

Abstract
Nicotinic ACh receptors (nAChRs) are the best studied members of the superfamily of pentameric ligand‐gated ion channels (pLGICs). Neuronal nAChRs regulate neuronal excitability and neurotransmitter release in the nervous system and form either homo‐ or hetero‐pentameric complexes with various combinations of the 11 neuronal nAChR subunits (α2–7, α9, α10 and β2–4) known to exist in humans. In addition to their wide distribution in the nervous system, neuronal nAChRs have been also found in immune cells and many peripheral tissues. These nAChRs are important drug targets for neurological and neuropsychiatric diseases (e.g. Alzheimer's, schizophrenia) and substance addiction (e.g. nicotine), as well as in a variety of diseases such as chronic pain, auditory disorders and some cancers. To decipher the functional mechanisms of human nAChRs and develop efficient and specific therapeutic drugs, elucidation of their high‐resolution structures is needed. Recent studies, including the X‐ray crystal structures of the near‐intact α4β2 nAChR and of the ligand‐binding domains of the α9 and α2 subunits, have advanced our knowledge on the detailed structure of the ligand‐binding sites formed between the same and different subunits and revealed many other functionally important interactions. The aim of this review is to highlight some of the structural and functional findings of these studies and to compare them with recent breakthrough findings on other pLGIC members and earlier data from their homologous ACh‐binding proteins.
Linked Articles
This article is part of a themed section on Nicotinic Acetylcholine Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.11/issuetoc
Abbreviations
- AChBP
ACh‐binding protein
- cryo‐EM
cryo‐electron microscopy
- DHβE
dihydro‐β‐erythroidine
- ECD
extracellular domain
- HS
high sensitivity
- LS
low sensitivity
- nAChR
nicotinic ACh receptor
- pLGICs
pentameric ligand‐gated ion channels
- TM
transmembrane
- α‐Bgtx
α‐Bungarotoxin
Introduction
http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76 are the prototypic members of the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=697 family, also including the 5‐HT3 receptor, GABAA/C receptor, glycine (Gly) receptor and some invertebrate and prokaryotic receptors (Albuquerque et al., 2009; Nemecz et al., 2016). These receptors are also called Cys‐loop receptors, due to the existence of 13–14 conserved residues flanked by linked cysteines at the N‐terminal domain of each subunit; this disulfide bridge is, however, absent in the prokaryotic members. They form cation‐selective channels of five homologous subunits, each comprising an N‐terminal extracellular domain (ECD) of 210–250 amino acids, bearing the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=294 or ligand‐binding sites, a transmembrane (TM) domain of four α‐helices and a large cytoplasmic loop (110–270 amino acids). nAChRs are classified into muscle and neuronal receptors, with the latter being widely distributed in the peripheral and central nervous systems, regulating neuronal excitability and neurotransmitter release (Millar and Gotti, 2009; Yakel, 2010; Engel et al., 2015). Neuronal nAChRs are also found in the immune system and in various peripheral tissues (Wessler and Kirkpatrick, 2008; Beckmann and Lips, 2013). To date, 11 neuronal nAChR subunits have been characterized in humans (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=463–http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=468, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=469, α10, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=472–β4), forming either homopentamers (α7 or α9) or heteropentamers of various combinations (e.g. http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=465, α7β2, α2β2, α9α10, α4β2), with each subtype presenting distinct pharmacological and electrophysiological properties (Millar and Gotti, 2009; Taly et al., 2009). In all neuronal nAChRs, the ligand‐binding sites are formed between the ECDs of an α subunit and an adjacent β or α subunit. The most abundant and widely distributed neuronal nAChRs are the α4β2 and α7 subtypes, being important drug targets as they are implicated in several disorders of the CNS, including Alzheimer's and Parkinson's diseases, schizophrenia, depression, anxiety, attention deficit hyperactivity disorder and smoking addiction (Taly et al., 2009; Quik et al., 2011; Dineley et al., 2015).
Our knowledge of the overall structure of nAChRs firstly came from the cryo‐electron microscopy (cryo‐EM) model of the Torpedo fish nAChR (Unwin, 2005) and recently from the structures of other pLGIC members, such as the invertebrate glutamate‐gated chloride channel (GluCl) (Hibbs and Gouaux, 2011), the human β3 GABAA receptor (Miller and Aricescu, 2014), the mouse 5‐HT3 receptor (Hassaine et al., 2014), the human α3 Gly receptor (Huang et al., 2015), the zebrafish α1 Gly receptor (Du et al., 2015) and two bacterial pLGICs (Hilf and Dutzler, 2008; Bocquet et al., 2009). Interestingly, the above structures revealed the whole range of possible states of the channels (closed, open and desensitized), providing mechanistic insights into gating transition and desensitization. In addition, the ligand‐binding site of nAChRs was revealed in higher resolution by studies on ACh‐binding proteins (AChBPs), the structural surrogates of the nAChR‐ECDs, with which they share 15–25% identities (Brejc et al., 2001; Celie et al., 2004), and by the crystal structures of the mouse muscle α1‐ECD (Dellisanti et al., 2007) and chimeric proteins made up from nAChR‐ECDs and AChBP regions (Li et al., 2011; Nemecz and Taylor, 2011). The first X‐ray crystal structures of neuronal nAChRs appeared only in the last couple of years; in chronological order, these are the wild‐type human neuronal α9‐ and α2‐ECDs (Zouridakis et al., 2014; Kouvatsos et al., 2016), in the presence of ligands (agonists and/or antagonists) elucidated at resolutions of 1.7 and 3.2 Å, respectively, and the heteromeric near‐intact α4β2 nAChR bound to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2585 at 3.9 Å (Morales‐Perez et al., 2016).
Here, we report structural and functional insights on neuronal nAChRs available from the recent studies on α9‐ and α2‐ECDs and the near‐intact α4β2 nAChR and make some comparisons with structural data derived earlier from AChBPs and from the recent breakthrough studies on other members of the Cys‐loop superfamily. The scope of this review is to discuss recent advances in our understanding of (i) the neuronal nAChR ligand‐binding sites formed between the same (i.e. α2/α2) and different subunits (i.e. α4/β2, α9α10), (ii) the initial structural key events following agonist binding and (iii) the interactions between the ligand‐binding domain and the TM domain, coupling agonist binding to gating.
Overall architecture of neuronal nAChRs
The overall structure of nAChRs resembles a cylinder with pseudo‐pentameric symmetry between the five subunits with an ion‐conducting pore along the major axis (Unwin, 2005; Morales‐Perez et al., 2016) (Figure 1A–C). Each subunit comprises a large ECD with an N‐terminal α‐helix, 10 β‐strands (β1–β10), forming a β‐sandwich core stabilized by several inner hydrophobic residues, and a number of functionally important loops A–F, forming the ACh‐binding sites between specific subunits, as will be discussed later (Figure 1D). Interestingly, the N‐terminal α‐helix of the Torpedo nAChR (Unwin, 2005) adopts a different orientation from the α‐helix of the neuronal α4β2 nAChR (Morales‐Perez et al., 2016) and of the neuronal α9‐ and α2‐ECDs (Zouridakis et al., 2014; Kouvatsos et al., 2016). However, in the crystal structure of muscle α1‐ECD (Dellisanti et al., 2007), the corresponding α‐helix superimposes very well the helices of the above neuronal subunits. Whether this discrepancy reflects functional differences between the Torpedo and mammalian nAChRs or is due to experimental limitations remains elusive. The TM domain comprises four helices (M1–M4), perpendicularly spanning the membrane, packed in three concentric circles. The M2 helices form the inner circle or pore‐lining region; the M1 and M3 form the intermediate circle, which is stabilized by extensive intra‐ and inter‐subunit interactions wrapping the M2 helices bundle; and the M4 helices form a more loosely packed outer circle at the periphery of the TM domain (Morales‐Perez et al., 2016). The relative tilts and lateral shifts of the TM helices indicate whether the receptor is in a closed, open or desensitized state (Hassaine et al., 2014; Miller and Aricescu, 2014; Du et al., 2015; Huang et al., 2015; Morales‐Perez et al., 2016). The intracellular domain or M3–M4 loop varies in length between nAChR subunits and all other members of pLGICs, being mostly hydrophilic and probably extensively disordered. Interestingly, in the structures of the 5‐HT3 receptor (Hassaine et al., 2014) and α4β2 nAChR (Morales‐Perez et al., 2016), the post‐M3 domain of the intracellular loop seems to form an α‐helical segment called MX (Figure 1A, D), while in the cryo‐EM structure of the Torpedo nAChR, a similar segment, called MA, was found prior to the M4 helix (Unwin, 2005). Despite their significant physiological role in trafficking and assembly of pLGICs (Kracun et al., 2008; Han et al., 2013; Zuber and Unwin, 2013), the presence of these large cytoplasmic loops seems to be a major bottleneck for structural studies of full‐length receptors; interestingly, the available structures of eukaryotic pLGICs were derived only after extensive truncations of these domains, mainly inspired by their prokaryotic homologues (Hilf and Dutzler, 2008; Bocquet et al., 2009).
Figure 1.
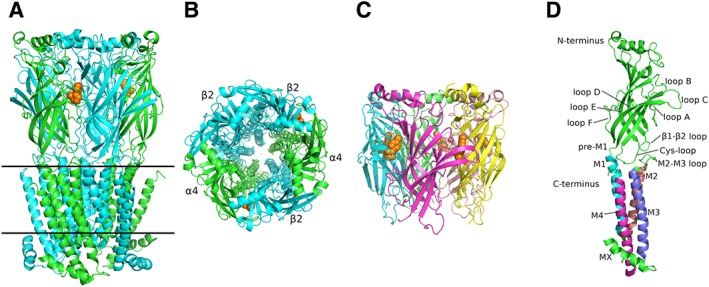
Overall architecture of the α4β2 nAChR and α2‐ECD. (A) View of α4β2 parallel to the plasma membrane (PDB ID: 5KXI) (Morales‐Perez et al., 2016). α subunits are shown in green and β in cyan, while nicotine is in orange spheres. Solid lines indicate the approximate limits of the membrane. (Β) View of α4β2 along the channel axis. Colour coding as in (A). (C) Side‐view of α2‐ECD (PDB ID: 5FJV) (Kouvatsos et al., 2016). Each of the α2 subunit is coloured differently and epibatidine is shown in orange spheres. (D) The protomer of the human α4 subunit participating in α4β2 nAChR (PDB ID: 5KXI). The critical domains, characteristic of pLGICs, are shown. The ECD and the intracellular helix (MX) are coloured in green, while each TM helix is in different colour. The coordinates of all the structures depicted were retrieved from Protein Data Bank (http://www.wwpdb.org), and PyMol (http://www.pymol.org) was used to generate the figures.
Ligand‐binding site
The ligand‐binding site of neuronal nAChRs is located at the interface between the ECDs of two adjacent subunits. These parts of the binding site are referred to as the principal or (+) side, conferred by an α subunit, and the complementary or (−) side, conferred by a β or α subunit. The binding site is mainly formed by six loops designated as loops A–F (Figure 1D). Loops A, B and C are situated on the principal side, whereas loops D, E and F are localized on the complementary side, as was initially shown in AChBPs (Brejc et al., 2001). The ligand‐binding site is surrounded and partially formed by several conserved aromatic residues along the various members of the Cys‐loop receptors family and the homologous AChBPs, which build the often termed ‘aromatic cage’.
Until recently, structural information on the ligand‐binding sites of neuronal nAChRs was only available from the X‐ray crystal structures of AChBPs (Brejc et al., 2001; Celie et al., 2004; Hansen et al., 2005) and their chimeras with nAChR domains (Li et al., 2011; Nemecz and Taylor, 2011). Engineered AChBPs towards specific nAChR subunits have greatly advanced our knowledge regarding structural issues and their correlation with the function of the corresponding nAChRs (Li et al., 2011; Nemecz et al., 2011; Shahsavar et al., 2015). This strategy has been very effective over the past few years, as in several cases, the mutants designed retained the AChBP solubility characteristics for ease of expression, purification and crystallization, and simultaneously depicted the nAChR‐binding sites with increased accuracy. The high identity of α7‐AChBP chimeras to α7 (up to 64%), crystallized either in apo or in agonist‐ or antagonist‐bound states (Li et al., 2011; Nemecz and Taylor, 2011), revealed important structural features of the α7 nAChR with plausible functional importance, as well as critical ligand–receptor interactions (Figure 2A). A more minimalistic approach, involving only three single‐point mutations at the (−) side of AChBP, was followed later to resemble the α4/α4 binding site of nAChRs (Shahsavar et al., 2015). Its crystal structures with two α4β2 agonists, NS3920 and NS3573, revealed their specific interactions with the α4/α4 binding site, while functional studies showed the contribution of these ligands to the activation of the α4β2 nAChR via the α4/α4 binding site (Shahsavar et al., 2015) (Figure 2B).
Figure 2.
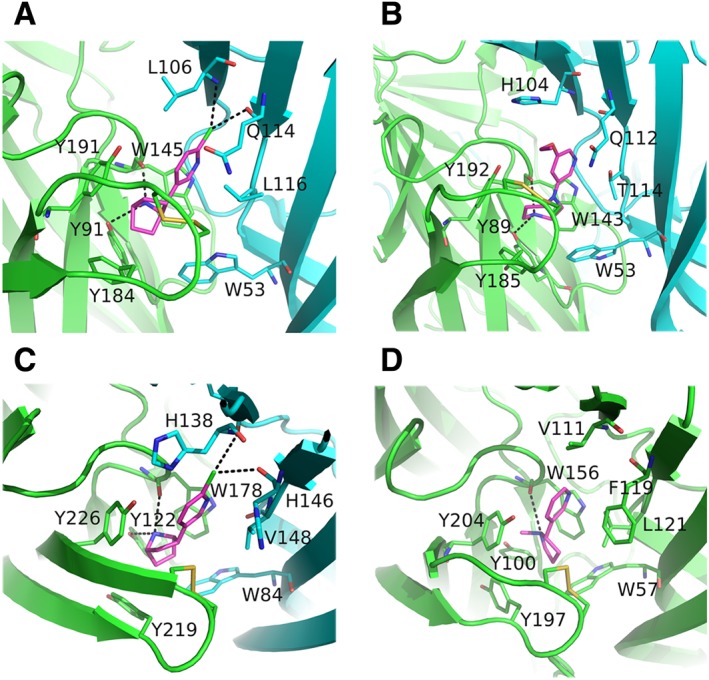
Close views of wild‐type or chimeric nAChR ligand‐binding sites. (A) The α7‐AChBP bound to epibatidine (PDB ID: 3SQ9) (Li et al., 2011). (B) Engineered AChBP towards α4/α4 bound to NS3920 (PDB ID: 4UM3) (Shahsavar et al., 2015). (C) The α2‐ECD bound to epibatidine (PDB ID: 5FJV) (Kouvatsos et al., 2016). (D) The α4β2 nAChR bound to nicotine (PDB ID: 5KXI) (Morales‐Perez et al., 2016). The principal sides are shown in green, the complementary in cyan and the agonists in magenta. Interactions are shown in black dashed lines. The coordinates of all the structures depicted were retrieved from Protein Data Bank (http://www.wwpdb.org), and PyMol (http://www.pymol.org) was used to generate the figures.
However, in the past couple of years, crystal structures of neuronal nAChR‐ECDs and of a near‐intact nAChR have emerged, shedding light on additional features absent from AChBPs (Zouridakis et al., 2014; Kouvatsos et al., 2016; Morales‐Perez et al., 2016). Interestingly, the structures of the full binding sites between α/α and α/β nAChR subunits are now fully elucidated in the pentameric assemblies of α2‐ECD (at 3.2 Å) and α4β2 nAChR (at 3.9 Å), respectively (Figure 2C, D). It is noteworthy that the latter structure also revealed the conformations of the β2/β2 and β2/α4 interfaces, which will be discussed later. The principal side contributes three highly conserved tyrosine residues located on loops A and C, and an invariant tryptophan residue on loop B to the aromatic cage, whereas the complementary side contributes a tryptophan located on loop D (Figure 2). Other nAChR residues involved in http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5348 and nicotine binding, inferred by the structures of α2‐ECD and α4β2 nAChR, respectively, are the cysteines located on the tip of loop C from the primary side and the hydrophobic residues of Val148 (α2 numbering) or Leu121 (β2 numbering) from the complementary side (Figure 2C, D). An important interaction occurring in the agonist‐bound nAChR resolved structures is a cation‐π interaction between a positively charged quaternary nitrogen of the ligand and the invariant tryptophan of loop B. This interaction was first revealed in AChBPs bound to several agonists and was considered as a molecular determinant for ligand binding and probably ligand orientation (Celie et al., 2004; Hansen et al., 2005). In addition, spectroscopic and crystallographic studies of AChBP complexes with benzylidene anabaseines revealed important interactions of the loop‐B tryptophan with the imine nitrogens of these ligands (Talley et al., 2006; Hibbs et al., 2009). It is worth noting that the tyrosine of loop A, despite its aromatic character, participates in ligand binding through hydrogen bonding, mediated by its hydroxyl group pointing to the ligand (Figure 2A–C). The tyrosines of loop C have also been shown to be essential for stabilizing several small ligands (Hansen et al., 2005), but also peptide toxins such as α‐conotoxins (Bourne et al., 2005) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3964 (α‐Bgtx) (Marinou and Tzartos, 2003; Dellisanti et al., 2007; Huang et al., 2013; Zouridakis et al., 2014).
The (+) side has been shown to have a dominant role in the orientation of bound ligands. For example, three structures with the agonist epibatidine have been published (AChBP, AChBP‐α7 chimera and α2‐ECD), and in all cases, unrestrained refinement has shown that epibatidine occupies the same space and essentially acquires the same orientation, despite the low conservation of the residues of the complementary sides (Hansen et al., 2005; Li et al., 2011; Kouvatsos et al., 2016). In addition, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4005 was co‐crystallized in the same orientation with AChBP (Hansen et al., 2005), an AChBP‐α7 chimera (Nemecz et al., 2011) and the (+) side of the monomeric α9‐ECD (Zouridakis et al., 2014); similarly, α‐Bgtx adopted the same orientation when bound to either the pentameric AChBP‐α7 chimera (Huang et al., 2013) or the monomeric α1‐ECD (Dellisanti et al., 2007) and α9‐ECD (Zouridakis et al., 2014). Finally, nicotine adopts the same orientation in its complexes with AChBP (Celie et al., 2004) and α4β2 nAChR (Morales‐Perez et al., 2016), involving conserved residues of the (+) binding sites.
Given the high degree of identity among α subunits, especially between the loops involved in the (+) side of the binding site, the differentiations on the (−) side have been assumed as determinants of ligand selectivity (Rucktooa et al., 2012). Tryptophan of loop D, the sole conserved aromatic residue in the complementary nAChR subunits, has been shown to be critical for the high‐affinity binding of epibatidine (Hansen et al., 2005) and α‐Bgtx to AChBP (Hansen et al., 2004). All other non‐conserved residues of the complementary side confer selectivity for ligands. For example, it was recently shown that three hydrophilic residues, His142, Gln150 and Thr152, on the complementary side of the α4 subunit and the hydrophobic Val136, Phe144 and Leu146 on corresponding positions of β2 comprise most of the differences between the core of α4/α4 and α4/β2 binding sites respectively. These substitutions are responsible for differences in both agonist‐binding affinities (Ahring et al., 2015) and agonist sensitivities (Harpsoe et al., 2011) between the two sites. Also, in the case of α7 nAChR, the importance of Glu57, which in all other subunits is lysine or arginine and is located just above the invariant tryptophan of loop D, was shown for the selective binding of an anthelmintic agent (Bartos et al., 2009). In addition, the sequence‐variable loop F has been shown to be a key determinant of high‐affinity binding and selectivity of pinnatoxins to nAChR subtypes and AChBPs (Bourne et al., 2015). Perhaps, the most divergent nAChR subunits, in terms of the components' composition of the complementary side, are the α9 and α10 subunits, for which detailed discussion will follow.
Structural rearrangements of the ECD upon ligand binding and their functional importance
The first conformational changes upon agonist binding were clearly shown when comparing the structures of AChBPs bound to nAChR agonists and antagonists. The most profound change is on the conformation of loop C, which upon binding of agonists makes significant inward movements to embrace them, whereas adopting an extended conformation upon antagonist binding (Brams et al., 2011). The most marked rearrangements of loop C occurred in the complexes of AChBPs with α‐conotoxin‐ImI and epibatidine (Hansen et al., 2005), where loop C swung as much as 11 Å between these two extreme positions. The same observation was made when comparing the X‐ray crystal structures of the agonist‐bound α2‐ECD (Kouvatsos et al., 2016) and α4β2 nAChR (Morales‐Perez et al., 2016) with the α‐Bgtx‐bound α1‐ECD (Dellisanti et al., 2007) and α9‐ECD (Zouridakis et al., 2014). Functional studies on the muscle‐type nAChR have shown that the closure of loop C upon ACh binding disrupts a conserved salt‐bridge between β7 and β10 strands, triggering a cascade of events leading to channel opening (Mukhtasimova et al., 2005). The structures of the free and agonist‐bound AChBPs revealed the salt‐bridge disruption by the closure of loop C upon agonist binding, which brought the conserved tyrosine of loop C in close proximity with the highly conserved Lys139 on the β7 strand (AChBP numbering) and weakened its interaction with the also conserved Asp194 on the β10 strand. Indeed, the same observation was made in the structures of α2‐ECD (Kouvatsos et al., 2016) and α4β2 nAChR (Morales‐Perez et al., 2016) bound to epibatidine or nicotine respectively (Figure 3A, B).
Figure 3.
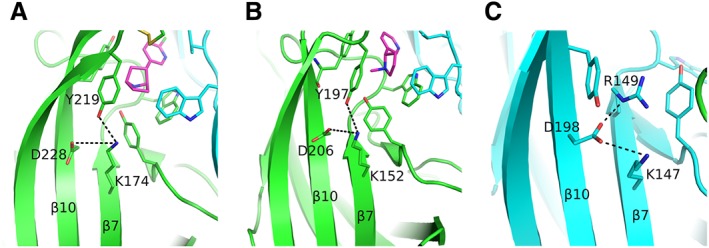
Rearrangements upon agonist binding. (A) The epibatidine‐bound α2 subunit showing the interaction of loop‐C Tyr219 with the β7‐strand Lys174, probably weakening the interaction between the residues of β7 and β10 strands. The (+) side is shown in green, the agonist in magenta and the (−) subunit in cyan. (B) Similarly for the α4 subunit bound to nicotine. Colours as in (A). (C) The β2‐subunit Asp198 on β10‐strand acquires a rotamer never observed before in α subunits. It is further stabilized by interactions with two positively charged residues of β7 strand. The β2 subunit is shown in cyan. α4 and β2 subunits were retrieved from PDB ID: 5KXI (Morales‐Perez et al., 2016) and α2 subunit from PDB ID: 5FJV (Kouvatsos et al., 2016). The coordinates of all the structures depicted were retrieved from Protein Data Bank (http://www.wwpdb.org), and PyMol (http://www.pymol.org) was used to generate the figures.
Another network of interactions between elements of the lower part of the ECD, called the membrane‐facing network, was found in a series of recently determined crystal structures, which in the case of the muscle nAChR has been shown by functional studies to contribute to the signal transduction (Mukhtasimova and Sine, 2013). More specifically, this network interconnects the invariant arginine at the very end of β10 strand with conserved negatively charged residues of Cys‐loop, β1–β2 loop and loop F (Figure 4). The high‐resolution monomeric structure of α9‐ECD (up to 1.7 Å) revealed these interactions in substantial accuracy, while the existence of the membrane‐facing network was also shown adequately in the pentameric structure of α2‐ECD and in the structures of α4β2 nAChR, GABAA receptors (Miller and Aricescu, 2014), 5‐HT3 receptors (Hassaine et al., 2014) and glycine receptors (Du et al., 2015; Huang et al., 2015), despite their much lower resolutions (2.97–3.90 Å) (Figure 4). Interestingly, in full receptors, this network is sandwiched between two aromatic conserved residues of loop F and Cys‐loop, and its location seems to be indicative of the state of the channel, as can be observed when comparing the closed, open and desensitized of the glycine receptor (Du et al., 2015) (Figure 4B). The network is well superimposed between the open and desensitized states, whereas in the closed state a rigid movement towards the channel pore is observed (Figure 4B). However, if one compares different receptors, co‐localization of this network is also observed between different states of the channels (e.g. closed 5‐HT3 receptor and desensitized α4β2 nAChR), while divergence between receptors of a similar state is noticed (e.g. α4β2 nAChR and GABAA receptor). It is also possible that this network facilitates the inter‐subunit motions and therefore the transitions among the various functional states (Miller and Aricescu, 2014; Du et al., 2015).
Figure 4.
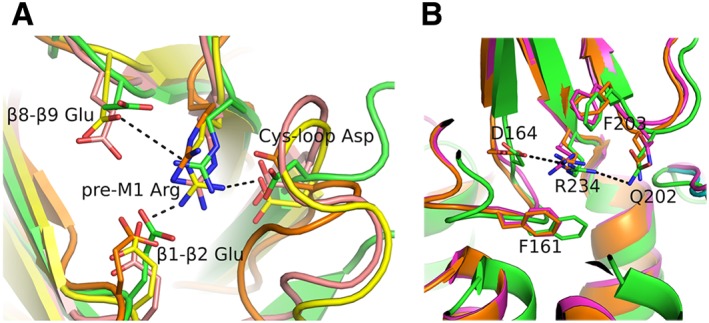
Membrane‐facing networks. (A) Close view of the interactions between structural elements at the lower part of the ECDs, viewed from the bottom of the ECD. These interactions are present in most of the resolved structures of pLGICs [α9‐ECD in green, PDB ID: 4D01 (Zouridakis et al., 2014); GABAA receptor in pink, PDB ID: 4COF (Miller and Aricescu, 2014); 5‐HT3 receptor in yellow, PDB ID: 4PIR (Hassaine et al., 2014); GLIC in orange, PDB ID: 3EAM (Bocquet et al., 2009)]. The invariant arginine at the end of β10 strand or pre‐M1 loop interconnects Cys‐loop, β1–β2 loop and in most cases β8–β9 loop. (B) Side view of the superimposed structures of the glycine receptor determined in closed (PDB ID: 3JAD), open (PDB ID: 3JAE) and desensitized (PDB ID: 3JAF) states (Du et al., 2015), shown in green, magenta or orange respectively. The interaction network in (B) is shown in equatorial orientation, while the aromatic residues that sandwich the charged residues of the network are in axial positions. Representative interactions are shown in black dashed lines. The coordinates of all the structures depicted were retrieved from Protein Data Bank (http://www.wwpdb.org), and PyMol (http://www.pymol.org) was used to generate the figures.
In conclusion, the closure of loop C triggers a cascade of events starting from the ligand‐binding site, propagating to the membrane‐facing network and finally ending down to the TM helices opening the gate (Purohit et al., 2007; Calimet et al., 2013; Sauguet et al., 2014).
Further functional insights from the structures of α4β2 nAChR and α2‐ and α9‐ECDs
α4/β2, α2/α2 and α4/α4 binding sites
The α2 subunit, which is not known to form functional homo‐pentamers, is incorporated in heteropentameric neuronal nAChRs mainly with β2 or β4 subunits and along with the α4 and α7 subunits is one of the more abundantly expressed nAChR subunits in primate brain (Han et al., 2000). Similar to the α4 subunit, it has been shown that when α2 is co‐expressed with the β2 subunit in Xenopus laevis oocytes, two subtypes of α2β2 nAChR are formed with either low or high ACh sensitivity [low sensitivity (LS) or high sensitivity (HS) respectively] (Khiroug et al., 2004; Dash et al., 2014). In the case of α4β2 nAChRs, the LS and HS subtypes display different ligand specificity, unitary conductance and desensitization kinetics (Nelson et al., 2003). It has been clearly demonstrated that these differences arise from the altered stoichiometry, since the LS subtype has, in addition to the two α4/β2 ligand‐binding sites, another one at the α4/α4 interface (Mazzaferro et al., 2011). It was recently shown that α2β2 nAChRs also exist in two stoichiometries, and in a similar fashion to the α4β2 nAChR, the LS and HS subtypes have stoichiometries of (α2)3(β2)2 or (α2)2(β2)3, respectively, with the former presenting an additional α2/α2 binding site to the previously known α2/β2 (Dash et al., 2014; Kouvatsos et al., 2016).
The recent crystal structure of the α4β2 nAChR involves its HS subtype in complex with the agonist nicotine (Morales‐Perez et al., 2016). Nicotine, which is known to up‐regulate the expression of the α4β2 HS subtype and also displays a ~100‐fold higher affinity for this subtype compared with the LS one (Nelson et al., 2003), was bound in the same orientation as was determined previously in its complex with AChBP (Celie et al., 2004), despite the significant differences of the complementary sides between these proteins. Also, the epibatidine bound to the pentameric α2‐ECD adopted the same orientation as in its complex with AChBP (Hansen et al., 2004). These observations underlie once again the dominant role of the principal side in ligand binding (Zouridakis et al., 2014). The lower affinities of nicotine for the α4/α4 and α2/α2 binding sites compared with the α4/β2 and α2/β2 sites may be attributed to the more polar character of the (−) sides of α4 or α2 compared with the (−) side of β2, which does not favour the accommodation of aromatic or hydrophobic groups.
Notably, the ECDs of α2 and α4 subunits present 77% identity and 91% similarity, the highest values among the nAChR subunits; thus, the structure of the α2/α2 interface elucidated by the crystal structure of α2‐ECD can be considered as the closest surrogate of the α4/α4 binding site. Indeed, the principal sides of α2 and α4 are almost identical, with the exception of two residues on loop C, which nevertheless do not alter either the hydrophilicity or the charge of the region (Lys for Arg and Asp for Glu). However, their (−) sides are more distant compared with their overall differentiations, which may partly explain the variation in inhibition potency by the specific antagonist dihydro‐β‐erythroidine (DHβE) between the α2β2 and α4β2 nAChRs (Khiroug et al., 2004). The inhibition potency of DHβE in α4β2 nAChR is higher by threefold compared with α2β2, while Khiroug et al. (2004) showed that this difference is sufficient to distinguish the various nAChR populations in stratum oriens interneurons. Whether this difference arises due to the presence of unorthodox binding sites between two α subunits has not been resolved, but it has been clearly demonstrated that DHβΕ displays high‐affinity competitive antagonism for the α4/α4 binding site and that inhibits ACh activation via that site (Mazzaferro et al., 2011). It is of note that the crystal structure of DHβE bound to AChBP shows its hydrophilic carbonyl facing loop D, while its hydrophobic multicyclic domain faces loop E, which is the loop that the two subunits differ the most, with that of α4 being more hydrophilic than that of the α2 subunit.
β2/α4 and β2/β2 interfaces
Functional studies over the years have shown that the β2 subunit does not offer the principal side for ACh or other nAChR ligands, despite its high sequence identity in loops involved in ligand binding. The crystal structure of the α4β2 nAChR revealed molecular‐level details that offer a full explanation of this deficiency (Morales‐Perez et al., 2016). The presence of arginine at the bottom of the β2/α4 and β2/β2 interfaces, along with the absence of the loop‐C Tyr192, precludes the binding of nicotine, firstly due to extensive changes in the rotamers of the ligand‐binding residues and secondly due to alterations in the charge distribution of the putative binding cavity. Arg149 (β2 numbering), which is glycine in α2 and α4 subunits, intrudes the binding side and coordinates with an unprecedented manner two conserved aromatic residues of the binding cavity (Figure 5). The loop‐C Tyr196 adopts a downwards conformation occupying the space where in orthodox binding sites Tyr192 lies, while the loop‐A Tyr95 recedes towards the side walls of the binding side overlapping spatially with the indole group of loop‐B tryptophan residue in orthodox binding sites. As a result, the guanidinium group of Arg149 is being sandwiched by these two tyrosines, while the loop‐B Trp151 adopts a previously unobserved conformation towards the β4–β5 loop of the complementary β2 or α4 subunit. Interestingly, this rotamer poses the indole ring of Trp151 to occupy space available only in the α4β2 nAChR and α2 homopentameric structures, in contrast to all AChBPs, where that cavity is unavailable and is being occupied by the β4–β5 loop (Figure 5).
Figure 5.
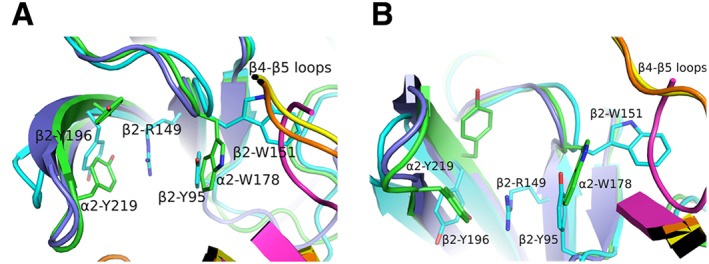
Comparison of subunit interfaces. (A) Superposition of β2/α4 [in cyan or orange, respectively; PDB ID: 5KXI (Morales‐Perez et al., 2016)], α2/α2 [in green or yellow, respectively; PDB ID: 5FJV (Kouvatsos et al., 2016)] and nicotine‐bound AChBP [in purple or magenta, respectively; PDB ID: 1UW6 (Celie et al., 2004)]. The lack of one tyrosine in loop C of the β2 subunit allows the radical rotation of its Tyr196 to occupy space that in α subunits is occupied by the other tyrosine (e.g. α2‐Tyr219). β2‐Tyr196 along with the β2‐Tyr95 from loop A stabilize the β2‐Arg149 that rams the cavity. This is possible only after β2‐Tyr95 recedes towards the complementary subunit, occupying the space where in α subunits the loop‐B tryptophan (e.g. α2‐Trp178) is found. As a result, β2‐Trp151 presents an extreme rotational movement towards β4–β5 loop. Notably, the α2‐ECD pentameric structure shows that this rotamer of loop‐B tryptophan is also possible in α subunits, but not in AChBPs where this space is occupied by β4–β5 loop. (B) The same as (A) rotated by 90o. The coordinates of all the structures depicted were retrieved from Protein Data Bank (http://www.wwpdb.org), and PyMol (http://www.pymol.org) was used to generate the figures.
Moreover, in the α4β2 nAChR structure, where both agonist‐bound and ligand‐free interfaces exist (corresponding to α4/β2 and β2/β2 or β2/α4, respectively), a subtle difference in the rotameric conformation of the highly conserved β10 aspartic acid is observed and which could be ascribed to the positioning of β2 Arg149 (Figure 3C). The β2 Asp198 adopts a rotamer that allows a closer interaction with both Arg149 and Lys145, whereas in the α4 subunit, the corresponding Asp206 is found farther from the corresponding lysine, existing in another rotameric form (Figure 3B). Similarly, in the agonist‐bound α2‐ECD, the equivalent Asp228 has the same orientation as in the case of α4 (Figure 3A), as was previously discussed.
α9‐ and α10‐containing binding sites
The α9α10 nAChR is an atypical nAChR heteropentamer, since it is composed only of α subunits (Elgoyhen et al., 1994; 2001; Sgard et al., 2002). Furthermore, it displays a very distinct pharmacological profile that fits neither the muscarinic nor the nicotinic classification scheme of ACh receptors (Verbitsky et al., 2000; Elgoyhen et al., 2001) and shares pharmacological properties with other members of the Cys‐loop family (Rothlin et al., 1999; 2003). In addition, nicotine and other nicotinic agonists, like cytisine and epibatidine, behave as antagonists of the α9 and α9α10 nAChRs, contrary to other nAChRs (Verbitsky et al., 2000; Elgoyhen et al., 2001). α9α10 nAChRs are found in sympathetic neurons, in the inner ear, skin keratinocytes and immune cells (e.g. lymphocytes), being a potential target for the therapy of diverse diseases, such as chronic pain, auditory disorders and breast and lung cancers (Elgoyhen et al., 2009; McIntosh et al., 2009; Wu and Lukas, 2011; Romero et al., 2017).
It has been shown that mammalian α9 subunits also form functional homomeric α9 receptors with similar efficacy regarding ACh to that of the heteromeric α9α10 nAChR (Elgoyhen et al., 1994, 2001). In contrast, rat and human α10 subunits do not form functional channels when expressed heterologously (Elgoyhen et al., 2001; Sgard et al., 2002). Based on these data, it was originally proposed that α10 might serve as a β‐subunit of heteromeric receptors, contributing to the (−) side of the agonist‐binding site (Elgoyhen and Katz, 2012). It has also been demonstrated that the α9α10 nAChR exists in both stoichiometries of (α9)2(α10)3 (Plazas et al., 2005) and (α9)3(α10)2 (Indurthi et al., 2014), with the latter presenting an additional LS α9/α9 binding site, as in the case of α4β2 nAChRs.
A recent study based on site‐directed mutagenesis, protein expression, electrophysiology and molecular docking showed that in addition to α9, the α10 subunit also contributes to the principal component of the binding site (Boffi et al., 2017). Thus, four different binding sites seem to be plausible in α9α10 nAChRs: the α9/α9, α9/α10, α10/α9 and the α10/α10. Moreover, this study demonstrated that the contribution of α9 and α10 to the complementary component of mammalian α9α10 nAChR is non‐equivalent, since mutation of the conserved tryptophan residue of loop D on α10 subunits did not impair the binding of ACh or α‐Bgtx, in contrast to the same mutation in the α9 subunit. The dominant role of the primary side of α10 subunit on activation of the α9α10 ion channel was also displayed previously by Azam et al. (2015), with the use of mutated residues on loops C and E of α10 and α9 subunits, respectively, to initially determine the binding site that affected the potency of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4008 potency (Azam et al., 2015). It was shown that mutations of non‐aromatic residues on the α10 (+) side increased the EC50 of ACh by 20‐ to 40‐fold, similarly to a mutation on the α9 (−) side, which decreased the sensitivity by more than 30‐fold.
Antagonism of α9α10 nAChRs by classical nAChR agonists
Even more profoundly than in the complementary side of α4, an uncommon accumulation of charged residues on the (−) sides of α9 and α10 subunits exists. The crystal structure of the α9‐ECD revealed that Arg59 and Asp121 emerging from loops D and E, respectively, form a salt bridge (Zouridakis et al., 2014), while these charged residues alter radically the physicochemical properties of the α9 (−) side. Furthermore, molecular dynamic calculations showed that this interaction was retained for most of the time in the modelled α9 homopentamers as well as in α9α10 heteropentamers (Figure 6), occupying a relative large space in the binding cavities, and that most probably the presence of these residues could interfere with the loop‐C closure (Azam et al., 2015). It should also be noted that in the case of α10, an additional arginine residue (Arg119) exists in its complementary side (Figure 6C).
Figure 6.
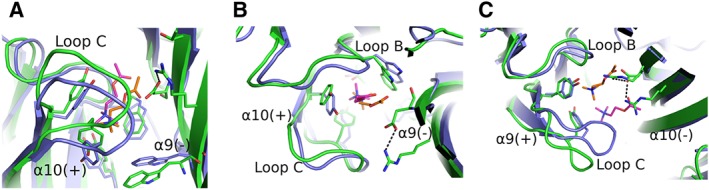
Models of α9/α10 and α10/α9 binding sites. (A–C) Superpositions of the ACh‐bound AChBP crystal structure [AChBP in blue; ACh in orange; PDB ID: 3WIP (Olsen et al., 2014)] with models of the ACh‐bound α9α10 binding sites (α9 and α10 in green; ACh in magenta) (Azam et al., 2015). (A) Side‐view of the α10/α9 interface, showing a similar binding mode for ACh with that in AChBP, although ACh and loop C are shifted upwards. (B) The same as in (A), rotated by 90o, also showing a lateral shift of loops B and C of α10(+) side. The stable salt bridge from the α9(−) side is also shown. (C) Top‐view of the α9/α10 interface, showing an extreme shift of ACh outwards, causing an equal shift of α9(+) loop C. A second arginine from α10(−) side penetrates the binding cavity, forming an uncommon charged environment. All interactions are shown in black dashed lines. The coordinates of all the structures depicted were retrieved from Protein Data Bank (http://www.wwpdb.org), and PyMol (http://www.pymol.org) was used to generate the figures.
In the light of these findings, it is plausible that the mode of engulfment of a ligand by loop C, determining its function as an agonist or antagonist, may differ significantly in α9α10 nAChRs, compared with the other nAChR subtypes, which could explain the conversion of classical agonists to antagonists in the case of α9‐containing nAChRs. More specifically, the presence of arginine residues in the (−) side of α9 or α10 could perturb the access of the quaternary ammonium of ACh to the binding pocket (Figure 6). This resembles what has been recently described in the crystal structure of the α4β2 nAChR, where three hydrophobic residues on the β2(−) side are replaced by polar ones on the α4(−) side. It has been suggested that this difference in chemical environment may be the reason for the lower sensitivity of the α4/α4 binding site to nicotine in the (α4)3(β2)2 stoichiometry (Morales‐Perez et al., 2016). It therefore seems that in the case of α9α10 nAChRs, the complementary side may also make contributions to the orientation of specific ligands, which in most cases is governed by the principal side.
In addition, a nicotine molecule bound to the α9α10 nAChR subtype with the expected orientation, as inferred from the structures of the nicotine‐bound AChBP (Celie et al., 2004) and α4β2 nAChR (Morales‐Perez et al., 2016), would probably have the hydrophobic pyridine exposed to the exceptionally charged complementary side of the α9α10 (similar when either α9 or α10 participates on the complementary side), indicating an alternative binding mode of nicotine, which could probably impose a less closed loop C conformation. Unfortunately, the above hypothesis has not been evaluated experimentally with electrophysiological recordings, since we and others have not achieved functional expression of α9/α10 nAChR mutants, bearing substitutions of the charged residues on the complementary side, in Xenopus oocytes.
Finally, in a recent study, the structure of AChBP determined with the α4β2 and α2β2 antagonist DHβE surprisingly revealed that the closure of the loop C was similar with that obtained with agonists, but also revealed a shift of loop C perpendicular to previously observed loop‐C movements, suggesting that DHbE may antagonize nAChRs via a different mechanism compared to prototypical antagonists and toxins (Shahsavar et al., 2012). In a similar fashion, in the case of α9α10 nAChRs, even a similar shift of the closed loop C due to a slightly different orientation of nicotine or epibatidine after their ‘repulsion’ from the (−) sides of the negative charged residues might account for their activity as antagonists.
Conclusions and future perspectives
During the past few years, there has been a remarkable accumulation of important structural and functional knowledge on neuronal nAChRs; notably the first crystal structures appeared on the ECDs of the α9 (Zouridakis et al., 2014) and α2 (Kouvatsos et al., 2016) subunits and on the near‐intact heteromeric α4β2 nAChR (Morales‐Perez et al., 2016).
The crystal structures of the monomeric α9‐ECD in its complexes with two antagonists revealed the interactions between the (+) side of α9 with antagonists at resolutions up to 1.7 Å, which is the highest reported yet for any member of the Cys‐loop receptor superfamily. The structure of α9‐ECD clearly showed a membrane‐facing network, previously shown to be functionally important in the muscle nAChR (Mukhtasimova and Sine, 2013), whose existence was also confirmed in the subsequent structures of the α2‐ECD and the α4β2 nAChR. Interestingly, α2‐ECD was crystallized in its pentameric complex with epibatidine, revealing the structure of the full α2/α2 binding site, previously suggested to exist in the LS subtype of α2β2 nAChRs (Dash et al., 2014) in a similar fashion to the α4β2 nAChR (Nelson et al., 2003). Given that the similarity of the α9‐ and α2‐ECDs with all other nAChR‐ECDs is far higher than that of AChBPs, the structures of α9‐ and α2‐ECDs should serve as a better template for modelling other nAChR‐ECDs of yet unknown structures. Also, chimeric constructs of the pentameric α2‐ECD, carrying the binding sites between other α or β nAChR subunits, should provide a more accurate approach to elucidate the crystal structures of other neuronal nAChR‐binding sites than using AChBPs. This strategy has already proven successful in the case of chimeric α7‐AChBPs (Li et al., 2011; Nemecz and Taylor, 2011), which also provided a much better template for computer‐based screening of novel ligands for α7 nAChR (Akdemir et al., 2012).
Finally, the crystal structure of the HS subtype of the near‐intact α4β2 nAChR, among others, revealed the structure of the α4/β2 binding site and the organization of the TM helices at the desensitized state of the channel (Morales‐Perez et al., 2016). Importantly, this study also introduced an invaluable methodology for the expression and purification of single stoichiometries of other complex heteromeric nAChRs, which is prerequisite for their crystallization. At the same time, pioneering structural studies for other members of the pLGIC family have emerged (Hassaine et al., 2014; Miller and Aricescu, 2014; Du et al., 2015; Huang et al., 2015), also after substitution of their large intracellular loops with short loops mainly inspired by their prokaryotic homologues.
The above advancements are expected to facilitate the structural studies of many other nAChRs, needed for the design of highly specific and effective drugs for individual subtypes. Indeed, the orthosteric ligand‐binding sites in nAChRs are highly conserved at their principal side and only subtle differences in residues of their complementary sides confer subtype‐selectivity to drugs, which may be revealed only by high‐resolution structural studies of the different nAChR ligand‐binding sites.
Of great importance would also be the elucidation of the topology and the structure of allosteric binding sites in nAChRs, which are reviewed by Wang and Lindstrom in this issue. Briefly, such sites have been identified on the ECD and TM domains of several nAChRs; moreover, several positive and negative allosteric modulators have been identified to bind to these sites potentiating or attenuating the efficacy of nAChR classical agonists respectively (Arias, 2010; Chatzidaki and Millar, 2015). Due to the low conservation of allosteric binding sites, the use of allosteric modulators specifically targeting distinct nAChR subtypes has gained ground for the future therapeutic approaches against nAChR‐related diseases (Chatzidaki and Millar, 2015). Chimeric AChBPs have provided an invaluable tool for the identification and structure elucidation of nAChR allosteric binding sites, as well as for high‐throughput drug screening for novel allosteric modulators (Spurny et al., 2015). Again, the use of chimeric α2‐ECDs could, however, be an even more accurate approach for such studies.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The research data of our research group discussed in this review article were supported by the EC FP7 grants NeuroCypres (202088) and REGPOT‐NeuroSign (264083), the Greek General Secretariat for Research and Technology Aristeia grant (1154) and a grant from Stavros Niarchos Foundation.
Giastas, P. , Zouridakis, M. , and Tzartos, S. J. (2018) Understanding structure–function relationships of the human neuronal acetylcholine receptor: insights from the first crystal structures of neuronal subunits. British Journal of Pharmacology, 175: 1880–1891. doi: 10.1111/bph.13838.
Contributor Information
Marios Zouridakis, Email: marzouri@gmail.com.
Socrates J Tzartos, Email: stzartos@gmail.com.
References
- Ahring PK, Olsen JA, Nielsen EO, Peters D, Pedersen MHF, Rohde LA et al. (2015). Engineered α4β2 nicotinic acetylcholine receptors as models for measuring agonist binding and effect at the orthosteric low‐affinity α4‐α4 interface. Neuropharmacology 92: 135–145. [DOI] [PubMed] [Google Scholar]
- Akdemir A, Edink E, Thompson AJ, Lummis SC, Kooistra AJ, de Graaf C et al. (2012). Identification of novel alpha7 nicotinic receptor ligands by in silico screening against the crystal structure of a chimeric alpha7 receptor ligand binding domain. Bioorg Med Chem 20: 5992–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89: 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The concise guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias HR (2010). Positive and negative modulation of nicotinic receptors. Adv Protein Chem Struct Biol 80: 153–203. [DOI] [PubMed] [Google Scholar]
- Azam L, Papakyriakou A, Zouridakis M, Giastas P, Tzartos SJ, McIntosh JM (2015). Molecular interaction of α‐conotoxin RgIA with the rat α9α10 nicotinic acetylcholine receptor. Mol Pharmacol 87: 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Price KL, Lummis SC, Bouzat C (2009). Glutamine 57 at the complementary binding site face is a key determinant of morantel selectivity for α7 nicotinic receptors. J Biol Chem 284: 21478–21487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann J, Lips KS (2013). The non‐neuronal cholinergic system in health and disease. Pharmacology 92: 286–302. [DOI] [PubMed] [Google Scholar]
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M et al. (2009). X‐ray structure of a pentameric ligand‐gated ion channel in an apparently open conformation. Nature 457: 111–114. [DOI] [PubMed] [Google Scholar]
- Boffi JC, Marcovich I, Gill‐Thind JK, Corradi J, Collins T, Lipovsek MM et al. (2017). Differential contribution of subunit interfaces to α9α10 nicotinic acetylcholine receptor function. Mol Pharmacol 91: 250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y, Talley TT, Hansen SB, Taylor P, Marchot P (2005). Crystal structure of a Cbtx‐AChBP complex reveals essential interactions between snake alpha‐neurotoxins and nicotinic receptors. EMBO J 24: 1512–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y, Sulzenbacher G, Radić Z, Aráoz R, Reynaud M, Benoit E et al. (2015). Marine macrocyclic imines, pinnatoxins A and G: structural determinants and functional properties to distinguish neuronal α7 from muscle α1(2)βγδ nAChRs. Structure 23: 1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brams M, Pandya A, Kuzmin D, van Elk R, Krijnen L, Yakel JL et al. (2011). A structural and mutagenic blueprint for molecular recognition of strychnine and d‐tubocurarine by different cys‐loop receptors. PLoS Biol 9: e1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB et al. (2001). Crystal structure of an ACh‐binding protein reveals the ligand‐binding domain of nicotinic receptors. Nature 411: 269–276. [DOI] [PubMed] [Google Scholar]
- Calimet N, Simoes M, Changeux JP, Karplus M, Taly A, Cecchini M (2013). A gating mechanism of pentameric ligand‐gated ion channels. Proc Natl Acad Sci U S A 110: E3987–E3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celie PH, van Rossum‐Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK (2004). Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41: 907–914. [DOI] [PubMed] [Google Scholar]
- Chatzidaki A, Millar NS (2015). Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol 97: 408–417. [DOI] [PubMed] [Google Scholar]
- Dash B, Lukas RJ, Li MD (2014). A signal peptide missense mutation associated with nicotine dependence alters α2*‐nicotinic acetylcholine receptor function. Neuropharmacology 79: 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellisanti CD, Yao Y, Stroud JC, Wang ZZ, Chen L (2007). Crystal structure of the extracellular domain of nAChR alpha1 bound to alpha‐bungarotoxin at 1.94 A resolution. Nat Neurosci 10: 953–962. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Pandya AA, Yakel JL (2015). Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci 36: 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Lu W, Wu S, Cheng Y, Gouaux E (2015). Glycine receptor mechanism elucidated by electron cryo‐microscopy. Nature 526: 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S (1994). α9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79: 705–715. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J (2001). α10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A 98: 3501–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Katz E, Fuchs PA (2009). The nicotinic receptor of cochlear hair cells: a possible pharmacotherapeutic target? Biochem Pharmacol 78: 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Katz E (2012). The efferent medial olivocochlear‐hair cell synapse. J Physiol Paris 106: 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG, Shen XM, Selcen D, Sine SM (2015). Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol 14: 461. [DOI] [PubMed] [Google Scholar]
- Han L, Talwar S, Wang Q, Shan Q, Lynch JW (2013). Phosphorylation of α3 glycine receptors induces a conformational change in the glycine‐binding site. ACS Chem Nerosci 4: 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han ZY, Le Novere N, Zoli M, Hill JA, Champtiaux N, Changeux JP (2000). Localization of nAChR subunit mRNAs in the brain of Macaca mulatta . Eur J Neurosci 12: 3664–3674. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y (2005). Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J 24: 3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SB, Talley TT, Radic Z, Taylor P (2004). Structural and ligand recognition characteristics of an acetylcholine‐binding protein from Aplysia californica . J Biol Chem 279: 24179–24202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpsoe K, Ahring PK, Christensen JK, Jensen ML, Peters D, Balle T (2011). Unraveling the high‐ and low‐sensitivity agonist responses of nicotinic acetylcholine receptors. J Neurosci 31: 10759–10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, Hovius R et al. (2014). X‐ray structure of the mouse serotonin 5‐HT3 receptor. Nature 512: 276–281. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Sulzenbacher G, Shi J, Talley TT, Conrod S, Kem WR et al. (2009). Structural determinants for interaction of partial agonists with acetylcholine binding protein and neuronal alpha7 nicotinic acetylcholine receptor. EMBO J 28: 3040–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E (2011). Principles of activation and permeation in an anion‐selective Cys‐loop receptor. Nature 474: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R (2008). X‐ray structure of a prokaryotic pentameric ligand‐gated ion channel. Nature 452: 375–379. [DOI] [PubMed] [Google Scholar]
- Huang S, Li SX, Bren N, Cheng K, Gomoto R, Chen L et al. (2013). Complex between α‐bungarotoxin and an α7 nicotinic receptor ligand‐binding domain chimaera. Biochem J 454: 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Chen H, Michelsen K, Schneider S, Shaffer PL (2015). Crystal structure of human glycine receptor‐alpha3 bound to antagonist strychnine. Nature 526: 277–280. [DOI] [PubMed] [Google Scholar]
- Indurthi DC, Pera E, Kim HL, Chu C, McLeod MD, McIntosh JM et al. (2014). Presence of multiple binding sites on α9α10 nAChR receptors alludes to stoichiometric‐dependent action of the α‐conotoxin, Vc1.1. Biochem Pharmacol 89: 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khiroug SS, Khiroug L, Yakel JL (2004). Rat nicotinic acetylcholine receptor α2β2 channels: comparison of functional properties with α4β2 channels in Xenopus oocytes. Neuroscience 124: 817–822. [DOI] [PubMed] [Google Scholar]
- Kouvatsos N, Giastas P, Chroni‐Tzartou D, Poulopoulou C, Tzartos SJ (2016). Crystal structure of a human neuronal nAChR extracellular domain in pentameric assembly: ligand‐bound alpha2 homopentamer. Proc Natl Acad Sci U S A 113: 9635–9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kracun S, Harkness PC, Gibb AJ, Millar NS (2008). Influence of the M3‐M4 intracellular domain upon nicotinic acetylcholine receptor assembly, targeting and function. Br J Pharmacol 153: 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SX, Huang S, Bren N, Noridomi K, Dellisanti CD, Sine SM et al. (2011). Ligand‐binding domain of an alpha7‐nicotinic receptor chimera and its complex with agonist. Nat Neurosci 14: 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinou M, Tzartos SJ (2003). Identification of regions involved in the binding of α‐Bungarotoxin to the human α7 neuronal nicotinic acetylcholine receptor using synthetic peptides. Biochem J 372: 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzaferro S, Benallegue N, Carbone A, Gasparri F, Vijayan R, Biggin PC et al. (2011). Additional acetylcholine (ACh) binding site at α4/α4 interface of (α4β2)2α4 nicotinic receptor influences agonist sensitivity. J Biol Chem 286: 31043–31054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M (2009). α9 nicotinic acetylcholine receptors and the treatment of pain. Biochem Pharmacol 78: 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar NS, Gotti C (2009). Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 56: 237–246. [DOI] [PubMed] [Google Scholar]
- Miller PS, Aricescu AR (2014). Crystal structure of a human GABAA receptor. Nature 512: 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales‐Perez CL, Noviello CM, Hibbs RE (2016). X‐ray structure of the human α4β2 nicotinic receptor. Nature 538: 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N, Free C, Sine SM (2005). Initial coupling of binding to gating mediated by conserved residues in the muscle nicotinic receptor. J Gen Physiol 126: 23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N, Sine SM (2013). Nicotinic receptor transduction zone: invariant arginine couples to multiple electron‐rich residues. Biophys J 104: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J (2003). Alternate stoichiometries of α4β2 nicotinic acetylcholine receptors. Mol Pharmacol 63: 332–341. [DOI] [PubMed] [Google Scholar]
- Nemecz A, Prevost MS, Menny A, Corringer PJ (2016). Emerging molecular mechanisms of signal transduction in pentameric ligand‐gated ion channels. Neuron 90: 452–470. [DOI] [PubMed] [Google Scholar]
- Nemecz A, Taylor P (2011). Creating an alpha7 nicotinic acetylcholine recognition domain from the acetylcholine‐binding protein: crystallographic and ligand selectivity analyses. J Biol Chem 286: 42555–42565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JA, Balle T, Gajhede M, Ahring PK, Kastrup JS (2014). Molecular recognition of the neurotransmitter acetylcholine by an acetylcholine binding protein reveals determinants of binding to nicotinic acetylcholine receptors. PLoS ONE 9: e91232–e91232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plazas PV, Katz E, Gomez‐Casati ME, Bouzat C, Elgoyhen AB (2005). Stoichiometry of the α9α10 nicotinic cholinergic receptor. J Neurosci 25: 10905–10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Mitra A, Auerbach A (2007). A stepwise mechanism for acetylcholine receptor channel gating. Nature 446: 930–933. [DOI] [PubMed] [Google Scholar]
- Quik M, Bordia T, Huang L, Perez X (2011). Targeting nicotinic receptors for Parkinson's disease therapy. CNS Neurol Disord Drug Targets 10: 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero HK, Christensen SB, Di Cesare ML, Gajewiak J, Ramachandra R, Elmsliec KS et al. (2017). Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy‐induced neuropathic pain. Proc Natl Acad Sci U S A 114: E1825–E1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin CV, Katz E, Verbitsky M, Elgoyhen AB (1999). The α9 nicotinic acetylcholine receptor shares pharmacological properties with type A γ‐aminobutyric acid, glycine, and type 3 serotonin receptors. Mol Pharmacol 55: 248–254. [DOI] [PubMed] [Google Scholar]
- Rothlin CV, Lioudyno MI, Silbering AF, Plazas PV, Casati ME, Katz E et al. (2003). Direct interaction of serotonin type 3 receptor ligands with recombinant and native α9α10‐containing nicotinic cholinergic receptors. Mol Pharmacol 63: 1067–1074. [DOI] [PubMed] [Google Scholar]
- Rucktooa P, Haseler C, Van Elk R, Smit AB, Gallagher T, Sixma TK (2012). Structural characterization of binding mode of smoking cessation drugs to nicotinic acetylcholine receptors through study of ligand complexes with acetylcholine‐binding protein. J Biol Chem 287: 23283–23293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L, Shahsavar A, Poitevin F, Huon C, Menny A, Nemecz A et al. (2014). Crystal structures of a pentameric ligand‐gated ion channel provide a mechanism for activation. Proc Natl Acad Sci U S A 111: 966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgard F, Charpantier E, Bertrand S, Walker N, Caput D, Graham D et al. (2002). A novel human nicotinic receptor subunit, α10, that confers functionality to the α9‐subunit. Mol Pharmacol 61: 150–159. [DOI] [PubMed] [Google Scholar]
- Shahsavar Α, Ahring PK, Olsen JA, Krintel C, Kastrup JS, Balle T et al. (2015). Acetylcholine‐binding protein engineered to mimic the α4‐α4 binding pocket in α4β2 nicotinic acetylcholine receptors reveals interface specific interactions important for binding and activity. Mol Pharmacol 88: 697–707. [DOI] [PubMed] [Google Scholar]
- Shahsavar A, Kastrup JS, Nielsen EO, Kristensen JL, Gajhede M, Balle T (2012). Crystal structure of Lymnaea stagnalis AChBP complexed with the potent nAChR antagonist DHβE suggests a unique mode of antagonism. PLoS One 7: e40757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurny R, Debaveye S, Farinha A, Veys K, Vos AM, Gossas T et al. (2015). Molecular blueprint of allosteric binding sites in a homologue of the agonist‐binding domain of the alpha7 nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A 112: E2543–E2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley TT, Yalda S, Ho KY, Tor Y, Soti FS, Kem WR et al. (2006). Spectroscopic analysis of benzylidene anabaseine complexes with acetylcholine binding proteins as models for ligand‐nicotinic receptor interactions. Biochemistry 45: 8894–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP (2009). Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov 8: 733–750. [DOI] [PubMed] [Google Scholar]
- Unwin N (2005). Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol 346: 967–989. [DOI] [PubMed] [Google Scholar]
- Verbitsky M, Rothlin CV, Katz E, Elgoyhen AB (2000). Mixed nicotinic‐muscarinic properties of the α9 nicotinic cholinergic receptor. Neuropharmacology 39: 2515–2524. [DOI] [PubMed] [Google Scholar]
- Wessler I, Kirkpatrick CJ (2008). Acetylcholine beyond neurons: the non‐neuronal cholinergic system in humans. Br J Pharmacol 154: 1558–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lukas RJ (2011). Naturally expressed nicotinic acetylcholine receptor subtypes. Biochem Pharmacol 82: 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakel J (2010). Advances and hold‐ups in the study of structure, function and regulation of Cys‐loop ligand‐gated ion channels and receptors. J Physiol 588: 555–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouridakis M, Giastas P, Zarkadas E, Chroni‐Tzartou D, Bregestovski P, Tzartos SJ (2014). Crystal structures of free and antagonist‐bound states of human alpha9 nicotinic receptor extracellular domain. Nat Struct Mol Biol 21: 976–980. [DOI] [PubMed] [Google Scholar]
- Zuber B, Unwin N (2013). Structure and superorganization of acetylcholine receptor‐rapsyn complexes. Proc Natl Acad Sci U S A 110: 10622–10627. [DOI] [PMC free article] [PubMed] [Google Scholar]