

Summary
Current treatment options for human stroke are limited mainly to the modestly effective infusion of tissue plasminogen activator (tPA), with additional improvement of functional independence and higher rates of angiographic revascularization observed after mechanical thrombectomy. However, new therapeutic strategies that address post‐stroke immune‐mediated inflammatory responses are urgently needed. Recent studies in experimental stroke have firmly implicated immune mechanisms in the propagation and partial resolution of central nervous system damage after the ischaemic event. A new‐found anti‐inflammatory role for regulatory B (Breg) cells in autoimmune diseases sparked interest in these cells as potential immunomodulators in stroke. Subsequent studies identified interleukin‐10 as a common regulatory cytokine among all five of the currently recognized Breg cell subsets, several of which can be found in the affected brain hemisphere after induction of experimental stroke in mice. Transfer of enriched Breg cell subpopulations into both B‐cell‐depleted and wild‐type mice confirmed their potent immunosuppressive activities in vivo, including recruitment and potentiation of regulatory T cells. Moreover, Breg cell therapy strongly reduced stroke volumes and treatment outcomes in ischaemic mice even when administered 24 hr after induction of experimental stroke, a treatment window far exceeding that of tPA. These striking results suggest that transfer of enriched Breg cell populations could have therapeutic value in human stroke, although considerable clinical challenges remain.
Keywords: B cells, brain, cell therapy, neuroinflammation, regulation/suppression
Abbreviations
- µMT−/−
B‐cell knockout mice
- Breg
regulatory B
- CIA
collagen‐induced arthritis
- CNS
central nervous system
- EAE
experimental autoimmune encephalomyelitis
- GFP
green fluorescent protein
- IL‐10
interleukin‐10
- MCAO
middle cerebral artery occlusion
- MZ
marginal zone
- NOD
non‐obese diabetic mice
- T2‐MZP
transitional 2 marginal‐zone precursor
- tPA
tissue plasminogen activator
- Treg
regulatory T
- WT
wild‐type
Stroke and the immune response
Stroke is the fifth leading cause of death and the leading cause of disability in the USA, affecting up to 795 000 individuals a year.1 Worldwide, 15 million individuals experience a stroke, resulting in 5 million deaths and another 5 million individuals left with permanent disabilities. Despite intensive efforts to find better therapeutics, the only drug approved to treat ischaemic stroke is recombinant tissue plasminogen activator (tPA), which must be administered within 4·5 hr of the stroke event but fails to treat haemorrhagic stroke. tPA was given US Food and Drug Administration approval in 1996.2 More recent studies using mechanical thrombectomy + tPA have shown additional benefit, providing better functional outcomes, lower mortality and more successful recanalization,3, 4 but do not address immune‐mediated tissue damage.
Stroke research has become increasingly focused on the immune response to brain injury after a stroke. Animal models of stroke have clearly demonstrated that approaches that modulate the immune system after induction of experimental stroke can be neuroprotective. Splenectomy, which removes a large reservoir of potentially damaging immune cells, reduces infarct volume when performed 2 weeks before induction of experimental stroke.5, 6, 7, 8, 9 After stroke induction, however, the spleen undergoes a massive reduction in size and cellularity that mirrors the onset of immunosuppression in mice,10 rats11 and stroke patients,12, 13, 14 which may account for increased risk of life‐threatening infections.
Evaluation of cellular and humoral immune responses after induction of experimental stroke has identified both pathological and protective cytokines and cell types. Studies using immune cell‐specific knockout mice have revealed which cells are detrimental and even which cells are protective after stroke. Mice that lack lymphocytes (Rag−/− or SCID mice) have significantly smaller infarcts compared with wild‐type (WT) mice.15, 16 The depletion of T cells, both CD4+ and CD8+ subsets, significantly reduced infarct volumes, whereas depletion of B cells had no effect on infarct volumes 24 hr after stroke induction.15 This suggests that T cells, but not B cells, might contribute to post stroke neural injury. When B cells were examined more specifically, it was found that B‐cell knockout mice (μMT−/−) had larger infarcts than WT mice,17 suggesting that B cells could be playing a protective role in limiting detrimental neuroinflammation. In particular, a B‐cell subset, regulatory B (Breg) cells, has been shown to be protective in experimental stroke.
Regulatory B cells in autoimmune diseases
Regulatory B cells are a subset of B cells that secrete interleukin‐10 (IL‐10) and have anti‐inflammatory effects on T cells, macrophages, natural killer cells and dendritic cells. Research on Breg cells has been hampered by the lack of a single cell‐specific marker or a signature transcription factor such as FoxP3 in the case of regulatory T (Treg) cells. To date, there are at least seven different subsets of Breg cells, including two immature Breg cell types and five mature Breg cell types. All Breg cells secrete IL‐1018, 19 and most are found within the spleen, with the exception of plasma cells that are found in the lymph nodes. As is illustrated in Fig. 1, splenic Breg cell subtypes include immature transitional 2 marginal‐zone precursor (T2‐MZP) cells (CD21hi CD23hi CD24hi) and plasmablasts (CD138+ CD44hi) as well as mature marginal zone (MZ) cells (CD21hi CD23−), B10 (CD5+ CD1dhi) cells, B‐1a (CD5+) cells, and Tim‐1 (TIM‐1+) cells, whereas the lymph nodes contain only mature plasma (CD138+ MHC‐IIlo B220+) cells.20 In addition to not being located within the spleen with the other Breg cells, plasma cells also produce IL‐35, another anti‐inflammatory cytokine that suppresses lymphocyte proliferation.21 Breg cells have also been shown to increase the number of Treg cells, including both CD4+ FoxP3+ and CD8+ CD122+ Treg cell subsets (Fig. 1).
Figure 1.
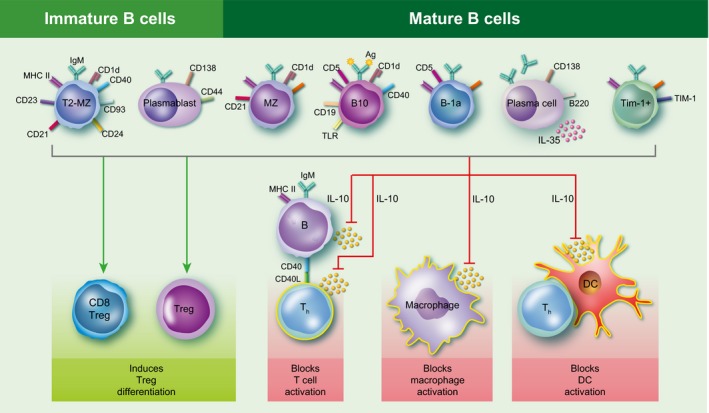
Regulatory B‐cell types and functions. Regulatory B (Breg) cells consist of immature and mature B‐cell subsets. The two immature Breg cell subsets include transitional 2 marginal zone precursor (T2‐MZP) cells (CD21hi CD23hi CD24hi) and plasmablasts (CD138+ CD44hi), whereas the five mature Breg cell subsets include marginal zone (MZ) cells (CD21hi CD23−), B10 cells (CD5+ CD1dhi), B‐1a cells (CD5+), Tim‐1 cells (TIM‐1+) and plasma cells (CD138+ MHC‐II lo B220+). All Breg cells produce the anti‐inflammatory cytokine interleukin‐10 (IL‐10), which can suppress T‐cell, macrophage and dendritic cell activation. In addition, plasma cells also produce IL‐35, another anti‐inflammatory cytokine that suppresses lymphocyte proliferation. Breg cells have also been shown to increase the number of regulatory T (Treg) cells, including the CD8+ CD122+ Treg cell subset.
Breg cells have been extensively studied in autoimmune diseases including animal models of multiple sclerosis [experimental autoimmune encephalomyelitis (EAE)], rheumatoid arthritis [collagen‐induced arthritis (CIA)], type 1 diabetes [non‐obese diabetic (NOD) mice], and systemic lupus erythematous (NZB/W F1 mice). Breg cells were shown to play an important protective role in all of the above‐mentioned autoimmune diseases22 by dampening pro‐inflammatory T cells and enhancing the expansion of Treg cells.23 In CIA, specifically IL‐10‐producing B cells were found to be protective, whereas B cells from IL‐10−/− mice did not provide any protection against CIA. Moreover, transfer of different Breg subsets into CIA‐affected mice demonstrated that only the T2‐MZP Breg subpopulation could protect the recipient mice from CIA disease progression.24 In systemic lupus erythematous, susceptible NZB/W F1 mice were found to have increased disease progression when B cells were depleted at 4 weeks of age but not if B cells were depleted at later time‐points.25 This observed exacerbation of lupus‐like disease was associated with expansion of B10 Breg cells that occurred at ~ 4 weeks of age as was found in C57BL/6 mice.25, 26 In diabetes, transfer of activated B cells into disease‐susceptible NOD mice reduced the incidence of diabetes, whereas this effect was not present in B cells from IL‐10−/− mice. Furthermore, transfer of activated B cells into NOD mice correlated with CD4+ T‐cell polarization towards an anti‐inflammatory Th2 activation state, with reduced activation of pro‐inflammatory Th1 cells.27
In the EAE model of multiple sclerosis, B‐cell depletion before disease induction resulted in increased clinical severity, whereas reconstitution with B cells from WT but not IL‐10−/− mice conferred protection,28, 29 a pattern similar to that observed in other autoimmune diseases. Other studies demonstrated that B cells played a role in EAE disease initiation but that Breg cells, particularly B10 cells, could down‐regulate activation of encephalitogenic T cells and reduce ongoing disease severity.26 Our own studies demonstrated that the strongly protective effect of oestrogen pretreatment on EAE induction30, 31, 32 was mediated in large part by IL‐10‐producing Breg cells. We found that oestrogen‐treated B‐cell‐deficient μMT−/− mice were not protected from EAE,33 whereas transfer of WT IL‐10‐producing B cells before oestrogen administration could protect the recipient WT mice from subsequent EAE induction.34 Moreover, oestrogen treatment was found to induce B10 (CD5+ CD1dhi), Tim‐1+ Breg cells and (CD138+ CD44hi) Breg cell plasmablasts in the EAE protected mice.35
It is this protective role in autoimmune diseases, particularly EAE, that prompted investigation into whether Breg cells could also modulate the immune system after stroke to protect the brain from increased neuroinflammation and neural injury.
Regulatory B cells in stroke
It is now well established that human stroke results in multi‐organ systemic disease rather than being restricted solely to the infarcted region of the brain. In animals, focal brain ischaemia induces systemic changes in the immune system that can lead to immune dysfunction. Although the immune system can become suppressed in response to pathogens, it becomes highly pro‐inflammatory towards the injured brain. The spleen, a reservoir of immune cells, is affected by cerebral ischaemia in mice, rats and stroke patients.5, 10, 11, 12 Splenic activation leads to expulsion of immune cells into the blood that then migrate to the brain.11 These immune cells probably exacerbate the evolving brain infarct, enhancing neural injury. However, data suggest that some types of immune cells may have favourable effects on the injured brain and provide a type of natural central nervous system (CNS) protection.
Recent studies have identified a small but powerful subset of IL‐10‐producing CD5+ CD1dhi Breg cells (B10) that can limit CNS inflammation and clinical signs of neurological disease in rodent experimental models.36 Depletion of B cells, including the B10 subset (B‐cell‐deficient mice or depletion with anti‐CD22 monoclonal antibody) causes increased disease severity in EAE models and transfer of the CD5+ CD1dhi (B10) Breg subset can provide protection against EAE disease induction. B‐cell‐deficient mice have a significant increase in cortical infarction after middle cerebral artery occlusion (MCAO), implicating protective regulatory effects by B cells (Fig. 2a). Transfer of 50 million B cells intraperitoneally into B‐cell knockout mice 24 hr before MCAO reduced infarct volume, yet B cells transferred from IL‐10−/− mice did not affect infarct size.17 The ability of the B10 subset to limit CNS injury probably resides in these cells' marked secretion of IL‐10 (as discussed above), a well‐recognized anti‐inflammatory cytokine that is effective against most forms of CNS damage. These findings strongly support the need to further evaluate the role of Breg cells in stroke.
Figure 2.
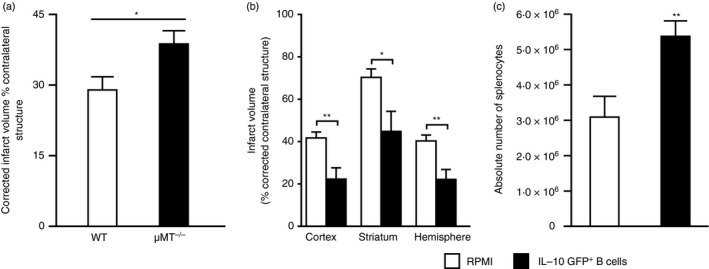
B‐cell‐deficient μ MT −/− mice and regulatory B (Breg) cells in stroke. (a) Infarct volume, corrected for the presence of oedema, at 48 hr reperfusion after 60 min middle cerebral artery occlusion (MCAO) given as bar graphs are visualized (mean ± SEM). Statistical analysis was performed with the Student's t‐test. There was a significant difference of infarct volumes between wild‐type (WT) and μ MT −/− mice; *P < 0·05. Groups: WT (n = 15); μ MT −/− (n = 12). (b) Intravenous transfer of 5 million IL‐10‐GFP + B‐cells reduced infarct volume in μ MT −/− B‐cell‐deficient mice 48 hr following 60 min of MCAO compared with intravenous transfer of RPMI vehicle (no cells). *P ≤ 0·05; **P ≤ 0·01 (Student's t‐test). (c) Forty‐eight hours after MCAO, mononuclear cells were isolated from spleens of RPMI‐1640 or IL‐10‐GFP + B‐cell‐recipient μ MT −/− mice and were analysed for total cell counts via haemocytometer. Values represent mean numbers (± SEM) of indicated cell subsets from 10 or 11 mice in each group, from at least three separate experiments. (a) Republished with permission of the Society for Neuroscience, from Ren, et al.;17 permission conveyed through Copyright Clearance Center, Inc. (b,c) Republished with permission by Bodhankar et al.,36 pp. 379, 382, © Springer Science+Business Media New York 2013. With the permission of Springer.
In mice there is a drastic loss of immune cells in the spleen accompanied by a pronounced increase in Treg cells within days of the ischaemic insult. In normal mice, Treg cells limit inflammation and inhibit autoimmune diseases.37, 38, 39, 40 Based on our observed increases in surviving CD3+ CD4+ CD25+ FoxP3+ Treg cells,10 it seemed that these cells were relatively resistant to apoptosis or other mechanisms that act to reduce viable immune splenocyte numbers. However, within the literature there are mixed results regarding the role of Treg cells after stroke and their ability to limit inflammation within the ischaemic brain.41 A number of reports suggested a protective role of Treg cells in stroke,42 whereas our earlier study using conditional FoxP3 knockout mice demonstrated CD4+ CD25+ FoxP3+ Treg cells did not contribute significantly in limiting infarct volume after MCAO.43 Alternatively, Breg cell subsets might play a crucial regulatory role that limits post CNS injury and neuroinflammation.
In mice, total spleen and blood cell numbers are reduced ~ 90% by 96 hr after MCAO. However, B cells still constitute ~ 30% of the remaining mononuclear cells.10 Infarct volumes were increased after MCAO in B‐cell‐deficient mice and a report in stroke patients showed worse clinical outcome when patients had lower circulating levels of B cells.44 Enter the timely reports of Breg cells45, 46, 47 and the newly described regulatory IL‐10‐secreting B‐cell subset, called B10.48 These Breg cells were found to have powerful inhibitory effects on other experimental inflammatory diseases, including EAE and murine lupus26, 49, 50 as described above. A key function of B10 cells is their secretion of IL‐10, an anti‐inflammatory cytokine that has been studied extensively in stroke.51 Indeed, IL‐10‐deficient mice developed larger infarcts after permanent focal ischaemia.52 Conversely, administration of exogenous IL‐10 to the lateral ventricle,53 intraperitoneally in combination with hypothermia,54 by adenoviral vectors,55 after induction of mucosal tolerance by IL‐10‐producing myelin oligodendrocyte glycoprotein‐reactive T cells56 or by transgenic over‐expression of IL‐1057 all reduced infarct volumes. Moreover, IL‐10 prevented neuronal damage induced by excitotoxicity in vitro.52 Clinically, early worsening of stroke was associated with lower IL‐10 plasma levels in patients.58 Conversely, excessive circulating levels of IL‐10 may predispose to increased infections.59 Taken together, these findings suggest that local secretion of IL‐10 may be preferable to systemic delivery.
We found that total CD19+ B cells were modestly increased in the CNS and periphery 48 hr after MCAO. Of key importance, the CD5+ CD1dhi B10 cell subset was highly enriched in the ipsilateral versus contralateral hemispheres of MCAO mice at this early time‐point (36·7% versus 13·4% of CD19+ B cells), so establishing the presence of this potentially important regulatory B‐cell subtype in the affected CNS infarcted region during stroke.60 Moreover, MCAO also induced an increased percentage of IL‐10‐secreting B cells in blood, demonstrating the availability of circulating Breg cells for entry into the CNS. Although these MCAO induced Breg cells may have limited CNS damage, they clearly were not sufficient to prevent it. Hence, passive transfer studies were carried out to increase Breg cell numbers before or just after MCAO induction.
Stereotaxic delivery of B cells, including IL‐10‐producing Breg cells, to the affected hemisphere significantly decreased the infarct volume.60 To more efficiently track the Breg cells, IL‐10‐producing cells that expressed green fluorescent protein (GFP) were used as donor cells. Hence, B cells were isolated from splenocytes obtained from IL‐10 GFP reporter mice and were primed with lipopolysaccharide for 48 hr to enhance IL‐10 production. Five million IL‐10‐producing B cells were transferred intravenously to μMT−/− mice and 24 hr later recipients were subjected to 60 min of MCAO via intraluminal filament (Fig. 3) followed by 48 hr of reperfusion. Compared with vehicle‐treated controls, the IL‐10+ B‐cell‐replenished μMT−/− mice had reduced infarct volumes (Fig. 2b) and fewer infiltrating T cells and activated monocytes (CD11b+ CD45hi) in the affected brain hemisphere. Hence, the improvement in stroke outcome could be attributed to significant reduction in infiltrating immune cells into the brain and/or the reduction in pro‐inflammatory mediators. Breg‐mediated reduction in infarct volumes reduced ischaemia‐related splenic atrophy, resulting in increased spleen cell numbers (Fig. 2c) accompanied by lower levels of pro‐inflammatory mediators and reduced activation status (CD69 and CD44 expression by CD4+ T cells) of the surviving splenic T cells and monocytes. This observation implicated a novel connection between the ischaemic lesion in brain and evolving inflammatory changes in distant peripheral immune cell populations. Not only could the IL‐10+ B cells attenuate pro‐inflammatory responses, but they also significantly enhanced other Treg cell subsets (ie. CD4+ Foxp3+ and CD8+ CD122+ Treg cells) in the periphery. These novel observations were the first to implicate the IL‐10‐producing capacity of B cells as a major regulatory mediator in stroke.36
Figure 3.
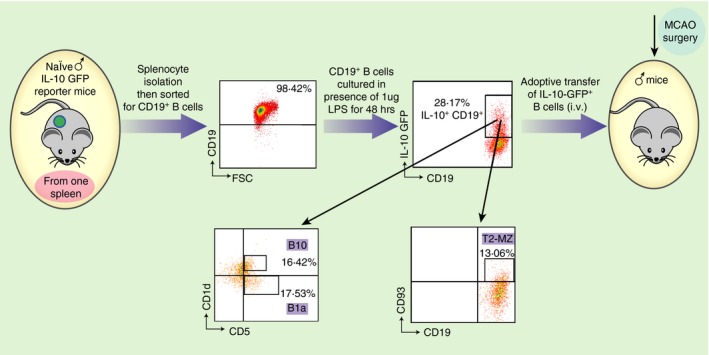
Experimental design. Schematic representation of enrichment of IL‐10+ GFP + CD19+ B cells from spleens of IL‐10‐GFP reporter mice, cultured in the presence of 1 μg/ml lipopolysaccharide (LPS) for 48 hr and adoptive transfer of 5 million IL‐10‐GFP + B cells into mice via intravenous injections. Purified B cells obtained from IL‐10‐GFP reporter mice were further characterized for regulatory B‐cell sub‐populations (B10, B1a and T2‐MZ) after 48 hr of culture in the presence of LPS by flow cytometry. Republished with permission by Bodhankar et al.,36 p. 379, © Springer Science+Business Media New York 2013. With the permission of Springer.
Interleukin‐10‐producing Breg cells potentially could impart significant benefit for stroke patients in the clinic. Hence, therapeutic strategies were explored for the treatment of MCAO in mice. Interleukin‐10‐producing Breg cells were transferred into B‐cell‐sufficient WT C57BL/6J mice. Five million IL‐10‐producing B cells, obtained from IL‐10 GFP reporter mice were transferred intravenously to WT mice 24 hr before 60 min of MCAO followed by 96 hr of reperfusion. The IL‐10+ B cells that were transferred into the mice were composed of primarily B10 cells, B‐1a cells and T2‐MZP cells (16·42%, 17·53% and 13·06%, respectively) (Fig. 3). Compared with vehicle‐treated controls, the IL‐10+ B‐cell‐pretreated WT mice had reduced infarct volumes (Fig. 4a). Reduction in infarct volumes also promoted a reduction in ischaemia‐related splenic atrophy (Fig. 5a) accompanied by lower pro‐inflammatory (tumour necrosis factor and IL‐17 production) milieu and lower activation status (CD69 expression by CD4+ T cells and CD80 expression by CD11b+ monocytes). Further evaluations demonstrated that the IL‐10+ B‐cell‐treated mice had decreased infiltration of T cells and activated microglia/monocytes (CD11b+ CD45hi) and a significant reduction in inflammatory milieu in the ischaemic hemispheres versus those in vehicle‐treated mice. Not only could the Breg cells attenuate pro‐inflammatory responses and increase anti‐inflammatory status, but they also significantly up‐regulated regulatory subsets (CD4+ Foxp3+ and CD8+ CD122+ Treg cells and CD19+ CD1dhi Breg cells) in the spleen, indicating their role in immunomodulatory mechanisms, post‐stroke (Fig. 6).
Figure 4.
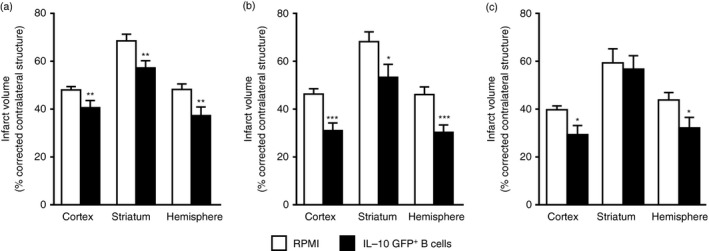
Infarct volumes after regulatory B (Breg) cell treatment of experimental stroke. (a) Intravenous transfer of 5 million IL‐10+ B cells given 24 hr before surgery to induce middle cerebral artery occlusion (MCAO) reduced infarct volume in wild‐type (WT) mice (n = 10), 96 hr following 60 min of MCAO compared with intravenous transfer of RPMI‐1640 vehicle (no cells) (n = 12). **P < 0·01. (b) Intravenous transfer of 5 million IL‐10+ B cells 4 hr after surgery to induce MCAO reduced cortical and hemispheric (total) infarct volume in WT mice (n = 13), 96 hr following 60 min of MCAO compared with intravenous transfer of RPMI‐1640 vehicle (no cells) (n = 14). *P < 0·05; ***P < 0·001. (c) Intravenous transfer of 5 million IL‐10+ B cells given 24 hr after induction of MCAO reduced infarct volume in C57BL/6J (WT) mice 96 hr after 1 hr of MCAO compared with intravenous transfer of RPMI Vehicle (no cells). *P < 0·05. (a,b) Republished with permission by Bodhankar et al.,61 p. 63, © Springer Science+Business Media New York 2013, With permission of Springer. (c) Bodhankar et al.,62 p. 915, © Springer Science+Business Media New York 2014. With permission of Springer.
Figure 5.
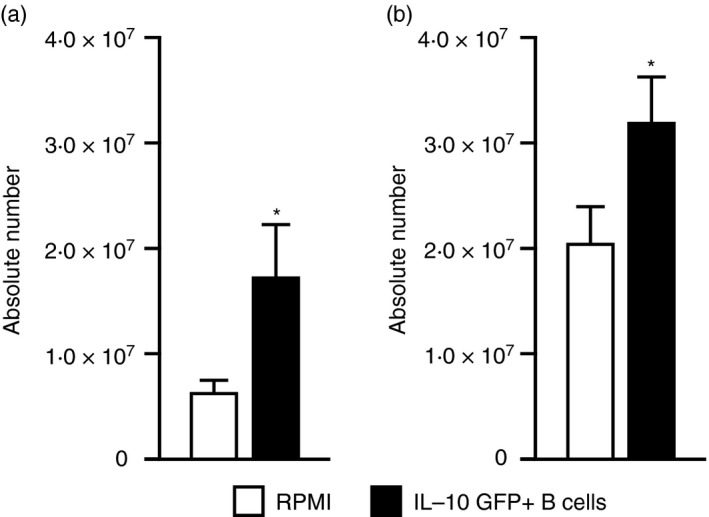
Splenocyte numbers after regulatory B (Breg) cell pre‐ and post‐treatment of experimental stroke. (a) Ninety‐six hours after middle cerebral artery occlusion (MCAO), mononuclear cells were isolated from spleens of RPMI‐1640 or IL‐10‐GFP + B‐cell recipient wild‐type (WT) mice treated 24 hr before MCAO and analysed for total cell counts via a haemocytometer. Values represent mean numbers (± SEM) of indicated cell subsets from 16 or 17 mice in each group, from at least five separate experiments. *P < 0·05 (b) Ninety‐six hours after MCAO, mononuclear cells were isolated from spleens of RPMI‐1640 or IL‐10‐GFP + B‐cell recipient WT mice treated 24 hr after MCAO and analysed for total cell counts via haemocytometer. Values represent mean numbers (± SEM) of indicated cell subsets with from 28 to 30 mice in each group, from at least eight separate experiments. *P < 0·05, **P < 0·01 and ***P < 0·001 (a) Republished with permission by Bodhankar et al.,61 p. 63, © Springer Science+Business Media New York 2013, with the permission of Springer. (b) Bodhankar et al.,62 p. 916, © Springer Science+Business Media New York 2014. With the permission of Springer.
Figure 6.
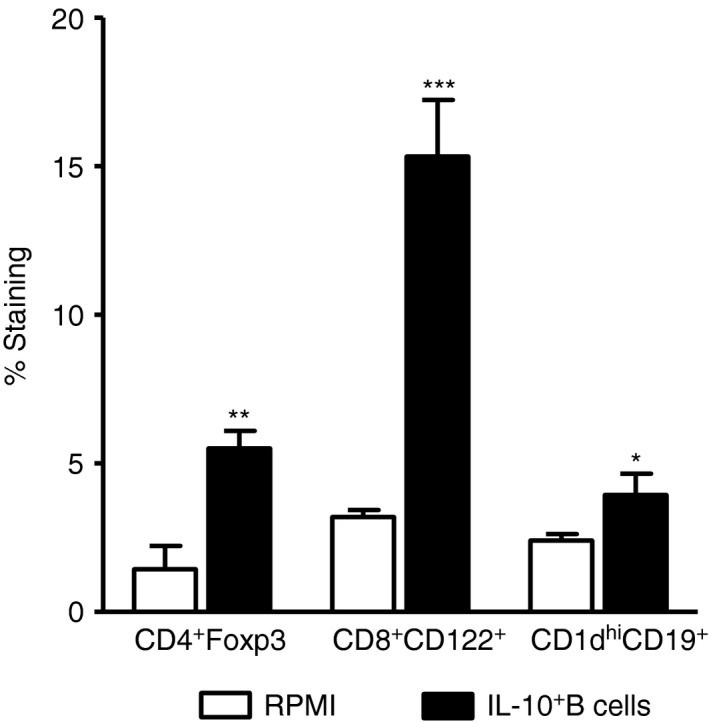
Changes in regulatory cells in the spleen after regulatory B (Breg) cell pretreatment of middle cerebral artery occlusion (MCAO). Splenocytes from RPMI‐1640 and IL‐10‐GFP + B‐cell transferred wild‐type (WT) recipient mice were harvested 96 hr after MCAO and assessed for expression of FoxP3+ CD4+ T cells, CD8+ CD122+ T cells and CD1dhi CD19+ Breg cells. Data are representative of two independent experiments with spleens processed from six RPMI‐pretreated and seven IL‐10+ B‐cell‐pretreated mice (mean ± SEM). Significant differences between the groups were determined using Student's t‐test (*P ≤ 0·05, **P ≤ 0·01 and ***P ≤ 0·001). Republished with permission by Bodhankar et al.,61 p. 67, © Springer Science+Business Media New York 2013, With the permission of Springer.
Further studies successfully demonstrated a therapeutic role of IL‐10+ B cells in stroke. IL‐10+ B cells transferred to WT mice 4 hr after MCAO significantly reduced infarct volume compared with vehicle‐treated recipient mice (Fig. 4b). Remarkably, transfer of IL‐10+ Breg cells 4 hr after MCAO induction in WT recipient mice reduced infarct volumes more than transfers given 24 hr before MCAO. These results clearly indicate the potent immunoregulatory role of the IL‐10+ B cells as they potentially regulate the native B‐cell population to generate a more favourable milieu to alleviate stroke outcome.61
A subsequent study established an effective therapeutic time‐point as late as 24 hr after MCAO in B‐cell‐sufficient male WT mice. IL‐10+ Breg‐cell‐treated mice had significantly reduced infarct volumes (Fig. 4c) and improved neurological deficits versus vehicle‐treated control mice after 60 min occlusion and 96 hr of reperfusion. The MCAO‐protected Breg‐cell‐recipient mice had less splenic atrophy (Fig. 5b) and reduced numbers of activated, inflammatory T cells, decreased infiltration of T cells and a less inflammatory milieu in the ischaemic hemispheres compared with vehicle‐treated control mice.62 These immunoregulatory changes occurred in concert with the predominant appearance of IL‐10 secreting CD8+ CD122+ Treg cells in both the spleen (Fig. 7) and the MCAO‐affected brain hemisphere (Fig. 8).62 This study demonstrates a major neuroprotective role for IL‐10+ B cells in treating MCAO in male WT mice at a time‐point as late as 24 hr post‐stroke leading to the generation of an additional highly potent regulatory subset (IL‐10+ CD8+ CD122+ Treg cells), which in turn appears to play a dominant role in attenuating pro‐inflammatory reactions generated upon reperfusion‐based cerebral injury.
Figure 7.
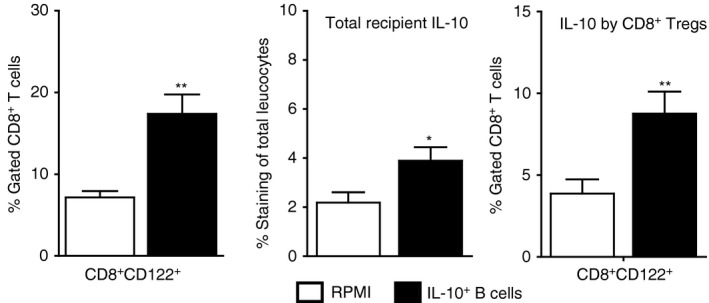
CD8+ CD122+ cell changes in the spleen after 24‐hr regulatory B (Breg) cell treatment of middle cerebral artery occlusion (MCAO). Splenocytes were isolated from RPMI‐1640 or IL‐10‐GFP + B‐cell recipient wild‐type (WT) mice 96 hr after MCAO and assessed for frequency of CD8+ CD122+ T cells (Left panel). Data are representative of five independent experiments (mean ± SEM). Total interleukin‐10 (IL‐10) producing GFP − cells were identified using IL‐10 APC monoclonal antibody for detection by flow cytometry (Center panel) and for the expression of IL‐10 (GFP −) on gated CD8+ CD122+ T cells (Right panel). Data are representative of five independent experiments with spleens processed from nine or ten individual mice (mean ± SEM). *P < 0·05; **P < 0·01. Republished with permission by Bodhankar et al.,62 p. 918, © Springer Science+Business Media New York 2014. With the permission of Springer.
Figure 8.

CD8+ CD122+ cell changes in the brain after 24 hr regulatory B (Breg) cell treatment of middle cerebral artery occlusion (MCAO). Brain leucocytes from ischaemic hemispheres of RPMI‐1640 and IL‐10‐GFP + B‐cell‐transferred wild‐type (WT) recipient mice were harvested 96 hr after MCAO and assessed for frequency of CD8+ CD122+ T cells. (Left panel) Data are representative of five independent experiments (mean ± SEM). Total interleukin 10 (IL‐10) producing GFP − cells were identified using IL‐10 APC monoclonal antibody for detection by flow cytometry (Center panel). Expression of IL‐10 (GFP −) was assessed on gated CD8+ CD122+ T cells (Right panel). Data are representative of five independent experiments with spleens processed from nine or ten individual mice (mean ± SEM). Significant differences between the groups were determined using Student's t‐test and are indicated (*P ≤ 0·05 and **P ≤ 0·01). Republished with permission by Bodhankar et al.,62 p. 918, © Springer Science+Business Media New York 2014. With the permission of Springer.
Conclusion
Regulatory B cells have been demonstrated to play a protective role in treating stroke, due largely to their ability to secrete IL‐10, which modulates the post‐stroke immune response and to up‐regulate and recruit IL‐10‐secreting Treg cells into the injured brain. Local delivery of IL‐10 and recruitment of Treg cells within the infarcted brain regions can then reduce pro‐inflammatory cells and cytokines that mediate neural injury. Additionally, Breg cells have the ability to extend the current therapeutic window for treating stroke from 4·5 hr to 24 hr. Treatment with Breg cells consistently reduces infarct volumes and improves neurological function compared with WT mice after experimental stroke, so demonstrating unique therapeutic potential. Continued research into the role of Breg cells in limiting post stroke immune‐mediated neural injury will be required to ensure that Breg cells do not further suppress protective immune responses known to be impaired after stroke. Moreover, depending upon the source and proliferative potential of transferred Breg cells to stroke patients, it may be useful to introduce ‘safety switches’ to allow selective elimination of the cells63 as well as methods to prevent the possibility of cytokine release syndrome.64 Finally, given the potential protective versus infective functions of the microbiome on stroke outcomes,65, 66 modulating the effects of microbial products on Breg cell functions may need to be considered to achieve successful long‐term stroke therapy.
Disclosures
There are no conflicts of interest.
Acknowledgements
The authors thank Gail Kent for assistance with manuscript preparation and submission. This work was supported by American Heart Association grant 17GRNT33220001 and NIH/NINDS R01NS076013, R01NS080890 and R01NS075887 (Halina Offner). Dr. Vandenbark is the recipient of a Senior Research Career Scientist award (#1IK6BX004209) from the Department of Veterans Affairs. This material is the result of work supported with resources and the use of facilities at the VA Portland Health Care Center in Portland, Oregon. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
References
- 1. Writing Group M , Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ et al Heart disease and stroke statistics‐2016 update: a report from the American Heart Association. Circulation 2016; 133:e38–360. [DOI] [PubMed] [Google Scholar]
- 2. Zivin JA. Acute stroke therapy with tissue plasminogen activator (tPA) since it was approved by the U.S. Food and Drug Administration (FDA). Ann Neurol 2009; 66:6–10. [DOI] [PubMed] [Google Scholar]
- 3. Badhiwala JH, Nassiri F, Alhazzani W, Selim MH, Farrokhyar F, Spears J et al Endovascular thrombectomy for acute ischemic stroke: a meta‐analysis. JAMA 2015; 314:1832–43. [DOI] [PubMed] [Google Scholar]
- 4. Mistry EA, Mistry AM, Nakawah MO, Chitale RV, James RF, Volpi JJ et al Mechanical thrombectomy outcomes with and without intravenous thrombolysis in stroke patients: a meta‐analysis. Stroke 2017; 48:2450–6. [DOI] [PubMed] [Google Scholar]
- 5. Ajmo CT Jr, Vernon DO, Collier L, Hall AA, Garbuzova‐Davis S, Willing A et al The spleen contributes to stroke‐induced neurodegeneration. J Neurosci Res 2008; 86:2227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jin R, Zhu X, Liu L, Nanda A, Granger DN, Li G. Simvastatin attenuates stroke‐induced splenic atrophy and lung susceptibility to spontaneous bacterial infection in mice. Stroke 2013; 44:1135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seifert HA, Leonardo CC, Hall AA, Rowe DD, Collier LA, Benkovic SA et al The spleen contributes to stroke induced neurodegeneration through interferon‐γ signaling. Metab Brain Dis 2012; 27:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dotson AL, Wang J, Saugstad J, Murphy SJ, Offner H. Splenectomy reduces infarct volume and neuroinflammation in male but not female mice in experimental stroke. J Neuroimmunol 2015; 278:289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee ST, Chu K, Jung KH, Kim SJ, Kim DH, Kang KM et al Anti‐inflammatory mechanism of intravascular neural stem cell transplantation in haemorrhagic stroke. Brain 2008; 131:616–29. [DOI] [PubMed] [Google Scholar]
- 10. Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A et al Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol 2006; 176:6523–31. [DOI] [PubMed] [Google Scholar]
- 11. Seifert HA, Hall AA, Chapman CB, Collier LA, Willing AE, Pennypacker KR. A transient decrease in spleen size following stroke corresponds to splenocyte release into systemic circulation. J Neuroimmune Pharmacol 2012; 7:1017–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sahota P, Vahidy F, Nguyen C, Bui TT, Yang B, Parsha K et al Changes in spleen size in patients with acute ischemic stroke: a pilot observational study. Int J Stroke 2013; 8:60–7. [DOI] [PubMed] [Google Scholar]
- 13. Vahidy FS, Parsha KN, Rahbar MH, Lee M, Bui TT, Nguyen C et al Acute splenic responses in patients with ischemic stroke and intracerebral hemorrhage. J Cereb Blood Flow Metab 2016; 36:1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chiu NL, Kaiser B, Nguyen YV, Welbourne S, Lall C, Cramer SC. The volume of the spleen and its correlates after acute stroke. J Stroke Cerebrovasc Dis 2016; 25:2958–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon‐γ in ischemic stroke. Circulation 2006; 113:2105–12. [DOI] [PubMed] [Google Scholar]
- 16. Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA et al T‐ and B‐cell‐deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab 2007; 27:1798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ren X, Akiyoshi K, Dziennis S, Vandenbark AA, Herson PS, Hurn PD et al Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci 2011; 31:8556–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H et al Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol 2007; 178:6092–9. [DOI] [PubMed] [Google Scholar]
- 19. Duddy ME, Alter A, Bar‐Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol 2004; 172:3422–7. [DOI] [PubMed] [Google Scholar]
- 20. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity 2015; 42:607–12. [DOI] [PubMed] [Google Scholar]
- 21. Egwuagu CE, Yu CR. Interleukin 35‐producing B cells (i35‐Breg): a new mediator of regulatory B‐cell functions in CNS autoimmune diseases. Crit Rev Immunol 2015; 35:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tedder TF. Introduction: regulatory B‐cell special issue‐making all the pieces fit. Int Immunol 2015; 27:467–70. [DOI] [PubMed] [Google Scholar]
- 23. Yang M, Rui K, Wang S, Lu L. Regulatory B cells in autoimmune diseases. Cell Mol Immunol 2013; 10:122–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Evans JG, Chavez‐Rueda KA, Eddaoudi A, Meyer‐Bahlburg A, Rawlings DJ, Ehrenstein MR et al Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol 2007; 178:7868–78. [DOI] [PubMed] [Google Scholar]
- 25. Haas KM, Watanabe R, Matsushita T, Nakashima H, Ishiura N, Okochi H et al Protective and pathogenic roles for B cells during systemic autoimmunity in NZB/W F1 mice. J Immunol 2010; 184:4789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late‐phase immunopathogenesis. J Immunol 2010; 185:2240–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hussain S, Delovitch TL. Intravenous transfusion of BCR‐activated B cells protects NOD mice from type 1 diabetes in an IL‐10‐dependent manner. J Immunol 2007; 179:7225–32. [DOI] [PubMed] [Google Scholar]
- 28. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL‐10. Nat Immunol 2002; 3:944–50. [DOI] [PubMed] [Google Scholar]
- 29. Ray A, Mann MK, Basu S, Dittel BN. A case for regulatory B cells in controlling the severity of autoimmune‐mediated inflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neuroimmunol 2011; 230:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bebo BF Jr, Fyfe‐Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low‐dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol 2001; 166:2080–9. [DOI] [PubMed] [Google Scholar]
- 31. Ito A, Bebo BF Jr, Matejuk A, Zamora A, Silverman M, Fyfe‐Johnson A et al Estrogen treatment down‐regulates TNF‐α production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine knockout mice. J Immunol 2001; 167:542–52. [DOI] [PubMed] [Google Scholar]
- 32. Seifert HA, Benedek G, Nguyen H, Kent G, Vandenbark AA, Offner H. Estrogen protects both sexes against EAE by promoting common regulatory cell subtypes independent of endogenous estrogen. Metab Brain Dis 2017; 32:1747–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bodhankar S, Wang C, Vandenbark AA, Offner H. Estrogen‐induced protection against experimental autoimmune encephalomyelitis is abrogated in the absence of B cells. Eur J Immunol 2011; 41:1165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang J, Lapato A, Bodhankar S, Vandenbark AA, Offner H. Treatment with IL‐10 producing B cells in combination with E2 ameliorates EAE severity and decreases CNS inflammation in B cell‐deficient mice. Metab Brain Dis 2015; 30:1117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Benedek G, Zhang J, Bodhankar S, Nguyen H, Kent G, Jordan K et al Estrogen induces multiple regulatory B cell subtypes and promotes M2 microglia and neuroprotection during experimental autoimmune encephalomyelitis. J Neuroimmunol 2016; 293:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL‐10‐producing B‐cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis 2013; 28:375–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maloy KJ, Powrie F. Regulatory T cells in the control of immune pathology. Nat Immunol 2001; 2:816–22. [DOI] [PubMed] [Google Scholar]
- 38. Mason D, Powrie F. Control of immune pathology by regulatory T cells. Curr Opin Immunol 1998; 10:649–55. [DOI] [PubMed] [Google Scholar]
- 39. Roncarolo MG, Levings MK. The role of different subsets of T regulatory cells in controlling autoimmunity. Curr Opin Immunol 2000; 12:676–83. [DOI] [PubMed] [Google Scholar]
- 40. Shevach EM. Regulatory T cells in autoimmmunity*. Annu Rev Immunol 2000; 18:423–49. [DOI] [PubMed] [Google Scholar]
- 41. Liesz A, Suri‐Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S et al Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 2009; 15:192–9. [DOI] [PubMed] [Google Scholar]
- 42. Liesz A, Hu X, Kleinschnitz C, Offner H. Functional role of regulatory lymphocytes in stroke: facts and controversies. Stroke 2015; 46:1422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ren X, Akiyoshi K, Vandenbark AA, Hurn PD, Offner H. CD4+FoxP3+ regulatory T‐cells in cerebral ischemic stroke. Metab Brain Dis 2010; 26:87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Urra X, Cervera A, Villamor N, Planas AM, Chamorro A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience 2009; 158:1174–83. [DOI] [PubMed] [Google Scholar]
- 45. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood 2008; 112:1570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lund FE. Cytokine‐producing B lymphocytes‐key regulators of immunity. Curr Opin Immunol 2008; 20:332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol 2010; 10:236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell‐dependent inflammatory responses. Immunity 2008; 28:639–50. [DOI] [PubMed] [Google Scholar]
- 49. Rafei M, Hsieh J, Zehntner S, Li M, Forner K, Birman E et al A granulocyte–macrophage colony‐stimulating factor and interleukin‐15 fusokine induces a regulatory B cell population with immune suppressive properties. Nat Med 2009; 15:1038–45. [DOI] [PubMed] [Google Scholar]
- 50. Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K et al Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol 2010; 184:4801–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Planas AM, Gorina R, Chamorro A. Signalling pathways mediating inflammatory responses in brain ischaemia. Biochem Soc Trans 2006; 34:1267–70. [DOI] [PubMed] [Google Scholar]
- 52. Grilli M, Barbieri I, Basudev H, Brusa R, Casati C, Lozza G et al Interleukin‐10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur J Neurosci 2000; 12:2265–72. [DOI] [PubMed] [Google Scholar]
- 53. Spera P, Elison J, Feuerstein G, Barone F. IL‐10 reduces rat brain injury following focal stroke. Neurosci Lett 1998; 251:189–92. [DOI] [PubMed] [Google Scholar]
- 54. Dietrich WD, Busto R, Bethea JR. Postischemic hypothermia and IL‐10 treatment provide long‐lasting neuroprotection of CA1 hippocampus following transient global ischemia in rats. Exp Neurol 1999; 158:444–50. [DOI] [PubMed] [Google Scholar]
- 55. Ooboshi H, Ibayashi S, Shichita T, Kumai Y, Takada J, Ago T et al Postischemic gene transfer of interleukin‐10 protects against both focal and global brain ischemia. Circulation 2005; 111:913–9. [DOI] [PubMed] [Google Scholar]
- 56. Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL. Neuroprotection by IL‐10‐producing MOG CD4+ T cells following ischemic stroke. J Neurol Sci 2005; 233:125–32. [DOI] [PubMed] [Google Scholar]
- 57. de Bilbao F, Arsenijevic D, Moll T, Garcia‐Gabay I, Vallet P, Langhans W et al In vivo over‐expression of interleukin‐10 increases resistance to focal brain ischemia in mice. J Neurochem 2009; 110:12–22. [DOI] [PubMed] [Google Scholar]
- 58. Vila N, Castillo J, Davalos A, Esteve A, Planas AM, Chamorro A. Levels of anti‐inflammatory cytokines and neurological worsening in acute ischemic stroke. Stroke 2003; 34:671–5. [DOI] [PubMed] [Google Scholar]
- 59. Chamorro A, Amaro S, Vargas M, Obach V, Cervera A, Torres F et al Interleukin 10, monocytes and increased risk of early infection in ischaemic stroke. J Neurol Neurosurg Psychiatry 2006; 77:1279–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen Y, Bodhankar S, Murphy SJ, Vandenbark AA, Alkayed NJ, Offner H. Intrastriatal B‐cell administration limits infarct size after stroke in B‐cell deficient mice. Metab Brain Dis 2012; 27:487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. Treatment of experimental stroke with IL‐10‐producing B‐cells reduces infarct size and peripheral and CNS inflammation in wild‐type B‐cell‐sufficient mice. Metab Brain Dis 2014; 29:59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bodhankar S, Chen Y, Lapato A, Vandenbark AA, Murphy SJ, Saugstad JA et al Regulatory CD8+CD122+ T‐cells predominate in CNS after treatment of experimental stroke in male mice with IL‐10‐secreting B‐cells. Metab Brain Dis 2015; 30:911–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy‐Nasser A, Martinez C et al Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365:1673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M et al Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014; 124:188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M et al Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med 2016; 22:516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wen SW, Wong CHY. An unexplored brain–gut microbiota axis in stroke. Gut Microbes 2017; 8:601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]