

Abstract
Substantial data from preclinical studies have revealed the biphasic effects of statins on cardiovascular angiogenesis. Although some have reported the anti‐angiogenic potential of statins in malignant tumors, the underlying mechanism remains poorly understood. The aim of this study is to elucidate the mechanism by which simvastatin, a member of the statin family, inhibits tumor angiogenesis. Simvastatin significantly suppressed tumor cell‐conditioned medium‐induced angiogenic promotion in vitro, and resulted in dose‐dependent anti‐angiogenesis in vivo. Further genetic silencing of hypoxia‐inducible factor‐1α (HIF‐1α) reduced vascular endothelial growth factor and fibroblast growth factor‐2 expressions in 4T1 cells and correspondingly ameliorated HUVEC proliferation facilitated by tumor cell‐conditioned medium. Additionally, simvastatin induced angiogenic inhibition through a mechanism of post‐transcriptional downregulation of HIF‐1α by increasing the phosphorylation level of AMP kinase. These results were further validated by the fact that 5‐aminoimidazole‐4‐carboxamide ribonucleotide reduced HIF‐1α protein levels and ameliorated the angiogenic ability of endothelial cells in vitro and in vivo. Critically, inhibition of AMPK phosphorylation by compound C almost completely abrogated simvastatin‐induced anti‐angiogenesis, which was accompanied by the reduction of protein levels of HIF‐1α and its downstream pro‐angiogenic factors. These findings reveal the mechanism by which simvastatin induces tumor anti‐angiogenesis, and therefore identifies the target that explains the beneficial effects of statins on malignant tumors.
Keywords: AMPK, anti‐angiogenesis, HIF‐1α, simvastatin, statin
Abbreviations
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleotide
- AMPK
AMP kinase
- ATCC
American type culture collection
- CM
conditioned medium
- DMEM
Dulbecco's modified Eagle's medium
- FGF‐2
fibroblast growth factor‐2
- HIF‐1a
hypoxia‐inducible factor‐1α
- HUVEC
human umbilical vascular endothelial cell
- IHC
immunohistochemistry
- LKB1
liver kinase B1
- PBS
phosphate‐buffered saline
- SD
standard deviation
- SFM
serum‐free medium
- TCM
tumor cell‐conditioned medium
- TRITC
tetraethyl rhodamine isothiocyanate
- VEGFR
vascular endothelial growth factor receptor
- VEGF
vascular endothelial growth factor
- α‐SMA
alpha smooth muscle actin
1. INTRODUCTION
Angiogenesis, the process of developing new vascular lumen from the pre‐existing vessels, has been deeply implicated in the progression and development of many diseases.1, 2 It has been well documented that angiogenesis is essential for malignant tumors advancing from the dormant stage to a highly metastatic phenotype.3 Previously published articles have provided substantial evidence for the necessity of targeting tumor angiogenesis.4, 5 During the last decade, anti‐angiogenic drugs have been designed to chiefly target the VEGF/VEGFR‐2 signaling axis.4 However, this therapeutic strategy has been greatly challenged for its indiscriminate targeting of both healthy and tumoral VEGF/VEGFR‐2 pathways.6 Thus, it is urgent to develop new drugs or novel therapeutic strategy for overcoming the challenge.
Simvastatin, a member of the statin family, has been widely used to lower lipid levels and reduce cardiovascular risk.7 Due to its high efficacy, the mechanisms underlying simvastatin‐induced cardiovascular protection have increasingly attracted researchers’ attention. It has been reported that simvastatin showed disparate effects on hypoxia and inflammation‐induced angiogenesis.8 In addition, low and high doses of simvastatin had biphasic effects on endothelial proliferation and migration.9 Although tumor angiogenesis has some similarity with inflammatory angiogenesis, the effects of simvastatin on tumor angiogenesis and the associated underlying mechanisms remain poorly understood.
Recently, HIF‐1α, a critical factor for mediating angiogenesis, has been reported to be deeply involved in simvastatin‐induced antitumor activities.10 However, less is known about whether simvastatin induces tumor anti‐angiogenesis and how HIF‐1α is affected by the upstream signaling. We previously reported that metformin, a drug that activates AMPK, suppressed breast tumor angiogenesis by targeting the HIF‐α/VEGF signaling pathway.11 Similar to metformin, simvastatin is capable of inhibiting tumor proliferation by activating AMPK.12 Together with this evidence, we therefore propose AMPK‐mediated HIF‐1α inhibition to be a meaningful mechanism by which simvastatin induces tumor anti‐angiogenesis. Thus, the aim of this study is to investigate the definite effect of simvastatin on tumor angiogenesis, and to reveal the involvement of the AMPK‐HIF‐1α pathway.
2. MATERIALS AND METHODS
2.1. Cells and reagents
4T1 (murine breast carcinoma cell), MDA‐MB‐231 (human triple‐negative breast cancer cell), and HUVECs were obtained from ATCC (Manassas, VA, USA). Cells were cultured with DMEM containing 10% FBS at 37°C in an incubator.
Compound C (No. 11967), AICAR (No. 10010241), and simvastatin (No. 10010344) were purchased from Cayman Chemical. The siRNA control and siRNA HIF‐1α were obtained from Thermo Fisher Scientific. Rabbit anti‐CD31 antibody (ab28364) was purchased from Abcam. Alexa Fluor 488‐ and 546‐conjugated antibodies were commercially provided by Life Technologies.
2.2. Tumor cell‐conditioned medium
Briefly,11 tumor cells were initially cultured in 10% FBS DMEM supplemented with simvastatin, AICAR, compound C, siRNA control, siRNA‐HIF‐1α, or the combined pretreatment for 12 or 24 hours. After washing with PBS washing three times, cells were cultured in SFM for 24 hours. Then the TCM was collected and centrifuged to remove cell debris. The number of live cells in each corresponding dish was finally counted to make a correction of TCM loading.
2.3. Tube formation assay
To mimic in vivo angiogenesis, in vitro HUVEC‐mediated tube formation assay was carried out.11 First, we used 100 μL VEGF‐reduced Matrigel (Basement Membrane Matrix; BD Bioscience) to precoat the whole bottom of a 96‐well plate. The Matrigel was allowed to polymerize at 37°C for approximately 30 minutes, then 15 000 HUVECs were seeded and cultured in SFM or TCM at 37°C. Twelve hours later, the formed capillary‐like network was recorded by using a microscope. The length of formed endothelial networks was measured by using the software of ImageJ 2X (NIH, Bethesda, MD, USA).
2.4. Proliferation assay
Cellular viability of HUVECs was determined by using CCK‐8 (Dojindo, Japan) following the manufacturer's instructions. At the end of culture, the absorbance at 450 nm of each well was measured using a microplate reader. Finally, the cellular viability relative to the SFM group was calculated to observe the proliferative ability of HUVECs treated with different interventions.
2.5. Animal models
BALB/c mice, 6‐8 weeks old, were selected for further in vivo experiments. Briefly, the 4T1 orthotopic model was established by injection of 5 × 105 4T1 cancer cells (in 100 μL) into the fat pad of the fourth breast of BALB/c mice. Seven days later, the average tumor volume reached approximately 100 mm3. All tumor‐bearing mice were randomly divided into different groups with eight mice per cohort. Mice were treated with simvastatin (15 or 5 mg/kg), AICAR (400 mg/kg), or concomitant compound C (20 mg/kg) in the context of simvastatin through i.p. injection. Treatment lasted for 2 weeks, and tumor volumes were calculated every 2‐3 days by using the formula 1/2 × (length × width2).
2.6. Immunoblotting
Briefly, the protein concentration of each sample was determined by the BCA protein assay. To determine the protein levels of HIF‐1α (Abcam), p‐AMPK (Cell Signaling Technology), total AMPK (Cell Signaling Technology), VEGF (ProteinTech), FGF‐2 (ProteinTech), and GAPDH (ProteinTech), 80 μg protein was loaded and subsequently separated by 8%‐12% SDS‐PAGE. After transferring the protein to a PVDF membrane, antibodies were diluted and incubated with the membrane at 4°C for 24 hours, followed by the corresponding HRP‐conjugated secondary antibody (ProteinTech). Finally, the Western blotting results were recorded by using a chemiluminescence imaging system (Bio‐Rad).
2.7. Reverse transcription‐PCR
To detect mRNA levels of HIF‐1α, VEGF, and FGF‐2, a standardized RT‐PCR was carried out. After extraction of mRNA of tumor cells, the RT reaction was immediately undertaken to obtain the corresponding cDNA, according to the manufacturer's instructions. The cDNA fragment was amplified by PCR using the following primers: HIF‐1α (forward, AGTGGTATTATTCAGCACGAC; reverse, AGTGGTATTATTCAGCACGAC), VEGF (forward, GTGCAGGCTGCTGTAACGAT; reverse, GGGATTTCTTGCGCTTTCGT), and FGF‐2 (forward, GGCTGCTGGCTTCTAAGTGT; reverse, TCTGTCCAGGTCCCGTTTTG). The intensity of each band was measured by using ImageJ2X software for mRNA quantification.
2.8. Immunohistochemistry and immunofluorescence
To observe changes in the perfusion status of tumors, TRITC‐Lectin (Vector Laboratories) was i.v. injected into the tail veins of mice 15 minutes before they were killed.13 The wholly extracted tumor tissues were prepared for further immunofluorescent and IHC staining as previously described.11, 13 For IHC staining, the paraffin‐embedded tumor tissues were cut into 3‐4‐μm sections; for immunofluorescence staining, tumor tissues fixed by 4% paraformaldehyde were cut into approximately 6‐μm sections. Tissue sections were double‐stained with CD31 (Abcam) and α‐SMA (Boster Bio) antibodies to detect the change in vascular pericyte coverage, which indicates vascular maturity.
2.9. Statistical analysis
All quantitative data are represented as mean ± SD. One‐way anova and Student's t‐test were used to detect the significant difference between two or multiple groups. Statistical analysis was carried out by using GraphPad Prism 5 software (GraphPad, San Diego, CA, USA). Significance is indicated by *P < .05, **P < .01, and ***P < .001; ns indicates that there was no statistically significant difference between groups.
2.10. Ethics statement
All experimental procedures were approved by the Committee of Institutional Animal Care and Use of Xi'an Jiaotong University (Xi'an, China). Methods were carried out were in accordance with the approved guideline.
3. RESULTS
3.1. Simvastatin inhibited tumor angiogenesis in vivo in a dose‐dependent manner
To investigate the effects of simvastatin on tumor angiogenesis, we established an orthotopic breast cancer model by injection of 4T1 tumor cells into murine breast fat pad. Tumor‐bearing BALB/c mice received daily simvastatin treatment at a dose of 5 or 15 mg/kg/day for 14 days. Characterization of CD31+ vessels showed that vessels in untreated 4T1 tumors showed an aberrantly high ability of vascular sprouting (Figure 1A,B). This phenomenon suggests that 4T1 breast cancer has a similar characterization to cancers of the angiogenic phenotype. Simvastatin greatly reduced the number of vessels in the 4T1 tumors, whilst also increasing vascular pericyte coverage and improving the function of tumor vasculature (Figure 1A‐D). As angiogenesis is critical for tumor growth, we next focused on the effect of simvastatin on in vivo tumor growth. As shown in Figure 1E, both doses of simvastatin significantly suppressed 4T1 tumor growth, and the higher dose did appear more effective. These results indicate that simvastatin has the potential to inhibit tumor angiogenesis and growth in vivo.
Figure 1.

Inhibition of tumor angiogenesis by simvastatin through a paracrine‐related mechanism. A, 4T1 tumors from simvastatin‐treated and control mice were double‐immunostained for CD31 (green) and α‐smooth muscle actin (α‐SMA) (red), and B, quantified for microvessel density and vessels with pericyte coverage (n = 8). White arrows indicate vascular sprouts; yellow arrows indicate vascular pericyte. Scale bar = 100 μm. C, Micrographs of TRITC‐Lectin‐perfused and CD31‐stained vessels in control and simvastatin‐treated 4T1 tumors. TRITC‐Lectin was injected into the tail vein of mice 15 min before they were killed. Scale bar = 100 μm. D, Quantification of Lectin‐perfused CD31+ vessels (% total CD31+ vessels) in 4T1 tumors (n = 6). E, Representative images showing in vivo growth curve of 4T1 tumors from mice untreated or treated with simvastatin (SMV) (n = 8). F, G, SMV pretreatment (0.5 μmol/L) reduced the tube formation ability of HUVECs, which was promoted by the tumor cell‐conditioned medium of 4T1 tumor cells (n = 6). HUVECs were cultured in serum free medium (SFM) or conditioned medium (CM) of 4T1 cells, untreated (Con) or pretreated with SMV. H, I, SMV pretreatment greatly inhibited the proliferative ability of HUVECs (EC) cultured in (H) 4T1 or (I) MDA‐MB‐231 conditioned medium (CM) (n = 6). Quantitative data are indicated as mean ± SD. *P < .05; ***P < .001. ns, no statistically significant difference (P > .05)
3.2. Simvastatin suppressed in vitro tumor angiogenesis by a paracrine mechanism
In our previous study,11 breast tumor cells promoted endothelial cell‐mediated angiogenesis by secretion of pro‐angiogenic factor. We next evaluated whether simvastatin has potential to restrain paracrine mechanism‐related angiogenic promotion using a range of doses 5‐80 mg/day. As the plasma concentration of simvastatin was estimated to be <0.5 μmol/L in blood of patients,14, 15 we thus used 0.1 and 0.5 μmol/L for further in vitro experiments. We first collected the CM of 4T1 and MDA‐MB‐231 cells pretreated with 0.5 μmol/L simvastatin. Consistent with our previous study,11 CM of untreated 4T1 and MDA‐MB‐231 cells greatly promoted proliferative and tube‐formation abilities of HUVECs (Figure 1F‐I). Critically, this CM‐induced angiogenic promotion was greatly abrogated by simvastatin pretreatment (Figure 1G‐I), suggesting that simvastatin can negatively affect tumor cell paracrine mechanism‐related angiogenesis. Further morphological characterization showed that the same dose of simvastatin had no obvious effect on HUVEC morphology (data not shown). Thus, these results are more inclined to support the fact that the tumor cell‐mediated paracrine effect is closely correlated to simvastatin‐associated angiogenic inhibition.
3.3. Simvastatin reduced expression levels of HIF‐1α and its downstream pro‐angiogenic factors
Hypoxia‐inducible factor‐1α is a well‐documented factor that induces both physiological and pathological angiogenesis.16, 17 We therefore investigated the effect of simvastatin on HIF‐1α protein levels. As shown in Figure 2A, both doses (0.1 and 0.5 μmol/L) of simvastatin treatment resulted in a great reduction of HIF‐1α protein in normoxia (Figure 2A). The inhibition of HIF‐1α protein levels became more apparent as the concentration increased from 0.1 to 0.5 μmol/L, reflecting the same inhibitory trend with in vivo tumor angiogenesis. We next examined mRNA levels of HIF‐1α and its downstream factors (Figure 2B). The results of RT‐PCR showed that simvastatin exhibited a great decrease in mRNA levels of both VEGF and FGF‐2, while having no obvious effect on mRNA levels of HIF‐1α (Figures 2B, S1). Consistent with our in vitro results, simvastatin (15 mg/kg/day) significantly decreased the signal intensities of HIF‐1α, VEGF, and FGF‐2 in sections of 4T1 tumors (Figure 2C‐F). Furthermore, simvastatin‐treated tumors had significantly lower protein levels of both VEGF and FGF2 than control (Figure 2G). Overall, these results suggest that HIF‐1α‐induced paracrine pro‐angiogenic factors are deeply involved in simvastatin‐induced suppression of tumor angiogenesis.
Figure 2.
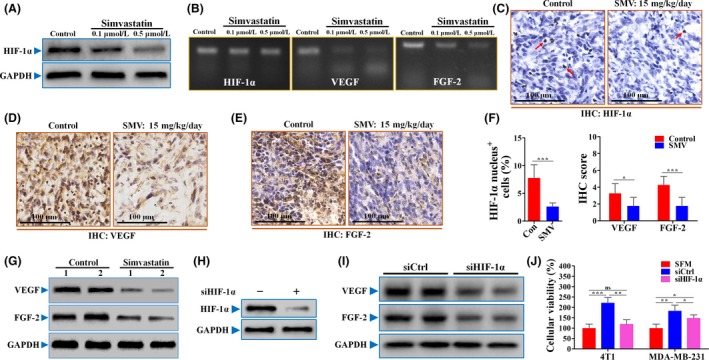
Suppression of hypoxia‐inducible factor‐1α (HIF‐1α)‐induced pro‐angiogenic factors was greatly attributed to simvastatin (SMV)‐induced angiogenic inhibition. A, Immunoblotting for protein level of HIF‐1α in 4T1 cells untreated or treated with 0.1 or 0.5 μmol/L SMV for 12 h. GAPDH was used as a control for protein loading. B, Representative images for showing the mRNA levels of HIF‐1α, vascular endothelial growth factor (VEGF), and fibroblast growth factor‐2 (FGF‐2) in untreated or SMV‐treated 4T1 cells. For immunohistochemical (IHC) detection, 4T1 tumor‐bearing BALB/c mice were treated with PBS (Con) or 15 mg/kg/day SMV for 14 d. IHC detection of HIF‐1α (C), VEGF (D), and FGF‐2 (E) in sections of 4T1 tumors. Scale bar = 100 μm. Red arrows in panel (C) indicate HIF‐1α nucleus+ cells. F, Percentage of HIF‐1α nucleus+ cells (% total cells; left) and quantification of IHC scores of VEGF and FGF‐2 of 4T1 tumors (right; n = 8). G, Western blotting for detecting expression levels of VEGF and FGF‐2 in tissues of control and SMV‐treated 4T1 tumors. Protein expressions of HIF‐1α (H), and VEGF and FGF‐2 (I) of 4T1 cells in response to SMV treatment after HIF‐1α knockdown. J, Cellular viability of HUVECs cultured with serum‐free medium (SFM) or 4T1‐conditioned or MDA‐MB‐213‐conditioned medium (n = 6). Both 4T1 and MDA‐MB‐231 cells were treated with siRNA‐Control (siCtrl) or siRNA‐HIF‐1α (siHIF‐1α) for 24 h. After washing with PBS, the conditioned medium was collected 24 h later. Quantitative data are indicated as mean ± SD. *P < .05; **P < .01; ***P < .001. ns, no statistically significant difference (P > .05). Con, control
3.4. Hypoxia‐inducible factor‐1α was essential for tumor angiogenesis promoted by paracrine pro‐angiogenic factors
To further investigate the involvement of HIF‐1α, we genetically knocked down HIF‐1α expression in both 4T1 and MDA‐MB‐231 cells by using siRNA. Compared with siRNA‐control (Figure 2H), siRNA‐HIF‐1α greatly reduced the HIF‐1α protein level. In addition, HIF‐1α knockdown resulted in a great reduction of VEGF and FGF‐2 (Figure 2G), indicating that HIF‐1α is required for tumor cell‐derived pro‐angiogenic factors. To further confirm this conclusion, 4T1 and MDA‐MB‐231 tumor cells were pretreated with siRNAs. Consistently, 4T1‐CM‐induced promotion of HUVEC proliferation was almost completely abrogated by siRNA‐HIF‐1α pretreatment (Figure 2J). In the MDA‐MB‐231 cell line, siRNA‐HIF‐1α pretreatment showed a partial but significant abrogation of CM‐induced proliferative promotion (Figure 2J). These results provide evidence to suggest that HIF‐1α plays an essential role in tumor angiogenesis promoted by tumor cell‐derived pro‐angiogenic factors.
3.5. Simvastatin inhibited HIF‐1α‐induced expressions of pro‐angiogenic factors through activation of AMPK
Recently, AMPK activation by agents, such as metformin, has been reported to be applicable to restrain the HIF‐1α protein level.11, 18 This evidence indicates that AMPK could be an upstream regulator of HIF‐1α. Therefore, we next focused on the effect of simvastatin on AMPK activation. Simvastatin greatly increased the level of phosphorylated AMPK protein (active form) in a dose‐dependent manner (Figure 3A), while not affecting the total AMPK protein level.
Figure 3.
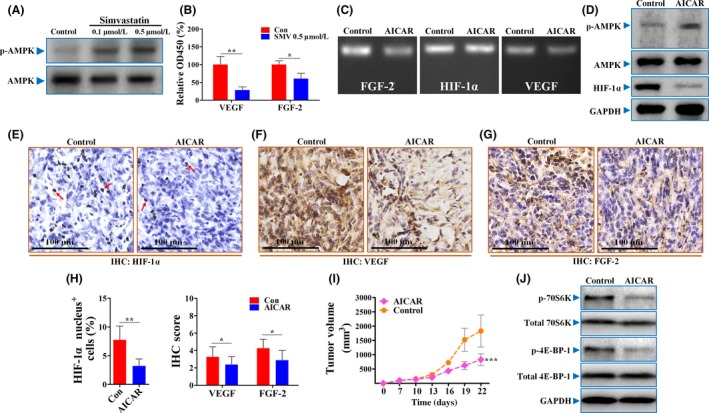
Simvastatin (SMV) reduced hypoxia‐inducible factor‐1α (HIF‐1α) protein levels by activation of AMP kinase (AMPK). A, Representative Western blot analysis showing AMPK/phosphorylated (p‐)AMPK protein levels in 4T1 tumor cells treated with PBS or different doses of SMV. B, Quantifications of secreted vascular endothelial growth factor (VEGF) and fibroblast growth factor‐2 (FGF‐2) proteins in supernatants of 4T1 cells by ELISA (n = 6). C, Representative images showing the mRNA levels of HIF‐1α, VEGF, and FGF‐2 in 4T1 cells untreated or treated with 1 mmol/L 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR). D, Immunoblotting for p‐AMPK/AMPK/HIF‐1α in AICAR‐treated or ‐untreated 4T1 cells. GAPDH was used as a control for protein loading. Immunohistochemical (IHC) detection of HIF‐1α (E), VEGF (F), and FGF‐2 (G) in sections of 4T1 tumors. Red arrows in panel (E) indicate HIF‐1α nucleus+ cells. Scar bar = 100 μm. H, Percentage of HIF‐1α nucleus+ cells (% total cells; left) and IHC quantification of VEGF and FGF‐2 in sections of 4T1 tumors treated with PBS (Con) or 400 mg/kg AICAR (right; n = 8). I, Representative images showing growth curve of 4T1 tumors from mice untreated or treated with AICAR (n = 8). J, Immunoblotting for both total and phosphorylation levels of 70S6K and 4E‐BP‐1, two direct targets of mTOR, in AICAR‐treated or ‐untreated 4T1 cells. Quantitative data are indicated as mean ± SD. *P < .05; **P < .01; ***P < .001. ns, no statistically significant difference (P > .05)
To further investigate whether AMPK activation has a role in influencing HIF‐1α‐promoted angiogenesis, we next focused on the effect of AICAR, an AMPK activator. As expected, secretion levels of both VEGF and FGF‐2 were significantly lower in supernatants of AICAR‐treated 4T1 cells than that of control (Figure 3B). Treatment with AICAR decreased the mRNA levels of both VEGF and FGF‐2 in 4T1 tumor cells (Figures 3C, S2), but had no effect on HIF‐1α. Further Western blot analysis showed that AICAR greatly increased the phosphorylation level of AMPK protein (Figure 3D), which was accompanied by decreased HIF‐1α expression. Consistently, the percentage of HIF‐1α nucleus+ cells and intensities of downstream VEGF and FGF‐2 were significantly lower in AICAR‐treated 4T1 tumors than controls (Figure 3E‐H). Additionally, AICAR exerted a similar inhibitory effect on tumor growth to simvastatin (Figure 3I).
We also focused on mTOR, which is a downstream factor of AMPK and regulates protein synthesis of HIF‐1α. As shown in Figure 3J, AICAR treatment greatly reduced the phosphorylation levels of both 4E‐BP‐1 and 70S6K, two direct targets of mTOR, but did not affect their total protein levels. Taken together, our results suggest that simvastatin‐induced angiogenic inhibition depends on AMPK activation‐induced inhibition of the mTOR/HIF‐1α signaling pathway.
3.6. Sprouting ability of vessels in vivo restrained by AICAR
As HIF‐1α is critical for tumor angiogenesis, we then focused on the effects of AICAR on in vivo tumor angiogenesis and the sprouting ability of tumor vessels. As shown in Figure 4A,B, 4T1 tumors from mice treated with AICAR showed a significantly lower microvessel density than those from untreated mice. The vascular sprouting ability, which is frequently evaluated by the number of sprouts, is one important indicator for evaluating tumor angiogenesis. Immunofluorescence for CD31 (Figure 4C,D) showed that vessels of 4T1 untreated tumors had more vascular sprouts (per vessel; Figure 4C, white arrows), which originated from the pre‐existing vessels. This angiogenic phenomenon could not be easily found in the AICAR group, indicating the potential of AICAR‐induced AMPK activation on angiogenic inhibition.
Figure 4.
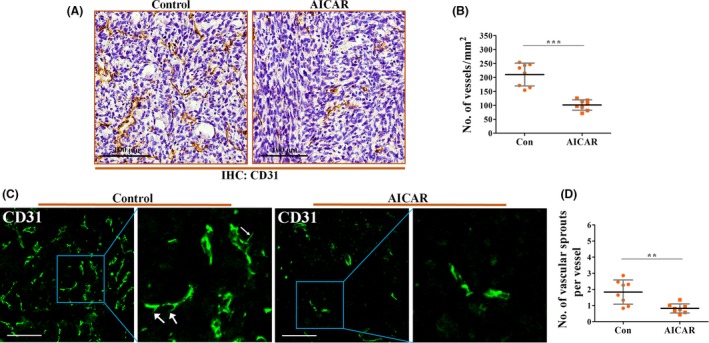
5‐Aminoimidazole‐4‐carboxamide ribonucleotide (AICAR), a specific AMP kinase activator, suppressed tumor angiogenesis in vivo. 4T1 tumor‐bearing mice were treated with PBS (Control; Con) or 400 mg/kg AICAR for 14 d. Tumor tissues were extracted and prepared for further immunohistochemical (IHC) and immunofluorescence detection. A, Representative IHC analysis for CD31+ vessels 4T1 orthotopic tumors. Scare bar = 100 μm. B, Quantification of the number of CD31+ vessels in AICAR or PBS‐treated 4T1 tumors (n = 8). C, Immunofluorescent staining showing the morphology of CD31+ vessels. White arrows indicate the vascular sprout in sections of 4T1 tumors from the control group. AICAR inhibited the sprouting ability of tumor vessels. D, Quantification of vascular sprouts (per vessel) in sections of 4T1 tumors from control or AICAR treatment groups (n = 8). Quantitative data are indicated as mean ± SD. **P < .01; ***P < .001. ns, no statistically significant difference (P > .05)
3.7. Abrogation of TCM‐mediated promotion of proliferation and tube formation of HUVECs by AICAR
Many studies have reported that activation of AMPK promoted the angiogenic phenotype in the breast cancer model.19, 20 Herein, AICAR, an AMPK activator, significantly abrogated HUVEC tube formation facilitated by the CM of both 4T1 and MDA‐MB‐231 cells (Figure 5A‐D). This AICAR‐induced abrogation was almost complete, which was indicated by the lack of significant difference between the SFM and AICAR groups. Consistently, AICAR pretreatment restrained the viability of HUVECs cultured in 4T1‐CM to a comparable level of that cultured in SFM (Figure 5). We also found that there was a significant difference in HUVEC viability between SFM and CM of MDA‐MB‐231 pretreated with AICAR. This result suggests that 4T1 cells are more responsive to AICAR pretreatment in promoting HUVEC proliferation than the MDA‐MB‐231 cell line. This AICAR‐induced inhibition of tumor angiogenesis was further validated by the fact that AICAR‐treated tumor cells secreted less VEGF and FGF‐2 to the medium than that of control (Figure 5F). Moreover, we also observed that AICAR had no obvious effect on cellular morphology of HUVECs (data not shown). These results indicated that AMPK activation by AICAR could inhibit breast tumor angiogenesis through a paracrine‐related mechanism.
Figure 5.
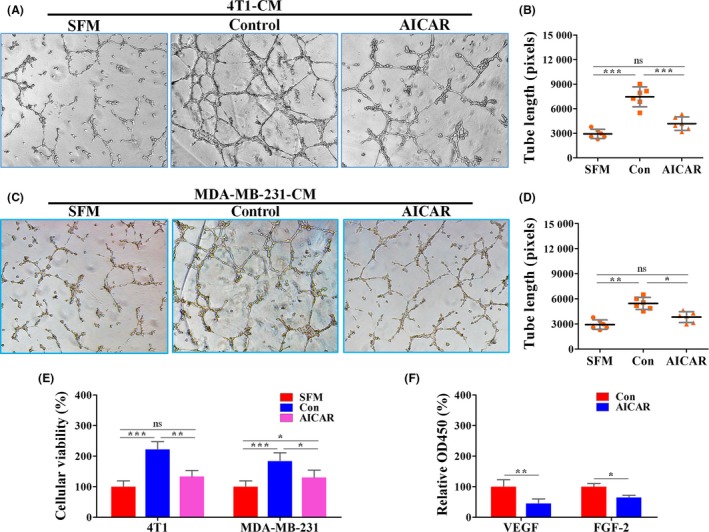
5‐Aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) inhibited the pro‐angiogenic effects of tumor cell‐conditioned medium. 4T1 and MDA‐MB‐231 cells were pretreated with 1 mmol/L AICAR for 24 h. After washing with PBS, the conditioned medium (CM) was collected 24 h later for further culture of HUVECs. A, C, Representative images showing HUVEC‐mediated tube formation promoted by 4T1‐CM (A) and MDA‐MB‐231‐CM (C). B, D, Quantification of tube length of HUVECs in response to 4T1‐CM (B) and MDA‐MB‐231‐CM (D) (n = 6). E, Representative images showing the cellular viability of HUVECs treated with serum‐free medium (SFM) or CM of tumor cells pretreated with AICAR. F, Quantifications of secreted vascular endothelial growth factor (VEGF) and fibroblast growth factor‐2 (FGF‐2) proteins in supernatants of 4T1 cells by ELISA (n = 6). Quantitative data are indicated as mean ± SD. *P < .05; **P < .01; ***P < .001. Con, control; ns, no statistically significant difference (P > .05)
3.8. Combined AMPK inhibition ameliorated simvastatin‐induced suppression of HIF‐1α and tumor angiogenesis
It has been reported that hypoxic activation of HIF‐1α was inhibited by compound C,21 a selective inhibitor of AMPK. Combining the above results, we next focused on whether compound C is capable of abrogating simvastatin‐induced angiogenic inhibition. Simultaneous compound C treatment showed an almost complete abrogation of simvastatin‐induced suppression of tumor angiogenesis and growth (Figure 6A‐C). This result was further supported by IHC characterization that simvastatin‐induced inhibition of HIF‐1α and its downstream angiogenic factors was significantly abrogated by combined treatment with compound C (Figure 6D‐G).
Figure 6.
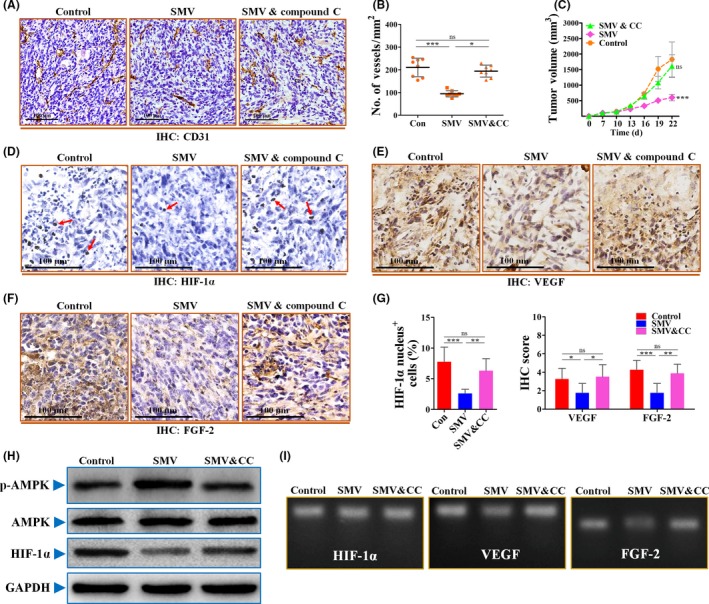
Inhibition of AMP kinase (AMPK) activation by compound C abrogated the inhibitory effects of simvastatin (SMV) on hypoxia‐inducible factor‐1α (HIF‐1α)‐induced tumor angiogenesis. A, Representative images for showing immunohistochemical (IHC) analysis of CD31+ vessels in 4T1 tumors from mice treated with PBS (Control; Con), 15 mg/kg SMV, or combined treatment with 15 mg/kg SMV and 20 mg/kg compound C (CC). Scale bar = 100 μm. B, Quantification of microvessel density (no. of vessels/mm2) in 4T1 tumors (n = 8). C, Representative images showing growth curve of 4T1 tumor from mice untreated or treated with SMV or combination treatment of SMV and CC (n = 8). D‐F, Representative images of immunostaining for HIF‐1α (D), vascular endothelial growth factor (VEGF) (E), and fibroblast growth factor‐2 (FGF‐2) (F) in 4T1 tumor sections of control, SMV, or SMV and CC combination groups. Red arrows in panel (D) indicate HIF‐1α nucleus+ cells. Scar bar = 100 μm. G, Percentage of HIF‐1α nucleus+ cells (% total cells; left) and quantification of IHC scores of VEGF and FGF‐2 in sections of 4T1 tumors (right; n = 8). H, Immunoblotting for phosphorylated p‐AMPK, total AMPK, and HIF‐1α of 4T1 cells treated with PBS (Control), 0.5 μmol/L SMV, or combination of 0.5 μmol/L SMV and 5 μmol/L CC. GAPDH was used as a control for protein loading. I, RT‐PCR for determining the mRNA levels of HIF‐1α, VEGF, and FGF‐2 of 4T1 tumor cells. Quantitative data are indicated as mean ± SD. *P < .05; **P < .01; ***P < .001. ns, no statistically significant difference (P > .05)
To further investigate whether compound C abrogates simvastatin‐induced angiogenic inhibition, we next detected the expression levels of p‐AMPK, AMPK, HIF‐1α, VEGF, and FGF‐2 in vitro. As shown in Figure 6H, combined simvastatin and compound C resulted in comparable protein levels of p‐AMPK and HIF‐1α to the control, indicating that compound C offsets the effects of simvastatin on the AMPK‐HIF‐1α signaling axis. Furthermore, simultaneous compound C treatment restored the mRNA and secretion levels of VEGF and FGF‐2 of 4T1 cells, while having no apparent effect on HIF‐1α (Figures 6I, S3). Taken together, our results provide evidence for the anti‐angiogenic potentials of simvastatin and reveal the involvement of the AMPK‐HIF‐1α signaling pathway (Figure 7).
Figure 7.

Schematic diagram of simvastatin‐induced anti‐angiogenesis. Simvastatin treatment increases the phosphorylation level of AMP kinase (AMPK) in tumor cells, thus inhibiting pro‐angiogenic factor (including vascular endothelial growth factor [VEGF] and fibroblast growth factor‐2 [FGF‐2])‐induced tumor angiogenesis by reducing tumoral hypoxia‐inducible factor‐1α (HIF‐1α) protein levels. AICAR, 5‐aminoimidazole‐4‐carboxamide ribonucleotide
4. DISCUSSION
In the current report, we showed that simvastatin inhibited breast tumor angiogenesis facilitated by tumor cell‐derived VEGF and FGF‐2, two pro‐angiogenic factors. Underlying these activities is the ability of simvastatin to reduce tumoral HIF‐1α protein levels by activating the upstream AMPK (Figure 7).
Although a number of studies have been increasingly focused on the antitumor activities of statins,12, 22, 23 their anti‐angiogenic potential is largely unknown. In fact, statin is the drug that was primarily designed for treatment of metabolic disorders and cardiovascular diseases.24 It has been reported that statins showed pro‐angiogenic potentials in ischemic disorders,25, 26 while having anti‐angiogenic effects on inflammation.27 This discrepancy in pathological angiogenesis might be closely associated with the biphasic effects of statins on HIF‐1α.26, 28 However, less is known about their effects on tumor angiogenesis. Recently, Li and colleagues reported the anti‐angiogenesis activities of simvastatin in human colorectal cancers.29 Consistently, our results further confirm the anti‐angiogenic potential of simvastatin by using a murine orthotopic breast cancer model with a focus on its underlying mechanism.
For many years, the cholesterol lowering effects of statins were considered to have contributed to most of its beneficial activities.30 In the last decade, both clinical and preclinical studies have revealed several cholesterol‐lowering independent mechanisms underlying statins’ cardiovascular protection.31 Kureishi and colleagues reported that simvastatin promoted angiogenesis in ischemic tissues in normocholesterolemic animals.32 It appears that, regardless of whether statins inhibit HMG‐CoA reductase in the liver or not, these agents could exert their cardiovascular protection or antitumor effects by directly acting on the targeted cells. Here, we report a direct activation of tumoral AMPK signaling by simvastatin, which impedes downstream HIF‐1α‐induced angiogenesis. Activation of AMPK could be a result of the nature of simvastatin, which is unrelated to cholesterol lowering, but closely related to direct targeting of signaling pathways in tumor cells. This direct action on tumoral signaling pathways has also been observed in other statins,33, 34 such as atorvastatin and lovastatin.35 Hence, direct anti‐tumor activities may be not specific to a particular statin.
The current anti‐angiogenic therapies have been greatly challenged by their targeting of normal vasculature.6 The VEGF inhibitors are associated with increased risks of various cardiovascular pathologies, such as hypertension, cardiovascular ischemia, and thromboembolism.36 Unlike these VEGF inhibitors, simvastatin had no significant effect on increasing the risks of fatal and non‐fatal cardiovascular pathologies.37 Thus, the discrepancy between these drugs in terms of cardiovascular complications suggests that statins might be safer than direct VEGF inhibitors for clinical applications. Considering the results reported in this paper, statins, like simvastatin, could be one of the choices that should be considered for targeting tumor angiogenesis.
AMP kinase has emerged as a target for treating metabolic syndromes, malignancies, and cardiovascular diseases.21, 38, 39 Recently, simvastatin has been reported to induce tumor cell cycle arrest by AMPK activation.12 Consistently, we found that simvastatin treatment resulted in an elevated protein level of phosphorylated AMPK in breast tumors, whereas total protein levels of AMPK were not changed. This evidence is further supported by previously published results that the AMPK activator, metformin (a widely prescribed biguanide for type 2 diabetes), showed potential antitumor activities through multiple mechanisms.40, 41, 42 One of the frequently observed mechanisms activating AMPK is an increase in the expression of upstream LKB1.43 Although LKB1 expression was not detected in this paper, we could not exclude the involvement of LKB1 in mediating the activation of AMPK by simvastatin. Further research should be focused on assessing the requirement of LKB1.
Activation of AMPK greatly contributes to simvastatin‐induced anti‐angiogenesis. Due to aberrant activation of oncogenes or loss of tumor suppressor genes, tumor cells have the potential to facilitate angiogenesis through the secretion of multiple pro‐angiogenic factors, such as VEGF and FGF‐2. HIF‐1α is one of the most critical transcription factors that mediate oncogene‐induced tumor angiogenesis.44 In addition, HIF‐1α facilitates tumor angiogenesis when the suppressor gene is lost.45 When activated, AMPK is able to restrain the function of the mTOR complex, which regulates the protein synthesis of HIF‐1α.46 This mechanism helps to explain why statin‐induced AMPK activation suppresses tumor angiogenesis involving HIF‐1α.47 As AMPK activation has multiple activities,39, 48, 49 simvastatin‐induced AMPK activation in tumor cells should not be simply limited to inhibition of tumor angiogenesis.
In summary, the present data suggest that targeting HIF‐1α‐induced expression of pro‐angiogenic factors by simvastatin in breast tumor cells resulted in strong anti‐angiogenesis through activation of AMPK. As inhibition of pro‐angiogenic factors has vascular normalization effects on the tumor vasculature,50 further studies should explore whether statins remodel tumor vasculature.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This work was financially supported by grants from the National Natural Science Foundation of China (Grant Nos. 81272342, 81472747, 81602638, and 81602502), the Keypoint Research and Invention Program of Shaanxi Province (Nos. 2017SF‐017 and 2016SF196), the Nature Science Foundation of Shaanxi Province (No. 2016JM8125), and the College Scientific Research Foundation of Xi'an Jiaotong University (No. xjj2016104).
Wang J‐C, Li X‐X, Sun X, et al. Activation of AMPK by simvastatin inhibited breast tumor angiogenesis via impeding HIF‐1α‐induced pro‐angiogenic factor. Cancer Sci. 2018;109:1627‐1637. https://doi.org/10.1111/cas.13570
Funding information
National Natural Science Foundation of China (81272342, 81472747, 81602638, and 81602502); Keypoint Research and Invention Program of Shaanxi Province (2017SF‐017 and 2016SF196); Nature Science Foundation of Shaanxi Province (2016JM8125); College Scientific Research Foundation of Xi'an Jiaotong University (xjj2016104)
J.‐C. Wang and X.‐X. Li contributed equally to this work.
Contributor Information
Jun Feng, Email: 1223134642@qq.com.
Pei‐Jun Liu, Email: liupeijun@mail.xjtu.edu.cn.
REFERENCES
- 1. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249‐257. [DOI] [PubMed] [Google Scholar]
- 2. Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol Symp Proc. 2000;5:40‐46. [DOI] [PubMed] [Google Scholar]
- 3. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15‐18. [DOI] [PubMed] [Google Scholar]
- 4. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335‐2342. [DOI] [PubMed] [Google Scholar]
- 5. Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653‐660. [DOI] [PubMed] [Google Scholar]
- 6. Shojaei F. Anti‐angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 2012;320:130‐137. [DOI] [PubMed] [Google Scholar]
- 7. Greenwood J, Mason JC. Statins and the vascular endothelial inflammatory response. Trends Immunol. 2007;28:88‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu XY, Daghini E, Chade AR, et al. Disparate effects of simvastatin on angiogenesis during hypoxia and inflammation. Life Sci. 2008;83:801‐809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105:739‐745. [DOI] [PubMed] [Google Scholar]
- 10. Licarete E, Sesarman A, Rauca VF, Luput L, Patras L, Banciu M. HIF‐1alpha acts as a molecular target for simvastatin cytotoxicity in B16.F10 melanoma cells cultured under chemically induced hypoxia. Oncol Lett. 2017;13:3942‐3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J, Li G, Wang Y, et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF‐1alpha/VEGF secretion axis. Oncotarget. 2015;6:44579‐44592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang S‐T, Ho HJ, Lin J‐T, Shieh J‐J, Wu C‐Y. Simvastatin‐induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017;8:e2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang JC, Li GY, Li PP, et al. Suppression of hypoxia‐induced excessive angiogenesis by metformin via elevating tumor blood perfusion. Oncotarget. 2017;8:73892‐73904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsubaki M, Takeda T, Kino T, et al. Statins improve survival by inhibiting spontaneous metastasis and tumor growth in a mouse melanoma model. Am J Cancer Res. 2015;5:3186‐3197. [PMC free article] [PubMed] [Google Scholar]
- 15. Backman JT, Kyrklund C, Kivisto KT, Wang JS, Neuvonen PJ. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin Pharmacol Ther. 2000;68:122‐129. [DOI] [PubMed] [Google Scholar]
- 16. Semenza GL. Hypoxia‐inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med. 2001;7:345‐350. [DOI] [PubMed] [Google Scholar]
- 17. Kyzas PA, Stefanou D, Batistatou A, Agnantis NJ. Hypoxia‐induced tumor angiogenic pathway in head and neck cancer: an in vivo study. Cancer Lett. 2005;225:297‐304. [DOI] [PubMed] [Google Scholar]
- 18. Treins C, Murdaca J, Obberghen EV, Giorgetti‐Peraldi S. AMPK activation inhibits the expression of HIF‐1α induced by insulin and IGF‐1. Biochem Biophys Res Comm. 2006;342:1197‐1202. [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Tang G, Zhang Z, Wang Y, Yang GY. Metformin promotes focal angiogenesis and neurogenesis in mice following middle cerebral artery occlusion. Neurosci Lett. 2014;579:46‐51. [DOI] [PubMed] [Google Scholar]
- 20. Phoenix KN, Vumbaca F, Claffey KP. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERalpha negative MDA‐MB‐435 breast cancer model. Breast Cancer Res Treat. 2009;113:101‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emerling BM, Viollet B, Tormos KV, Chandel NS. Compound C inhibits hypoxic activation of HIF‐1 independent of AMPK. FEBS Lett. 2007;581:5727‐5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolfe AR, Debeb BG, Lacerda L, et al. Simvastatin prevents triple‐negative breast cancer metastasis in pre‐clinical models through regulation of FOXO3a. Breast Cancer Res Treat. 2015;154:495‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kanugula AK, Gollavilli PN, Vasamsetti SB, et al. Statin‐induced inhibition of breast cancer proliferation and invasion involves attenuation of iron transport: intermediacy of nitric oxide and antioxidant defence mechanisms. FEBS J. 2014;281:3719‐3738. [DOI] [PubMed] [Google Scholar]
- 24. Skaletz‐Rorowski A, Kureishi Y, Shiojima I, Walsh K. The pro‐ and antiangiogenic effects of statins. Semin Vasc Med. 2004;4:395‐400. [DOI] [PubMed] [Google Scholar]
- 25. Chade AR, Zhu X, Mushin OP, Napoli C, Lerman A, Lerman LO. Simvastatin promotes angiogenesis and prevents microvascular remodeling in chronic renal ischemia. FASEB J. 2006;20:1706‐1708. [DOI] [PubMed] [Google Scholar]
- 26. Nishimoto‐Hazuku A, Hirase T, Ide N, Ikeda Y, Node K. Simvastatin stimulates vascular endothelial growth factor production by hypoxia‐inducible factor‐1alpha upregulation in endothelial cells. J Cardiovasc Pharmacol. 2008;51:267‐273. [DOI] [PubMed] [Google Scholar]
- 27. Katsumoto M, Shingu T, Kuwashima R, Nakata A, Nomura S, Chayama K. Biphasic effect of HMG‐CoA reductase inhibitor, pitavastatin, on vascular endothelial cells and angiogenesis. Circ J. 2005;69:1547‐1555. [DOI] [PubMed] [Google Scholar]
- 28. Hisada T, Ayaori M, Ohrui N, et al. Statin inhibits hypoxia‐induced endothelin‐1 via accelerated degradation of HIF‐1alpha in vascular smooth muscle cells. Cardiovasc Res. 2012;95:251‐259. [DOI] [PubMed] [Google Scholar]
- 29. Li G, Zheng J, Xu B, Ling J, Qiu W, Wang Y. Simvastatin inhibits tumor angiogenesis in HER2‐overexpressing human colorectal cancer. Biomed Pharmacother. 2017;85:418‐424. [DOI] [PubMed] [Google Scholar]
- 30. Lefer AM, Scalia R, Lefer DJ. Vascular effects of HMG CoA‐reductase inhibitors (statins) unrelated to cholesterol lowering: new concepts for cardiovascular disease. Cardiovasc Res. 2001;49:281‐287. [DOI] [PubMed] [Google Scholar]
- 31. Werner N, Nickenig G, Laufs U. Pleiotropic effects of HMG‐CoA reductase inhibitors. Basic Res Cardiol. 2002;97:105‐116. [DOI] [PubMed] [Google Scholar]
- 32. Kureishi Y, Luo Z, Shiojima I, et al. The HMG‐CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou TY, Zhuang LH, Hu Y, et al. Inactivation of hypoxia‐induced YAP by statins overcomes hypoxic resistance tosorafenib in hepatocellular carcinoma cells. Sci Rep. 2016;6:30483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Araújo FA, Rocha MA, Mendes JB, Andrade SP. Atorvastatin inhibits inflammatory angiogenesis in mice through down regulation of VEGF, TNF‐α and TGF‐β1. Biomed Pharmacother. 2010;64:29‐34. [DOI] [PubMed] [Google Scholar]
- 35. Horiuchi A, Kikuchi N, Osada R, et al. Overexpression of RhoA enhances peritoneal dissemination: RhoA suppression with Lovastatin may be useful for ovarian cancer. Cancer Sci. 2008;99:2532‐2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Touyz RM, Lang NN, Herrmann J, van den Meiracker AH, Danser AHJ. Recent advances in hypertension and cardiovascular toxicities with vascular endothelial growth factor inhibition. Hypertension. 2017;70:220‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Criner GJ, Connett JE, Aaron SD, et al. Simvastatin for the prevention of exacerbations in moderate‐to‐severe COPD. N Engl J Med. 2014;370:2201‐2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun W, Lee T‐S, Zhu M, et al. Statins activate AMP‐activated protein kinase in vitro and in vivo. Circulation. 2006;114:2655‐2662. [DOI] [PubMed] [Google Scholar]
- 39. Li H, Lee J, He C, Zou M‐H, Xie Z. Suppression of the mTORC1/STAT3/Notch1 pathway by activated AMPK prevents hepatic insulin resistance induced by excess amino acids. Am J Physiol Endocrinol Metab. 2014;306:E197‐E209. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40. Troncone M, Cargnelli SM, Villani LA, et al. Targeting metabolism and AMP‐activated kinase with metformin to sensitize non‐small cell lung cancer (NSCLC) to cytotoxic therapy; translational biology and rationale for current clinical trials. Oncotarget. 2017;8:57733‐57754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu X, Romero IL, Litchfield LM, Lengyel E, Locasale JW. Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab. 2016;24:728‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pearce EL, Walsh MC, Cejas PJ, et al. Enhancing CD8 T‐cell memory by modulating fatty acid metabolism. Nature. 2009;460:103‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP‐activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015;356:165‐170. [DOI] [PubMed] [Google Scholar]
- 44. Liu LZ, Jing Y, Jiang LL, et al. Acacetin inhibits VEGF expression, tumor angiogenesis and growth through AKT/HIF‐1alpha pathway. Biochem Biophys Res Comm. 2011;413:299‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chowdhury AR, Long A, Fuchs S, Rustgi A, Avadhani NG. Mitochondrial stress induced p53 attenuates HIF‐1α activity by physical association and enhanced ubiquitination. Oncogene. 2017;36:397‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2010;12:21‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012;2:778‐790. [DOI] [PubMed] [Google Scholar]
- 48. Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eid AA, Ford BM, Block K, et al. AMP‐activated protein kinase (AMPK) negatively regulates Nox4‐dependent activation of p53 and epithelial cell apoptosis in diabetes. J Biol Chem. 2010;285:37503‐37512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Can Res. 2004;64:3731‐3736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials