

Abstract
Background and Purpose
Tobacco smoke contains many classes of carcinogens and although nicotine is unable to initiate tumourigenesis in humans and rodents, it promotes tumour growth and metastasis in lung tumours by acting on neuronal nicotinic ACh receptors (nAChRs). The aim of this study was to identify molecularly, biochemically and pharmacologically which nAChR subtypes are expressed and functionally activated by nicotine in lung cancer cell lines.
Experimental Approach
We used A549 and H1975 adenocarcinoma cell lines derived from lung tumours to test the in vitro effects of nicotine, and nAChR subtype‐specific peptides and compounds.
Key Results
The two adenocarcinoma cell lines express distinctive nAChR subtypes, and this affects their nicotine‐induced proliferation. In A549 cells, nAChRs containing the α7 or α9 subunits not only regulate nicotine‐induced cell proliferation but also the activation of the Akt and ERK pathways. Blocking these nAChRs by means of subtype‐specific peptides, or silencing their expression by means of subunit‐specific siRNAs, abolishes nicotine‐induced proliferation and signalling. Moreover, we found that the α7 antagonist MG624 also acts on α9–α10 nAChRs, blocks the effects of nicotine on A549 cells and has dose‐dependent cytotoxic activity.
Conclusions and Implications
These results highlight the pathophysiological role of α7‐ and α9‐containing receptors in promoting non‐small cell lung carcinoma cell growth and intracellular signalling and provide a framework for the development of new drugs that specifically target the receptors expressed in lung tumours.
Linked Articles
This article is part of a themed section on Nicotinic Acetylcholine Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.11/issuetoc
Abbreviations
- Abs
antibodies
- MLA
methyllycaconitine
- nAChR
nicotinic ACh receptor
- NSCLC
non‐small cell lung
- q‐PCR
quantitative real‐time PCR
- αBgtx
α‐bungarotoxin
Introduction
Lung cancer is the leading cause of cancer‐related deaths worldwide, and cigarette smoking is related to 90% of all deaths due to lung cancer (Siegel et al., 2013).
Tobacco smoke contains many classes of carcinogens (Hecht and Hoffmann, 1988) and although nicotine (the addictive and most active component of tobacco smoke) is unable to initiate tumourigenesis in humans and rodents, it promotes tumour growth and metastases by inducing cell‐cycle progression, cell migration and invasion, angiogenesis and the evasion of apoptosis in a variety of tumour cells (reviewed in Egleton et al., 2008 ; Schaal and Chellappan, 2014; Grando, 2014; Mucchietto et al., 2016).
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2585 and its metabolites are highly lipophilic compounds that bind and activate a family of ligand‐gated cation channels the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76 that are widely expressed in the central and peripheral nervous systems (Zoli et al., 2015). It was originally believed that nAChRs are only present in the nervous system, but it has now been shown that they are expressed in numerous cell types. In particular, they are expressed in airway epithelial cells, which synthesize, store, process and secrete ACh (reviewed in Wessler and Kirkpatrick, 2008; Spindel, 2016). When lung cancer arises from airway epithelial cells, they continue to express nAChRs, and their growth is stimulated by nicotine (reviewed in Tournier and Birembaut, 2010). Many lung cancers also synthesize ACh, which interacts with the nAChRs and muscarinic ACh receptors on lung cancer cells as an autocrine growth factor (Song et al., 2003, 2008). Cancer growth and development can therefore be stimulated by both endogenous ACh and exogenous (nicotine) ligands of nAChRs (reviewed in Schuller, 2009; Spindel, 2016).
It has been shown that http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=466&familyId=76&familyType=IC, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=468&familyId=76&familyType=IC and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=469&familyId=76&familyType=IC nACh receptors are expressed in various types of tumours (Grando, 2014; Mucchietto et al., 2016).
The nicotinic α5 subunit product of the CHRNA5 gene can only form functional channels when it is associated with the α4 and β2 or α3 and β4 subunits (Gotti et al., 2009; Albuquerque et al., 2009; Improgo et al., 2010). Genetic studies have shown that the non‐synonymous single nucleotide polymorphism rs16969968 of the CHRNA5 gene is associated with lung cancer and nicotine dependence (reviewed in Bierut et al., 2008; Improgo et al., 2010). However, as non‐smokers bearing the rs16969968 polymorphism are also at increased risk of lung cancer, their disease may be directly related to the polymorphism (Hung et al., 2008). CHRNA5 mRNA levels were 30 times higher in lung adenocarcinoma tissue than in normal lung tissue, but no differences were found between the cancer and normal samples in the expression for other genes of chromosome 15 outside the CHRNA5/A3/B4 gene cluster (Falvella et al., 2009).
Various studies have shown that α7 is the main nAChR subtype mediating the proliferative and angiogenic effects of nicotine in lung cancer cells (reviewed in reviewed in Schaal and Chellappan, 2014; Grando, 2014). The α7 subunit is encoded by the 10‐exon CHRNA7 gene located on chromosome 15q14, which gives rise to a transcript that is translated into a protein of approximately 57 kDa. CHRNA7 is partially duplicated in the human genome and forms a hybrid gene with the novel FAM7A gene (CHRFAM7A, α7dup) whose transcript codes for a α7dup protein that has a molecular weight of 46.2 kDa and is unique to humans (Sinkus et al., 2015).
Published data have shown that the expression of CHRFAM7A in oocytes acts as a dominant negative regulator of α7 nAChR activity by means of a mechanism involving a reduction in the number of functional α7 nAChRs incorporated into the oocyte surface (de Lucas‐Cerrillo et al., 2011). The α7‐containing nAChRs on neurons and astrocytes function as ligand‐gated ion channels, have very high levels of calcium permeability, are blocked by the antagonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3964 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4005 (MLA) (Albuquerque et al., 2009) and form homo‐ (α7) or hetero‐oligomeric (α7β2) cation channels (Moretti et al., 2014).
The CHRNA9 gene encodes a plasma membrane protein that forms homo‐ (α7) or hetero‐ (α9‐α10) oligomeric cation channels that are also highly permeable to calcium, blocked by α‐Bgtx and MLA and have an atypical mixed nicotinic–muscarinic pharmacological profile (Elgoyhen et al., 1994; Verbitsky et al., 2000).
Lee et al. (2010) by RT‐PCR analysis have found that α9 nAChRs are ubiquitously expressed in many epithelial, lung and breast cancer cell lines, most of which also express α5 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=470&familyId=76&familyType=IC nAChR subunits. α9 nAChRs are present in primary tumour and non‐malignant breast tissue obtained from patients, but their expression is higher in breast cancer cells than the surrounding normal tissue. Silencing α9 nAChR expression in breast cancer cells reduces their proliferation and tumourigenic potential in in vitro and in vivo assays (Lee et al., 2010).
In this study, we identified molecularly, biochemically and pharmacologically which nAChR subtypes are expressed and functionally activated by nicotine in adenocarcinoma cell lines.
Methods
Cell lines
Human non‐small cell lung adenocarcinoma (NSCLC) A549 and H1975 cell lines were obtained from the American Type Culture Collection. The human neuroblastoma cell line SH‐SY5Y was obtained from the European collection of authenticated cell culture.
NSCLC cells and the neuroblastoma were grown in RPMI medium (Lonza) supplemented with 10% FBS, 1% penicillin–streptomycin and 1% L‐glutamine (complete medium). The cells were maintained in an environment at 37°C containing 5% CO2. Each cell line was treated with a different protocol in order to optimize the correct conditions for the experiments.
Human samples
Specimens of six histopathologically confirmed lung adenocarcinoma and adjacent normal tissue (normal lung parenchyma) were obtained from patients who had undergone lung lobectomy (Dr Dragani, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan).
Total RNA extraction and reverse transcription
Total RNAs were extracted using the RNeasy Mini kit and accompanying QIAshredder (Qiagen), according to the manufacturer's instructions. Briefly, a maximum of 9 × 106 cells was collected by centrifugation and lysed with 600 μL of lysis buffer, containing β‐mercaptoethanol (10 μL·mL−1 lysis buffer). The lysate was homogenized by means of QIAshredder column centrifugation for 2 min at maximum speed. For human samples, about 30 mg of tissue were disrupted and homogenized in 1.8 mL of lysis buffer, by a rotor‐stator homogenizer until it was uniformly homogeneous. To avoid DNA contamination, samples on‐column were incubated with DNAse I for 15 min and RNA was eluted with 50 μL of RNase‐free water. The total amount of eluted RNA was determined by a spectrophotometer at 260 nm, and its purity was evaluated using the 260/280 ratio; 0.5–1 μg per sample was reverse transcribed using the GoScript™ Reverse Transcriptase (Promega), according to information provided by the company.
Quantitative real‐time PCR (q‐PCR)
Gene expression analyses were performed by a q‐PCR assay using the ABI Prism Thermocycler QuantStudio 5. The target sequences were amplified from 50 ng of cDNA in the presence of TaqMan® Gene expression master mix (Life Technologies, Inc.).
The TaqMan® primer and probe assays used were human CHRNA2 (ID #Hs00181237_m1), CHRNA3 (ID #Hs01088199_m1), CHRNA4 (ID #Hs00181247_m1), CHRNA5 (ID #Hs00181248_m1), CHRNA6 (ID #Hs00610233_m1), CHRNA7 (ID #Hs01063373_m1), CHRFAM7A (ID #Hs04189909_m1), CHRNA9 (ID #Hs00214034_m1), CHRNA10 (ID #Hs00220710_m1), CHRNB2 (ID #Hs00181267_m1), CHRNB3 (ID #Hs00181269_m1) and CHRNB4 (ID #Hs00609520_m1). GAPDH (ID #Hs99999905_m1) or 18S (ID #Hs99999901_s1) was used as endogenous control, as described in the figure legend. The 2−ΔCT or 2−CT method was used to calculate the results, as described in the figure legends, thus allowing the normalization of each sample to the endogenous control, and comparison with the calibrator for each experiment (set to a value of 1).
Antibody production and characterization
For the detection of nAChR subunits, we used affinity‐purified, subunit‐specific, polyclonal antibodies (Abs), produced in rabbit against the human α7 and α5 subunits. For each of these subunits, Abs against peptides derived from the C‐terminal (COOH) or intracytoplasmic loop (Cyt) of human nAChR subunit sequences were produced as previously described (Grady et al., 2009). The Abs against the human α5 subunit were made against the COOH peptide (PVHIGNANK) and against the Cyt peptide (cgDRYFTQKEETESGSGPKSSRNTLEA). The Abs against the human α7 subunit were made against a COOH (SAPNFVEAVSKDFA) and Cyt peptide (ACSPTHDEHLLHGGQPPEGDPDL).
In order to exclude any cross reactivity between nAChR subunits, anti‐α7 human subunit Abs were also tested by means of immunoprecipitation studies and Western blotting in HEK293 cells transfected with the human α2β4‐, α4β2‐, α4β4 or α3β4‐nAChR subtypes (Supporting Information Figure S1B) or in SH‐SY5Y cells transfected with human α7‐nAChRs (Supporting Information Figure S1A). The α5 Abs were tested using HEK cells transfected with the α3β4 and α3β4α5 human subunits (Supporting Information Figure S1B).
Purification of α‐bungarotoxin‐binding nAChRs
In order to identify the subunit composition of the α7‐ and α9‐containing receptors in A549 and H1975 cells, the receptors were purified by means of affinity purification on α‐Bgtx covalently bound to Sepharose beads. α‐Bgtx receptors from SH‐SY5Y cells were purified as a positive control. In each purification experiment, 1.2 × 108 A549 or H1975 cells and 4 × 107 for SH‐SY5Y cells were homogenized in 10 mL of 10 mM Na phosphate, pH 7.4, 137 mM NaCl, 4 mM KCl and 2 mM PMSF (in order to covalently inactivate serine protease activity), and the homogenates were diluted to 50 mL and centrifuged at 60 000 g at 4°C for 1.5 h. The entire membrane homogenization, dilution and centrifugation procedure was then repeated, and the resulting pellets were collected and rapidly rinsed. The washed pellets were then resuspended in 2 mL of the same buffer, further supplemented with 10 μg·mL−1 of each of the following protease inhibitors: leupeptin, bestatin, pepstatin A and aprotinin. Triton X‐100 at a final concentration of 2% was added to the washed membranes, which were extracted for 2 h at 4°C. The extracts were centrifuged at 60 000 g, at 4°C for 1.5 h, recovered, and an aliquot of the supernatants was collected for protein measurement using the BCA protein assay (Pierce Biotechnology, Inc., Rockford, IL, USA), with BSA as the standard. Extracts (2 mL) of A549 and H1975 cells containing similar amounts of protein and the extract of SH‐SY5Y cells containing approximately five times less protein were incubated with 200 μL of Sepharose‐α‐Bgtx (concentration of coupled toxin 1 mg·mL−1 of gel) and shaken overnight at 4°C. The following day, the beads were centrifuged, the supernatant was recovered and the resins were washed four to six times by resuspension followed by centrifugation. After being washed, the Sepharose‐α‐Bgtx beads with bound nAChRs (purified αBgtx‐binding receptors) were incubated with one to two volumes of Laemmli sample buffer (125 mM Tris phosphate, 4% SDS, 20% glycerol, 0.02% bromophenol blue and 10% 2‐mercaptoethanol pH 6.8) and boiled for 2 min. The supernatant was then recovered by centrifugation.
Immunoprecipitation of nAChRs containing the α5 and/or α7 subunits
The immunoprecipitation of receptors containing the α5 and α7 subunits was determined on two human adenocarcinoma cell lines (H1975 and A549) and on a human neuroblastoma cell line (SH‐SY5Y).
Affinity‐purified Abs against the α5 or α7 subunits were covalently immobilized on agarose‐protein A beads at a concentration of 4 mg·mL−1 of wet resin. Immunoprecipitation was then performed by adding 20 μL of agarose‐protein A beads with bound, affinity‐purified Abs or control IgG to the 2% TritonX‐100 cell extracts. After overnight incubation, immunoprecipitates were recovered by centrifugation and washed three times with PBS containing 0.1% Triton X‐100 and then loaded on a SDS gel.
Immunoblotting and densitometric quantification of Western blotting bands
The nAChR subunit contents of immunoprecipitated tissue extracts, affinity‐purified α‐Bgtx receptor complexes or cells transfected with different subtypes were analysed by Western blotting. For the extracts loaded before the purification 10 μg of proteins were loaded, whereas for the α‐Bgtx‐purified receptors a constant volume (40 μL) that, depending on the cell line, may represent 1/3 or 1/4 of the total recovered Laemmli sample of buffer‐eluted receptors was loaded onto a 9% acrylamide (Biorad, Hercules, CA, USA) gel and subjected to SDS‐PAGE. After SDS‐PAGE, proteins were electrophoretically transferred to nitrocellulose membranes with 0.45 mm diameter pores (Schleicher and Schuell, Dassel, Germany). The blots were blocked overnight in 4% non‐fat milk in Tris‐buffered saline (TBS), washed in a buffer containing 4% non‐fat milk and 0.3% Tween 20 in TBS, incubated for 2 h with the primary antibody (1–2.5 mg·mL−1) and then incubated with the appropriate peroxidase conjugated secondary Abs (Sigma‐Aldrich. St Louis, MO, USA). After 10 washes, peroxidase was detected using a chemiluminescent substrate (Pierce, Rockford, IL, USA).
In the case of Abs directed against ERK, p‐ERK, Akt and p‐Akt proteins, we used commercial Abs. The blots were blocked overnight followed by incubation with the primary antibody at 1:1000 dilution: anti‐p44/42 MAPK and phospho‐p44/42 (Thr202 and Tyr204) were purchase from Cell Signaling Technology, anti‐Akt BD from Transduction Laboratories and Akt (PS473) mAb from Life Technologies. The blots were then incubated for 1 h with the appropriate secondary antibody (anti‐rabbit Ly‐Cor IRDye800RD; anti‐mouse Ly‐Cor IRDye680RD). After another series of washes, the membranes were dried overnight in the dark at RT. The IR signal was measured using an Odyssey CLx – Infrared Imaging System. The signal intensity of the Western blot bands was quantified using iStudio software. The OD ratio was calculated by taking the OD of the control saline as 100%. The data are expressed as mean values ±SEM of at least three separate experiments using each antibody.
Proliferation assay
A total of 68 × 103 cells per well of A549 and H1975 cells were seeded in 24‐well plates and rendered quiescent by means of serum deprivation for 72 h. After this time, serum was added to the culture and the cells were treated with nicotine or the other drugs for 48 h.
The capacity of antagonists to block the effect of nicotine was assessed by pre‐incubating the cells for 30 min with appropriate concentrations of specific antagonists before adding nicotine and leaving the antagonists present throughout the treatment period.
At the end of the treatment, the cells were washed once with PBS 1X, trypsinized, suspended in 500–800 μL of medium, and manually counted using a haemocytometer (Burker camera) and microscope.
Viability assay
A total of 68 × 103 cells per well of A549 and H1975 cells were plated in 24‐well plates and rendered quiescent by means of serum deprivation for 72 h. After this time, serum was added to the culture and the cells were treated with nicotine, nicotinic antagonists or other compounds synthesized by the groups of Professor M. Pallavicini and Professor De Amici (University of Milan) for 48 h in complete medium.
Cell viability was assessed using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega). The assay is based on the reduction of MTS tetrazolium compound [3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H–tetrazolium] by viable cells to generate a coloured formazan product that is soluble in cell culture media. This conversion is carried out by NAD(P)H‐dependent dehydrogenase enzymes in metabolically active cells. The formazan dye produced is quantified by measuring the absorbance at 490 nm and is directly proportional to the number of living cultured cells.
At the end of the treatment, the cells were washed and 50 μL of MTS solution (Promega) was added to each well. The plates were incubated for 2 h, and absorbance was read using a spectrophotometer at a wavelength of 490 nm.
The OD of each plate was measured in triplicate and normalized by taking the OD of untreated cells as 100%.
ERK and Akt pathways activation
To evaluate the activation of ERK and Akt signalling pathways, the NSCLC cell lines were seeded in 24‐well plates (8 × 104 cells per well) and starved overnight before being treated with nicotine in the presence or absence of the antagonists or of the compound to be tested.
The signal detection was obtained by Western blotting analyses with Abs directed against ERK, p‐ERK, Akt and p‐Akt.
siRNA transfection
Sequences of siRNA, obtained from Dharmacon (GE Healthcare), were used to knock down the endogenous nAChR α7 and α9 subunits.
For each subunit (α7 and α9), four different siRNAs were analysed along with a non‐targeting scrambled siRNA sequence that was used as a control in the transfection experiments.
A549 cells were plated to 70–80% confluency in a six‐well plate (4 × 105 cells per well) and transfected in Opti‐MEM with 75 pmol of siRNA (scrambled or subunit‐specific siRNA) (GE Healthcare, Buckinghamshire, UK) using Lipofectamine 3000 reagent (Invitrogen) in RPMI with 10% FBS and 1% of L‐glutamine minus penicillin/streptomycin. Five hours after transfection, the cells were trypsinized, counted and plated in a 24‐well plate (8–10 × 103 cells per well for cell proliferation, and 8 × 104 cells per well for Akt and ERK signalling), and the medium was replaced by the complete medium that also contained penicillin/streptomycin. The cells were collected at different times (48–72 h) in order to isolate their mRNA and determine the mRNA and protein levels of the subunits.
The siRNAs that were most active in decreasing the expression of mRNA and subunit proteins were used for the cell proliferation and signalling experiments. To study the effect of nicotine, siRNA‐transfected A549 cells were rendered quiescent for 72 h using serum‐free RPMI. Subsequently, complete medium with or without nicotine (100 nM) was added for 48 h, and then the cells were counted to assess their proliferation.
To evaluate the activation of ERK and Akt signalling pathways, A549 cells, transfected with siRNA, were starved for 24 h and then treated with or without nicotine (100 nM) for different times (from 5 min to 1 h). The cells were then examined by means of Western blotting.
Two‐electrode voltage clamp recording of α7‐ and α9a10‐nAChR function
Detailed methods for conducting the electrophysiology of Xenopus oocytes and the pharmacological assessment of the activity of the MG624 compound on heterologously expressed α7 and α9‐a10 nAChR subtypes have been described previously (Hone et al., 2009). Briefly, stages IV–V oocytes were injected with equal ratios of capped cRNA encoding human nAChR subunits prepared using the mMessage mMachine in vitro transcription kit (Ambion, Austin, TX) and incubated at 17°C in ND96 (96.0 mM NaCl, 2.0 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2 and 5 mM HEPES, pH 7.5) containing antibiotics (100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin, 100 μg·mL−1 amikacin sulfate, 160 μg·mL−1 sulfamethoxazole and 32 μg·mL−1 trimethoprim). One to 5 days after injection, Xenopus oocytes expressing nAChR subtypes were voltage clamped at −70 mV with an Axoclamp 900A amplifier (Molecular Devices, Sunnyvale, CA, USA) and exposed to ACh and compounds as described previously (Hone et al., 2009).
Briefly, the oocyte chamber consisting of a cylindrical well (~30 μL in volume) was gravity perfused at a rate of ~2 mL·min−1 with ND‐96 buffer (96.0 mM NaCl, 2.0 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5 mM HEPES, pH 7.1–7.5, supplemented with 0.1 mg·mL−1 BSA). The oocyte was subjected once a minute to a 1 s pulse of 10 μM ACh [α9‐α10 nAChR or 200 μM ACh (α7 nAChR)]. MG624 was bath‐applied, during which the buffer flow was stopped, and allowed to incubate with the oocyte for 5 min, after which the ACh pulse was resumed.
ROS production
ROS generation in A549 cells was assessed by using 2′,7′‐diclhorofluorescin di‐acetate (DCFH‐DA), a fluorogenic dye that measures intracellular hydroxyl, peroxyl and other ROS activity. DCFH‐DA is deacetylated in cells by esterases and becomes highly fluorescent when oxidized by ROS.
Briefly, A549 cells were seeded in a black 96‐well plate (1.2 × 104 cells per well), washed with HBSS buffer (HEPES 25 mM, NaCl 120 mM, KCl 5.4 mM, CaCl2 dihydrate 0.8 mM, NaHCO3 25 mM, glucose 15 mM) and incubated with 20 μM DCFH‐DA for 45 min at 37°C.
After the incubation, DCFH‐DA was removed and the cells were treated with different concentrations of the MG624 compound for 24 h.
ROS levels were measured during the treatment period at emission wavelengths of 485–535 nm. The results were normalized using the protein value of each sample.
Data and statistical analyses
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All the experiments were performed in triplicate or quadruplicate, and the results are expressed as mean values ±SEM.
The statistics of the data from cell proliferation and Western blotting studies were performed in at least five separate experiments, expressed as mean values ±SEM and analysed by means of Student's t‐test for comparisons of two groups or one‐way ANOVA followed by a Bonferroni post hoc test (parametric data) or by Kruskal–Wallis test followed by Dunn's post hoc test (non‐parametric data). The accepted level of significance was P < 0.05. All of the statistical analyses were made using Prism software, version 5 (GraphPad).
Chemicals and drugs
Nicotine and α‐Bgtx were purchased from Tocris Bioscience (Bristol, UK). All other reagents (PMSF, proteases inhibitors, ACh chloride, atropine and BSA, chemicals) or compound (MLA) were from Sigma‐Aldrich.
Drugs
Triethyl‐(β‐4‐stilbenoxy‐ethyl) ammonium (MG624) was synthesized from the group of Prof Marco Pallavicini (Department of Medicinal Chemistry ‘Pietro Pratesi’, Milan), and 3‐methoxy‐1‐oxa‐2,7‐diaza‐7,10‐ethanospiro[4.5]dec‐2‐ene sesquifumarate (ICH3) was a generous gift of Prof Marco De Amici from the same department. The α‐conotoxins were synthesized as described previously (Ellison et al., 2006; Whiteaker et al., 2007; Azam and McIntosh, 2012).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Nicotinic receptors are expressed in NSCLC cell lines and tumour lung tissue
Relative q‐PCR studies
We used relative q‐PCR analysis to compare the expression of the α2–α10 and β2–β4 nAChR subunits and CHRFAM7A in A549 and H1975 adenocarcinoma cell lines. No α2, α3, α6 or β3 transcript was expressed in either cell line (data not shown) but they expressed similar levels of α5 transcripts. The level of CHRFAM7A, α9, β2 and β4 transcripts was higher in the A549 cells. H1975 cells did not express α7 but had a higher α10 transcript level than the A549 cells (Figure 1A).
Figure 1.
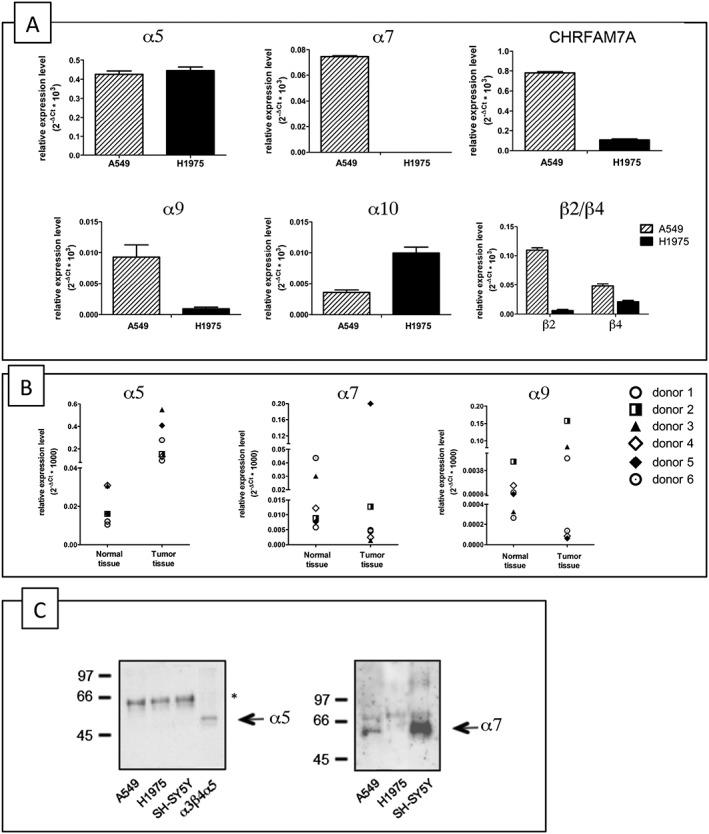
mRNA and protein expression level of different nAChR subunits in adenocarcinoma A549 and H1975 cell lines and human samples. (A) q‐PCR analysis of the human adenocarcinoma cell lines A549 and H1975. Data are shown as relative expression ±SD, normalized to the endogenous GAPDH expression, according to the 2−ΔCt method. (B) q‐PCR analysis of α5, α7 and α9 nAChR subunits in human normal lung parenchyma and lung adenocarcinoma tissues. The symbols indicate the four donors. The data are shown as relative expression levels normalized to endogenous 18S expression, in accordance with the 2−ΔCt method. (C) Western blot analysis of the α‐Bgtx affinity receptors purified from A549, H1975 and SH‐SY5Y cells. Proteins were separated on 9% acrylamide SDS gels, transferred to nitrocellulose and probed with Abs directed against the α5 (left) or α7 (right) subunits. Extract from cells transfected with α5 subunit (α3α5β4) was used as control for α5 labelling. The asterisk indicates an aspecific band.
As the major difference between the cell lines in terms of nicotinic subunit transcripts were those for α7 and α9 nAChRs, we investigated whether this variability reflects their different expression in human lung adenocarcinoma tissue. To this end, we analysed the levels of the mRNA of these subunits in lung adenocarcinoma tissue specimens (histopathologically confirmed to be stage 2) taken from six patients, for whom adjacent normal tissue was also available.
Figure 1B shows that the expression of α7 and α9 transcripts varied widely in the normal tissue specimens but not that of α5, which was always very low. However, in comparison with the normal samples, the level of α5 subunit transcripts was higher in all six tumour samples, and α9 subunit transcript levels were higher in three, whereas α7 subunit transcript levels were not significantly increased in any of them.
These results confirmed that the A549 and H1975 cell lines are good models for studying the role of nAChR subunits in the proliferation of lung adenocarcinoma as they reflect the same variability found in human tumour tissue.
Biochemical characterization of native nAChRs
In order to confirm the expression of nAChR subunits in the NSCLC cell lines at protein level, we carried out immunoprecipitation and Western blotting studies, using Abs against various nAChR subunits whose specificity was checked and is shown in Supporting Information Figure S1. The positive control was the SH‐SY5Y neuroblastoma cell line, which has high levels of receptors containing the α3, α5, α7, β2 and β4 subunits (Riganti et al., 2005).
Western blot analysis of extracts obtained from A549, H1975 and SH‐SY5Y cells using 2% Triton X‐100, and immunoprecipitated with anti‐α5, or anti‐α7 Abs, or control IgG bound to protein A, showed that the anti‐α5 Abs recognized a peptide of 50 KDa in all three cell lines (Supporting Information Figure S1C) that was absent when the extracts were immunoprecipitated using anti‐α7 or control IgG (Supporting Information Figure S1C, lanes b and c).
As α7‐ and α9‐containing receptors bind and are blocked by α‐Bgtx in native neuronal tissues and heterologous systems, and Sepharose‐α‐Bgtx resin has been used to affinity purify α7‐containing receptors from different neuronal tissues (Gotti et al., 2009), we affinity‐purified α‐Bgtx receptors from A549 and H1975 cells and neuroblastoma SH‐SY5Y cells. Figure 1C shows that the α7 Abs recognized a peptide of approximately 57 KDa in the receptors purified from A549 and SH‐SY5Y cells, but not in those purified from H1975 cells. The lack of α7 labelling of the α‐Bgtx receptors on H1975 cells is in agreement with the q‐PCR findings.
Our anti‐α7 Abs are directed against a peptide in the cytoplasmic loop of the α7 subunit between M3 and M4 and also recognize the CHRFAM7A (α7dup) protein (see Supporting Information Figure S1A, panel e). However, as shown in Figure 1C α7dup was not present in the α‐Bgtx receptors purified from the A549 or SH‐SY5Y cells as we only found labelling at 57 KDa, which corresponds to the molecular weight of the α7 subunit.
As α5 protein is highly expressed in all three cell lines, we checked whether this subunit was present in the α‐Bgtx receptors. To this end, we probed the purified α‐Bgtx receptors with anti‐α5 Abs, but although the anti‐α5 Abs recognized the α5 subunit in the positive control (α3β4α5 transfected HEK cells), they did not recognize it in the α‐Bgtx receptors purified from A549 or SH‐SY5Y cells. The band (marked with *) recognized by the Abs in all three samples is aspecific, as shown in the Supporting Information.
Nicotine increases A549 cell proliferation and viability
In preliminary experiments, we checked whether it was important to synchronize the cells in order to see the effects of nicotine. To this end, we deprived A549 and H1975 cells of serum for 24, 48 or 72 h in separate experiments and found that the best synchronization was after 72 h, with approximately 80% of A549 and H1975 cells in the Go/Gi phase as measured by means of FACS analysis after labelling the cells with propidium iodide. In all the proliferation and viability experiments of NSCLC cells, we first synchronized A549 and H1975 cells by means of serum deprivation and then treated them with increasing nicotine concentrations from 10 nM to 100 μM. As shown in Figure 2A, 48 h exposure to nicotine increased A549 cell proliferation (measured by counting cells, n = 8) and viability (measured by means of an MTS assay, n = 8) in a statistically significant manner, but not the proliferation (n = 8) or viability of H1975 cells (n = 8) (Figure 2B).
Figure 2.
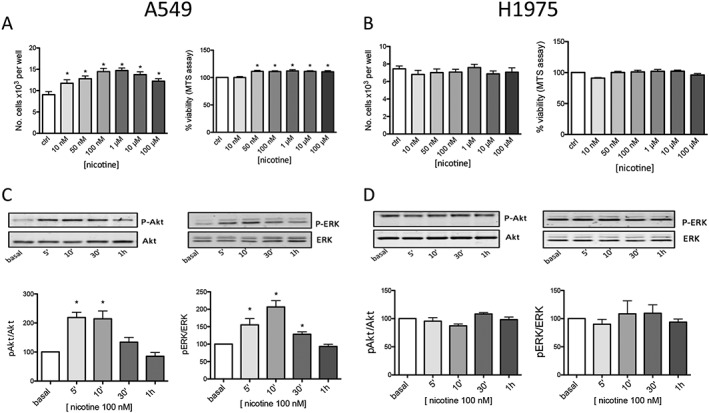
Effects of chronic nicotine exposure on A549 and H1975 cell viability, proliferation and signalling. (A, B) Chronic (48 h) exposure to the indicated concentrations of nicotine increased A549 cells viability and number of A549 (A) but not H1975 (B) cells. Results are the average of eight proliferation and viability experiments for A549 and H1975 cells performed in triplicate. The analysis was made using one‐way ANOVA followed by a post hoc Bonferroni test (cell count) or Dunn's test (cell viability) (*P < 0.05). A549 (C) and H1975 (D) cells were treated with 100 nM nicotine for the indicated times, and the incubation was blocked by adding sample buffer. Proteins were separated on 9% acrylamide SDS gels, transferred to nitrocellulose and probed with Abs directed against Akt and p‐Akt ERK and p‐ERK as described in Methods. The Western blot results are expressed as the p‐Akt/Akt and p‐ERK/ERK ratios, taking the ratio of control samples as 100 at the indicated time of nicotine stimulation (0, 5, 10, 30 and 60 min). The graphs show the mean values ±SEM obtained in eight different experiments performed in duplicate or triplicate. A representative blot at the corresponding times is shown above the graph. The Western blotting data were statistically analysed using one‐way ANOVA followed by Dunn's test [*P < 0.05 vs. untreated (basal) cells].
We also studied the time‐kinetics of the proliferation of A549 cells by 100 nM nicotine (Supporting Information Figure S2). One‐way ANOVA followed by a post hoc Bonferroni test did not show a significant difference between ctrl and nicotine at 24 h, but a clearly significant difference at 48 h (n = 5).
As nicotine 100 nM showed a significant effect and falls within the range of the blood nicotine levels of habitual smokers (50–1000 nM), we used this dose for most of the follow‐up experiments.
We also tested a selective α7 agonist (ICH3, previously identified by our group, Dallanoce et al. (2011), and we found it increased A549 cell viability at concentrations >100 nM (data not shown).
The lack of effect of nicotine on H1975 cells suggests that nicotine‐mediated effects are due to specific α7‐containing or α9‐containing subtype(s) that are little or not expressed in the H1975 cells.
Nicotine stimulates activation of intracellular ERK and Akt pathways in A549 cells
Previous work by other groups has shown that nicotine increases the cell proliferation of a number of tumour cells by activating the ERK pathway or inhibits cell apoptosis by activating the Akt pathway.
We used Western blotting to analyse the possible nicotine‐induced activation of the ERK and Akt pathways in A549 and H1975 cell lines. As shown in Figure 2 lower part, 100 nM nicotine increased the p‐Akt and p‐ERK levels in the A549 cells (n = 8), but not in the H1975 cells (n = 8). This activation was time‐dependent: nicotine‐stimulated phosphorylation peaked 5 and 10 min after nicotine stimulation and decreased permanently after 30–60 min. Nicotine did not affect the protein levels of total Akt or ERK.
α7‐ and α9‐containing nAChR subtypes are involved in the effects of nicotine on A549 cells
As we found that A549 cells express α7 and α9 mRNAs, we evaluated the involvement of receptors containing these subunits in the proliferative effect of nicotine using subtype‐specific antagonists.
We first tested the α‐Bgtx and MLA selective nAChR antagonists of homomeric and/or heteromeric receptors containing α7 or α9 subunits (Elgoyhen et al., 1994; Verbitsky et al., 2000) in order to investigate the nicotine‐induced proliferation of A549 cells. The cells were pretreated with 1 μM α‐Bgtx (n = 8) or 500 nM MLA (n = 8) for 30 min, followed by 100 nM nicotine for 48 h.
We found that α‐Bgtx and MLA abolished the nicotine‐induced proliferation of A549 cells as measured by means of counting assay (Figure 3A). Both peptides also prevented nicotine‐induced p‐Akt and the p‐ERK activation (n = 6) (Figure 3B, C).
Figure 3.
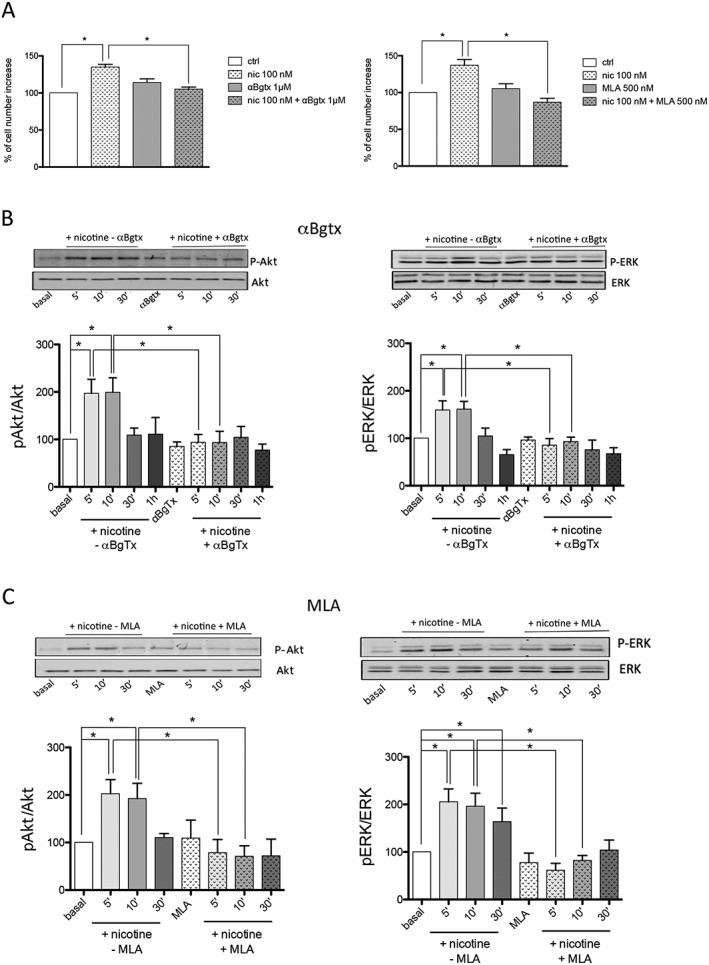
α‐Bgtx and MLA inhibit nicotine‐induced A549 cell proliferation and signalling activation. (A) Effect (expressed as percentage) of the 100 nM nicotine treatment for 48 h in the presence or absence of 1 μM α‐Bgtx (left) or 500 nM MLA (right) on A549 cell number. The graphs show the mean values ±SEM obtained in eight different experiments performed in triplicate. The analysis was made using one‐way ANOVA followed by Dunn's test (*P < 0.05). (B–C) Western blot analysis of Akt and ERK pathway activation in A549 cells exposed to nicotine in the presence or absence of 1 μM α‐Bgtx (B) or 500 nM MLA (C) (n = 6). The cells were pre‐incubated or not with 1 μM α‐Bgtx or 500 nM MLA for 30 min and then treated with 100 nM nicotine for the indicated times in the absence (−) or presence (+) of the antagonists. The Western blot results are expressed as the p‐Akt/Akt and p‐ERK/ERK ratios, taking the ratio of control samples as 100. The graphs show the mean values ±SEM obtained in six different experiments performed in duplicate or triplicate. A representative blot at the corresponding times is shown above each graph. The Western blotting data were statistically analysed using one‐way ANOVA followed by Dunn's test (*P < 0.05).
In order to discriminate the subtypes involved, the same proliferation and signalling experiments were further carried out using an antagonist peptide selective for the α9‐containing subtype, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9472 (Romero et al., 2017). RGIA4 at 1 μM (n = 8) completely abolished the proliferation of A549 cells induced by 48 h treatment with 100 nM nicotine (Figure 4A) and, when administered alone, had no effect on basal cell proliferation. When tested for the activation of intracellular pathways, RGIA4 blocked the nicotine‐activated downstream cascades of p‐Akt and p‐ERK (n = 8) (Figure 4C).
Figure 4.
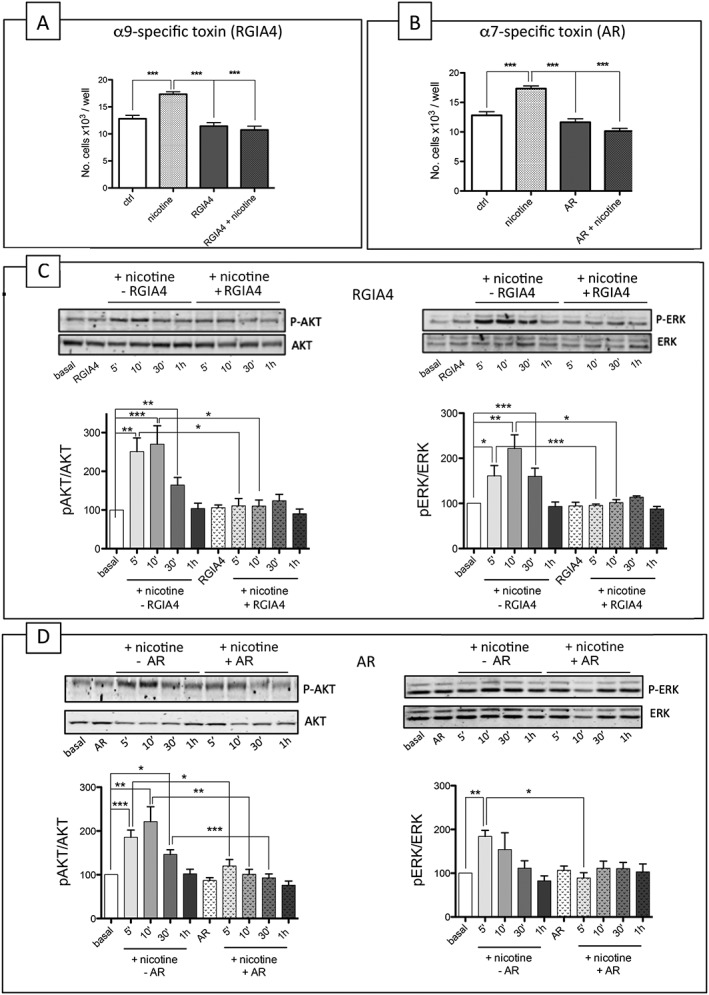
The α9 subtype‐selective peptide RGIA4 and the α7 subtype‐selective peptide ArIB [V11L; V16D] (AR) block the nicotine‐induced proliferation and intracellular signalling of A549 cells. (A–B) The effect of treatment with 100 nM nicotine for 48 h, in the presence or absence of 1 μM RGIA4 (A) (n = 8) and AR (B) (n = 8), on the number of A549 cells. The results are the average of eight experiments performed in triplicate and are expressed as the number of proliferating cells in each well. The statistical analysis was made using one‐way ANOVA followed by a post hoc Bonferroni test (*P < 0.05). (C) Western blot analysis of ERK and Akt pathway activation in A549 cells in the presence or absence of 1 μM RGIA4 (n = 8). The method was the same as that described in part B of Figure 3 except for the fact that 1 μM RGIA4 was used. The Western blotting data were statistically analysed using one‐way ANOVA followed by Dunn's test (*P < 0.05,). (D) Western blot analysis of ERK and Akt pathway activation in A549 cells in the presence or absence of 1 μM AR peptide (n = 8). The method was the same as that described in part B of Figure 3 except for the fact that 1 μM peptide AR was used. The Western blotting data were statistically analysed using one‐way ANOVA followed by Dunn's test (*P < 0.05).
The α7‐specific antagonist peptide http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4007 (V11L; V16D) (AR) 1 μM (Whiteaker et al., 2007; Romero et al., 2017) significantly blocked nicotine‐induced proliferation (Figure 4B) and decreased the nicotine‐induced activation of p‐Akt (Figure 4D) after 10 min of nicotine stimulation, but had no significant effect on the nicotine‐induced modulation of the ERK pathway.
The inhibition of the nicotine‐induced activation of ERK was stronger when the α‐Bgtx or MLA toxins were used instead of the subtype‐specific peptide (AR). This suggests that α7‐ and α9‐containing receptors act synergistically to activate this pathway and that blocking the α7‐containing receptor alone is not enough to block the effect of nicotine.
Gene silencing studies
The effect of siRNAs on α7‐containing nAChRs
In preliminary experiments, we screened four different siRNAs (Dharmacon) using q‐PCR and Western blotting.
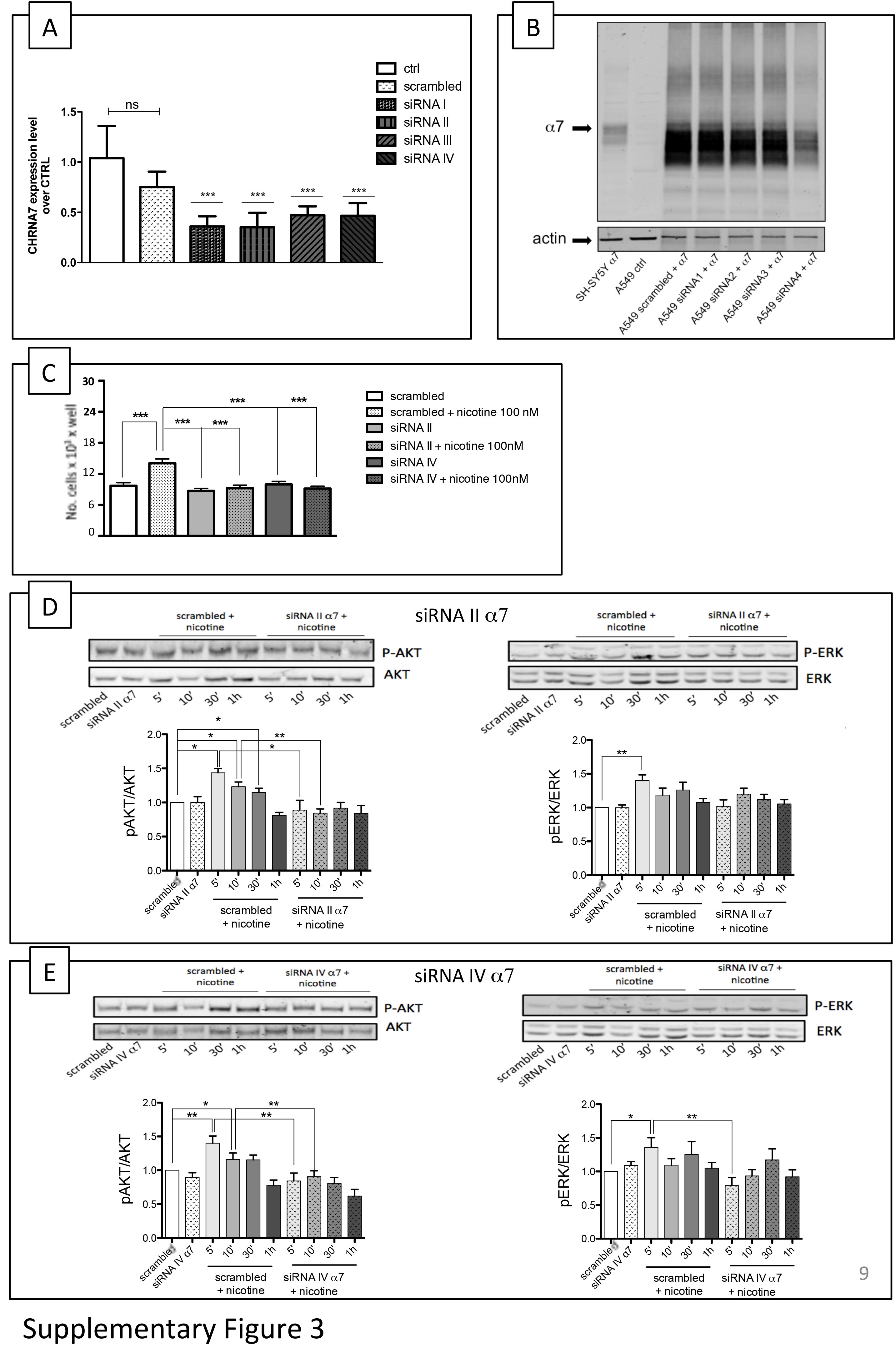
In order to evaluate the ability to knock down the gene of interest, A549 cells were transfected for 48 h with scrambled siRNA (used as a negative control) or one of the four siRNAs by targeting specific CHRNA7 sequences (Supporting Information Figure S3A).
q‐PCR analysis showed that, in comparison with untreated control cells, scrambled siRNA had no significant effect on α7 mRNA levels, but all four α7 siRNAs knocked down more than 50% of CHRNA7 gene transcript: siRNA I (64%), siRNA II (65%), siRNA III (53%) and siRNA IV (54%).
Due to the low α7 protein level present in A549 cells, we could not detect the endogenous α7 protein using our α7‐specific Abs, and so, in order to investigate the effect of the different siRNAs on α7 expression, we co‐transfected α7 cDNA into A549 cells for 48 h and then measured the level of knock down by means of Western blotting with the anti‐α7 Abs. We found that the siRNAs that were most potent in knocking down the transfected α7 protein were siRNA II and siRNA IV (Supporting Information Figure S3B).
We next analysed the effects of scrambled siRNA, siRNA II and siRNA IV on the nicotine‐induced stimulation of cell proliferation. To this end, the cells were first transfected with scrambled siRNA or siRNAs II or IV, starved by means of FBS deprivation and then treated with 100 nM nicotine for 48 h (n = 7). Transfection with the scrambled siRNA did not affect basal proliferation or the pro‐proliferative effect of 100 nM nicotine (Supporting Information Figure S3C); on the contrary, transfection with siRNAs II and IV abolished nicotine‐induced proliferation, without changing basal proliferation.
Knocking down α7 protein with siRNA II and siRNA IV (n = 7) blocked the nicotine activation of p‐Akt at 5 and 10 min and also decreased the p‐ERK activation induced by nicotine stimulation (Supporting Information Figure S3D, E) although the decrease of p‐ERK was only significant when siRNA IV was used (Supporting Information Figure S3D, E). These data suggest that α7‐containing receptors are mainly involved in Akt pathway activation and are not essential for mediating the effect of nicotine on the ERK pathway in these cells.
The effect of siRNAs on α9‐containing nAChRs
Our preliminary q‐PCR studies showed that the α9 subunit is expressed in A549 cells, and so, we analysed the ability of four α9‐directed siRNAs to knockdown the α9 subunit. Using the same protocol as that used to study the effect of α7 siRNAs, we found that all four siRNAs down‐regulated α9 mRNA, but the most potent were siRNAs II (91.5%), III (90.5%) and IV (84.9%) (Figure 5A). Unfortunately, we could not use Western blotting to assess the effect of α9 siRNAs on the overexpressed α9 subunit because no α9 subunit‐specific Abs are available.
Figure 5.
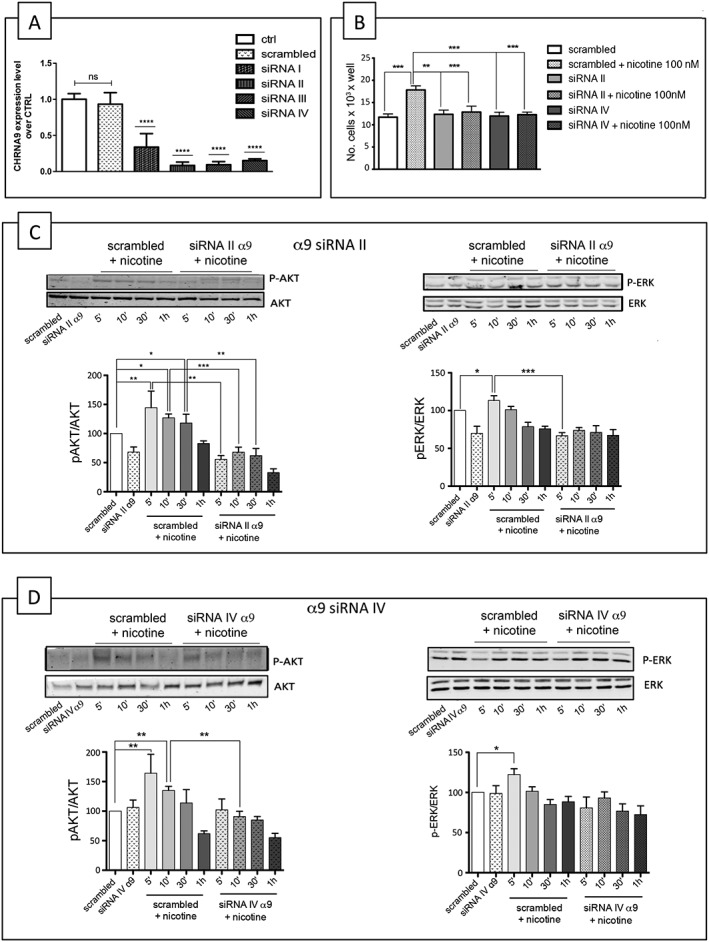
siRNA knockdown of the α9 subunit reduces the nicotine‐induced proliferation and activation of p‐ERK and p‐Akt in A549 cells. (A) q‐PCR analysis of α9 subunit mRNA level in A549 cells transfected for 48 h with the indicated α9 siRNAs. Data are shown as relative expression ±SD of three independent experiments performed in triplicate and are expressed with respect to the level of α9 subunit mRNA level in the cells transfected with scrambled siRNA set as 1, according to the 2−ΔΔCt method. *P < 0.05, statistically significant with respect to control (ctrl) cells (one‐way ANOVA, post‐Tukey's test). ns: not significant. (B) Effect of knocking down the α9 subunit on nicotine‐induced cell proliferation. A549 cells transfected with siRNA II or siRNA IV were treated for 48 h with 100 nM nicotine and then counted. (C) Western blot analysis of ERK and Akt pathway activation in A549 cells in the presence or absence of 100 nM nicotine, with or without α9 siRNA II. (D) Western blot analysis of ERK and Akt pathway activation in A549 cells in the presence or absence of 100 nM nicotine, with or without α9 siRNA IV. The Western blot results are expressed as the p‐Akt/Akt and p‐ERK/ERK ratios, taking the ratio of cells treated with scrambled siRNA as 100. The statistical analysis of nicotine‐induced proliferation and ERK and Akt pathway activation in the presence of scrambled siRNA or α9 siRNAs was made in seven independent experiments performed in triplicate using one‐way ANOVA followed by Bonferroni's test (*P < 0.05 vs. untreated cells) or Dunn's test.
Figure 5B shows that siRNAs II and siRNA IV both inhibited nicotine‐induced cell growth (n = 7), and Figure 5C shows that siRNA II reduced the nicotine‐induced activation of p‐Akt at 5, 10 and 30 min, whereas siRNA IV only significantly reduced the Akt stimulation at 10 min. Moreover siRNA II reduced p‐ERK activation after 5 min, but siRNA IV had no significant effect on p‐ERK (Figures 5C, D).
MG624 also acts on the α9–α10 subtype and blocks the effects of nicotine on A549 cells
We have previously characterized a 4‐oxystilbene MG624 compound as a selective α7 antagonist (Gotti et al., 1998), and we have now characterized its effect on the α9–α10 subtype expressed in oocytes. MG624 reduced ACh activation of the α9–α10 subtype in a dose‐dependent manner with IC50 values of 12.4 nM (5.6–18 nM), whereas the IC50 value for the α7 subtype tested in parallel was 41.5 nM (21–53 nM) (Figure 6A).
Figure 6.
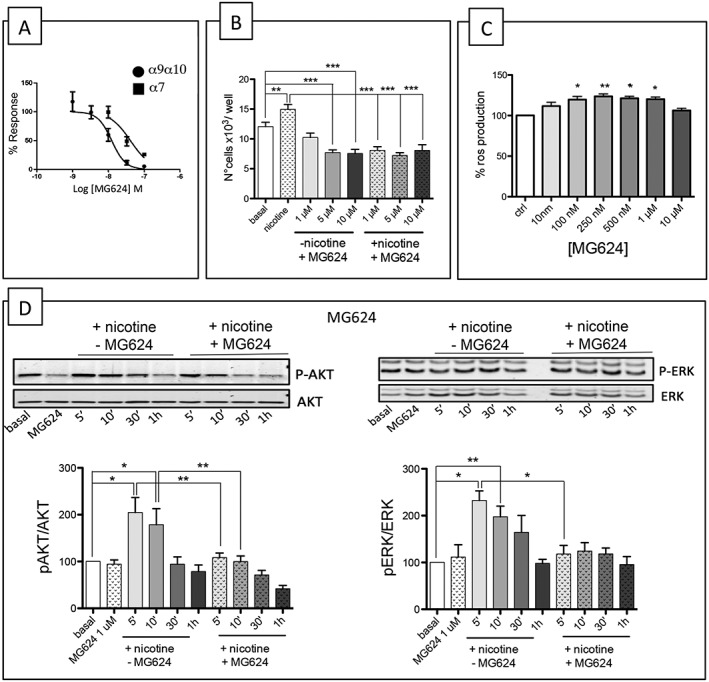
Characterization of the effect of MG624. (A) Effects of MG624 on Xenopus oocytes expressing α9–α10‐ and α7‐containing nicotinic receptors. Oocytes were injected with mRNA encoding the human α9–α10 or α7 subunits. Concentration–effect curves of MG624 inhibition of ACh‐induced ion currents. Within each group, all responses were normalized to an initial control stimulation (see Methods). The data points represent mean values ±SEM. Drug potency and efficacy parameters were calculated using nonlinear least‐squares curve fitting to the Hill equation. (B) MG624 decreases A549 proliferation. MG624 (1, 5 and 10 μM) abolished the chronic (48 h) nicotine‐induced increase in the number of A549 cells (n = 5). The analysis was made using one‐way ANOVA followed by a post hoc Bonferroni's test (cell count) (*P < 0.05). (C) MG624 increases ROS production. A549 cells were seeded in black 96‐well plates, pretreated with the indicated concentrations of MG624 for 24 h and then incubated with 25 μM DCFH‐DA for 30 min at 37°C. Fluorescence was measured at 485 nmex/530 nmem using a microplate reader. The results are expressed as the increase in fluorescence units·mg−1 of cell proteins taking the production of ROS in untreated cells as 100%. The analysis was made using one‐way ANOVA followed Dunn's test (*P < 0.05). (D) MG624 blocks the nicotine‐induced activation of p‐ERK and p‐Akt. Western blot analysis of ERK and Akt pathway activation upon nicotine stimulation in A549 cells with or without MG624 (1 μM) (n = 5).
As it has been recently reported that this compound blocks angiogenesis and the pro‐proliferative effects of nicotine on airway smooth muscle cells (Brown et al., 2012), we also tested its effect on the nicotine (100 nM) activation of A549 cells in the presence of three different doses of MG624 (1, 5 and 10 μM) for 48 h and counted the cells manually (n = 5). Our results showed that the lowest concentration of MG624 (1 μM) was not toxic but blocked nicotine‐induced proliferation, but the higher concentrations (5 and 10 μM) administered alone decreased the viability of control cells. For this reason, the following experiments were conducted using MG624 at the concentration of 1 μM.
Finally, we analysed the effect of MG624 on intracellular ERK and Akt signalling by treating A549 cells with nicotine (100 nM) for different times (from 5 min to 1 h) alone or in combination with MG624 (1 μM) (n = 5). Western blotting analyses showed that MG624 significantly decreased the nicotine‐induced activation of Akt after 5 and 10 min of nicotine stimulation and the nicotine‐induced activation of ERK at 5 min (Figure 6D).
MG624 also increases ROS production in A549 cells
It has recently been reported that resveratrol and stilbene derivatives are cytotoxic in vitro (Miki et al., 2012; Kong et al., 2016; Sassi et al., 2014 ) and selectively induce the mainly necrotic death of fast‐growing tumour cells when supplied in the low μM range.
As MG624 shares the stilbene scaffold and a para positioned ω‐onium alkyloxy group with the phosphonium butyl derivatives of resveratrol, we tested whether it can also affect ROS production. A549 cells were treated with increasing concentrations of MG624 (10 nM to 10 μM) for 24 h, and ROS production was measured using the DCFH‐DA probe. The results showed that MG624 increases ROS production at concentrations of 50, 100, 250 and 500 nM (Figure 6C) but not at highest concentration (1 μM, 10 μM) (n = 5). We believe that after 24 h incubation with 1 or 10 μM MG624, the cytotoxic effect that we have discussed above (measured after 48 h) is already effective and interferes with the production of ROS.
Discussion
The experiments described in this paper showed that nicotinic receptors containing the α7 and/or α9–α10 subunits are central in mediating the nicotine‐induced increase in the proliferation of A549 cells. They also showed that nicotine, acting on α7‐ and/or α9‐containing receptors, stimulates the Akt and ERK cell signal transduction pathways. We finally determined that MG624, which was previously shown to be an α7 antagonist, also acts on α9–α10‐containing receptors. Low μM concentrations block nicotine‐induced proliferation and intracellular signalling, and higher concentrations can be cytotoxic for A549 cells.
These findings and the fact that nicotine is active on a cell lines expressing the α7 and α9‐containing receptors support the idea that inhibiting nicotinic receptors containing these subunits may be a valid therapeutic approach for lung cancer.
In agreement with data reported by others (Song et al., 2008; Falvella et al., 2009), our q‐PCR studies showed that the α5 subunit transcript was always expressed in the two adenocarcinoma cell lines and the adenocarcinomas at much higher levels than in normal lung tissue. The level of the α9 subunit transcript was higher in three out of four tumour samples; and the α7 subunit transcript was not significantly increased in the tumour samples.
However, as it is not easy to interpret data obtained using q‐PCR because there is not always a correlation between mRNA and expressed subunit protein levels, we tried to identify the receptors expressed in the adenocarcinoma cell lines biochemically and pharmacologically using direct affinity purification and immunoprecipitation, which have been validated for the nAChRs present in the CNS (Gotti et al., 2009; Zoli et al., 2015). As shown in Supporting Information Figure S1, we carefully checked the ability of our Abs to recognize the native subtypes and found that we could identify the α7 present in A549 cells only after affinity purification on α‐Bgtx resin. The α‐Bgtx affinity‐purified receptors contain the α7 subunit, but no α7dup isoform, even though its mRNA is highly expressed, and it can be recognized by our Abs. This is important because α7 and α7dup subunits can co‐exist in the same receptors in heterologous systems (de Lucas‐Cerrillo et al., 2011; Wang et al., 2014). As α‐Bgtx also binds α9‐containing receptors, we cannot rule out the possibility that α‐Bgtx‐purified A549 receptors include α9‐receptors.
Western blotting also showed that, in agreement with the lack of α7 subunit transcript, there are no α7‐containing receptors in the H1975 adenocarcinoma cell line.
Genome wide association studies have indicated that α5‐containing nAChRs are closely associated with the risk of lung cancer (Mucchietto et al., 2016), but the underlying mechanisms are not yet well known. The published data are contradictory; Krais et al. (2011) found that using antagonists or RNA interference to modulate the activity of CHRNA5 in non‐transformed bronchial cells and lung cancer cell lines significantly increased migration in both normal and tumour cells, but Sun and Ma (2015) blocked the nicotine‐stimulated activation of α5‐containing nAChRs and suppressed cell migration and invasion by treating adenocarcinoma A549 cells with α5 subunit‐specific siRNA.
A549 cells express a high level of α5 subunit mRNA and protein, but we have no α5‐selective antagonists that can be used to study the role of α5‐containing receptors in nicotine‐induced proliferation. As a previous study by our lab suggested that an α7α5 subtype may be present in lung cancer cells (Chini et al., 1992) and as present study demonstrate that α‐Bgtx‐sensitive receptors are involved in the pro‐proliferative effects of nicotine on A549 cells, we investigated whether α‐Bgtx receptors purified from A549 cells contain the α5 subunit. However, probing the purified receptors with anti‐α5 Abs did not reveal any specific labelling, indicating that α‐Bgtx‐purified A549 receptors do not contain the α5 subunit.
Treating A549 and H1975 cells with nicotine concentrations in the range of those detected in the serum of smokers (10−8–10−7 M) induced the proliferation of A549 but not H1975 cells, thus suggesting the involvement of cell‐specific nAChRs in nicotine‐mediated effects on A549 cells. α‐Bgtx and MLA toxins acting on α7 and α9 receptors, and subtype‐specific α7 (AR) and α9 (RgIA4) toxins all blocked the nicotine‐induced proliferation of A549 cells. The involvement of the α7 subtype in nicotine‐induced proliferation was confirmed by the fact that a very low concentration of the subtype‐selective α7 agonist ICH3 also increased cell proliferation.
The concentration of nicotine that increases cell proliferation is very low, much lower than that necessary to activate α7 receptors in electrophysiological experiments. We therefore believe that, although nAChRs are ion channels mediating the influx of Na+ and Ca2+ and the efflux of K+, their activation (and that of α9 receptors) by low nicotine concentration in A549 cells may elicit non‐ionic signalling events that regulate the phosphorylation and dephosphorylation of target proteins that mediate some of the effects of nicotine (see review by Schaal and Chellappan, 2014). Similar results have been seen in immune cells in which, although they have α7 receptors on their outer membranes, no ACh‐dependent currents have been recorded (reviewed in Treinin et al., 2017).
We therefore analysed which intracellular pathways are involved in the nicotine‐induced proliferation of A549 cells. Nicotine stimulation of nAChRs triggered a number of protein kinase‐signalling cascades, thus simultaneously altering gene expression and inducing cellular changes. We confirmed that nicotine increases p‐ERK/ERK and p‐Akt/Akt ratios after a very short time of exposure (5–10 min) and that the α7/α9 toxins (MLA and α‐Bgtx), the α9‐selective peptide RGIA4 and the α7‐selective peptide AR all prevented the nicotine‐induced p‐Akt/Akt activation.
The nicotine‐induced proliferation and intracellular signalling was further confirmed by silencing experiments that replicated the results obtained with the α7‐ or α9‐specific peptides. Furthermore, our data suggest that α7‐ and α9‐containing receptors act synergistically to activate the ERK pathway because, although subtype‐specific peptides or silencing of the α7 and α9 genes reduce ERK activation, the reduction is much less than that induced by the toxins acting on both subtypes (α‐Bgtx or MLA). This is probably due to the fact that both receptors have to be activated in order to reach the cytoplasmic calcium concentration that triggers the activation of this pathway.
Some recent evidence indicates that nAChRs are expressed on the mitochondrial outer membrane of lung cells (mt‐nAChRs) (Kalashnyk et al., 2012; Gergalova et al., 2012), are non‐covalently connected to voltage‐dependent anion channels and control cytochrome C release by inhibiting the opening of mitochondrial permeability transition pores (Saxena et al., 2011). Although the specificity of many Abs against nAChRs has been questioned and the presence of mt‐nAChR is still an open question, the fact that the nicotine‐induced proliferation and intracellular signalling of A549 cells is blocked by peptides that are not cell permeable and can only act at the plasma membrane indicates that the effects of nicotine are due to the activation of nAChRs in the plasma membrane.
Stimulated by data showing that MG624 potently suppresses the proliferation of primary human microvascular endothelial cells of the lung (HMEC‐Ls), has robust anti‐angiogenic activity in matrigel, rat aortic ring and rat retinal explant assays and suppresses the in vivo angiogenesis of NCi‐H69 SCLC tumours in the chorioallantoic membrane and nude mice model (Brown et al., 2012), we characterized its anti‐proliferative effects further. MG624 is highly selective for α7 nAChRs in chicken, rat, monkey and human brain membranes (Gotti et al., 1998; 2000), and the findings of this study demonstrate that very low concentrations (nM range) also block the ACh‐induced current in oocytes expressing the α9–α10 subtype (Figure 6A). We also found that relatively low concentrations of MG624 potently inhibit nicotine‐induced cell viability and proliferation and decrease the nicotine‐induced activation of the pERK and pAkt pathways.
However, in addition to being a competitive α7 antagonist, its stilbene structure makes MG624 very similar to resveratrol and ptero‐stilbene, which can pass through the plasma membrane and increase ROS production (reviewed in Biasutto et al., 2011). We therefore investigated whether it exerts some of its effects in the same manner. ROS play contradictory roles in tumourigenesis: they play a causal role in tumour development and progression by inducing DNA mutations, genomic instability and aberrant pro‐tumourigenic signalling, but high levels can also be toxic to cancer cells and potentially induce cell death (Glasauer and Chandel, 2014). We found that MG624 stimulated ROS production at relatively low concentrations (100–250 nM) and so it is possible that its anti‐proliferative effect is due to synergistic mechanisms (α7/α9–α10 nAChR blockade and increased ROS production). Given these findings and the fact that no non‐peptidic antagonist is available that can specifically target α7/α9–α10 nAChR subtypes in vivo, MG624 may be a lead structure for the development of new therapeutic agents for human lung cancers expressing high levels of the α7 and/or α9 subunits.
Author contributions
V.M., F.F., F.C. and C.G. participated in the design of the experiments; V.M., F.F., M.M. and S.P. conducted the experiments; R.B., A.M. and S.D.L. made the q‐PCR and the siRNA experiments; C.B. and M.P. made the MG624 and its derivatives; C.D. and M.McI. synthesized and functionally characterized the subtype‐specific peptides and MG624; V.M., F.F., F.C. and C.G. wrote or contributed to the writing of the paper.
Conflict of interest
Conotoxin peptides, including those referenced in this paper, have been patented by the University of Utah with J.M.M. listed as an inventor. The authors declare no other conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Characterisation of the subunit specificity of the antibodies.
Figure S2 Time course of the effect of 100 nM nicotine exposure on A549 cells proliferation.
Figure S3 siRNA knockdown of the α7 subunit in A549.
{kind=link}
Acknowledgements
V.M. is a recipient of a fellowship from the Fondazione Confalonieri, Milan. F.F. is a recipient of a fellowship from the Fondazione Vollaro, Milan. This work was supported by the CNR Research Project on Aging, the CNR project PRONAT, the IN‐CNR InterOmics project and by grants from the Fondazione Monzino (Milano) and from the Fondazione Vollaro (Milano). We thank Dr Dragani (Fondazione IRCCS Istituto Nazionale dei Tumori, Milano) for the generous gift of human samples. We thank Prof Marco De Amici (Department of Pharmaceutical Science, Università degli Studi di Milano, Milano) for the generous gift of ICH3.
Mucchietto, V. , Fasoli, F. , Pucci, S. , Moretti, M. , Benfante, R. , Maroli, A. , Di Lascio, S. , Bolchi, C. , Pallavicini, M. , Dowell, C. , McIntosh, M. , Clementi, F. , and Gotti, C. (2018) α9‐ and α7‐containing receptors mediate the pro‐proliferative effects of nicotine in the A549 adenocarcinoma cell line. British Journal of Pharmacology, 175: 1957–1972. doi: 10.1111/bph.13954.
References
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89: 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, McIntosh JM (2012). Molecular basis for the differential sensitivity of rat and human alpha9alpha10 nAChRs to alpha‐conotoxin RgIA. J Neurochem 122: 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasutto L, Szabo I, Zoratti M (2011). Mitochondrial effects of plant‐made compounds. Antioxid Redox Signal 15: 3039–3059. [DOI] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X et al (2008). Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry 165: 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KC, Lau JK, Dom AM, Witte TR, Luo H, Crabtree CM et al (2012). MG624, an alpha7‐nAChR antagonist, inhibits angiogenesis via the Egr‐1/FGF2 pathway. Angiogenesis 15: 99–114. [DOI] [PubMed] [Google Scholar]
- Chini B, Clementi F, Hukovic N, Sher E (1992). Neuronal‐type alpha‐bungarotoxin receptors and the alpha 5‐nicotinic receptor subunit gene are expressed in neuronal and non‐neuronal human cell lines. PNAS 89: 1572–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallanoce C, Magrone P, Matera C, Frigerio F, Grazioso G, De Amici M et al (2011). Design, synthesis, and pharmacological characterization of novel spirocyclic quinuclidinyl‐Delta2‐isoxazoline derivatives as potent and selective agonists of alpha7 nicotinic acetylcholine receptors. ChemMedChem 6: 889–903. [DOI] [PubMed] [Google Scholar]
- de Lucas‐Cerrillo AM, Maldifassi MC, Arnalich F, Renart J, Atienza G, Serantes R et al (2011). Function of partially duplicated human alpha77 nicotinic receptor subunit CHRFAM7A gene: potential implications for the cholinergic anti‐inflammatory response. J Biol Chem 286: 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egleton RD, Brown KC, Dasgupta P (2008). Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. TIPS 29: 151–158. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S (1994). Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79: 705–715. [DOI] [PubMed] [Google Scholar]
- Ellison M, Haberlandt C, Gomez‐Casati ME, Watkins M, Elgoyhen AB, McIntosh JM et al (2006). Alpha‐RgIA: a novel conotoxin that specifically and potently blocks the alpha9alpha10 nAChR. Biochemistry 45: 1511–1517. [DOI] [PubMed] [Google Scholar]
- Falvella FS, Galvan A, Frullanti E, Spinola M, Calabro E, Carbone A et al (2009). Transcription deregulation at the 15q25 locus in association with lung adenocarcinoma risk. Clin Cancer Res 15: 1837–1842. [DOI] [PubMed] [Google Scholar]
- Gergalova G, Lykhmus O, Kalashnyk O, Koval L, Chernyshov V, Kryukova E et al (2012). Mitochondria express alpha7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: study on isolated mitochondria. PLoS One 7: e31361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasauer A, Chandel NS (2014). Targeting antioxidants for cancer therapy. Biochem Pharmacol 92: 90–101. [DOI] [PubMed] [Google Scholar]
- Gotti C, Balestra B, Moretti M, Rovati GE, Maggi L, Rossoni G et al (1998). 4‐Oxystilbene compounds are selective ligands for neuronal nicotinic alphaBungarotoxin receptors. Br J Pharmacol 124: 1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Carbonnelle E, Moretti M, Zwart R, Clementi F (2000). Drugs selective for nicotinic receptor subtypes: a real possibility or a dream? Behav Brain Res 113: 183–192. [DOI] [PubMed] [Google Scholar]
- Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I et al (2009). Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol 78: 703–711. [DOI] [PubMed] [Google Scholar]
- Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L et al (2009). Rodent habenulo‐interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the alpha3beta4* and alpha3beta3beta4* subtypes mediate acetylcholine release. J Neurosci 29: 2272–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grando SA (2014). Connections of nicotine to cancer. Nat Rev Cancer 14: 419–429. [DOI] [PubMed] [Google Scholar]
- Hecht SS, Hoffmann D (1988). Tobacco‐specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis 9: 875–884. [DOI] [PubMed] [Google Scholar]
- Hone AJ, Whiteaker P, Christensen S, Xiao Y, Meyer EL, McIntosh JM (2009). A novel fluorescent alpha‐conotoxin for the study of alpha7 nicotinic acetylcholine receptors. J Neurochem 111: 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D et al (2008). A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452: 633–637. [DOI] [PubMed] [Google Scholar]
- Improgo MR, Scofield MD, Tapper AR, Gardner PD (2010). From smoking to lung cancer: the CHRNA5/A3/B4 connection. Oncogene 29: 4874–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnyk OM, Gergalova GL, Komisarenko SV, Skok MV (2012). Intracellular localization of nicotinic acetylcholine receptors in human cell lines. Life Sci 91: 1033–1037. [DOI] [PubMed] [Google Scholar]
- Kong Y, Chen G, Xu Z, Yang G, Li B, Wu X et al (2016). Pterostilbene induces apoptosis and cell cycle arrest in diffuse large B‐cell lymphoma cells. Sci Rep 6: 37417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krais AM, Hautefeuille AH, Cros MP, Krutovskikh V, Tournier JM, Birembaut P et al (2011). CHRNA5 as negative regulator of nicotine signaling in normal and cancer bronchial cells: effects on motility, migration and p63 expression. Carcinogenesis 32: 1388–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Huang CS, Chen CS, Tu SH, Wang YJ, Chang YJ et al (2010). Overexpression and activation of the alpha9‐nicotinic receptor during tumorigenesis in human breast epithelial cells. J Natl Cancer Inst 102: 1322–1335. [DOI] [PubMed] [Google Scholar]
- Miki H, Uehara N, Kimura A, Sasaki T, Yuri T, Yoshizawa K et al (2012). Resveratrol induces apoptosis via ROS‐triggered autophagy in human colon cancer cells. Int J Oncol 40: 1020–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti M, Zoli M, George AA, Lukas RJ, Pistillo F, Maskos U et al (2014). The novel alpha7beta2‐nicotinic acetylcholine receptor subtype is expressed in mouse and human basal forebrain: biochemical and pharmacological characterization. Mol Pharmacol 86: 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucchietto V, Crespi A, Fasoli F, Clementi F, Gotti C (2016). Neuronal acetylcholine nicotinic receptors as new targets for lung cancer treatment. Curr Pharm Des 22: 2160–2169. [DOI] [PubMed] [Google Scholar]
- Riganti L, Matteoni C, Di Angelantonio S, Nistri A, Gaimarri A, Sparatore F et al (2005). Long‐term exposure to the new nicotinic antagonist 1,2‐bisN‐cytisinylethane upregulates nicotinic receptor subtypes of SH‐SY5Y human neuroblastoma cells. Br J Pharmacol 146: 1096–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero HK, Christensen SB, Di Cesare ML, Gajewiak J, Ramachandra R, Elmslie KS et al (2017). Inhibition of alpha9alpha10 nicotinic acetylcholine receptors prevents chemotherapy‐induced neuropathic pain. PNAS 114: E1825–E1E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassi N, Mattarei A, Azzolini M, Bernardi P, Szabo I, Paradisi C et al (2014). Mitochondria‐targeted resveratrol derivatives act as cytotoxic pro‐oxidants. Curr Pharm Des 20: 172–179. [DOI] [PubMed] [Google Scholar]
- Saxena G, Patro IK, Nath C (2011). ICV STZ induced impairment in memory and neuronal mitochondrial function: a protective role of nicotinic receptor. Behav Brain Res 224: 50–57. [DOI] [PubMed] [Google Scholar]
- Schaal C, Chellappan SP (2014). Nicotine‐mediated cell proliferation and tumor progression in smoking‐related cancers. Mol Cancer Res : MCR 12: 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller HM (2009). Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer 9: 195–205. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A (2013). Cancer statistics, 2013. Cancer J Clin 63: 11–30. [DOI] [PubMed] [Google Scholar]
- Sinkus ML, Graw S, Freedman R, Ross RG, Lester HA, Leonard S (2015). The human CHRNA7 and CHRFAM7A genes: a review of the genetics, regulation, and function. Neuropharmacology 96 ((Pt B)): 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Sekhon HS, Fu XW, Maier M, Jia Y, Duan J et al (2008). Activated cholinergic signaling provides a target in squamous cell lung carcinoma. Cancer Res 68: 4693–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Sekhon HS, Jia Y, Keller JA, Blusztajn JK, Mark GP et al (2003). Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res 63: 214–221. [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindel ER (2016). Cholinergic targets in lung cancer. Curr Pharm Des 22: 2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Ma X (2015). alpha5‐nAChR modulates nicotine‐induced cell migration and invasion in A549 lung cancer cells. Exp Toxicol Pathol 67: 477–482. [DOI] [PubMed] [Google Scholar]
- Tournier JM, Birembaut P (2010). Nicotinic acetylcholine receptors and predisposition to lung cancer. Curr Opin Oncol 23: 83–87. [DOI] [PubMed] [Google Scholar]
- Treinin M, Papke RL, Nizri E, Ben‐David Y, Mizrachi T, Brenner T (2017). Role of the alpha7 nicotinic acetylcholine receptor and RIC‐3 in the cholinergic anti‐inflammatory pathway. Cent Nerv Syst Agents Med Chem 17: 1–9. [DOI] [PubMed] [Google Scholar]
- Verbitsky M, Rothlin CV, Katz E, Elgoyhen AB (2000). Mixed nicotinic‐muscarinic properties of the alpha9 nicotinic cholinergic receptor. Neuropharmacology 39: 2515–2524. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xiao C, Indersmitten T, Leonard S, Lester HA (2014). The duplicated α7 subunits assemble and form functional nicotinic receptors with the full‐length α7. J Biol Chem 289: 26451–26463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessler I, Kirkpatrick CJ (2008). Acetylcholine beyond neurons: the non‐neuronal cholinergic system in humans. Br J Pharmacol 154: 1558–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, Christensen S, Yoshikami D, Dowell C, Watkins M, Gulyas J et al (2007). Discovery, synthesis, and structure activity of a highly selective alpha7 nicotinic acetylcholine receptor antagonist. Biochemistry 46: 6628–6638. [DOI] [PubMed] [Google Scholar]
- Zoli M, Pistillo F, Gotti C (2015). Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96: 302–311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Characterisation of the subunit specificity of the antibodies.
Figure S2 Time course of the effect of 100 nM nicotine exposure on A549 cells proliferation.
Figure S3 siRNA knockdown of the α7 subunit in A549.