

Abstract
Background and Purpose
Genomic analysis has shown many variants in both CHRNA4 and CHRNB2, genes which encode the α4 and β2 subunits of nicotinic ACh receptors (nAChR) respectively. Some variants influence receptor expression, raising the possibility that CHRNA4 variants may affect response to tobacco use in humans. Chronic exposure to nicotine increases expression of nAChRs, particularly α4β2‐nAChRs, in humans and laboratory animals. Here, we have evaluated whether the initial level of receptor expression affects the increase in expression.
Experimental Approach
Mice differing in expression of α4 and/or β2 nAChR subunits were chronically treated with saline, 0.25, 1.0 or 4.0 mg·kg−1·h−1nicotine. Brain preparations were analysed autoradiographically by [125I]‐epibatidine binding, immunoprecipitation and Western blotting.
Key Results
Immunochemical studies confirmed that most of the [3H]‐epibatidine binding corresponds to α4β2*‐nAChR and that increases in binding correspond to increases in α4 and β2 proteins. Consistent with previous reports, the dose‐dependent increase in nAChR in wild‐type mice following chronic nicotine treatment, measured with any of the methods, reached a maximum. Although receptor expression was reduced by approximately 50% in β2+− mice, the pattern of response to chronic treatment resembled that of wild‐type mice. In contrast, both α4+− and α4+−/β2+− exhibited relatively greater up‐regulation. Consistent with previous reports, α4β2α5‐nAChR did not increase in response to nicotine.
Conclusions and Implications
These results indicate that mice with reduced expression of the α4 nAChR subunit have a more robust response to chronic nicotine than mice with normal expression of this subunit.
Linked Articles
This article is part of a themed section on Nicotinic Acetylcholine Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.11/issuetoc
Abbreviations
- Abs
polyclonal antibodies
- nAChR
nicotinic ACh receptor
Introduction
Tobacco use, primarily through cigarette smoking, is estimated to contribute annually to 4 000 000 deaths worldwide and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2585 is regarded as the component responsible for eliciting dependence. The initial sites of action of nicotine and related compounds are the nicotinic ACh receptors (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76) for which http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=294 is the endogenous neurotransmitter. nAChR are ligand‐gated ion channels assembled from five homologous subunits. Physiological and pharmacological properties of various nAChR subtypes are determined by the subunit composition of the receptor (Albuquerque et al., 2009; Fasoli and Gotti, 2015).
Classical genetic analyses indicated that both genetic and environmental factors influence smoking behaviour (Hall et al., 2002). Among the genes implicated in aspects of human smoking behaviour are nAChR variants. The most thoroughly documented genetic variants are in the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=466&familyId=76&familyType=IC gene encoding the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=466 (Ware et al., 2011; Bierut et al., 2014). Genetic studies also support roles for both http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=465&familyId=76&familyType=IC and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=472&familyId=76&familyType=IC genes in aspects of tobacco use (Feng et al., 2004; Hutchison et al., 2007; Kamens et al., 2013; Esterlis et al., 2016; Melroy‐Greif et al., 2016). These genes encode the α4 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=472, which assemble into the most widely expressed subtype in the brain, the α4β2* nAChR (Millar and Gotti, 2009; Baddick and Marks, 2011) [* indicates potential additional subunits in the pentameric receptor (Lukas et al., 1999)].
Genetic factors also contribute to variation in responses of mice to nicotine. Inbred mouse strains differ in their initial response to nicotine administration for which 35–40% of the variance may be explained by differences in nAChR expression (Marks et al., 1989; Damaj et al., 2007). The development of genetically modified mice for which expression of either the α4 or β2 subunit (encoded by Chrna4 and Chrnb2, analogous to the human CHRNA4 and CHRNB2) has been deleted has established that both of these subunits are necessary for assembly of α4β2*‐nAChRs (Picciotto et al., 1995; Marubio et al., 1999; Ross et al., 2000; Whiteaker et al., 2006; Baddick and Marks, 2011). Expression of α4β2*‐nAChRs is reduced, essentially in a gene dose‐dependent fashion, in heterozygotes for either Chrna4 (Marks et al., 2007) or Chrnb2 (Marks et al., 2000). The role of α4β2*‐nAChRs in modulating aspects of nicotine‐mediated responses has been demonstrated following deletion of either the α4 (Marubio et al., 1999) or β2 nAChR subunit (Marubio et al., 1999; Tritto et al., 2004) (reduced responsiveness to nicotine) or replacement of α4 by hypersensitive variants (Tapper et al., 2007) (increased responsiveness to nicotine).
More than 4000 variants in human CHRNA4 and 2000 variants in CHRNB2 have been identified (http://NCBI/SNP database-CHRNA4 and CHRNB2). The variants are located throughout these genes, many in non‐coding regions. While variants in non‐coding regions would not necessarily be expected to alter receptor function, they may influence expression of subunit proteins. Indeed, expression of human α4β2*‐nAChRs varies widely (Breese et al., 1997, Breese et al., 2000, Cosgrove et al., 2012, D'Souza et al., 2012) and individuals differing in receptor expression could respond differently to chronic tobacco use. Exposure to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2585 or tobacco increases levels of α4β2*‐nAChR expression in rodents chronically exposed to nicotine (Marks et al., 1983; Schwartz and Kellar, 1983; Flores et al., 1992; Marks et al., 2011), human tobacco users (Benwell et al., 1988; Breese et al., 1997; Perry et al., 1999; Staley et al., 2006) and cultured cells expressing α4β2*‐nAChR natively or following transfection (Davila‐Garcia et al., 1999; Lomazzo et al., 2011; Zambrano et al., 2012). The current study was undertaken to investigate whether variation in the initial expression of α4β2*‐nAChRs (achieved by deletion or partial deletion of the α4 and/or β2 subunits) affects the pattern and/or extent of the up‐regulation of the receptors following chronic nicotine treatment.
Methods
Antibody production and characterization
Affinity‐purified, subunit‐specific, polyclonal antibodies (Abs) were used for the detection of nAChR subunits. The production of Abs complied fully with the guidelines established by the Italian Council on Animal Care and was approved by the Italian Government Decree No. 2/2010. Becuase the validity of the results presented in this paper strongly depend on the antibody used, we extensively characterized the specificity of the Abs for the different techniques that were used.
We used subunit‐specific Abs that were produced in rabbits against peptides derived from the C‐terminal (COOH) or intracytoplasmic loop (CYT) regions of the rodent (mouse and rat) or human subunit sequences, and affinity purified as previously described by Gotti et al. (2008) and Grady et al. (2009). For the β2 subunit, we used the Abs directed against the rodent COOH (cgLHPDHSAPSSK) and the human β2 CYT (RQREREGAGALFFREAPGADSCTy) peptides. For the α4 subunit, we used the Abs directed against the rat CYT (PTSSPTSLKARPSQLPVSDQASPC) and COOH (PPWLAACI) peptides. For the α5 subunit, we used Abs directed against the mouse CYT (DRYFTQREEAESGAGPKSRNTLEAALD) and COOH (cgPVHIGNTIK) peptides. The specificity of the Abs was tested by immunoprecipitation and by Western blotting using tissues from α4, α5 and β2 from wild type (++), heterozygote (+−) and null mutant (−−) mice, as previously described (Gotti et al., 2008). For the experiments reported here, we used Abs obtained from five rabbits immunized with human β2 CYT or rat CYT peptides, three rabbits immunized with the mouse α5 CYT peptide and two rabbits each immunized for the rodent β2, α4 and α5 COOH peptides.
Mice
All animal care and experimental procedures were reviewed and approved by the Institutional Animal Care and Utilization Committee of the University of Colorado Boulder and conformed to the guidelines of the NIH. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Mice were chosen as the experimental model owing to the availability of animals differing in the expression of nAChR subunits (null mutants for the α4 and β2 nAChR subunit expression), which allowed the examination of differences in receptor expression on the regulation of these receptors by chronic nicotine treatment. Mice expressing the α4 nAChR null mutation were originally obtained from John Drago, University of Melbourne, Australia (Ross et al., 2000). Mice expressing the β2 nAChR null mutation were originally obtained from Marina Picciotto, Yale University (Picciotto et al., 1995). Each line has been backcrossed with C57BL/6J mice for at least 10 generations. Wild type (++), heterozygote (+−) and null mutant (−−) mice were generated by mating the appropriate heterozygotes. These heterozote matings were used to generate all ++, +− and – mice used for the experiments in this study. Offspring were genotyped using DNA extracted from tail clippings as described previously (Salminen et al., 2004). Double heterozygote mice (α4+−/β2+−) were generated by mating α4 and β2 null mutants. All mating and housing occurred in the specific pathogen‐free colony at the Institute for Behavioural Genetics, University of Colorado Boulder. Before surgery, mice were housed with like sex littermates (two to five mice per cage) and maintained on a 12 h light/12 h dark cycle (lights on 07:00 h). Mice were allowed free access to food and water. Mice used for these experiments were between 60 and 120 days of age. The average weight of male mice was 27 g (range 19 to 36 g) and that of female mice was 22 g (range 17 to 28 g). A total of 621 mice were used for the experiments (329 males and 292 females. Eighty two were α4++, 88 were β2++, 97 were α4+−, 93 were β2+−, 90 were α4+−/β2+−, 65 were α4 and 105 were β2−−).
Chronic nicotine treatment
Methods previously described for continuous infusion (Marks et al., 1983; Marks et al., 2011) were followed with minor modifications. Mice were anaesthetised by intraperitoneal injection of a mixture of ketamine (100 mg·kg−1) and xylazine (10 mg·kg−1). The right jugular vein was exposed, and a cannula constructed of silastic tubing (0.30 mm inner diameter, 0.64 mm outer diameter) filled with sterile isotonic saline containing 0.3% sodium citrate was inserted 8 mm into the vein and anchored to the underlying tissue with surgical silk thread. The silastic tubing was attached to 22 gauge stainless steel tubing affixed to a nylon circle (1 cm diameter), which was attached to the back of the mouse between the scapulae. Following surgery, each mouse was injected with 0.1 mg·kg−1 buprenorphine and placed in a freshly bedded cage. The mouse was warmed and monitored repeatedly until recovery from the anaesthetic.
The day after surgery, the cannula was checked for free flow. The mouse was weighed and transferred to an infusion chamber (15 × 15 × 30 cm, l × w × h). Infusion chambers were constructed of black acrylic plastic with clear acrylic hinged plastic doors. Each infusion cage consisted of a bank of 12 infusion chambers arranged in two rows of six chambers. The infusion chamber was bedded with aspen wood chips and also included a cotton nesting block (nestlet). The stainless steel tubing was connected to polyethylene tubing connected to a Harvard Infusion pump that delivered isotonic sterile saline at a rate of 35 μL·h−1. The polyethylene tubing was connected to a swivel dripper that permitted the mouse free movement within its cage. Saline infusion was maintained for 2 days before beginning drug treatment. Four treatment groups were used: saline‐infused (controls), 0.25 mg·kg−1·h−1 nicotine, 1 mg·kg−1·h−1 nicotine and 4 mg·kg−1·h−1 nicotine. Liquid nicotine that had been redistilled was used for the chronic treatments. Nicotine solutions were neutralized with HCl before administration. Nicotine doses are calculated as free base. Mice were treated for 10 days after which time treatment was discontinued.
As the study included mice of seven genotypes and four nicotine treatment doses, there were 28 different treatment conditions. Based on previous studies, the original design specified a group size of six. Mice were assigned randomly by sex to each of the four treatment groups within each of the seven genotypes. However, it should be noted that at the completion of the experiments, the numbers of mice in each group varied somewhat and consequently differ from the ideal that treatment groups contain equal numbers. In part, this variability in group number was intentional as results for one group in particular (α4+−) were quite unusual. It was deemed necessary to add more animals to this group to examine whether this response was correct. Some variability in group numbers also occurred because occasional re‐genotyping changed the placement of mice originally assigned to a treatment group. On some occasions, occasional loss of mice during treatment may have skewed group sizes.
Autoradiographic measurement of [125I]epibatidine binding
Two hours after nicotine administration, mice were killed by cervical dislocation and decapitated. The brain was removed from the skull and frozen by immersion in isopentane (−30°C). The frozen brain was wrapped in aluminium foil and stored at −70°C until sectioning.
Preparation of tissue sections
On the day of sectioning, a brain was removed from the −70°C freezer and allowed to warm to the temperature of the cryostat (−14°C). The brain was subsequently mounted on the cryostat chuck with M‐1 Embedding Matrix (Anatomical Pathology, Pittsburgh, PA, USA). Subsequently, coronal sections (14 μ thick) were obtained using either a Leica CM 1850 cryostat/microtome (Leica, Nussloch, Germany) or an IEC Minotome (Damon, Corp., Needham, MA, USA) and thaw mounted on Fisher Suprafrost/Plus microscope slides. A series of 10 sets of sections was prepared from each brain to allow comparison of results for several different experiments on adjacent or near‐adjacent sections. Slides containing the brain sections were stored desiccated at −70°C until use.
[125I]‐Epibatidine autoradiography
Slides containing the tissue sections prepared from mice of each nicotine treatment group were warmed to room temperature in a desiccator. Slides were subsequently transferred to Bel‐Art slide racks that have been modified to hold 50 slides and rehydrated by incubation at 22°C for 15 min in isotonic buffer (NaCl, 144 mM; KCl, 2.2 mM, CaCl2, 2.0 mM, MgSO4, 1.0 mM; HEPES, 25 mM; pH = 7.5). The racks containing the rehydrated slides were subsequently transferred to the isotonic buffer containing 200 pM epibatidine. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3976‐(specific activity 2200 Ci·mmol−1) was mixed with unlabelled 5‐I‐epibatidine to yield a final specific activity of 110 Ci·mmol−1 (a 20‐fold dilution). A second series of slides was incubated with [125I]‐epibatidine in buffer that also included 50 nM http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5347 to selectively inhibit primarily α4β2*‐nAChR binding sites (Marks et al., 1998). A third set of slides with representative mice from each treatment group was incubated with the [125I]‐epibatidine in buffer that also included 10 μM nicotine to establish blanks. Samples were incubated for 2 h at 22°. Following the incubation, the slides were redistributed to slide racks containing 25 slides and washed as follows (all solutions at 4°C): Twice for 30 s in isotonic buffer, twice for 5 s in hypotonic buffer (0.1×) and twice for 5 s in 10 mM HEPES, pH = 7.5. The samples were air dried and stored overnight under vacuum at room temperature in a desiccator. The dried slides were apposed to Perkin‐Elmer Packard Super Resolution Cyclone Storage Phosphor Screens for 7–14 days to yield images for quantitation. Each Phosphor Screen was also simultaneously exposed to a series of tissue paste standards containing measured amounts of 125I to allow quantitation of the image intensity. Blanks determined by incubation in the presence of 10 μM nicotine did not differ significantly from film background.
Quantitation of signal intensity
Tissue paste samples prepared from whole brain homogenates and containing measured amounts of 125I were used to construct standard curves. The standards were prepared such that the standard with the highest amount of 125I contained approximately 30 000‐fold more 125I than the standard with the lowest amount of 125I. This wide range of 125I content in the standards assured that all signals measured for brain samples fell within the standard curve. The phosphor screens yield a linear relationship between signal intensity and tissue radioactivity content over several orders of magnitude. Signal intensity was measured using the Optiquant program (Perkin‐Elmer Packard) designed for use with the Cyclone Imager. The regression line calculated for the standard curve was used to convert the measured value of pixels mm−2 to the cpm·mg−1 wet weight from which signal intensity in fmol·mg−1 wet weight was calculated from the specific activity of the [125I]epibatidine (110 Ci·mmol−1). Brain regions were identified using a mouse brain atlas (Franklin Keith and Paxinos, 1997). Several measurements were made in each brain region of every mouse, and the average of these measurements defined the signal intensity for each region of each subject.
Blinding during analysis
In order to maintain accurate records, it was necessary to label the printout of each scan with identifiers for each animal including genotype, dose and mouse coding number (to allow re‐genotyping if needed). However, that information was not taken into account during measurement of signal intensity on each screen until the analysis was complete. Measurements were made in a defined order on each screen. First, the signal intensity of the standards was measured. Subsequently, signal from each mouse was measured for the brain areas proceeding from the anterior sections to the posterior sections of that mouse. That pattern was repeated for sections of each mouse on screen. After data collection and calculation, values for each experiment were entered into the appropriate data sheets. In order to achieve as much consistency as possible for the imaging studies, frequently but not always, microscope slides with samples from each of the treatment groups for a single genotype were included on a screen (there is space to accommodate slides from four different treatments). As noted above, each screen also included a set of 125I standards.
Immunochemical analysis of nAChR subunits
Following chronic treatment and 2 h withdrawal, mice were killed by cervical dislocation, their brains removed, frozen by immersion in isopentane (−30°C), wrapped in aluminium foil and stored at −70°C before transfer to Milan, Italy.
Tissue homogenates
The particulate fractions from whole mouse brains were washed copiously with 0.32 M sucrose 10 mM−1 TRIS buffer and homogenized in the same sucrose/Tris buffer using an Ultraturrax homogenizer and glass‐Teflon tissue grinder.
Triton X‐100 extracts
The particulate fractions from whole mouse brains of the various treatment groups were washed by centrifugation in the Tris–HCl 50 mM, NaCl 120 mM, KCl 5 mM, MgCl2 1 mM, CaCl2 2.5 mM, pH 7.5 buffer by centrifugation. The resulting pellets were suspended in the same buffer containing 2% Triton X‐100. Extraction was performed for 2 h at 4°C as previously described (Moretti et al., 2010).
Protein content of the membranes and 2% Triton X‐100 extracts was measured using the bicinchonic acid protein assay (Pierce, Rockford, IL, USA) with bovine serum albumin as the standard.
[3H]‐Epibatidine binding studies
After incubating, the samples from each of the different groups with 1 μM http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3964 for 2 h (in order to block binding of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5348 to α7‐nAChR sites), binding experiments were carried out by incubating aliquots of the membranes or the extracts with 1 nM [3H]epibatidine for 2 h at 20°C. Non‐specific binding (average of 5–10% of total binding) was determined in parallel samples containing 100 nM unlabelled epibatidine. At the end of the incubation, the samples were filtered on a Millipore apparatus using a glass GFC filter soaked in 0.5% polyethylenimine, washed with 15 mL of buffer solution (sodium phosphate, 10 mM, pH 7.4 and 50 mM NaCl) and counted in a Tri‐Carb 2100TR β‐counter (Perkin Elmer).
Extracts were incubated overnight (14 h) with 1 nM [3H]‐epibatidine at 4°C. Following the overnight incubation, receptors were captured using DEAE‐Sepharose™ Fast flow (GE Healthcare, Uppsala, Sweden). The bound receptors were eluted with 1 N NaOH and after addition of the scintillation mixture (filter count, GE Healthcare, Uppsala, Sweden) counted in a Tri‐Carb 2100TR β‐counter (Perkin Elmer) counter. Non‐specific binding (averaging 5–10% of total binding) was determined in parallel samples containing 100 nM unlabelled epibatidine.
Immunoprecipitation of [3H]‐epibatidine‐labelled receptors by subunit‐specific antibodies
The extracts (100–150 μL) obtained from whole brain were incubated with 1 μM α‐bungarotoxin followed by incubation with 1 nM [3H]‐epibatidine and then incubated overnight with a saturating concentration of affinity purified anti‐subunit IgG (anti‐α4, ‐α5 or ‐β2) (10 μg) bound to Sepharose‐ProteinA (GE Healthcare, Italy). Immunoprecipitates were recovered by centrifugation (5 min at 2500 × g). The level of Ab immunoprecipitation was expressed as fmol of immunoprecipitated receptors per mg protein and is the mean ± SEM of two to three replicates for each individual sample.
Immunoblotting and densitometric quantification of Western blot bands
The nAChR subunits were analysed by Western blotting as described previously (Gotti et al., 2008; Grady et al., 2009). In brief, for whole brain, homogenates of the different groups were loaded, separated by means of SDS‐polyacrylamide gel electrophoresis using 9% acrylamide and electrophoretically transferred to nitrocellulose membranes with 0.45 mm pores (Schleicher and Schull ll, Dassel, Germany).
The blots were blocked overnight with 4% non‐fat milk powder in Tris‐buffered saline, washed with buffer containing 4% non‐fat milk and 0.3% Tween20 in Tris‐buffered saline and incubated for 2 h with the primary antibody directed against the α4 and β2 CYT peptides at the concentration of 5 μg·mL−1. Blots were then incubated for 1 h with the appropriate secondary antibody (anti‐rabbit Ly‐Cor IRDye800RD). After six washes of 5 min each in Tris‐buffered saline with 0.3% Tween 20, the membranes were dried overnight in the dark at room temperature. The infrared signal was measured using an Odyssey CLx – Infrared Imaging System. The signal intensity of the Western blot bands was quantified using iStudio software.
The response of both the α4 and β2 subunits to nicotine treatment for mice of each genotype was calculated as the ratio between the signal for the nicotine‐treated mice and saline‐treated mice of the same genotype. The optical density of the saline‐treated mice of each genotype was set at 100%. The data are expressed as mean values ± S.E.M. of 68 (homogenates) independent preparations. Each preparation was analysed two to three times in separate experiments for each antibody to obtain the mean value for each preparation.
The mean values of the saline‐treated mice for the α4++ and β2++ genotypes were as follows: α4 subunit (1 ± 0.13; 1 ± 0.12), β2 subunit (1 ± 0.11; 1 ± 0.11). For the α4+− and β2+− genotypes were as follows: α4 subunit (1 ± 0.12; 1 ± 0.12), β2 subunit (1 ± 0.12; 1 ± 0.13). For α4+−/β2+− genotype was as follows: α4 subunit (1 ± 0.12), β2 subunit (1 ± 0.11).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Considering past experience with studies such as those described in the current study, a minimum group size of n = 5 was chosen, and upon final analysis, most groups exceeded this minimum number. However, some differences in group sizes occurred. Each group size is specified in the legends for the table and figures. Some group differences occurred owing to reassignment of samples following re‐genotyping, while others were intentional to verify unusual or unexpected results. SPSS version 21 was used for statistical analysis, and P < 0.05 was used throughout the analyses. Three‐way ANOVA with independent variables nicotinic genotype, nicotine treatment dose and brain region as the independent variables and cytisine‐sensitive [125I]‐epibatidine binding as the dependent variable was conducted. Subsequently, two‐way ANOVAs were performed for each brain region with nicotinic genotype and nicotine treatment dose as the independent variables. One‐way ANOVAs were then performed for each brain region and nicotinic genotype to assess response to chronic nicotine treatment.
Materials
[125I]‐Epibatidine (2200 Ci·mmol−1) and (±)‐[3H]‐epibatidine (66 Ci·mmol−1) were obtained from Perkin‐Elmer NEN, Boston, MA, USA. NaCl, KCl, MgSO4, CaCl2, Na2HPO4, NaH2PO4, nicotine and cytisine were obtained from Sigma Chemical Co., St. Louis, MO, USA. α‐Bungarotoxin was purchased from Tocris Bioscience (Bristol, UK). Ketamine, xylazine, acepromazine and buprenorphine were obtained from MWI Veterinary Supply. HEPES and NaHEPES products of BDH and silastic tubing a product of Dow Chemical were obtained through VWR International. Nylon mesh and 22 gauge stainless steel tubing were obtained from Small Parts, Inc. Aspen wood chip bedding is a product of Harlen Tecklad (Madison, WI, USA), and cotton nestlets are a product of Ancare Corp. (Bellmore, NY, USA). 5‐I‐Epibatidine was a generous gift of Dr Kenneth Kellar, Georgetown University.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Epibatidine binding following deletion or partial deletion of the α4 or β2 nAChR genes
Deletion and/or partial deletion of α4 and/or β2 nAChR subunits reduces cytisine‐sensitive [125I]‐epibatidine binding in a gene‐dose‐dependent manner (Supporting Information Fig. S1). Significant binding persists in several brain regions known to contain high expression of non‐α4β2*‐nAChR.
Supporting Information Fig. S2 summarizes the effect of deletion of α4 and/or β2 nAChR genes on expression of α4, β2 and α5 nAChR proteins measured by immunoprecipitation with subunit selective antibodies. Immunoprecipitation of [3H]‐epibatidine binding sites from whole brain extracts with anti α4‐ or β2‐selective antibodies parallels autoradiographic results, namely a gene‐dose‐dependent reduction in binding. Furthermore, deletion of either α4 or β2 eliminates [3H]‐epibatidine binding sites precipitable by anti α5‐selective antibody indicates that virtually all α5*‐nAChR in mouse brain are α4β2α5‐nAChR.
[125I]‐Epibatidine binding following chronic nicotine treatment
Effects of chronic nicotine treatment of mice differing in initial expression of α4 and/or β2* nAChR subunits on cytisine sensitive [125I]‐epibatidine binding are shown in Supporting Information Table S1.
To briefly summarize results from this section, chronic treatment with nicotine by continuous intravenous delivery elicits up‐regulation in cytisine‐sensitive [125I]‐epibatidine binding measured autoradiographically in wild‐type, heterozygote and double heterozygote mice. The pattern of response to chronic nicotine treatment in α4++, β2++ and β2+− mice is very similar. However, mice with lower expression of the α4 nAChR subunit (α4+− and α4+−/β2+−) tend to exhibit greater relative increases in cytisine‐sensitive [125I]‐epibatidine binding than either wild‐type or β2+/− mice. Little or no binding or response to nicotine treatment was observed in either null mutant.
[125I]‐Epibatidine binding following chronic nicotine treatment in mice differing in expression of α4 and/or β2 nAChR subunits
Cytisine‐sensitive [125I]‐epibatidine binding sites was initially analysed by three‐way ANOVA to evaluate the effects of genotype, nicotine treatment dose and brain region as independent variables. Significant overall effects of genotype (F6, 6178 = 1166.34), nicotine treatment dose (F3, 6178 = 33.62) and brain region (F43, 6178 = 510.88) were detected. Furthermore, all three two‐way interactions were significant indicating that deletion or partial deletion of α4 or β2 nAChR genes differentially affected binding site density in the various brain regions as well as response of these sites to chronic nicotine treatment (genotype by nicotine dose: F18, 6178 = 11.49; genotype by brain region: F258, 6178 = 11.49; and nicotine dose by brain region: F129, 6178 = 2.17).
Effects of deletion of the α4 and β2 subunits were further examined in 44 brain regions by two‐way ANOVA (genotype and nicotine treatment dose as independent variables).
Significant dose‐dependent increases in [125I]‐epibatidine binding were observed in 32 of 44 brain regions, consistent with previous reports (Marks et al., 1983; Marks et al., 2004; Marks et al., 2011). Brain areas for which no significant overall effect of nicotine treatment was observed were clustered in four thalamic nuclei, the habenular‐interpeduncular nucleus pathway (three nuclei) and in four areas related to the visual pathway.
In order to evaluate the overall effects of chronic nicotine treatment as a function of nicotine treatment dose and genotype, scattergrams were constructed comparing the changes from control binding for mice differing in α4 and/or β2 nAChR gene expression (Figure 1). Each data point represents the calculation for an individual brain region as ratio of the binding for nicotine‐treated mice to that of saline‐treated mice. The line of unit slope that bisects each panel is expected if response to chronic nicotine treatment for each mutant is the same as that for wild‐types. These analyses omitted results for medial habenula, fasiculus retroflexus and interpeduncular nucleus.
Figure 1.
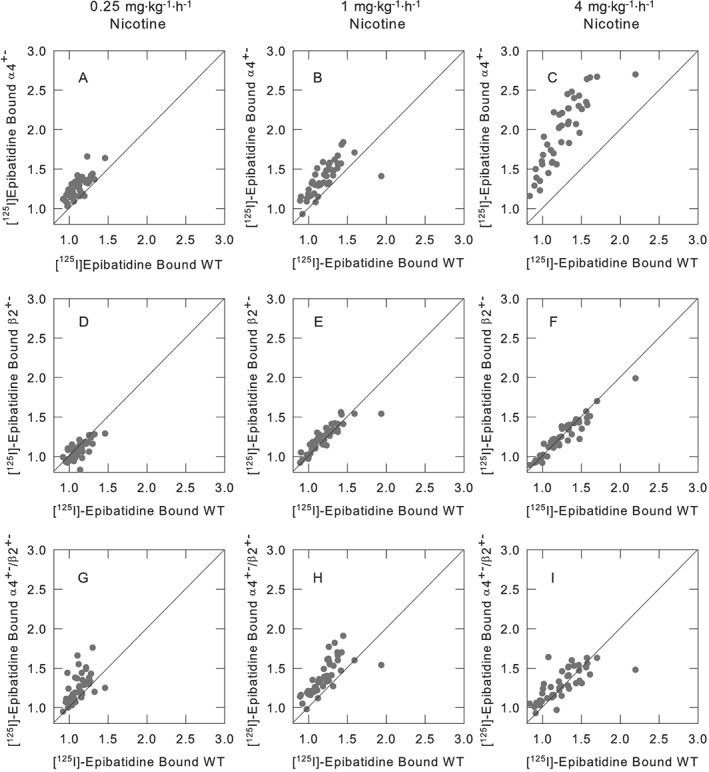
Relative effects of chronic nicotine treatment in mice differing in α4β2*‐expression, compared with the effects in wild‐type mice. nAChR binding sites were measured by quantitative autoradiography of [125I]epibatidine binding for wild‐type, α4+−, β2+− or α4+−/β2+− mice that had been chronically treated with saline, 0.25, 1.0 or 4.0 mg·kg−1·h−1 nicotine for 10 days. Panels (A), (B) and (C) compare the binding for α4+− mice to that of wild‐type. Panels (D), (E) and (F) compare the binding for β2+− to that of wild‐type, and panels (G), (H) and (I) compare the binding for α4+−/β2+− to that of wild‐type for each of the brain regions listed in Supporting Information Table S1 to demonstrate the effect of partial gene deletion of the α4 and/or β2 nAChR subunits. The panels illustrate the relative changes in the density of [125I]epibatidine binding following chronic nicotine of α4+−, β2+− or α4+−/β2+− mice to the changes measured for wild‐type mice. Each point has been normalized to the binding measured for the saline infused of the same genotype. Lines of unit slope are included.
Figure 1A–C compares relative expression of cytisine‐sensitive [125I]‐epibatidine binding sites for α4+− to that for α4++ following chronic treatment with 0.25, 1.0 or 4.0 mg·kg−1·h−1 nicotine respectively. Initial binding in α4+− mice was approximately half (overall average 54.2 ± 1.2%) that for wild‐types. A larger increase in the density of cytisine‐sensitive [125I]‐epibatidine binding sites was noted for α4+− mice than for α4++ mice following treatment with each dose of nicotine (overall increases for α4++ and α4+−, respectively – 0.25 mg·kg−1·h−1 nicotine 13.7 ± 2.2% and 27.4 ± 2.1%; 1.0 mg·kg−1·h−1 nicotine 21.5 ± 3.8% and 38.2 ± 3.1%; and 4.0 mg·kg−1·h−1 nicotine 25.8 ± 5.1% and 95.2 ± 6.8%).
Figure 1D–F compares relative expression of cytisine‐sensitive [125I]‐epibatidine binding sites for β2++ and β2+− following treatment with 0.25, 1.0 or 4.0 mg·kg−1·h−1 nicotine respectively. Binding in saline‐treated β2+− mice was approximately half (overall average 56.6 ± 1.6%) that in wild‐types. Data points for relative regional changes for β2++ and β2+− mice cluster around the line of unit slope showing very similar overall responses to chronic nicotine treatment (overall average increases for β2++ and β2+− mice, respectively – 0.25 mg·kg−1·h−1 nicotine 10.8 ± 1.5% and 8.8 ± 1.6%; 1.0 mg·kg−1·h−1 nicotine 18.4 ± 4.9% and 24.1 ± 2.5%; and 4.0 mg·kg−1·h−1 nicotine 23.9 ± 3.5% and 24.1 ± 3.6%).
Figure 1G–I compares relative expression of cytisine‐sensitive [125I]‐epibatidine binding sites in α4+−/β2+− mice to that of the average of the two wild‐types following chronic treatment with nicotine (0.25, 1.0 and 4.0 mg·kg−1·h−1). Cytisine‐sensitive [125I]‐epibatidine binding site density in α4+−/β2+− mice was one‐third (overall average 33.7 ± 1.2%) that of wild‐type mice. Responses of α4+−/β2+− mice differed somewhat from responses of either β2+− or α4+−. As illustrated in Figure 1G, H, α4+−/β2+− mice show relatively larger increases in cytisine‐sensitive [125I]‐epibatidine binding sites following treatment with 0.25 mg·kg−1·h−1 nicotine (12.3 ± 1.8% vs. 25.5 ± 2.6% increase for wild‐types vs. α4+−/β2+−, respectively) or 1.0 mg·kg−1·h−1 nicotine (19.9 ± 3.0% vs. 38.8 ± 3.3% increase for wild‐types vs. α4+−/β2+−, respectively). However, increases noted following treatment with 4.0 mg·kg−1·h−1 nicotine were only slightly higher for α4+−/β2+− mice (Panel 1I, 24.8 ± 4.1% vs. 29.4 ± 3.0% increase for wild‐types vs. α4+−/β2+−, respectively).
[3H]‐Epibatidine binding sites immunoprecipitated by anti‐nAChR antibodies following chronic nicotine treatment of mice differing in expression of α4 and/or β2 nAChR genes
The preceding section described effects of deletion or partial deletion of α4 and/or β2 nAChR genes on response to chronic nicotine treatment on cytisine‐sensitive [125I]‐epibatidine binding (primarily α4β2*‐nAChR binding sites). Immunochemical experiments were undertaken to investigate effects on subunit protein expression. These studies were conducted using whole brain extracts.
Figure 2 illustrates effects of chronic nicotine treatment on expression of [3H]‐epibatidine binding sites in mice differing in expression of α4 and/or β2 nAChR subunits. Briefly, immunoprecipitation with antibodies directed against α4 or β2 nAChR subunits confirms that increased [3H]‐epibatidine binding sites are predominately α4β2*‐nAChR for wild‐type, α4+−, β2+− and α4+−/β2+− mice. Although most of α5 subunit is contained in α4β2α5‐nAChR, no increase in [3H]‐epibatidine binding sites precipitated by anti‐α5 antibody was observed following nicotine treatment.
Figure 2.
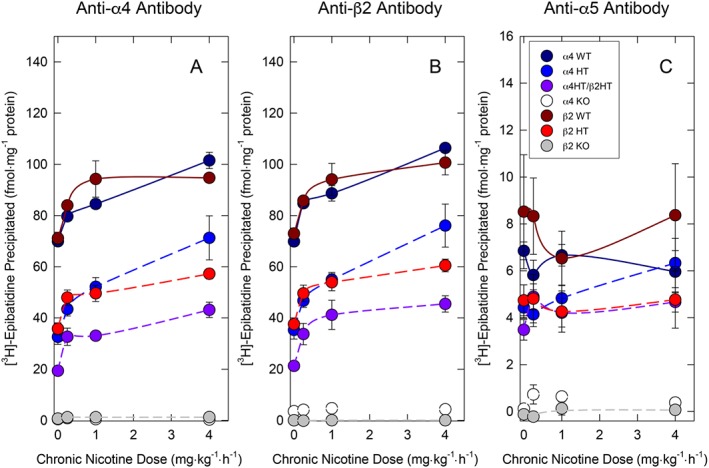
Effect of deletion or partial deletion of the α4 and/or β2 nAChR subunits on response to chronic nicotine treatment measured by immunoprecipitation. nAChR binding sites were measured by quantitative immunoprecipitation of [3H]epibatidine binding sites by anti‐α4, anti‐β2 or anti‐α5 antibodies in whole brain extracts for α4++, β2++, α4+−, β2+−, α4+−/β2+−, α4−− or β2−−for mice that had been chronically treated with saline, 0.25, 1.0 or 4.0 mg·kg−1·h−1 nicotine for 10 days. Points are the mean ± SEM for the number of mice indicated in the following table. Numbers are presented as number α4 antibody, number β2 antibody, number α5 antibody.
| Nicotine dose | α4++ | β2++ | α4+− | β2+− | α4+−/β2+− | α4−− | β2−− |
| Saline | 7,7,7 | 6,7,5 | 8,7,6 | 7,7,6 | 8,8,8 | 6,6,6 | 5,5,5 |
| 0.25 mg·kg−1·h−1 | 7,7,7 | 6,5,5 | 8,9,8 | 6,6,6 | 6,6,6 | 6,6,6 | 6,7,7 |
| 1.0 mg·kg−1·h−1 | 6,7,5 | 5,6,5 | 9,9,7 | 7,7,6 | 8,8,8 | 8,8,5 | 7,7,7 |
| 4.0 mg·kg−1·h−1 | 7,8,6 | 6,6,5 | 8,9,7 | 7,6,6 | 7,7,7 | 7,7,7 | 5,5,5 |
Effects of chronic nicotine treatment on [3H]‐epibatidine binding sites immunoprecipitated by anti‐α4 antibody or anti‐β2 antibody are illustrated in Figure 2A, B respectively. Similar to results obtained for the autoradiographic analyses presented in Supporting Information Table S1, two‐way ANOVA revealed significant main effects of genotype (anti‐α4: F6,164 = 492.90; anti‐β2: F6,173 = 414.29), nicotine treatment dose (anti‐α4: F3,164 = 53.30; anti‐β2: F3,173 = 51.71) and significant genotype by nicotine treatment interaction (anti‐α4: F18,164 = 4.83; anti‐β2: F18,173 = 4.45) that were virtually identical for the anti‐α4 and anti‐β2 antibodies. Chronic nicotine elicited dose‐dependent increases in these sites in wild‐type, α4+−, β2+− and α4+−/β2+− mice. The effect of chronic nicotine treatment in mice of these genotypes was confirmed by one‐way ANOVAs for both anti‐α4 and anti‐β2 antibodies. Significant increases from control were noted following treatment with each nicotine dose for all five genotypes that still expressed α4β2*nAChR. Deletion of either the α4 or β2 nAChR genes virtually eliminated [3H]‐epibatidine binding sites. Moreover, chronic nicotine treatment produced no detectable increase in immunoprecipitated [3H]‐epibatidine binding sites in either null mutant.
Effects of chronic nicotine treatment on [3H]‐epibatidine binding sites immunoprecipitated by anti‐α5 antibody are illustrated in Figure 2C. Significant differences among the genotypes (F6, 145 = 53.04) were detected with both α4−− and β2−− exhibiting few sites immunoprecipitated by anti‐α5 antibody. No significant effects of nicotine treatment (F3, 145 = 0.49) or genotype by nicotine interaction (F18, 145 = 0.66) were detected.
Western blot analysis of α4 and β2 nAChR subunits as a function of nicotine treatment and genotype
As immunoprecipitation experiments demonstrated an increase in α4β2‐nAChR subtype in whole brains of α4++, β2++, α4+−, β2+− and α4+−/β2+− mice accounted for the increase of [3H]‐epibatidine binding sites, the effect of nicotine treatment on expression of α4 and β2 subunits was analysed using quantitative Western blotting shown in Figure 3. Results were normalized to those of saline‐treated mice within each genotype and subunit probe.
Figure 3.
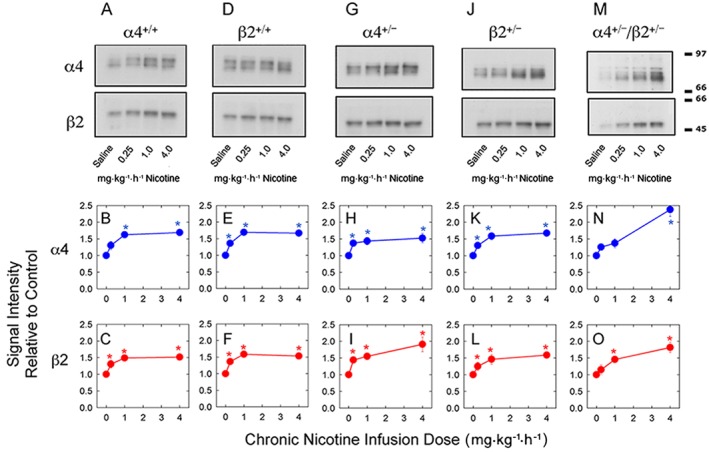
Quantitative Western blotting to measure expression of α4 and β2 subunit protein following chronic nicotine treatment of mice differing in nAChR expression. Relative levels of α4 and β2 nAChR subunit proteins were measured by quantitative Western blotting for α4++, β2++, α4+−, β2+− and α4+−/β2+− mice chronically treated for 10 days saline, 0.25, 1.0 or 4.0 mg·kg−1·h−1 nicotine as described in the Methods section. The top panels show representative blots. The middle panels (red points and lines) show the relative effects of chronic nicotine treatment on α4 subunit levels. The lower panels (blue points and lines) show the relative effects of chronic nicotine treatment on β2 subunit levels. All data points have been normalized to the expression level measured for saline‐treated mice. Errors for the saline‐treated mice are summarized in the Methods section and all fall within the symbols. The mean values of the saline‐treated mice for the α4++ and β2++ genotypes were α4 subunit (1 ± 0.13; 1 ± 0.12) and β2 subunit (1 ± 0.11; 1 ± 0.11); for the α4+− and β2+− genotypes were α4 subunit (1 ± 0.12; 1 ± 0.12) and β2 subunit (1 ± 0.12; 1 ± 0.13); and for α4+−/β2+− genotype were α4 subunit (1 ± 0.12) and β2 subunit (1 ± 0.11). Each point is the mean ± SEM of six to eight separate mice. Numbers in the table are the number of mice analysed. Each mouse has been tested at least three times with different anti‐ α4 or anti‐β2 CYT Abs.
| Nicotine dose | α4++ | β2++ | α4+− | β2+− | α4+−/β2+− | α4−− | β2−− |
| Saline | 7 | 7 | 8 | 7 | 8 | 7 | 7 |
| 0.25 mg·kg−1·h−1 | 7 | 6 | 8 | 6 | 8 | 7 | 6 |
| 1.0 mg·kg−1·h−1 | 7 | 7 | 8 | 7 | 7 | 7 | 7 |
| 4.0 mg·kg−1·h−1 | 8 | 7 | 8 | 7 | 8 | 7 | 7 |
Briefly, quantitative Western blotting for nAChR subunits detected with anti‐α4 or anti‐β2 selective antibodies demonstrates that both subunits increase following chronic nicotine treatment with a tendency for greater increases in α4+− and α4+−/β2+− brains than for wild‐type or β2+− brains. Western blotting for α4 nAChR protein demonstrated significant effect of chronic nicotine treatment (F3, 160 = 36.37), but not of genotype (F4, 160 = 0.99). The significant genotype by nicotine treatment interaction (F12, 160 = 2.29) suggested a potential difference in the pattern of response to chronic nicotine treatment among the groups. Western blotting for β2 nAChR protein yielded results similar to those observed for α4 nAChR subunit protein: significant effect of nicotine treatment (F3,180 = 30.89), but not of genotype (F4,180 = 0.71) or genotype by nicotine treatment dose interaction (F12, 180 = 0.94).
Representative Western blots probed with either anti‐α4 or anti‐β2 antibodies are shown in Figure 3A, D for α4++ and β2++ mice respectively. Normalized subunit signal intensities as a function of nicotine treatment dose are presented in Figure 3B, E for α4++ and Figure 3C, F for β2++ mice, for the α4 and β2 nAChR subunit proteins respectively. The responses of α4++ and β2++ mice were very similar both for pattern of response to chronic nicotine treatment and for relative increases in both α4 and β2 subunit expression (compare Figure 3B with Figure 3E for α4 and Figure 3C with Figure 3F for β2; two‐way ANOVAs for α4 nAChR subunit protein: significant main effect of nicotine‐F3,44 = 16.65, but not of genotype‐F1,44 = 0.14 or nicotine by genotype interaction‐F3,44 = 0.16 and for the β2 nAChR subunit protein: significant main effect of nicotine‐F3,56 = 21.07 but not of genotype‐F1,56 = 0.003 or nicotine by genotype interaction‐F3,56 = 0.055). Subsequent analysis of the effect of nicotine treatment by one‐way ANOVA showed significant effects for both α4++ mice (for α4 signal, F3, 29 = 25.08; for β2 signal, F3, 30 = 11.46) and β2++ mice (for α4 signal, F3, 28 = 24.38; for β2 signal, F3, 28 = 18.23). Expression of both subunit proteins attained near maximal increases following treatment with 1.0 mg·kg−1·h−1 nicotine. The relative percentage increases in expression of α4 subunit protein following treatment with 4.0 mg·kg−1·h−1 nicotine (69 ± 7% and 67 ± 9% for α4++ and β2++ mice, respectively) were slightly, but not significantly, greater than the relative percentage increases in β2 subunit protein (52 ± 9% and 53 ± 9% for α4++ and β2++ mice, respectively). Representative Western blots probed with either anti‐α4 or anti‐β2 antibodies for α4+− mice are shown in Figure 3G and for β2+− mice in Figure 3J and α4 and β2 subunit signal intensity as a function of nicotine dose are presented in Figures 3H, I for α4+− mice and 3 K and 3 L for β2+− mice respectively.
For α4+− mice, statistically significant increase in the expression of α4 nAChR subunit protein as a function of nicotine treatment dose was observed (F3, 32 = 7.85). This increase appears saturable with a 58 ± 18% increase following treatment with 4.0 mg·kg−1·h−1 nicotine. This pattern is similar to that noted for wild‐type mice. A statistically significant increase in the expression of the β2 nAChR subunit protein as a function of nicotine treatment was also noted (F3, 32 = 14.91). However, expression of β2 continues to increase with nicotine treatment dose attaining a 91 ± 22% increase following treatment with 4.0 mg·kg−1·h−1 nicotine. The response to chronic nicotine treatment for β2+− mice was similar to that observed for wild‐type mice. Statistically significant increases in expression of α4 (F3, 37 = 18.00) and β2 (F3, 36 = 7.60) nAChR subunit proteins as a function of nicotine treatment dose were observed. These increases appear saturable with a 67 ± 9% increase for α4 subunit and a 49 ± 9% increase for β2 subunit following treatment with 4.0 mg·kg−1·h−1 nicotine. Representative Western blots probed with either anti‐α4 or anti‐β2 antibodies for α4+−/β+− mice are shown in Figure 3M, and α4 and β2 subunit signal intensity as a function of nicotine treatment dose is presented in Figure 3N, O respectively.
The response of α4+−/β+− mice to chronic nicotine treatment differed from those of α4++, β2++, α4+− and β2+− mice and was especially noticeable for α4 nAChR subunit protein (F3, 65 = 22.14). The 136 ± 21% increase in expression of α4 nAChR subunit protein observed following treatment with 4.0 mg·kg−1·h−1 nicotine was substantially greater than that observed for either wild‐types or heterozygotes. A significant increase in β2 nAChR subunit protein was also noted (F3, 65 = 10.95). Although the 81 ± 15% increase in expression of β2 nAChR subunit protein was not as large as that observed for α4 nAChR subunit protein, it was larger than that noted for wild‐type and β2+− mice and was comparable with that observed for α4+− mice.
Discussion
Effects of deletion or partial deletion of the α4 and β2 nAChR subunits on the expression of α4β2*‐nAChR and their response to chronic nicotine treatment are reported. Particularly, striking was a more robust relative up‐regulation of α4β2‐nAChR following chronic nicotine treatment in mice with reduced expression of the α4 subunit (α4+− and α4+−/β2+− mice).
Autoradiographic and immunochemical analysis of epibatidine binding: effect of α4 and/or β2 deletion or partial deletion
Consistent with previous reports for the β2 nAChR subunit (Picciotto et al., 1995; Baddick and Marks, 2011) or the α4 nAChR subunit (Marubio et al., 1999; Ross et al, 2000; Marks et al., 2007; Baddick and Marks, 2011) null mutant mice, autoradiographic analysis confirmed that deletion of either the α4 or β2 subunit eliminates virtually all cytisine‐sensitive [125I]‐epibatidine binding. Although effects of deletion of α4 or β2 nAChR subunits were generally similar, deletion of the β2 subunits had a greater effect on binding than deletion of α4 subunits in basal ganglia and visual tracts reflecting the presence of α6β2*‐nAChR and/or α3β2*‐nAChR (Whiteaker et al., 2000; Champtiaux et al., 2002; McClure‐Begley et al., 2012). Significant [125I]‐epibatidine binding persisted in medial habenula, fasciculus retroflexus and interpeduncular nucleus of knockout mice reflecting the limits of cytisine inhibition specificity in regions with high numbers of cytisine‐resistant sites (Marks et al., 1998; Perry et al., 2002).
As noted previously (Marks et al., 2000; Marks et al., 2007), partial deletion of α4 or β2 reduces cytisine‐sensitive [125I]‐epibatidine binding approximately 50% perhaps indicating that neither subunit is expressed in excess. Partial deletion of both α4 and β2 nAChR subunits (α4+−β2+− mice) reduced [125I]‐epibatidine binding below levels seen for either heterozygote. Immunoprecipitation of [3H]‐epibatidine binding sites following deletion or partial deletion of either α4 or β2 nAChR subunit paralleled results measured autoradiographically. Numbers of [3H]‐epibatidine binding sites precipitated by anti‐α4 and anti‐β2 antibodies were virtually identical. The immunoprecipitation data are similar to those obtained previously when protein expression was measured autoradiographically using radiolabeled monoclonal antibodies (Whiteaker et al., 2006).
Fewer [125I]‐epibatidine binding sites were precipitated by anti‐α5 antibody (~9%) than by anti‐α4 or anti‐β2 antibodies. Deletion of either α4 or β2 nAChR subunit essentially eliminated all [3H]‐epibatidine binding detected by anti‐α5 antibody indicating that in whole mouse brain, the α5 subunit occurs primarily in α4β2α5‐nAChR. The percentage decrease in of [3H]‐epibatidine binding sites precipitated by anti‐α5 antibody following partial deletion of α4 and/or β2 subunit was less than that measured with anti‐α4 or anti‐β2 antibodies. Thus, a greater percentage of α4β2*‐nAChR in α4+− and β2+− mice (~15%) and α4+−/β2+− mice (~18%) contain the α5 subunit. Predominant α4β2α5‐nAChR composition expressed in mouse brain contrasts with that for nAChR expression in the autonomic nervous system where α3β4α5‐nAChR or, to a lesser extent, α3β2α5‐nAChR, predominate (Wang et al., 1996; Garza et al., 2009). However, α5 is expressed in small numbers in non‐α4β2*‐nAChR subtypes in brain regions such as medial habenula and interpeduncular nucleus (Grady et al., 2009).
Chronic nicotine treatment
Chronic nicotine treatment up‐regulated cytisine‐sensitive [125I]‐epibatidine binding sites measured autoradiographically for wild‐types. Up‐regulation varied among brain regions as reported previously (Marks et al., 2004; Marks et al., 2011). Little difference in binding site density between brain regions of mice treated with 1 mg·kg−1·h−1 nicotine or 4 mg·kg−1·h−1 nicotine was observed. Response to chronic treatment corresponds to that reported for [125I]‐http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=5460 binding and α4 and β2 nAChR subunit protein expression measured with radiolabelled monoclonal antibodies (Marks et al., 2011). Quantitative immunoprecipitation and Western blot analyses measured for wild‐type mice also confirmed that chronic nicotine treatment increased subunit expression and reinforce previous observations that increased binding sites are α4β2*‐nAChR (Flores et al., 1992). Results obtained with anti‐α5 antibody for wild‐type mice are also consistent with previous reports that α5*‐nAChR do not markedly increase following chronic nicotine treatment (Mao et al., 2008; Gahring and Rogers, 2010). However, α5*nAChR in cell lines have been reported to up‐regulate following nicotine treatment (Kuryatov et al., 2008).
Deletion of either subunit virtually eliminated epibatidine binding measured autoradiographically or by immunoprecipitation. Nicotine treatment had no effect on these parameters in null mutants. Although expression of [125I]‐epibatidine binding measured autoradiographically and [3H]epibatidine binding precipitated by anti‐α4 or anti‐β2 antibodies was reduced by approximately 50% in β2+/− mice, their response to chronic nicotine treatment was qualitatively very similar to that for wild‐type mice as noted previously (McCallum et al., 2006). Furthermore, quantitative Western blotting for β2+− mice revealed a pattern of response to chronic nicotine treatment similar to that for wild‐type mice. These results indicate that, despite a shift in stoichiometry favouring higher expression of (α4)3(β2)2‐nAChR (Nelson et al., 2003; Moroni et al., 2006; Gotti et al., 2008), regulation of α4β2‐nAChR persisting in β2+− mice following chronic nicotine treatment is similar to that occurring for wild‐type mice.
The relative increase in [125I]‐epibatidine binding for α4+− mice following chronic nicotine treatment was significantly greater than that observed for wild‐type or β2+/− mice, especially following treatment with 4.0 mg·kg−1·h−1. Immunoprecipitation by both the anti‐α4 and anti‐β2 antibodies and Western blotting to measure α4 and β2 nAChR subunit proteins confirmed this difference. Relatively more receptors with the (α4)2(β2)3‐nAChR stoichiometry persist in α4+− mice (Nelson et al., 2003; Moroni et al., 2006; Gotti et al., 2008). Perhaps this subtype exhibits more relative up‐regulation following chronic nicotine treatment.
α4+−/β2+− mice also responded more robustly to chronic nicotine treatment than did wild‐type or β2+/− mice. Relatively greater increase in [125I]‐epibatidine binding was observed autoradiographically following treatment with 0.25 and 1.0 mg·kg−1·h−1. Immunoprecipitation experiments suggested more up‐regulation following treatment with 4.0 mg·kg−1·h−1 nicotine than autoradiographic studies did. Quantitative Western blotting also indicates that chronic nicotine treatment elicits a significantly higher percentage increase of both α4 and β2 subunits for α4+−β2+− mice than observed for mice of any other genotype.
As was the case with wild‐type mice and consistent with previous reports, little relative change in α5*‐nAChR expression (mostly α4β2α5‐nAChR) occurred following chronic nicotine treatment for any mice differing in initial expression of the α4 or β2 nAChR subunit proteins.
Interpretation
Expression of α4β2*‐nAChR varies among individuals. For example, SPECT scans of non‐smokers using 5‐[123I] A85380 to measure β2*‐nAChR reported at least a fourfold range in signal intensity (Cosgrove et al., 2012; D'Souza et al., 2012). A similar variability was reported in post mortem samples (Breese et al., 2000). Variations in binding site densities were also observed for up‐regulated sites in smokers. Furthermore, less up‐regulation of high affinity nAChR binding sites was noted for schizophrenic smokers than for non‐schizophrenic smokers despite the observation that schizophrenics were heavier smokers (Breese et al., 2000; D'Souza et al., 2012). Perhaps, differences in responses of controls and schizophrenic individuals could arise from variation in relative expression of α4 or β2 subunits.
As noted in the Introduction, genetic factors have been shown to influence aspects of tobacco smoking behaviour. Results of this study with a mouse model that provides lowered initial expression of α4 and/or β2 nAChR subunit proteins indicate that differences in expression, particularly for the α4 subunit, alter the relative extent of up‐regulation in response to chronic nicotine treatment. Variants in human genes, particularly CHRNA4, may affect initial levels of nAChR expression in a manner analogous to that observed for mice by partial gene deletion in the mouse model. Genetic variation in individual humans could result in differences in receptor regulation following tobacco use and subsequently affect dependence and withdrawal from chronic nicotine exposure.
Author contributions
M.M. and F.F. did the experimental design, immunoprecipitation and Western blotting experiments, quantification and statistical analyses; C.G. performed the experimental design, experiment quantitation and statistical analyses and contributed to the writing of the paper; M.J.M. carried out the experimental design, surgery, tissue harvest, tissue sectioning, autoradiography experiments and quantitation, statistical analyses, figure preparation, wrote the initial draft and manuscript editing.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effect of deletion or partial deletion of the α4 and/or β2 nAChR subunits examined autoradiographically.
Figure S2 Effect of deletion or partial deletion of the α4 and/or β2 nAChR subunits examined by immunoprecipitation.
Table S1 Cytisine‐sensitive [125I]‐epibatidine binding in mice differing in α4 and β2 expression following chronic nicotine treatment.
Acknowledgements
The research was supported by grants to M.J.M. from the National Institute on Drug Abuse, National Institutes of Health, USA (Grant numbers: R01 DA003194 and P30 DA015663) and by the CNR Research Project on Aging to C.G. The authors wish to thank Esteban Loetz, Erin Meyers, Daphne Baber, Penelope Herder and Nick Ortiz for assistance with genotyping, surgical procedures and drug treatments. The authors also thank Allan C. Collins and Sharon R. Grady for critical reading of the manuscript and helpful editorial comments.
Moretti, M. , Fasoli, F. , Gotti, C. , and Marks, M. J. (2018) Reduced α4 subunit expression in α4+− and α4+−/β2+− nicotinic acetylcholine receptors alters α4β2 subtype up‐regulation following chronic nicotine treatment. British Journal of Pharmacology, 175: 1944–1956. doi: 10.1111/bph.13896.
Contributor Information
Cecilia Gotti, Email: c.gotti@in.cnr.it.
Michael J Marks, Email: marksm@colorado.edu.
References
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89: 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddick CG, Marks MJ (2011). An autoradiographic survey of mouse brain nicotinic acetylcholine receptors defined by null mutants. Biochem Pharmacol 82: 828–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ, Anderson JM (1988). Evidence that tobacco smoking increases the density of (−)‐[3H]nicotine binding sites in human brain. J Neurochem 50: 1243–1247. [DOI] [PubMed] [Google Scholar]
- Bierut LJ, Johnson EO, Saccone NL (2014). A glimpse into the future – personalized medicine for smoking cessation. Neuropharmacology 76 (Pt B): 592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese CR, Lee MJ, Adams CE, Sullivan B, Logel J, Gillen KM et al. (2000). Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology 23: 351–364. [DOI] [PubMed] [Google Scholar]
- Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC et al. (1997). Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther 282: 7–13. [PubMed] [Google Scholar]
- Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L et al. (2002). Distribution and pharmacology of alpha 6‐containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci 22: 1208–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove KP, Esterlis I, McKee SA, Bois F, Seibyl JP, Mazure CM et al. (2012). Sex differences in availability of beta2*‐nicotinic acetylcholine receptors in recently abstinent tobacco smokers. Arch Gen Psychiatry 69: 418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaj MI, Fonck C, Marks MJ, Deshpande P, Labarca C, Lester HA et al. (2007). Genetic approaches identify differential roles for alpha4beta2* nicotinic receptors in acute models of antinociception in mice. J Pharmacol Exp Ther 321: 1161–1169. [DOI] [PubMed] [Google Scholar]
- Davila‐Garcia MI, Houghtling RA, Qasba SS, Kellar KJ (1999). Nicotinic receptor binding sites in rat primary neuronal cells in culture: characterization and their regulation by chronic nicotine. Brain Res Mol Brain Res 66: 14–23. [DOI] [PubMed] [Google Scholar]
- D'Souza DC, Esterlis I, Carbuto M, Krasenics M, Seibyl J, Bois F et al. (2012). Lower ss2*‐nicotinic acetylcholine receptor availability in smokers with schizophrenia. Am J Psychiatry 169: 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterlis I, Hillmer AT, Bois F, Pittman B, McGovern E, O'Malley SS et al. (2016). CHRNA4 and ANKK1 polymorphisms influence smoking‐induced nicotinic acetylcholine receptor upregulation. Nicotine Tob Res 18: 1845–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasoli F, Gotti C (2015). Structure of neuronal nicotinic receptors. Curr Top Behav Neurosci 23: 1–17. [DOI] [PubMed] [Google Scholar]
- Feng Y, Niu T, Xing H, Xu X, Chen C, Peng S et al. (2004). A common haplotype of the nicotine acetylcholine receptor alpha 4 subunit gene is associated with vulnerability to nicotine addiction in men. Am J Hum Genet 75: 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ (1992). A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up‐regulated by chronic nicotine treatment. Mol Pharmacol 41: 31–37. [PubMed] [Google Scholar]
- Franklin Keith BJ, Paxinos G (1997). The Mouse Brain in Stereotaxic Coordinates. Acamdemic Press: San Diego. [Google Scholar]
- Gahring LC, Rogers SW (2010). Nicotinic receptor subunit alpha5 modifies assembly, up‐regulation, and response to pro‐inflammatory cytokines. J Biol Chem 285: 26049–26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza A, Huang LZ, Son JH, Winzer‐Serhan UH (2009). Expression of nicotinic acetylcholine receptors and subunit messenger RNAs in the enteric nervous system of the neonatal rat. Neuroscience 158: 1521–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Moretti M, Meinerz NM, Clementi F, Gaimarri A, Collins AC et al. (2008). Partial deletion of the nicotinic cholinergic receptor alpha 4 or beta 2 subunit genes changes the acetylcholine sensitivity of receptor‐mediated 86Rb+ efflux in cortex and thalamus and alters relative expression of alpha 4 and beta 2 subunits. Mol Pharmacol 73: 1796–1807. [DOI] [PubMed] [Google Scholar]
- Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L et al. (2009). Rodent habenulo‐interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the alpha3beta4* and alpha3beta3beta4* subtypes mediate acetylcholine release. J Neurosci 29: 2272–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall W, Madden P, Lynskey M (2002). The genetics of tobacco use: methods, findings and policy implications. Tob Control 11: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison KE, Allen DL, Filbey FM, Jepson C, Lerman C, Benowitz NL et al. (2007). CHRNA4 and tobacco dependence: from gene regulation to treatment outcome. Arch Gen Psychiatry 64: 1078–1086. [DOI] [PubMed] [Google Scholar]
- Kamens HM, Corley RP, McQueen MB, Stallings MC, Hopfer CJ, Crowley TJ et al. (2013). Nominal association with CHRNA4 variants and nicotine dependence. Genes Brain Behav 12: 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Onksen J, Lindstrom J (2008). Roles of accessory subunits in alpha4beta2(*) nicotinic receptors. Mol Pharmacol 74: 132–143. [DOI] [PubMed] [Google Scholar]
- Lomazzo E, Hussmann GP, Wolfe BB, Yasuda RP, Perry DC, Kellar KJ (2011). Effects of chronic nicotine on heteromeric neuronal nicotinic receptors in rat primary cultured neurons. J Neurochem 119: 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK et al. (1999). International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 51: 397–401. [PubMed] [Google Scholar]
- Mao D, Perry DC, Yasuda RP, Wolfe BB, Kellar KJ (2008). The alpha4beta2alpha5 nicotinic cholinergic receptor in rat brain is resistant to up‐regulation by nicotine in vivo. J Neurochem 104: 446–456. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Burch JB, Collins AC (1983). Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J Pharmacol Exp Ther 226: 817–825. [PubMed] [Google Scholar]
- Marks MJ, McClure‐Begley TD, Whiteaker P, Salminen O, Brown RW, Cooper J et al. (2011). Increased nicotinic acetylcholine receptor protein underlies chronic nicotine‐induced up‐regulation of nicotinic agonist binding sites in mouse brain. J Pharmacol Exp Ther 337: 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Meinerz NM, Drago J, Collins AC (2007). Gene targeting demonstrates that alpha4 nicotinic acetylcholine receptor subunits contribute to expression of diverse [3H]epibatidine binding sites and components of biphasic 86Rb+ efflux with high and low sensitivity to stimulation by acetylcholine. Neuropharmacology 53: 390–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Rowell PP, Cao JZ, Grady SR, McCallum SE, Collins AC (2004). Subsets of acetylcholine‐stimulated 86Rb+ efflux and [125I]‐epibatidine binding sites in C57BL/6 mouse brain are differentially affected by chronic nicotine treatment. Neuropharmacology 46: 1141–1157. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Smith KW, Collins AC (1998). Differential agonist inhibition identifies multiple epibatidine binding sites in mouse brain. J Pharmacol Exp Ther 285: 377–386. [PubMed] [Google Scholar]
- Marks MJ, Stitzel JA, Collins AC (1989). Genetic influences on nicotine responses. Pharmacol Biochem Behav 33: 667–678. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Stitzel JA, Grady SR, Picciotto MR, Changeux JP, Collins AC (2000). Nicotinic‐agonist stimulated (86)Rb(+) efflux and [(3)H]epibatidine binding of mice differing in beta2 genotype. Neuropharmacology 39: 2632–2645. [DOI] [PubMed] [Google Scholar]
- Marubio LM, del Mar Arroyo‐Jimenez M, Cordero‐Erausquin M, Lena C, Le Novere N, de Kerchove d'Exaerde A et al. (1999). Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 398: 805–810. [DOI] [PubMed] [Google Scholar]
- McCallum SE, Collins AC, Paylor R, Marks MJ (2006). Deletion of the beta 2 nicotinic acetylcholine receptor subunit alters development of tolerance to nicotine and eliminates receptor upregulation. Psychopharmacology (Berl) 184: 314–327. [DOI] [PubMed] [Google Scholar]
- McClure‐Begley TD, Wageman CR, Grady SR, Marks MJ, McIntosh JM, Collins AC et al. (2012). A novel alpha‐conotoxin MII‐sensitive nicotinic acetylcholine receptor modulates [(3) H]‐GABA release in the superficial layers of the mouse superior colliculus. J Neurochem 122: 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melroy‐Greif WE, Stitzel JA, Ehringer MA (2016). Nicotinic acetylcholine receptors: upregulation, age‐related effects and associations with drug use. Genes Brain Behav 15: 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar NS, Gotti C (2009). Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 56: 237–246. [DOI] [PubMed] [Google Scholar]
- Moretti M, Mugnaini M, Tessari M, Zoli M, Gaimarri A, Manfredi I et al. (2010). A comparative study of the effects of the intravenous self‐administration or subcutaneous minipump infusion of nicotine on the expression of brain neuronal nicotinic receptor subtypes. Mol Pharmacol 78: 287–296. [DOI] [PubMed] [Google Scholar]
- Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I (2006). Alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long‐term exposure to nicotine. Mol Pharmacol 70: 755–768. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J (2003). Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol 63: 332–341. [DOI] [PubMed] [Google Scholar]
- Perry DC, Davila‐Garcia MI, Stockmeier CA, Kellar KJ (1999). Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther 289: 1545–1552. [PubMed] [Google Scholar]
- Perry DC, Xiao Y, Nguyen HN, Musachio JL, Davila‐Garcia MI, Kellar KJ (2002). Measuring nicotinic receptors with characteristics of alpha4beta2, alpha3beta2 and alpha3beta4 subtypes in rat tissues by autoradiography. J Neurochem 82: 468–481. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, Le Novere N et al. (1995). Abnormal avoidance learning in mice lacking functional high‐affinity nicotine receptor in the brain. Nature 374: 65–67. [DOI] [PubMed] [Google Scholar]
- Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK et al. (2000). Phenotypic characterization of an alpha 4 neuronal nicotinic acetylcholine receptor subunit knock‐out mouse. J Neurosci 20: 6431–6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC et al. (2004). Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol 65: 1526–1535. [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Kellar KJ (1983). Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science 220: 214–216. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JK, Krishnan‐Sarin S, Cosgrove KP, Krantzler E, Frohlich E, Perry E et al. (2006). Human tobacco smokers in early abstinence have higher levels of beta2* nicotinic acetylcholine receptors than nonsmokers. J Neurosci 26: 8707–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Marks MJ, Lester HA (2007). Nicotine responses in hypersensitive and knockout alpha 4 mice account for tolerance to both hypothermia and locomotor suppression in wild‐type mice. Physiol Genomics 31: 422–428. [DOI] [PubMed] [Google Scholar]
- Tritto T, McCallum SE, Waddle SA, Hutton SR, Paylor R, Collins AC et al. (2004). Null mutant analysis of responses to nicotine: deletion of beta2 nicotinic acetylcholine receptor subunit but not alpha7 subunit reduces sensitivity to nicotine‐induced locomotor depression and hypothermia. Nicotine Tob Res 6: 145–158. [DOI] [PubMed] [Google Scholar]
- Wang F, Gerzanich V, Wells GB, Anand R, Peng X, Keyser K et al. (1996). Assembly of human neuronal nicotinic receptor alpha5 subunits with alpha3, beta2, and beta4 subunits. J Biol Chem 271: 17656–17665. [DOI] [PubMed] [Google Scholar]
- Ware JJ, van den Bree MB, Munafo MR (2011). Association of the CHRNA5‐A3‐B4 gene cluster with heaviness of smoking: a meta‐analysis. Nicotine Tob Res 13: 1167–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, Cooper JF, Salminen O, Marks MJ, McClure‐Begley TD, Brown RW et al. (2006). Immunolabeling demonstrates the interdependence of mouse brain alpha4 and beta2 nicotinic acetylcholine receptor subunit expression. J Comp Neurol 499: 1016–1038. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, McIntosh JM, Luo S, Collins AC, Marks MJ (2000). 125I‐Alpha‐conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol Pharmacol 57: 913–925. [PubMed] [Google Scholar]
- Zambrano CA, Salamander RM, Collins AC, Grady SR, Marks MJ (2012). Regulation of the distribution and function of [(125)I]epibatidine binding sites by chronic nicotine in mouse embryonic neuronal cultures. J Pharmacol Exp Ther 342: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of deletion or partial deletion of the α4 and/or β2 nAChR subunits examined autoradiographically.
Figure S2 Effect of deletion or partial deletion of the α4 and/or β2 nAChR subunits examined by immunoprecipitation.
Table S1 Cytisine‐sensitive [125I]‐epibatidine binding in mice differing in α4 and β2 expression following chronic nicotine treatment.