essential for murine placental development11. Using yeast-two-hybrid screening, DNAJB6 was identified as a specific binder to keratin-1812. In addition, it was found to be brain-enriched13 and to inhibit polyQ aggregation, in Drosophila14,15 and in human cell lines13,16. The importance of DNAJB6 in placental development, and the failure in chorioallantoic attachment in knock-out mice, appear to relate to its role in keratin degradation17. Other roles assigned to DNAJB6 include inhibition of transcription factor activity through histone deacetylase recruitment in activated T-cells18, and a role in proteasomal turnover and autophagy17,19 and in cell cycle20,21, presumably through mediating keratin turnover and reorganization of cellular skeletal proteins during cell division. DNAJB6 is also highly expressed in myocytes, with mutations causing muscular dystrophy22 and amyloid formation, as shown in muscle biopsies23. There are several isoforms of DNAJB6, of which two are confirmed to be expressed, one nuclear (A) and the second cytosolic (B)24. The cytosolic isoform can also translocate into nucleus during heat shock25. The results reported here all relate to the cytosolic isoform of DNAJB6 (Uniprot #O75190-2).
According to recent results obtained by us and our collaborators26–30, DNAJB6 is a chaperone that may have a general role in preventing amyloid formation31, caused by peptides generated from full-length proteins during catabolic events. Indeed, expression network analyses show that DNAJB6 expression is strongly associated with catabolic processes (Fig. S7 in30). Aggregation of polyQ peptides is suppressed by DNAJB6, in human cell lines27 and in controlled assays with purified protein26. DNAJB6 can also suppress amyloid fibril formation by Aβ42, an aggregation-prone peptide associated with Alzheimer’s disease28, at remarkably low sub-stoichiometric levels, more efficiently than any other known chaperone. The molecular mechanism of the suppression of aggregation and amyloid fibril formation by the Aβ42 peptide is related to the inhibition of the primary nucleation step, as shown by kinetic analysis of the process28,29. Through site-specific mutations in the so-called S/T-rich region, a conserved and functionally important region rich in serine and threonine residues, we found that the potency of DNAJB6 to inhibit primary nucleation and suppress fibril formation by aggregation-prone polyQ peptides declined in proportion to the number of alanine substitutions30. The hydroxyl groups in the side chains may reduce the primary nucleation rate by competing with hydrogen bonding, which is necessary for formation of β-hairpins and amyloid fibrils32. The co-localization of many hydrogen-bonding groups in DNAJB6 will provide a synergistic or cooperative effect, which will gradually be lost with progressive S/T-to-A substitutions. We have also noted that although the ability of DNAJB6 to suppress amyloid fibril formation by polyQ peptides is mechanistically independent of the presence of Hsp7026, a dependency on Hsp70 can be detected under cellular conditions when DNAJB6 capacity is limited30. In cells, Hsp70 is presumably involved in the turnover and degradation of DNAJB6 and its bound peptides. Together, these findings imply that DNAJB6 can be used as a tool to facilitate the understanding of the aggregation process and its inhibition, as well as in the development of future treatment of amyloid diseases. Therefore, the mechanism of the highly efficient interactions of DNAJB6 with aggregation-prone peptides needs to be explored further.
The interactions of DNAJB6 with peptides depend on its structural organization, with unique features among DNAJ-proteins in terms of both oligomerization and the presence of the S/T-rich region. The size and polydisperse character of the DNAJB6 oligomers so far prevented structural studies by X-ray crystallography and NMR. In this work, we have combined homology modelling, chemical crosslinking mass spectrometry, small-angle X-ray scattering (SAXS) and negative stain electron microscopy (EM) to obtain a structural model of the monomer, dimer and higher order oligomeric forms of DNAJB6. The dimer model reveals a peptide-binding cleft lined with S/T-residues located at the interface between the two monomers in the DNAJB6 dimer. Detection of 14N-15N hybrid crosslinks shows that the DNAJB6 oligomers are dynamic entities that can exchange subunits and SAXS data indicate well folded DNAJB6 oligomers, forming a heterogeneous population with varying number of subunits, into which the structural model of the DNAJB6 dimers fit.
Results
Structural model of DNAJB6 monomer
A structural model of the DNAJB6 monomer was selected after homology modelling based on intra-molecular distance constraints obtained from crosslinking mass spectrometry. To ensure that crosslinks used as distance constraints were intra-subunit, only crosslinks that were detected in the band corresponding to monomers (Fig. 1A) were used to assess the five models returned by the Robetta server (Fig. 1B). The choice of the output models, according to the original publication33 on comparative modelling with RosettaCM, is that the final models which may be generated from different seed alignment) are collected and the best 10% of the models by energy is identified, then clustered and the center of the largest cluster where each model is weighted such that low-energy models have highest weight is selected. Out of the five models in the Robetta server output, Model #5 was the only model which satisfied the experimental data (Table 1). The crosslinking data were compared to the Cα-Cα distances between each pair of lysine residues within the five structural models were calculated by the MassAI software after uploading each pdb-file of the models. The crosslinker can span distances up to approximately 30 Å34, due to crosslinker length, lysine side chain dynamics, backbone motions, movement of flexible loops and N- and C-terminal regions. The lysine residues that were detected in crosslinks were either in the N-terminal domain (NTD, residues 1–71) or in the C-terminal domain (CTD, residues 190–241). The intra-domain crosslinks were found to be within the allowed maximum of the Cα–Cα distance of 30 Å for all 5 homology models, not unexpectedly since each domain is <10 kDa, with a diameter of approximately 15 Å, which positions most amine groups within a distance that is reachable by crosslinking. The inter-domain crosslinks, on the other hand, made it possible to distinguish between the structural models. Model #5 satisfied two out of four interdomain crosslinks and showed no crosslinked distances exceeding 40 Å, so called violating crosslinks, in contrast to the other four models. Thus, model #5 was selected as best-fit and used for further analyses and for building a dimer model. The structural model of the DNAJB6 monomer was evaluated (Fig. S1), and found to have stereochemical quality comparable to that observed for high-resolution experimental structures. The file ‘DNAJB6 monomer model_Robetta_5.pdb’ is enclosed in Supplementary Information.
Figure 1.

Chemical crosslinking mass spectrometry to select a best fit structural model of a DNAJB6 monomer. (A) Samples of DNAJB6 were crosslinked and bands corresponding to crosslinked monomer (enboxed) that contain only intra-subunit crosslinks were proteolytically digested, thereafter the peptide mixture was subjected to LC-MSMS and data analysis with the software MassAI to detect the crosslinked peptides. (B) The top five structural models from Rosetta modeling in cartoon presentation, and Robetta_5 was selected as best fit to the distance constraints (indicated as dashed lines in red) obtained from the crosslinking data in Table 1.
Table 1.
Crosslinks detected within DNAJB6 monomers.
| Precursor mass | Charge | Score | Crosslinked peptidesa | Xb | LysA | LysB | Distances (Å) in structural modelc | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | #2 | #3 | #4 | #5 | |||||||
| 2291.355 | 4+ | 51.2 | HASPEDIKKAYR + KLALK | (1) | K20 | K25 | 8.7 | 8.7 | 8.7 | 8.7 | 8.7 |
| 1245.768 | 3+ | 29.4 | KAYR + KLALK | (1) | K21 | K25 | 6.1 | 6.0 | 6.1 | 6.1 | 6.1 |
| 1973.062 | 4+ | 40.8 | WHPDKNPENK + KLALK | (1) | K34 | K25 | 11.9 | 11.9 | 11.9 | 12.0 | 11.9 |
| 2688.340 | 3+ | 53.1 | QVAEAYEVLSDAKK + DIYDKYGK | (1) | K60 | K67 | 10.5 | 10.5 | 10.5 | 10.4 | 10.7 |
| 1934.032 | 3+ | 39.1 | SISTSTKMVNGR + ALKW | (1) | K196 | K29 | 66.6 | 34.4 | 37.3 | 44.5 | 22.4 |
| 1978.025 | 3+ | 14.6 | KSLTINGK + KAYRK | (2) | K225 | K21 | 77.9 | 28.1 | 45.1 | 44.6 | 32.8 |
| 2443.217 | 3+ | 63.6 | VEVEEDGQLKSL + EVLSDAKK | (2) | K225 | K60 | 58.2 | 46.9 | 55.3 | 50.5 | 36.2 |
| 1978.024 | 3+ | 24.9 | KSLTINGK + DKYGKEGL | (2) | K225 | K70 | 51.8 | 56.2 | 50.5 | 47.7 | 28.7 |
a)All detected crosslinks are intra-subunit as described in Fig. 1, approved MSMS-spectra used to validate the crosslinks supplied in Fig. S5.
b)Crosslinker X was either BS3 (1) or BS2-G (2) which are very similar.
c)Calculated Cα-Cα distances for crosslinked lysine residues in the pdb files of the DNAJB6 Robetta structural models #1–5 (Fig. 1). The DNAJB6 monomer is composed of the N-terminal domain (NTD, residues 1–71), the C-terminal domain (CTD, residues 190–241), and a disordered middle domain (residues 72–189) and detected crosslinks in table are intra-domain (upper part) or inter-domain (lower part). Model #5 is satisfying two out of four inter-domain crosslinks with no crosslinked distances exceeding 40 Å, in contrast to the other four models.
The conformational flexibility of the DNAJB6 monomer was analyzed by normal mode analysis (NMA) simulations35. Comparison of predicted normal modes with transitions derived from multiple conformers of the same structures obtained by X-ray crystallography suggest that low-frequency normal modes are often functionally relevant36. Here, the low-frequency modes 7 to 11 from the structural model of the DNAJB6 monomer were analyzed to identify flexible regions, with all modes showing qualitatively similar results. As an example, mode 9 is shown in Fig. 2 and in the file ‘mode-9-movie.mov’ which is enclosed in Supplementary Information. A higher degree of flexibility in the loop regions is seen compared to the other regions of the protein. In particular, the loop between β-strands 1 and 2, with amino acid residues N199 and G200, possessed maximum conformational flexibility. This highly mobile loop may be involved in contacts between subunits within the DNAJB6 oligomer.
Figure 2.
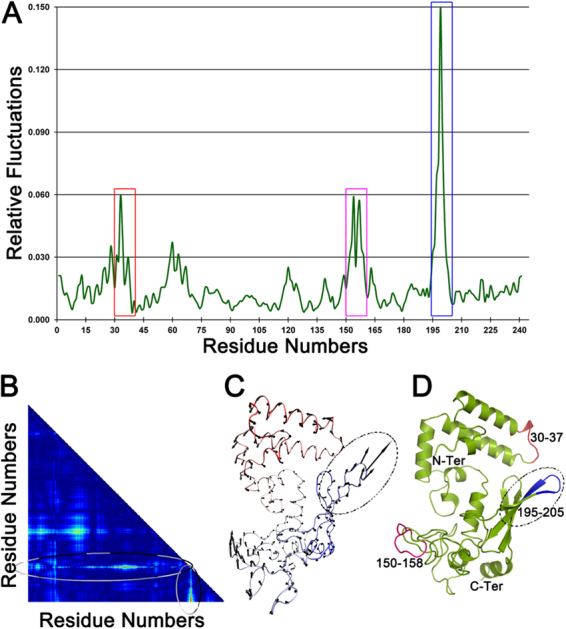
The conformational flexibility of DNAJB6 probed using normal mode analysis. The structural model of DNAJB6 monomer (selected as best fit, Fig. 1) was used for analysis. (A) The fluctuations of mode 9 reveal that the region 195–205 (within blue box) undergoes maximum conformational changes. (B) The regions with maximum (red) and minimum (blue) deviation represented using the contact map also reflect the previous observation. (C) The vector representation of the Cα displacements, with each vector represented as arrows along with their relative direction of motion and magnitude, also reveals the region with maximum displacement (within ellipse). The Cα trace is shown for reference colored from red (N-terminal) to blue (C-terminal). (D) A cartoon representation, indicating the loop region with the maximum fluctuation colored in blue, while the other two loops are colored red and pink.
Structural model of DNAJB6 dimer
The crosslinked samples were also analyzed without prior fractionation to restrict the analysis to intra-subunit crosslinks and, in order to also explore the interactions between DNAJB6 and Aβ42 that prevent its aggregation, crosslinking was performed also in the presence of Aβ42, and subsequently DNAJB6-DNAJB6 and DNAJB6-Aβ42 crosslinks were identified (Table 2). Concerning the DNAJB6-DNAJB6 crosslinks, although intra-subunit crosslinks are most frequently detected, most of the detected crosslinks are presumably both intra- and inter-subunit crosslinks. The obvious exception is the same-to-same-residue type of crosslinks, here K189-K189 and K232-K232, which can only be inter-subunit. These crosslinks are located in the CTD and therefore provide evidence for inter-subunit contacts in the CTD. Concerning the DNAJB6-Aβ42 crosslinks, four lysine residues in DNAJB6 (K189, K202, K225 and K232) were involved, suggesting that the CTD is involved not only in the inter-subunit contacts, but also in peptide binding.
Table 2.
Crosslinks detected within DNAJB6 oligomers and between DNAJB6 and Aβ42.
| Precursor mass | Charge | Score | Crosslinked peptides DNAJB6-DNAJB6a | LysA | LysB | #1–2b | Distancec(Å) |
|---|---|---|---|---|---|---|---|
| 3101.577 | 3+ | 52.9 | HASPEDIKKAYR + QVAEAYEVLSDAKK | K20 | K60 | 1 | 16.6 |
| 1708.898 | 3+ | 69.4 | HASPEDIKK + SDAKK | K20 | K60 | 2 | 16.6 |
| 2012.068 | 3+ | 78.3 | HASPEDIKK + KSISTSTK | K20 | K189 | 1,2 | 36.0 |
| 1750.981 | 3+ | 85.4 | HASPEDIKK + KITTK | K20 | K202 | 1,2 | 29.6 |
| 2532.388 | 3+ | 56.6 | HASPEDIKK + SLTINGKEQLL | K20 | K232 | 2 | 47.2 |
| 2587.334 | 4+ | 46.3 | WHPDKNPENKEEAER + KLALK | K34 | K25 | 2 | 12.0 |
| 2899.498 | 4+ | 57.3 | HPDKNPENKEEAER + KITTKR | K34 | K202 | 1 | 16.5 |
| 2116.153 | 3+ | 54.6 | KQVAEAY + TINGKEQLL | K47 | K232 | 1 | 36.4 |
| 2154.108 | 3+ | 57.1 | EVLSDAKKR + DIYDKY | K60 | K67 | 1 | 11.0 |
| 2275.082 | 3+ | 63.7 | GKEGLNGGGGGGSHF + KQVAEAY | K70 | K47 | 1 | 21.6 |
| 2439.121 | 3+ | 52.6 | GKEGLNGGGGGGSHF + DIYDKY | K70 | K67 | 1 | 5.4 |
| 2812.348 | 3+ | 61.4 | GKEGLNGGGGGGSHF + VEVEEDGQLKSL | K70 | K225 | 1 | 28.3 |
| 1839.014 | 4+ | 72.3 | KSISTSTK + KSISTSTK | K189 | K189 | 2,* | — |
| 2506.276 | 3+ | 54.6 | VEVEEDGQLKSL + HASPEDIKK | K225 | K20 | 1,2 | 33.1 |
| 3360.603 | 3+ | 65.2 | VEVEEDGQLKSL + WHPDKNPENKEEAER | K225 | K34 | 2 | 25.4 |
| 2371.237 | 3+ | 88.8 | VEVEEDGQLKSL + EVLSDAKK | K225 | K60 | 2 | 36.8 |
| 2483.232 | 3+ | 80.6 | VEVEEDGQLKSL + DIYDGYGK | K225 | K67 | 1,2 | 31.7 |
| 2492.265 | 3+ | 66.3 | VEVEEDGQLKSL + SISTSTKM | K225 | K196 | 1,2 | 14.5 |
| 2072.121 | 3+ | 79.5 | VEVEEDGQLKSL + KITTK | K225 | K202 | 2 | 12.3 |
| 2853.531 | 3+ | 77.8 | VEVEEDGQLKSL + SLTINGKEQLL | K225 | K232 | 1,2 | 16.1 |
| 2309.220 | 3+ | 67.4 | TINGKEQLLR + DIYDKYGK | K232 | K67 | 2 | 40.7 |
| 1198.116 | 3+ | 54.2 | TINGKEQLL + KITTK | K232 | K202 | 1 | 25.2 |
| 2679.516 | 3+ | 79.4 | SLTINGKEQLL + TINGKEQLLR | K232 | K232 | 1,2,* | — |
| Precursor mass | Charge | Score | Crosslinked peptidesaAβ42-DNAJB6 | LysA | LysB | ||
| 2437.218 | 3+ | 48.8 | HDSGYEVHHQKL + KSISTSTK | K16 | K189 | 2 | |
| 2384.301 | 3+ | 53.8 | EVHHQKL + VEVEEDGQLKSL | K16 | K225 | 2 | |
| 2348.265 | 3+ | 81.4 | FAEDVGSNKGAIIGLM + KITTK | K28 | K202 | 2 | |
| 3129.674 | 3+ | 59.3 | FAEDVGSNKGAIIGLM + SLTINGKEQLL | K28 | K232 | 2 |
aCrosslinks with BS3 within DNAJB6 can be either intra-subunit or inter-subunit, except the K189xK189 and K232xK232 crosslinks which must be inter-subunit.
bDatasets are: (#1) duplicate samples with crosslinked DNAJB6 oligomers, (#2) duplicate samples in which DNAJB6 oligomers and Aβ42. Approved MSMS-spectra used to validate the crosslinks are supplied in Fig. S6 (#1, 2). As indicated with asterisk (*), the inter-subunit crosslinks K189-K89 and K232-K232 are validated as inter-subunit also as 14N-15N hybrid crosslinks in a separate dataset with unlabeled (14N) and labeled (15N) DNAJB6, approved MSMS-spectra are supplied in Fig. S7.
cCalculated Cα-Cα distances for crosslinked lysine residues using the pdb file of the DNAJB6 Robetta structural model_5 described in Fig. 1.
The lysine residues in DNAJB6 involved in inter-subunit contacts and/or peptide binding are highlighted in the image of the DNAJB6 monomer in Fig. 3A. The crosslinked residues K189 and K232 were used as restraints in the docking of two DNAJB6 monomers to construct a DNAJB6 dimer. The docking program HADDOCK37 clustered 153 structures in 10 clusters, representing 76.5% of the water-refined models. The top cluster with a high HADDOCK score (−96.0 ± 4.8) shows a dimer with the CTD of each monomer contributing to the dimer interface (Fig. 3B), and with K189-K189 and K232-K232 at distances (dashed lines) 23.5 Å and 30.6 Å, respectively. Two symmetrically positioned β-hairpins, formed by the first two out of four β-stands in the CTD and the residues N199 and G200 in the loop between the strands, contain the conserved and functionally important S/T residues (high-lighted in pink). This clearly suggests that the S/T-residues are not buried but solvent-accessible and lining a central cleft formed at the interface between the two subunits, resulting in a possible site for peptide binding (Fig. 3C). The file ‘DNAJB6 dimer model_Haddock.pdb’ is enclosed in Supplementary Information.
Figure 3.

Structural model of DNAJB6 dimer. (A) The structural model of DNAJB6 monomer (selected as best fit, Fig. 1) in ribbon presentation (grey) with lysine residues as spheres: K189 (dark red), K232 (dark blue), K202 (light blue), K225 (light red). The inter-subunit crosslinks K189-K189 and K232-K232 (Table 2) were used as restraints in docking. (B) The DNAJB6 dimer model as cartoon, with the two subunits in yellow and blue, respectively. The lysine residues K189 and K232 (spheres, color coded according to each subunit) are shown with the inter-subunit crosslinks indicated with dashed lines, with distances K189-K189 = 23.5 Å and K232-K232 = 20.6 Å. The following residues are shown as sticks: N199, G200 (black); the S/T-residues S-STST aa 190, 192–195 (dark pink); other ST-residues in region aa 155–195 (light pink). The two symmetrically positioned β-hairpins (=β-strand 1 and 2 and the residues N199 and G200 in between) contain the functionally essential S/T residues. (C) The DNAJB6 dimer model in surface presentation color-coded as in (B), showing a central cleft between the domains which could serve as a peptide-binding cleft, surrounded by S/T-residues, S-STST aa 190, 192–195 (dark pink); other ST-residues in region aa 155–195 (light pink). (D) The DNAJB6 dimer model in spacefill presentation (grey) and one copy of Aβ42-peptide (red, with residues K16 and K28 as sticks). The image is obtained after HADDOCK docking of the Aβ42 (PDB ID 2NAO, shown as inset with the two K16 and K28 highlighted in red) to the DNAJB6 dimer model, using as restraints the crosslinked residues (K16 and K28 in Aβ42, K189, K232, K202 and K225 in one of the DNAJB6 dimer subunits).
That peptide binding indeed occurs in this peptide-binding cleft is supported by the detected DNAJB6-Aβ42 crosslinks (Table 2). There are three crosslinker-reactive primary amines in the Aβ42-peptide, one is the N-terminal amino group on the first residue and the other two are the primary amines in the side chains of the lysine residues K16 and K28 and only the latter two were crosslinked to DNAJB6. These crosslinks were used as restraints in another round of docking, using the above-mentioned pdb-file of the DNAJB6 dimer and, for Aβ42, the pdb-file 2NAO38. This resulted in a model showing a possible arrangement of how one copy of the Aβ42 peptide can be bound to the DNAJB6 dimer, as shown in Fig. 3D.
This peptide-binding cleft lined with the S/T-residues, which here presented data are suggesting for DNAJB6, represents a peptide-binding site in the CTD that is distinctly different compared to that in the crystal structures of other DNAJ-proteins, DNAJA1 and DNAJB18,39, recently suggested to be referred to as ‘double β-barrel J-proteins’5. Whereas DNAJB6 only has one they are composed of two β-barrel domains in the CTD. They also have no S/T-rich region, whereas this region is fully or partially conserved within a sub-group of DNAJ-proteins that we refer to as DNAJB6-like (DNAJB2, 3, 6–8), which cluster together in a phylogenetic tree (Fig. S2). The conserved S/T-rich region in the CTD, and other known features in DNAJ-proteins, such as the conserved Hsp70-binding HPD motif in the NTD and the muscular dystrophy-related phenylalanine residues in the GF-rich middle domain, are summarized in Fig. 4, highlighted and similarly color-coded in the sequence alignment and the DNAJB6 dimer model.
Figure 4.

Sequence alignment of DNAJB6-homologues with features highlighted in the structural model of the DNAJB6 dimer. (A) Sequence alignment of human DNAJB2, 3, 6–8. The S/T-rich region in DNAJB6 (aa 155–195) is underlined with its 18 S/T-residues highlighted (pink), corresponding S/T-residues in other sequences highlighted similarly. Other highlights: the Hsp70-interaction HPD-motif (orange); the disease-causing F-residues (mutations F89I, F91I/L, F93I/L in muscular dystrophy); the mobile loop residues, identified in Fig. 2, N199 and G200 (black-shaded); the K225/K232-residues (red) suggested to regulate activity through deacetylation; the two UIM (ubiquitin interaction motifs) unique to DNAJB2 (grey-shaded). Alignments were performed at http://www.ebi.ac.uk/Tools/msa/clustalo/. The last residues in CTR of DNAJB2 and DNAJB7, longer than the other ones, are not shown. (B) The structural model of the DNAJB6 dimer in Fig. 3 is here shown from two angles, with S/T-residues as cartoon, and other residues as sticks highlighted with same color-code as in the alignment.
The crosslinks K189-K189 and K232-K232 that were used as restraints in docking the monomers to dimer were readily identified as inter-subunit crosslinks since they are same-to-same-residue type of crosslinks. Since these two crosslinks are very important in constructing the DNAJB6 dimer model, their identity as inter-subunit crosslinks were also verified by mixed isotope crosslinking to rule out the possibility that they could have been incorrectly assigned. We have previously showed that this approach is useful for proof of inter-subunit crosslinks in homo-oligomeric proteins40. Hybrid crosslinks, formed when a labeled (15N) peptide is crosslinked to an unlabeled (14N) peptide, are detected by our software MassAI (MS/MS-spectra supplied in Fig. S7. The K189-K189 and K232-K232 crosslinks were detected among other hybrid crosslinks in a sample of DNAJB6 oligomers (14N-15N, mixed 1:1) incubated 1 h before adding the crosslinker. However they were not detected in a control sample where unlabeled and labeled oligomers were crosslinked separately and mixed 1:1 before LC-MSMS step. The detection of 14N-15N hybrid crosslinks shows that the DNAJB6 oligomers are dynamic entities that exchange subunits on a time-scale of less than an hour.
Structural model of DNAJB6 oligomers
That the purified DNAJB6 protein is composed of oligomers with a varying number of subunits, is well folded and is soluble up to 10 mg/ml is concluded from data obtained when the DNAJB6 oligomers were fractionated into six sub-populations by online size-exclusion chromatography combined with small-angle X-ray scattering (Fig. 5A). None of the scattering profiles, all derived from the main peak in the chromatogram (Fig. 5B), show any sign of aggregation, otherwise detected as a steep rise in a non-linear Guinier region of the scattering curve. The parabolic appearance of the Kratky plot (Fig. 5C), which can be used to assess the degree of fold of the scattering particles41, confirms our earlier data from circular dichroism spectroscopy that DNAJB6 is well folded. Flexibility or a deviation from a typical globular shape is observed for the DNAJB6 oligomers, since there is a peak maximum at a slightly higher value than 1.1 at qRg = √3. The radius of gyration (Rg) and maximum dimension (Dmax), calculated from the P(r)-distribution curves (Fig. 5A, inset), ranged from 6.9 nm to 8.7 nm, and from 22.2 nm to 29.6 nm, respectively (Table 3). The estimated mass of the DNAJB6 oligomers calculated from SAXS data ranged from 0.8 MDa to 1.5 MDa, in line with our earlier data from dynamic light scattering26 and, since the monomeric mass of DNAJB6 is 26.9 kDa, corresponds to DNAJB6 oligomers with a varying number of subunits, ranging from 34 to 55.
Figure 5.

SAXS-data of DNAJB6. (A) Experimental online SEC-SAXS data with sample name numbers relating to retention times; the inset shows the P(R) distance distribution function of DNAJB6 calculated from the experimental SAXS data for estimation of Dmax (Table 3). (B) The size-exclusion chromatography profile for DNAJB6, SAXS data shown are for the fractions from collected the main peak (8–11 ml) in the chromatogram, the peaks at >15 ml had too low scattering intensity. (C) The corresponding dimensionless Kratky plot of the SAXS data in (A).
Table 3.
SEC-SAXS data.
| Sample | aRg, nm | aDmax, nm | aVp, nm3 | bMWp, kDa | bMWa, kDa | cSubunits |
|---|---|---|---|---|---|---|
| DNAJB640 | 8.73 ± 0.16 | 29.6 | 2368 | 1480 | 1450 | 55 |
| DNAJB641 | 8.50 ± 0.35 | 29.4 | 2114 | 1321 | 1240 | 49 |
| DNAJB642 | 7.70 ± 0.2 | 27.5 | 1849 | 1156 | 1090 | 43 |
| DNAJB643 | 7.43 ± 0.11 | 26.6 | 1669 | 1043 | 979 | 39 |
| DNAJB644 | 6.72 ± 0.25 | 22.5 | 1523 | 952 | 856 | 35 |
| DNAJB645 | 6.88 ± 0.19 | 22.2 | 1470 | 919 | 838 | 34 |
aRg, Radius of gyration, Dmax, maximum size, Vp, Porod volume, calculated as described in Methods.
bMWp, MWa, mass estimates calculated by dividing Vp by 1.6 and by the DAMMIF model excluded volume by 2, respectively.
cSubunits, number of subunits fitting the average MWp calculated with mass of the DNAJB6 26.9 kDa.
The DNAJB6 oligomers with varying number of subunits were also detected by negative stain EM, and one sub-population of DNAJB6 oligomers was selected for closer analysis (Fig. 6). Reference-free classification of boxed out particles showed classes of slightly elongated particles with major and minor axes lengths of approximately 160 Å and 120 Å, respectively. The asymmetric 3D map showed an oligomeric complex, which can be fit into a box of 332 × 332 × 332 Å3. A two-fold symmetry was imposed to generate a map that was compared to the symmetry independent reconstruction. A high degree of similarity between these maps suggested that the analyzed fraction of DNAJB6 is symmetrical, at least at this low resolution level. The reconstructed map with C2-symmetry was obtained at a resolution of ~20 Å measured at 0.143 Fourier shell correlation (Fig. S3) and the file ‘dnajb6_negstain_c2.mrc’ is enclosed in Supplementary Information. Considering a particle mass of 540 kDa, very approximately estimated from non-denaturing PAGE and that one DNAJB6 subunit has the mass 26.9 kDa, this sub-population of DNAJB6 oligomers selected for single particle analysis corresponds approximately to 20 subunits.
Figure 6.

DNAJB6 oligomers analyzed by negative stain electron microscopy. (A) Insert shows native PAGE analysis of DNAJB6 fractions (1–8) collected after GraFix separation,’Inp’ DNAJB6 before GraFix. (B) EM image of DNAJB6 oligomers collected from fraction 5 in (A). (C,D) 2D class averages and corresponding projections of 3D maps obtained using C1 (C) and C2 (D) symmetry. (E) Asymmetric density map of DNAJB6 from two mutually perpendicular directions. (F) A comparison of a density map where 2-fold symmetry was imposed on the unsymmetrized C1 map (in mesh) and one density map where processing was done using C2 symmetry (in surface rendered blue). The maps are depicted at 5σ threshold. Scale bar represents 20 Å. See also Fourier shell correlation in Fig. S3.
The DNAJB6 dimers fit into the electron density map of the DNAJB6 oligomer. A structural model of the DNAJB6 dimer is presented in Fig. 7, with a peptide-binding cleft lined with S/T-residues at the dimer interface and a tentatively outlined Aβ42 peptide, similar to the image presented in Fig. 3D after crosslink-assisted docking. The S/T-residues in closest proximity to the peptide (the S/T-residues 190 and 192–195) are highlighted in dark pink and the other S/T-residues in light pink. The DNAJB6 dimer can be turned to appear in a bent shape as a crescent with the S/T-residues 190 and 192–195 on the concave side, and fitted to the electron density map of the DNAJB6 oligomer. The length of the dimer extends from one end of the oligomer to the other, with the N-terminal domains exposed towards the poles and the C-terminal domains located in the equatorial plane of the oligomer, as outlined in Fig. S4. An approximate volume calculation in Chimera at a threshold which is close to the noise-level yields a map volume of approximately 700 000 Å3, in which there would be space for approximately 10 dimers in total, i.e. 20 subunits. It should be noted that this is just one possible oligomer model, based on only one subfraction of DNAJB6 oligomers.
Figure 7.

Structural model of DNAJB6 dimer model in relation to DNAJB6 oligomer. The DNAJB6 dimer model generated as described in Fig. 3 here presented in space-fill (grey), with a tentatively outlined Aβ42 peptide (green) at the dimer interface in a peptide-binding cleft, which may interact with aggregation-prone peptides in various conformations (e.g. one peptide, a pair of peptides or one peptide in hairpin conformation), and which is surrounded by the S/T-residues 190 and 192–195 close to the cleft (dark pink) and the other S/T-residues further away from the cleft (light pink). The conserved S/T-residues are required to suppress fibril formation, presumably due to hydrogen-bonding between the hydroxyl groups in the side chains of the S/T-residues and the aggregation-prone peptides30. In the lower part of the figure it is outlined how the DNAJB6 dimer can be turned to appear in a bent shape as a crescent with the S/T-residues 190 and 192–195 on the concave side and that such DNAJB6 dimers can be fitted to the DNAJB6 electron density map, generated as described in Fig. 6. In Fig. S4 is further described how the DNAJB6 dimers fit into the DNAJB6 oligomers.
Discussion
We here present a structural model of oligomeric DNAJB6. It is based on modelling to obtain first a monomer and then a dimer based on restraints from crosslinking, on SAXS-data showing that the preparation is well folded and on EM-data showing an oligomer at low resolution into which the dimers can be fitted. Only crosslinks that are validated by manual inspection of the MSMS-spectra were used as restraints in modelling. Crosslinked samples with only one or two proteins are not that complex and there is a redundancy of observations of each crosslinks. The ones that are presented in Tables 1 and 2 have been observed many times, with strict selection criteria imposed to avoid any false positive crosslinks.
DNAJB6 dimer with a peptide binding site lined with S/T-residues
There are several reasons to expect that the DNAJB6 is composed of dimeric building blocks. Our recombinantly expressed and purified DNAJB6 protein shows not only the band corresponding to the mass of the 26.9 kDa monomer in SDS-PAGE, but also a band corresponding to the mass of a dimer at 60 kDa which is confirmed to contain DNAJB6 by mass spectrometry28. Furthermore, homologues for which the structure has been determined are dimeric, for example human DNAJB1 (PDB # 2QLD/3AGX)7,8, yeast DNAJB1 (1C3G/2B26)9,42, and yeast DNAJA1 (PDB # 1NLT/1XAO)39,43. The complete structure recently determined for a more distantly related homologue from T. thermophilus (PDB #4J80)44, also is composed of dimeric building blocks.
The structural model of the DNAJB6 monomer (Fig. 1) was obtained by modelling, and selection of best-fit based on crosslink constraints (Table 1) within the monomer. Since there is no suitable template for direct comparative modeling of DNAJB6, the Rosetta modelling was based on a protocol that determines the regions of a protein chain that align to different templates and find regions that may fold into globular units, which does not require a full template with high overall sequence identity. A structural model of the DNAJB6 dimer (Fig. 3) was then obtained by docking of two monomers with a software, in which additional information on the proximity of the protein interfaces is used to drive the docking process. As protein interface information, we used the lysine residues 189 and 232 in neighboring monomers (Table 2) as restraints. There is an ensemble of solutions as output from the HADDOCK server. Among the clusters with best energy the second best cluster was considerably less good (score −47.9 +/− 6.2) than the best (score −96.0 +/− 4.8). The lowest score is the most likely solution, and also low standard deviation is a positive factor. The score is based on an average of the best 4 models of each cluster together with the associated standard deviation. The distance constraints imposed by the crosslinking were largely met in the resulting dimer model, with K189-K189 at 23.5 Å distance and K232-K232 at 31.3 Å distance. Estimation of the buried surface area (2258.5 Å2) provides a reasonable value expected for a protein dimer interface45. An evaluation of the interface with the PISA server46 showed 65 interfacial residues, with 13 hydrogen bonds mainly between residues in the regions 175–178 and 232–234. Some may be hot spot residues critical in their contribution to the binding free energy47 and can be targeted for site-specific mutagenesis to evaluate the model.
An essential implication of the DNAJB6 dimer model is that the functionally important S/T-residues are not buried in the monomer-monomer interface (Fig. 3B,C). Instead, they surround a central cleft formed between the two CTD domains, one from each monomer. We suggest that this region can serve as a possible peptide-binding cleft with two symmetrically positioned sites, each having on one side five S/T-residues S-STST (aa 190, 192–195, color-coded dark pink) and on the other side the other ST-residues (in region aa 155–195, color-coded light pink). The S/T-rich region (residues 155–195) has in total 18S/T-residues that are to a varying extent conserved among DNAJB6 homologues (Figs 4 and S2) and the suppression of fibril formation and the inhibition of the primary nucleation is specifically dependent on the S/T-rich region30.
We have earlier proposed that S/T-residues provide a multivalent hydrogen-bonding capacity, whereby hydroxyl groups can reduce the primary nucleation rate by competing with the hydrogen bonding required for formation of β-hairpins and amyloid fibrils32. The detected crosslinks suggest that the possible peptide-binding cleft lined with the conserved S/T-residues is where DNAJB6 interacts with Aβ42, as shown in the image obtained after docking of the peptide (Fig. 3D) and as outlined in Fig. 7. Even if the detected crosslinks are correctly assigned it is impossible to tell if the detected crosslinks originate from one or several Aβ42-peptides, and if Aβ42 is disordered, is a β-hairpin or in some other form. Still, with the simple assumption of one peptide monomer we performed docking and, although one must be aware of and not overinterpret the results based on this assumption, it is possible to conclude (i) how all the detected crosslinks in DNAJB6 are localized in one interaction area, the presumed peptide binding cleft with the S/T-residues, (ii) how the N-terminal end of the Aβ42 peptide, with its primary amine not detected in crosslinks with DNAJB6, appears to point away from the interaction area, and (iii) how K16 and K28 in Aβ42, if they were in one and the same peptide, could be localized in close proximity to the S/T-residues. In the model presented in Fig. 3D, the sequence STST (192–195) in DNAJB6 was close enough to the Aβ42 peptide so that S194 and T195 could form hydrogen bonds to side-chains of SNK (26–28) in the peptide whereas S192 and T193 were closer to peptide residues with less polar side-chains. Although this is of some interest to note, the assumption of one peptide monomer is presumably not fully correct since DNAJB6 presumably does not bind Aβ42 monomers, according to our previous kinetic calculations28,29. Instead DNAJB6 binds aggregated forms of Aβ42, possibly already stacked as initiating amyloids with hydrogen bonding. There may still be unsatisfied hydrogens bonds in oligomeric assemblies and edge strands in such aggregated forms of Aβ42 that could bind to the S/T-rich region in DNAJB6.
Peptide binding is indeed expected to typically occur in clefts or surface pockets of proteins48. In DNAJB1 two peptide binding sites were found in CTD domain I, one of which was involved in binding an octapeptide to the C-terminal region of Hsp70 and the second contributed to recognizing ‘non-native polypeptide substrates’, with both binding sites being very hydrophobic8. The structure of the CTD in DNAJB1, and the mode of peptide binding is presumably distinctly different from that in DNAJB6.
The sofar known target peptides of DNAJB6, polyQ and Aβ4226–30, share the common property of being very aggregation-prone, with a high propensity to form amyloid fibrils. A dimer structure of Aβ42 has been observed in Aβ42 fibrils by cryo-EM49 and a hairpin conformation, assumed to occur in the pathway of fibril formation50, has been structurally characterized after capture of Aβ42 in complex with an anti-aggregation antibody51 or with an engineered lipocalin52. The crosslinks between Aβ42 and DNAJB6 that we here detected (Table 2) showed that crosslinking had occurred to both primary amines in the side chains of the two lysine residues, K16 and K28. However, no crosslink was detected that involved the third primary amine of the Aβ42-peptide, which is the N-terminal amine of the first amino acid, which is usually crosslinked in most proteins and previously detected in crosslinks between Aβ42 peptides53. This strongly indicates that it is the C-terminal rather than the N-terminal part of the Aβ42-peptide that interacts with DNAJB6. The Aβ peptide is comprised of a charged N-terminal and a hydrophobic C-terminal region, with a highly hydrophobic central region, in which the residues 16–21 (KLVFFA) constitute the most aggregation-prone segment and are alone sufficient to cause formation of insoluble fibrils54. A time-scale crosslinking study could possibly catch conformational changes in Aβ42 and monitor the DNAJB6 interference with the Aβ42 fibril formation, and explore the interactions between DNAJB6 and peptides.
DNAJB6 oligomers with a varying number of subunits in dynamic subunit exchange
The dynamic character of DNAJB6 oligomers and the exchange of subunits is evident from the detection of inter-subunit 14N-15N hybrid crosslinks (Table 2, Fig. S7). The SAXS data suggest that the DNAJB6 oligomers are well folded and soluble (Fig. 5), with estimated subunit content in the range between 34 to 55 subunits, based on Porod volume calculations and the DAMMIF software (Table 3). Calculation of mass based on I(0), the sequence and the protein concentration55, requires a known protein concentration and was therefore not applicable after the online SEC-SAXS fractionation. The subpopulation of DNAJB6 oligomers analyzed by EM (Fig. 6) corresponds to 20 subunits, however the density map is obtained from just one of the smaller subpopulations of DNAJB6 oligomers, and it should be mentioned that negative stain EM often underestimates the volume. It can be concluded from both SAXS- and EM-data that DNAJB6 oligomers have a slightly elongated shape and a varying number of subunits, at least 20 and up to 55.
The oligomeric conformation of DNAJB6 may increase the local concentration of the peptide-binding interaction surfaces, may control peptide binding and release and may maintain continuous weak and transient interactions at sub-stoichiometric ratios. The oligomeric character of DNAJB6 resembles that of the small heat shock protein (sHsp) chaperones16. In spite of the importance of sHsps in cells, there are only a few high-resolution structures available, probably due to difficulties related to the dynamic character of the often polydisperese oligomers56, an inherent property that appears to be required for their ability to bind and sequester target proteins. There appears to be distinct structural and functional differences between on one hand the DNAJB1-like chaperones, recently suggested to have a dis-aggregase activity in complex with Hsp7057, and on the other hand DNAJB6-like chaperones. Within the sub-group of DNAJB6-like chaperones (Fig. S2), the S/T-rich region is present in DNAJB8 which is oligomeric, as DNAJB6, but expressed only in testes16. The S/T-rich region is found also in DNAJB2, but it is not known whether DNAJB2 is oligomeric or not. DNAJB2 is expressed in neuronal cells with ubiquitin interaction motifs that bring clients to the proteasome for degradation58. Some S/T-residues are conserved also in DNAJB3 and DNAJB7, which are less well known. Structural models will benefit and progress research on DNAJB6-like chaperones.
DNAJB6 dimers fitted into DNAJB6 oligomers
One of the questions of particular interest to address concerning the DNAJB6 oligomers is where the functionally S/T-rich region, required to suppress fibril formation30, is situated. Is it exposed on the outer surface of the DNAJB6 oligomers, or is it hidden inside? In our DNAJB6 oligomer model with dimers fitted into the density map (Figs 7 and S4), the CTDs forming the dimer interface are located in the equatorial plane. The residues N199 and G200, in the mobile loop that was identified in the NMA simulations to be highly flexible (Fig. 4), are located in a position where they could form contacts between the dimers. The NTDs with the conserved Hsp70-interaction HPD-motif (residues 30–32) are accessible from the oligomer surface, whereas the S/T-rich region appears to face the interior of the oligomers. It remains to be investigated how peptides can get access to the S/T-rich region and the presumed peptide-binding cleft, if it is the oligomeric form of DNAJB6 that interacts with the aggregation-prone peptides or if smaller subunits, for example dimers in equilibrium with the oligomeric forms, are involved in the interactions.
In conclusion, we here present structural data that suggest that DNAJB6 unlike many other DNAJ-proteins is able to form a heterogeneous ensemble of oligomers, with a varying number of well-folded DNAJB6 dimers as building blocks. A structural model of a dimer obtained by a combination of homology modeling, crosslinking and docking suggests that a peptide-binding cleft, lined with S/T-residues, is created at the interface between monomers. The structural insights presented here open up new possibilities for targeted mutagenesis and exploration of the dimer-oligomer equilibrium and the interaction sites for peptides, Hsp70 and deacetylases that may regulate its activity and provide a testable DNAJB6 structural model as a frame-work to inform future investigations.
Methods
Proteins and reagents
Recombinantly expressed human DNAJB6 (Uniprot # O75190-2) and the Aβ42 peptide was produced as previously described28. DNAJB6 protein isotope labeled with 15N was obtained by growing the bacterial host in minimal medium containing 15N-NH4Cl as nitrogen source, and purified as the unlabeled protein. The crosslinking reagents, DSSG (DiSulfoSuccinimidylGlutarate)/BS2G (Bis[Sulfosuccinimidyl]Glutarate) and BS3 (Bis[SulfoSuccinimidyl]Suberate), were purchased from Creative Molecules Inc. (Victoria, Canada) or Thermo Fischer Scientific (Rockford, USA).
Chemical crosslinking mass spectrometry
Chemical crosslinking and protease digestion
The DNAJB6 protein concentration during crosslinking was approximately 60 μM, and if the Aβ42 peptide was present it was at a 1:1 molar ratio of DNAJB6 to peptide. Crosslinkers were dissolved in distilled water to a concentration of 30 mM immediately before use, and added to samples to yield a 3 mM concentration during crosslinking (molar ratio crosslinker to protein approximately 50:1). After 15 min, the crosslinking reaction was quenched by a surplus of primary amine by adding 1 M Tris-(hydroxymethyl)-aminomethane to a final concentration of 20 mM. Samples were subjected to denaturing gel electrophoresis as described in40 and the band corresponding to crosslinked DNAJB6 monomer was excised and subjected to in-gel digestion (ratio 1:50 protease to protein) with a combination of trypsin and chymotrypsin (Promega, Madison, WI, USA) at 37 °C. Samples were acidified by adding 2 μl 10% trifluoroacetic acid (TFA). Samples that were not prefractionated by electrophoresis, in order to analyze not only intra-subunit crosslinks, were precipitated with freeze-cold acetone to remove excess reagents and to concentrate the proteins. Pellets were then dissolved in 25 mM ammonium bicarbonate buffer, digested into peptides by in-solution digestion and pretreated on 3 mm long Poros R2 reversed phase microcolumns59 before LC-MSMS.
Mass spectrometry
Peptides were subjected to reversed-phase nano-LC prior to mass spectrometric analyses in a LTQ-Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific, Rockford, USA) equipped with a nanoEasy spray ion source (Proxeon Biosystems, Odense, Denmark), essentially as described in59, by data-dependent collision-induced dissociation MS/MS scans excluding 2+ since crosslinked peptides generally are charged 3+ or more. For the samples with unlabeled and (14N) and labeled (15N) peptides the gradient was created by solvent A (0.1% (v/v) FA in water) and solvent B (0.1% (v/v) FA in 100% (v/v) acetonitrile) as follows: 5–30% for 40 min, 30%-50% for 20 min and 50–95% for 5 min and at 95% for 10 min. With MIPS enabled the 15N-labeled peptides, as well as hybrid-crosslinked peptides, are discriminated against the non-labeled peptides for fragmentation since they show pre-peaks in the MS-spectra as 15N-salts contain some 2% 14N, thus the 15N-labelled peptides do not fulfill the requirement for fragmentation since A + 2 is not smaller than A + 1, where A is the monoisotopic peak. Therefore to promote MS/MS fragmentation of 15N-labeled peptides the mono isotopic precursor selection (MIPS) was disenabled.
Data analysis
Raw-files, typically containing 20 000 scans each, from the Thermo Xcalibur software were converted to mgf-files with an in-house licensed version of the software Mascot Server (version 2.5, or earlier, Matrix Science Inc, Boston, USA, http://www.matrixscience.com) and Mascot Distiller (version 2.6, or earlier), in case of the 14N-15N hybrid crosslinks the settings in MS and MSMS processing were optimized as described in ref.60. The mgf-files were analyzed to detect crosslinks with the free MassAI software61,62 (http://www.massai.dk, version August 2015; and in case of the 14N-15N hybrid crosslinks, version February 2017). Filtering was performed to retain only the top 125 most intense peaks per scan, using the built-in MGF-filter. Crosslinked peptides were detected with the following search settings: fragmentation mode: CID, 10 ppm MS accuracy, 0.05 Da or 0.2 MS/MS accuracy, trypsin combined with chymotrypsin as enzyme, 2 allowed missed cleavages, Fixed modification: none. Variable modifications: the crosslinker in question, including deadend variant and/or internal crosslink, 3 allowed modifications per peptide. The searches were performed with the setting ‘Also xlink modified peptides’, without or with the filtering option ‘Crosslink peptide only if peptide is observed as deadend’ which reduces the number of false positives. All MSMS spectra of crosslinks were manually inspected and enclosed in Supplementary Information, and accepted only if following criteria were fulfilled: scan number marked in green (no other explanation to spectra), intensity >1000, fragment ions from both peptides, and no major peaks unexplained. The sequences used in data analysis refer to the amino acid sequence for human DNAJB6 isoform B (UniProt # O75190-2) and for the amino acid sequence of the Aβ42-peptide from human APP (Amyloid Precursor Protein, Uniprot # P05067) the sequence DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA, with and without the extra start methionine that was added during recombinant expression.
Structural modelling
Rosetta modelling for DNAJB6 structural model
To generate structural models of DNAJB6, the sequence was uploaded to several different servers, the LOOPP server (http://clsb.ices.utexas.edu/web/loopp_server.html)63, the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER)64 and the Robetta server (http://robetta.bakerlab.org)33. The data from LOOPP did not cover the whole sequence. I-TASSER and Robetta returned five models, each covering the whole sequence. None of the structural models from I-TASSER matched the crosslinks. The Robetta server is based on a protocol to determine the regions of a protein chain that align to templates and otherwise find regions that will fold into globular units (Output file states: Rosetta provides both ab initio and comparative models of protein domains. Comparative models are built from structures detected and aligned by HHSEARCH, SPARKS, and Raptor. Loop regions are assembled from fragments and optimized to fit the aligned template structures. De novo models are built using the Rosetta denovo protocol and the procedure is fully automated). The server returned several proposed structural models that matched the crosslinks to varying extent. The intra-subunit crosslinks identified by crosslinking mass spectrometry were used as distance constraints to filter the structural models and select the best-fit model. Quality evaluation of the structural model of the DNAJB6 monomer was made by uploading the pdb-file, enclosed in Supplementary Information, to https://swissmodel.expasy.org/qmean/ at the Expasy (Swiss Institute of Bioinformatics). The data show that the model has stereochemical quality comparable to that observed for experimental structures. The QMEAN4 score for the structural model is calculated as a linear combination from 4 statistical potential terms and transformed to a Z-score relating it to high resolution X-ray structures of similar size.
NMA simulation of DNAJB6 monomer
Normal mode analysis (NMA) simulations using the elastic network model on DNAJB6 were carried out using the ElNémo software35. NMA is used to probe motions in biological macromolecules, assuming that the protein behaves like a harmonic oscillator, from which one can obtain the normal modes and their corresponding frequencies and the majority of the conformations can be captured using the low-frequency normal modes. The structural model of the DNAJB6 monomer (model 5, from the Robetta server) with one Cα atom per block and 10 Å interaction distance cut-off criterion was used for the computation. The resulting low-frequency modes 7–11 were analyzed with the help of various softwares. The contact map was computed using the CMView software65, the vector representation and the dynamics movie were generated using the software VMD66.
Crosslink-assisted docking for structural model of DNAJB6 dimer
The inter-subunit crosslinks identified by crosslinking mass spectrometry were used as restraints to generate a structural model of a DNAJB6 dimer by docking. The HADDOCK (High Ambiguity Driven protein-protein DOCKing) program (http://haddock.chem.uu.nl/) is an information-driven flexible docking approach distinguished from ab initio docking methods in that it encodes information on protein interfaces to drive the docking process67. Docking was performed using the structural model of the DNAJB6 monomer (Fig. 1, model Robetta_5), with constraints applied for the residues K189 and K232. The interface of the DNAJB6 dimer model was evaluated using the PISA tool (http://www.ebi.ac.uk/pdbe/pisa/)46 available for calculations of structural and chemical properties of macromolecular surfaces and interfaces on structures.
SAXS data collection and analysis
The SAXS data were collected at the Max II beamline I911-SAXS68 at the MAX IV laboratory. The data was initially reduced and processed using an automatic pipeline of scripts developed at MAX-Lab and using the ATSAS tools69,70. Data were normalized to the intensity of the transmitted beam and buffer scattering was subtracted. Forward scattering I(0) and the radius of gyration Rg were estimated using the Guinier approximation71. Using Primus, we calculated the distance distribution functions P(R) of the scattering patterns and estimated the maximum particle dimensions Dmax. Primus also automatically calculates the excluded volume of the hydrated particle (Porod volume Vp)70 together with the distance distribution function. All data sets were modelled with DAMMIF and the ab initio model’s excluded volume Va was divided by two in order to estimate the mass70. We performed online size exclusion chromatography on DNAJB6 using an ÄKTA Pure (GE Healthcare, Uppsala, Sweden) FPLC. The sample, 250 μl 6.5 mg/ml DNAJB6, was applied to a Superdex 200 Increase 10/300 (GE Healthcare, Uppsala, Sweden) equilibrated with 20 mM phosphate buffer pH 8.0 and 150 mM NaCl. Flow rate was 0.2 ml/min and data acquired every minute.
Electron microscopy and single particle averaging
Gradient fixation
The polydispersity of the DNAJB6 oligomers prevented single-particle reconstruction on the samples earlier analyzed by negative stain EM26 hence in order to improve the specimen for EM studies we here fractionated the DNAJB6 oligomers using the GraFix procedure to reduce the sample complexity by a gradient fixation protocol72,73, in which the sample is subjected to weak intramolecular chemical crosslinking during density gradient ultracentrifugation. A 4 ml linear 5–30% (w/w) sucrose and 0–0.2% (w/w) glutaraldehyde gradient in buffer (20 mM NaPO4, 150 mM NaCl, pH 8.0) was prepared using a gradient master (Biocomp, Canada) mixer. In total, 200 µl of DNAJB6 solution (0.4 mg/ml) was added on top of the gradient and run for 16 h at 4 °C at a speed of 30,000 rpm in a Beckman SW 55 rotor. Fractions of approx. 250 µl were collected from the bottom of the tube using a fraction collector (Gilson, USA) and analyzed by blue native-PAGE on precast 4–16% Bis-Tris Gels (Life Technologies, Sweden) and scanned with an Epson Perfection V600 photo scanner.
Sample preparation and imaging
Samples (5 µl of each) from fractions 1–6 after GraFix were applied to glow-discharged continuous carbon-coated 400 mesh copper grids. The grids were subsequently blotted with filter paper, washed with two drops of milli-Q water and negatively stained with 2% (w/v) uranyl acetate. Fraction 5 obtained well-defined particles at relatively high concentration (~100 particles/image) and was selected for single-particle analysis. The sample was imaged using a JEM2100F electron microscope (JEOL, Japan) operated at 200 kV. Images were recorded on a DE-20 direct electron detector (Direct Electron, USA) at a magnification of ×30,000 and 0.9–1.7 µm defocus. The selected magnification resulted in a pixel size of 2.08 Å at the specimen level. Images were recorded using a frame rate of 20 frames/s and 2 s exposure time. The accumulated dose for the whole exposure was approximately 20 e−/Å2. A total of 40 images were recorded.
Image processing
For each exposure, the comprised frames were drift corrected using the DE_process_frames-2.7.1.py script74. The first five frames were excluded from the data due to large amounts of drift. The remaining frames (6–40) were processed using full frame alignment. The alignment procedure was iterated four times and the resulting drift-corrected images were imported to EMAN2 (version 2.12) for further processing75. First, defocus, particle separation and contrast were evaluated with e2evalimage.py. Two images were discarded due to high defocus values. Then, 4 344 particles were selected automatically from the remaining 38 images using e2boxer.py in swarm mode. False positives from the automated picking procedure, representing clusters of aggregated particles or staining artefacts, were discarded. For each image, the contrast transfer function (CTF) parameters were estimated using 160 × 160 pixels boxed out (particle containing) regions using e2ctf.py. Reference-free two-dimensional (2D) classification was performed using 3 717 phase-flipped particles with e2refine2d.py. 2D classes representing different orientations were selected for initial model generation using e2initialmodel.py, assuming asymmetric particles (sym = C1). The model with the best matching 2D class averages and model projections was selected as a starting model for three-dimensional (3D) refinement. First, a 3D refinement was performed with downscaled (4.16 Å/pixel) data aiming at low resolution (targetres = 25 Å) using e2refine_easy.py. A second round of 3D refinement was performed using the full-size (2.08 Å/pixel) data and the final map from the first 3D refinement as input, targeting medium resolution (targetres = 20 Å). The resolution reported was calculated at Fourier shell correlation (FSC) = 0.14376, following the gold-standard FSC procedure77 embedded in EMAN2. The presence of two-fold symmetry was tested by imposing this symmetry element along a chosen axis. Subsequently, reconstruction of a 3D map was made by applying C2 symmetry. Molecular graphics and analyses were performed with the UCSF Chimera package78 (http://www.rbvi.ucsf.edu/chimera).
Data availability
Datasets generated and analysed during the current study, which are not already included in this published article and its Supplementary Information files, are available from the corresponding author on reasonable request.
Electronic supplementary material
Acknowledgements
Recombinant expression of DNAJB6 protein was performed at the Lund Protein Production Platform, Lund University, Sweden (http://www.lu.se/lp3). The Crafoord Foundation is acknowledged for providing funding to CE to develop chemical crosslinking mass spectrometry as a complementary tool in structure biology, the Danish Council for Independent Research, Natural Sciences (grant no 0602-02380B) for providing funding to PH, and the Center for Innovative Medicine (CIMED), Karolinska Institute, for supporting EM infrastructure (HH).
Author Contributions
C.S., C.M., J.H., H.H. and C.E. designed the research. C.M. purified protein and C.M. and C.E. performed crosslinking, K.B. performed LC-MSMS, C.E., M.R., P.H. analyzed crosslink data. S.A.-K. and S.R. performed modelling and simulations. C.E. performed crosslink-assisted docking. G.R. and C.S. performed SAXS and C.S. processed and analyzed SAXS data. J.H. and H.H. performed and analyzed E.M. C.E., J.H., S.A.-K. and C.S. wrote the manuscript and all coauthors contributed to the final version.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
9/3/2021
The original online version of this Article was revised: In the original version of this Article, the “DNAJB6 monomer model_Robetta_5” file, the “DNAJB6 dimer model_Haddock” file and the “dnajb6_negstain_c2” file, which were included with the initial submission, was omitted. The Supplementary Information files now accompany the original Article.
Contributor Information
Christopher A. G. Söderberg, Email: christopher.soderberg@ri.se
Cecilia Emanuelsson, Email: Cecilia.Emanuelsson@biochemistry.lu.se.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-23035-9.
References
- 1.Walsh P, Bursać D, Law YC, Cyr D, Lithgow T. The J-protein family: modulating protein assembly, disassembly and translocation. EMBO Rep. 2004;5:567–571. doi: 10.1038/sj.embor.7400172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qiu XB, Shao YM, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006;63:2560–2570. doi: 10.1007/s00018-006-6192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langer T, et al. Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature. 1992;356:683–689. doi: 10.1038/356683a0. [DOI] [PubMed] [Google Scholar]
- 4.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craig EA, Marszalek J. How Do J-Proteins Get Hsp70 to Do So Many Different Things? Trends in Biochemical Sciences. 2017;42:355–368. doi: 10.1016/j.tibs.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai J, Douglas MG. A conserved HPD sequence of the J-domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding. J Biol Chem. 1996;271:9347–9354. doi: 10.1074/jbc.271.16.9347. [DOI] [PubMed] [Google Scholar]
- 7.Hu J, et al. The crystal structure of the putative peptide-binding fragment from the human Hsp40 protein Hdj1. BMC Struct. Biol. 2008;8:3. doi: 10.1186/1472-6807-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki H, et al. Peptide-Binding Sites As Revealed by the Crystal Structures of the Human Hsp40 Hdj1 C-Terminal Domain in Complex with the Octapeptide from Human Hsp70. Biochemistry. 2010;49:8577–8584. doi: 10.1021/bi100876n. [DOI] [PubMed] [Google Scholar]
- 9.Sha B, Lee S, Cyr DM. The crystal structure of the peptide-binding fragment from the yeast Hsp40 protein Sis1. Structure. 2000;8:799–807. doi: 10.1016/S0969-2126(00)00170-2. [DOI] [PubMed] [Google Scholar]
- 10.Seki N, et al. Cloning, tissue expression, and chromosomal assignment of human MRJ gene for a member of the DNAJ protein family. J. Hum. Genet. 1999;44:185–189. doi: 10.1007/s100380050139. [DOI] [PubMed] [Google Scholar]
- 11.Hunter PJ, Swanson BJ, Haendel MA, Lyons GE, Cross JC. Mrj encodes a DnaJ-related co-chaperone that is essential for murine placental development. Development. 1999;126:1247–1258. doi: 10.1242/dev.126.6.1247. [DOI] [PubMed] [Google Scholar]
- 12.Izawa I, et al. Identification of Mrj, a DnaJ/Hsp40 family protein, as a keratin 8/18 filament regulatory protein. J Biol Chem. 2000;275:34521–34527. doi: 10.1074/jbc.M003492200. [DOI] [PubMed] [Google Scholar]
- 13.Chuang J-Z, et al. Characterization of a Brain-enriched Chaperone, MRJ, That Inhibits Huntingtin Aggregation and Toxicity Independently. J. Biol. Chem. 2002;277:19831–19838. doi: 10.1074/jbc.M109613200. [DOI] [PubMed] [Google Scholar]
- 14.Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- 15.Fayazi Z, et al. A Drosophila ortholog of the human MRJ modulates polyglutamine toxicity and aggregation. Neurobiol Dis. 2006;24:226–244. doi: 10.1016/j.nbd.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Hageman J, et al. A DNAJB Chaperone Subfamily with HDAC-Dependent Activities Suppresses Toxic Protein Aggregation. Mol. Cell. 2010;37:355–369. doi: 10.1016/j.molcel.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Watson ED, Geary-Joo C, Hughes M, Cross JC. The Mrj co-chaperone mediates keratin turnover and prevents the formation of toxic inclusion bodies in trophoblast cells of the placenta. Development. 2007;134:1809–1817. doi: 10.1242/dev.02843. [DOI] [PubMed] [Google Scholar]
- 18.Dai YS, Xu J, Molkentin JD. The DnaJ-related factor Mrj interacts with nuclear factor of activated T cells c3 and mediates transcriptional repression through class II histone deacetylase recruitment. Mol Cell Biol. 2005;25:9936–9948. doi: 10.1128/MCB.25.22.9936-9948.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rose JM, Novoselov SS, Robinson PA, Cheetham ME. Molecular chaperone-mediated rescue of mitophagy by a Parkin RING1 domain mutant. Hum Mol Genet. 2011;20:16–27. doi: 10.1093/hmg/ddq428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, et al. The Hsp40 family chaperone protein DnaJB6 enhances Schlafen1 nuclear localization which is critical for promotion of cell-cycle arrest in T-cells. Biochem J. 2008;413:239–250. doi: 10.1042/BJ20071510. [DOI] [PubMed] [Google Scholar]
- 21.Dey S, Banerjee P, Saha P. Cell cycle specific expression and nucleolar localization of human J-domain containing co-chaperon Mrj. Mol. Cell. Biochem. 2009;322:137–142. doi: 10.1007/s11010-008-9950-y. [DOI] [PubMed] [Google Scholar]
- 22.Sarparanta J, et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet. 2012;44(450–5):S1–2. doi: 10.1038/ng.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruggieri, A. et al. DNAJB6 Myopathies: Focused Review on an Emerging and Expanding Group of Myopathies. Front. Mol. Biosci. 3 (2016). [DOI] [PMC free article] [PubMed]
- 24.Hanai R, Mashima K. Characterization of two isoforms of a human DnaJ homologue, HSJ2. Mol. Biol. Rep. 2003;30:149–153. doi: 10.1023/A:1024916223616. [DOI] [PubMed] [Google Scholar]
- 25.Andrews JF, et al. Cellular stress stimulates nuclear localization signal (NLS) independent nuclear transport of MRJ. Exp Cell Res. 2012;318:1086–1093. doi: 10.1016/j.yexcr.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mansson C, et al. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones. 2014;19:227–239. doi: 10.1007/s12192-013-0448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gillis J, et al. The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J Biol Chem. 2013;288:17225–17237. doi: 10.1074/jbc.M112.421685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mansson C, et al. Interaction of the molecular chaperone DNAJB6 with growing amyloid-beta 42 (Aβ42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J. Biol. Chem. 2014;289:31066–31076. doi: 10.1074/jbc.M114.595124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arosio P, et al. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 2016;7:10948. doi: 10.1038/ncomms10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kakkar, V. et al. TheS/T-Rich Motif in the DNAJB6 Chaperone Delays Polyglutamine Aggregation and the Onset of Disease in a Mouse Model. Mol. Cell 1–12 10.1016/j.molcel.2016.03.017 (2016). [DOI] [PubMed]
- 31.Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 32.Hoop CL, et al. Huntingtin exon 1 fibrils feature an interdigitated β-hairpin–based polyglutamine core. Proc. Natl. Acad. Sci. 2016;113:201521933. doi: 10.1073/pnas.1521933113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song Y, et al. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merkley ED, et al. Distance restraints from crosslinking mass spectrometry: Mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 2014;23:747–759. doi: 10.1002/pro.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suhre K, Sanejouand YH. ElNemo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:610–614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayward, S. & DeGroot, B. L. In Molecular modelling of proteins (ed. Kukol, A.) 89–106 (Humana Press, 2008).
- 37.de Vries SJ, van Dijk M, Bonvin AMJJ. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 38.Wälti MA, et al. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc. Natl. Acad. Sci. USA. 2016;113:E4976–84. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y, Li J, Jin Z, Fu Z, Sha B. The crystal structure of the C-terminal fragment of yeast Hsp40 Ydj1 reveals novel dimerization motif for Hsp40. J. Mol. Biol. 2005;346:1005–1011. doi: 10.1016/j.jmb.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 40.Rutsdottir G, et al. Structural model of dodecameric heat-shock protein Hsp21: Flexible N-terminal arms interact with client proteins while C-terminal tails maintain the dodecamer and chaperone activity. J. Biol. Chem. 2017;292:8103–8121. doi: 10.1074/jbc.M116.766816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacques DA, Trewhella J. Small-angle scattering for structural biology - Expanding the frontier while avoiding the pitfalls. Protein Sci. 2010;19:642–657. doi: 10.1002/pro.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Wu Y, Qian X, Sha B. Crystal structure of yeast Sis1 peptide-binding fragment and Hsp70 Ssa1 C-terminal complex. Biochem. J. 2006;398:353–360. doi: 10.1042/BJ20060618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Qian X, Sha B. The Crystal Structure of the Yeast Hsp40 Ydj1 Complexed with Its Peptide Substrate. Structure. 2003;11:1475–1483. doi: 10.1016/j.str.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Barends TRM, et al. Combining crystallography and EPR: Crystal and solution structures of the multidomain cochaperone DnaJ. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013;69:1540–1552. doi: 10.1107/S0907444913010640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xue LC, Dobbs D, Bonvin AMJJ, Honavar V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015;589:3516–3526. doi: 10.1016/j.febslet.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 47.Keskin O, Ma B, Nussinov R. Hot regions in protein-protein interactions: The organization and contribution of structurally conserved hot spot residues. J. Mol. Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 48.Blundell TL, Jhoti H, Abell C. High-Throughput Crystallography for Lead Discovery in Drug Design. Nat. Rev. Drug Discov. 2002;1:45–54. doi: 10.1038/nrd706. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt M, et al. Peptide dimer structure in an Aβ(1-42) fibril visualized with cryo-EM. Proc. Natl. Acad. Sci. USA. 2015;112:11858–11863. doi: 10.1073/pnas.1503455112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abelein A, et al. The hairpin conformation of the amyloid beta peptide is an important structural motif along the aggregation pathway. J Biol Inorg Chem. 2014;19:623–634. doi: 10.1007/s00775-014-1131-8. [DOI] [PubMed] [Google Scholar]
- 51.Hoyer W, Gronwall C, Jonsson A, Stahl S, Hard T. Stabilization of a beta-hairpin in monomeric Alzheimer’s amyloid-beta peptide inhibits amyloid formation. Proc Natl Acad Sci USA. 2008;105:5099–5104. doi: 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rauth, S. et al. High-affinity Anticalins with aggregation-blocking activity directed against the Alzheimer β-amyloid peptide. Biochem. J. 1563–1578 10.1042/BCJ20160114 (2016). [DOI] [PMC free article] [PubMed]
- 53.Cukalevski R, et al. The A beta 40 and A beta 42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chem. Sci. 2015;6:4215–4233. doi: 10.1039/C4SC02517B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stewart, K. L. & Radford, S. E. Amyloid plaques beyond Aβ: a survey of the diverse modulators of amyloid aggregation. Biophys. Rev. 10.1007/s12551-017-0271-9 (2017). [DOI] [PMC free article] [PubMed]
- 55.Jacques DA, Guss JM, Svergun DI, Trewhella J. Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:620–626. doi: 10.1107/S0907444912012073. [DOI] [PubMed] [Google Scholar]
- 56.Treweek TM, Meehan S, Ecroyd H, Carver JA. Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell. Mol. Life Sci. 2015;72:429–451. doi: 10.1007/s00018-014-1754-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nillegoda NB, et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature. 2015;524:247–251. doi: 10.1038/nature14884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Westhoff B, Chapple JP, van der Spuy J, Höhfeld J, Cheetham ME. HSJ1 Is a Neuronal Shuttling Factor for the Sorting of Chaperone Clients to the Proteasome. Curr. Biol. 2005;15:1058–1064. doi: 10.1016/j.cub.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 59.Gobom J, et al. Alpha-cyano-4-hydroxycinnamic acid affinity sample preparation. A protocol for MALDI-MS peptide analysis in proteomics. Anal. Chem. 2001;73:434–438. doi: 10.1021/ac001241s. [DOI] [PubMed] [Google Scholar]
- 60.Bernfur, K., Rutsdottir, G. & Emanuelsson, C. The chloroplast-localized small heat shock protein Hsp21 associates with the thylakoid membranes in heat-stressed plants. Protein Sci. 10.1002/pro.3213 (2017). [DOI] [PMC free article] [PubMed]
- 61.Rasmussen MI, Refsgaard JC, Peng L, Houen G, Hojrup P. CrossWork: software-assisted identification of cross-linked peptides. J Proteomics. 2011;74:1871–1883. doi: 10.1016/j.jprot.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 62.Peng L, Rasmussen MI, Chailyan A, Houen G, Hojrup P. Probing the structure of human protein disulfide isomerase by chemical cross-linking combined with mass spectrometry. J Proteomics. 2014;108:1–16. doi: 10.1016/j.jprot.2014.04.037. [DOI] [PubMed] [Google Scholar]
- 63.Vallat BK, et al. templates: Learning and benchmarks. 2010;76:930–945. doi: 10.1002/prot.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roy A, Kucukural A, Zhang Y. Function Prediction. Nat Protoc. 2011;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vehlow C, et al. CMView: Interactive contact map visualization and analysis. Bioinformatics. 2011;27:1573–1574. doi: 10.1093/bioinformatics/btr163. [DOI] [PubMed] [Google Scholar]
- 66.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;33:27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 67.Van Zundert GCP, et al. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 68.Labrador, A., Cerenius, Y., Svensson, C., Theodor, K. & Plivelic, T. The yellow mini-hutch for SAXS experiments at MAX IV Laboratory. J. Phys. Conf. Ser. In press, 72019 (2013).
- 69.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003;36:1277–1282. doi: 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 70.Petoukhov MV, et al. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guinier A. La diffraction des rayons X aux tres petits angles: applications a l’etude de phenomenes ultramicroscopiques. Ann. Phys. (Paris). 1939;12:161–237. [Google Scholar]
- 72.Kastner B, et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods. 2008;5:53–55. doi: 10.1038/nmeth1139. [DOI] [PubMed] [Google Scholar]
- 73.Stark, H. GraFix: Stabilization of fragile macromolecular complexes for single particle Cryo-EM. Methods in Enzymology481, (Elsevier Masson SAS 2010). [DOI] [PubMed]
- 74.Bammes BE, Chen DH, Jin L, Bilhorn RB. Visualizing and correcting dynamic specimen processes in TEM using a direct detection device. Microsc. Microanal. 2013;19:1320–1321. doi: 10.1017/S1431927613008593. [DOI] [Google Scholar]
- 75.Tang G, et al. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 76.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 77.Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nat Methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pettersen EF, et al. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets generated and analysed during the current study, which are not already included in this published article and its Supplementary Information files, are available from the corresponding author on reasonable request.