

Summary
Neurodegenerative diseases, the leading cause of morbidity and disability, are gaining increased attention as they impose a considerable socioeconomic impact, due in part to the ageing community. Neuronal damage is a pathological hallmark of Alzheimer's and Parkinson's diseases, amyotrophic lateral sclerosis, Huntington's disease, spinocerebellar ataxia and multiple sclerosis, although such damage is also observed following neurotropic viral infections, stroke, genetic white matter diseases and paraneoplastic disorders. Despite the different aetiologies, for example, infections, genetic mutations, trauma and protein aggregations, neuronal damage is frequently associated with chronic activation of an innate immune response in the CNS. The growing awareness that the immune system is inextricably involved in shaping the brain during development as well as mediating damage, but also regeneration and repair, has stimulated therapeutic approaches to modulate the immune system in neurodegenerative diseases. Here, we review the current understanding of how astrocytes and microglia, as well as neurons and oligodendrocytes, shape the neuroimmune response during development, and how aberrant responses that arise due to genetic or environmental triggers may predispose the CNS to neurodegenerative diseases. We discuss the known interactions between the peripheral immune system and the brain, and review the current concepts on how immune cells enter and leave the CNS. A better understanding of neuroimmune interactions during development and disease will be key to further manipulating these responses and the development of effective therapies to improve quality of life, and reduce the impact of neuroinflammatory and degenerative diseases.
Keywords: immune response, inflammation, microbiome, neuroprotection, repair
Abbreviations
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- APP
amyloid precursor protein
- AQP4
aquaporin 4
- Aβ
amyloid‐beta
- BBB
blood–brain barrier
- BSCB
blood–spinal cord barrier
- CNS
central nervous system
- COX‐2
cyclooxygenase‐2
- CP
choroid plexus
- CVD
cerebrovascular diseases
- EV
extracellular vesicles
- EVD
Ebola virus disease
- FTD
frontotemporal dementia
- GDNF
glial cell‐derived neurotrophic factor
- HD
Huntington's disease
- HIV
human immunodeficiency virus
- HSPs
heat‐shock proteins
- IFN‐γ
interferon gamma
- MHC
major histocompatibility complex
- MRI
magnetic resonance imaging
- MS
multiple sclerosis
- NF
neurofilament light
- NK
natural killer
- PD
Parkinson's disease
- PET
positron emission tomography
- PNND
paraneoplastic neurological disorders
- PrP
prion protein
- ROS
reactive oxygen species
- SMA
spinal muscular atrophy
- SPECT
single‐photon emission computed tomography
- TBI
traumatic brain injury
- TDP‐43
TAR DNA‐binding protein 43 kD
- TLRT
oll‐like receptor
- TNF‐α
tumour necrosis factor alpha
- TREM2
triggering receptor expressed on myeloid cells 2
- ZIKV
Zika virus
- ZO‐1
zonula occludens‐1
Introduction
The prevalence of neurodegenerative diseases highly depends on the country surveyed, yet the most prevalent disease globally is dementia, with an estimated incidence of 9·33% worldwide1 (Table 1). The increase in the incidence of dementia, as with many neurodegenerative diseases, is in part due to the ageing population,2 as an ageing brain, or one following peripheral infections or other insults is ‘primed’ to render the central nervous system (CNS) more susceptible to damage.3 Priming of innate immune responses in the CNS may thus explain the higher prevalence of epilepsy in developing countries4 where CNS infections5 with neurotropic viruses are more frequent. The neurotropic virus Zika is a good example of how such viral infections not only contribute to neurodegenerative diseases in the elderly, but also have a major impact during development.
Table 1.
Aetiology, immune involvement and incidence of neuroinflammatory diseases
| Disease | (Proposed) Aetiology | Innate immune response involvement | Adaptive immune response involvement | Incidence % or number/100 000 | Predicted change in prevalence | References |
|---|---|---|---|---|---|---|
| MS | Autoimmune viral | Microglial and macrophage activation, ↑ROS, complement, ↑innate receptors, ↑cytokines, ↑chemokines |
↑HSPs, ↑neurotrophins, antibodies and T‐cells to CNS antigens. Effective therapies target B‐cells |
9·64 | ↑2·4% per year | 6, 7 |
| AD, other dementias | AD – misfolded and aggregated tau and APP | Activated and dystrophic microglia, ↑TNF‐α, ↑IFN‐γ, ↑chemokines, ↑complement, ↑TLRs | ↑antibody and T‐cell response | 9·33% | ↑3·3% per year (triple by 2050) | 1, 2, 6 |
| PD | Selective loss of dopaminergic neurons in substantia nigra due to α‐syn‐ intraneuronal inclusions | ↑TLRs, ↑CD14, activated NK cells, microglial activation, ↑IL‐1β, ↑IL‐6, ↑TNF‐α | ↑T‐cells, ↑antibody response | 100–200 | Double in 25 years | 6, 8, 9 |
| HD | Expansion of CAG (Q) in huntingtin gene induces aberrant toxic protein | ↑microglial proliferation, ↑complement, | Not reported | 0·02–9·71 | ↑15–20% per decade | 6, 10 |
| SMA | Genetic defect in the SMN1 gene | ↑IL‐6, ↑IL‐1β | Not reported | 1–2 | Not reported | 6, 11 |
| ALS (MND) | Aberrant aggregated proteins due to mutations SOD1, TDP; C9orf72 or FUS genes | ↑complement, ↑CD14, ↑macrophages, ↑IL‐6, ↑TNF‐α | ↑CD4+, ↑CD8+ T‐cells | 1·9 | ↑69% in 25 years | 6, 12, 13 |
| Prion diseases | Infectious forms of misfolded aggregated forms of prion protein | ↑microglial activation, ↑IL‐1β, ↑IL‐6, ↑complement, ↑ROS, mast cells expressing PrP | B‐cells aid transport of PrP | Variable | Not reported | 6 |
| Stroke | Ischaemia (thrombosis, embolism, or systemic hypoperfusion) or haemorrhage (intra‐cerebral or subarachnoid) | ↑lymphopenia, ↑NK cells, ↑IL‐10 | ↑Th2 responses | 115 | ↑44% in 20 years | 14, 15, 26 |
| TBI | Open and head injury, deceleration injuries, chemical/toxic, hypoxia, tumours, | ↑pro‐inflammatory cytokines, ↑TLRs | ↑T‐cells, ↑B‐cells | 295 | Not reported | 6, 16 |
| HIV/AIDS | HIV encephalopathy, toxoplasmosis, PML | ↓IL‐27, ↓IFN‐γ, ↓CD4 cells, ↑IL‐4 | ↓T‐cells, immunosenescence | 0·8% | Depending on country | 17, 27 |
| Meningitis | Bacterial and viral infections | ↑IL‐6, ↑TNF‐α, ↑NK cells, ↑microglial activation | ↑T‐cells, ↑B‐cells | 0·2–1000 | Outbreak dependent | 18, 29 |
| Ageing | Natural event | ↑pro‐inflammatory cytokines, ↓NK cell function | ↓T‐cells | n/a | n/a | 28 |
| Epilepsy | Unprovoked seizures, febrile events, autoantibodies, | ↑pro‐inflammatory cytokines, ↑chemokines, ↑TLRs, ↑complement | ↑autoantibodies, T‐ and B‐cell activation | 45–81·7 | ↑ | 4, 5, 6 |
| Autism | Genetic and environmental | ↑pro‐inflammatory cytokines | ↓T‐cells | 425–760 | Variable | 19, 20, 30 |
| Depression | Multifactorial e.g. genetics, hormonal | Microglial activation, ↑ cytokines, ↑chemokines | ↑ T‐reg cells | 3% | ↑ | 21, 22, 31 |
| Schizophrenia | Multifactorial | Microglial activation, ↑ROS, ↑pro‐inflammatory cytokines, ↑chemokines, ↑TLRs, ↓NK cells | Not reported | 18·5 | Not reported | 23, 32, 33 |
| Bipolar disorder | Genetic and environmental risk factors | Microglial activation, ↑pro‐inflammatory cytokines, ↑complement, ↑TNF‐α | ↑T‐cell activation | 2·4% lifetime prevalence | Debated | 24, 25, 34 |
AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; APP, amyloid‐β precursor protein; FUS, fused in sarcoma; HD, Huntington's disease; HIV/AIDS, human immunodeficiency virus/acquired immunodeficiency syndrome; MS, multiple sclerosis; PD, Parkinson's disease; PML, progressive multifocal leucoencephalopathies; SMA, spinal muscular atrophy; SOD1, superoxide dismutase 1; SMN, survival of motor neuron protein; TBI, traumatic brain injury, TDP‐43, TAR DNA‐binding protein 43 kDa.
Despite different aetiologies (Table 1), a common feature of neurodegenerative diseases is chronic activation of innate immune cells within the CNS, and in other diseases such as multiple sclerosis (MS), the influx of peripheral immune cells across the blood–brain barrier (BBB). Old notions on how cells traffic into the CNS, despite an apparently immune‐privileged environment, have been challenged recently by the identification of lymphatic drainage, as well as by detailed studies on immune cell trafficking in the choroid plexus (CP).
Here, we review the recent advances in our understanding of how immune responses in the CNS contribute to susceptibility to neurodegenerative diseases, how immune responses change with ageing, and how therapies can be designed to augment reparative processes in the CNS.
Immune privilege and CNS barriers
The concept of immune privilege originated from Sir Peter Medawar's studies in the mid‐20th century showing that tissue grafts in the CNS were not rejected. It also takes into account the presence of the BBB, revealed by Paul Ehrlich's studies in the late 1800s showing that solutes and molecules were excluded from the brain. However, it is now clear that entry of compounds into the CNS occurs via capillary venules, while cell migration occurs at the post‐capillary venules and is controlled by adhesion molecules, cytokines and chemokines.35 Anatomically, the CNS is separated by three barriers: the BBB/blood–spinal cord barrier (BSCB); the blood–cerebrospinal fluid barrier at the CP (Fig. 1); and the arachnoid barrier. Differences in the structure of the BBB and BSCB, as well as differences in the cranial and spinal meninges, in white and grey matter, and other regional differences may explain the differential susceptibility of anatomical regions to neuroinflammmatory events. For example, the BSCB has reduced levels of zonula occludens‐1 (ZO‐1), occludin, VE cadherin and P‐gp, and fewer pericytes than the BBB,36 indicating that the spinal cord may well be more susceptible to inflammatory insults than the brain. The presence of barriers originally explained why CNS antigens in the brain were ignored by the peripheral immune response. However, this dogma has been challenged recently by the identification of the glymphatic system37 and rediscovery of lymphatic vessels in the dura mater38, 39 that are crucial to clear waste products such as amyloid‐beta (Aβ) peptides and tissue debris that accumulate during disease. Dysfunction of these barriers is well known to occur in neuroinflammatory disorders, including MS, Parkinson's disease (PD), Alzheimer's disease (AD), stroke, epilepsy and traumatic brain injury (TBI),40 and is associated with activated endothelial cells that display an altered phenotype and a decrease in tight junction proteins. These changes that are also observed during ageing41 may explain the increase in susceptibility to neuroinflammation and neurodegenerative disorders in the elderly. As well as playing a protective role in neuromyelitis optica (NMO), the BBB is also a target of immune responses where pathogenic autoantibodies to aquaporin 4 (AQP4) damage astrocytes that otherwise maintain BBB.
Figure 1.
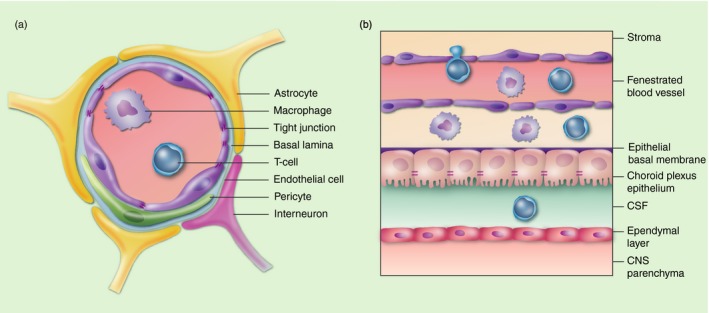
Blood–central nervous system (CNS) barriers. The blood–brain barrier (BBB) (a) and blood–spinal cord barrier (BSCB) (that resembles the BBB, see text for details) limit potential immune cells (shown in the lumen of the blood vessel), antibodies and soluble factors entering the CNS in health. Likewise, while the choroid plexus (CP) also limits cell migration, evidence suggests that regulatory T‐cells enter the brain via the CP (b) during health in order to ensure surveillance of the CNS (see text for details). CSF, cerebrospinal fluid.
Innate immunity
Innate immunity is the first line of defense in infection, but also plays a key role in tissue repair, clearance of apoptotic cells and cellular debris, as well as in response to tumours. While the key innate immune cells in the CNS are microglia and astrocytes, macrophages, natural killer (NK) cells and mast cells as well as oligodendrocytes and neurons all contribute to innate immune responses in the CNS. Pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs) include misfolded and aggregated proteins as in, for example, AD, amyotrophic lateral sclerosis (ALS), Huntington's disease (HD) and PD.42 Cellular receptors that recognize PAMPS and DAMPs, such as endogenous molecules, for example, heat‐shock proteins (HSPs), viral and bacterial antigens, and oxidized lipids, include the Toll‐like receptors (TLR), C‐type lectins and oxidized lipoprotein detectors and nuclear oligomerization domain‐like receptors (NLRs) that play a key role in the inflammasome. That innate receptors, for example, triggering receptor expressed on myeloid cells 2 (TREM2), are key to aiding clearance of dying cells, myelin debris and aggregated proteins may explain the association of a rare variant in TREM2 with AD, ALS and PD.42
Microglia, the principal resident innate immune cells of the CNS, have diverse functions. During development they shape neural circuits by pruning synapses as well as regulating cell death and elimination of waste products during inflammation or CNS damage. Differential activation of microglia is often classified as being either classical (M1) or alternative (M2), based on chemokine and cytokine expression in vivo.43 Switching between these polarizations is vital for remyelination and is affected by ageing, as shown by the use of parabiosis in mice.44 Microglia secrete pro‐inflammatory as well as anti‐inflammatory factors, which can either be beneficial or detrimental in neurodegenerative diseases,45 for example microglia depletion in a mouse model of AD reduced neuronal loss without affecting Aβ pathology.46 Microglia‐derived factors such as brain‐derived neurotrophic factor are important for learning and memory processes, processes that can be affected by maternal inflammation leading to disrupted behaviour and learning in later life.47 Microglia appear to have different transcriptomic profiles dependent on the region of the brain, ageing,48 neuropathological state45 and the microbiome.49
Monocytes are the blood‐borne precursors to macrophages and dendritic cells, and play a key role in innate immunity. While microglia, in contrast to other tissue‐resident macrophages, arise from yolk sac primitive macrophages, their distinct roles in CNS disorders are frequently hard to distinguish. The development of the CCR2‐red fluorescent protein knock‐in mouse has allowed researchers to better differentiate the infiltrating monocytes/macrophages and resident microglia in experimental diseases.50 In addition, the novel markers TMEM119 and P2Y12 have also helped differentiate microglia and macrophages,51 allowing the relative contribution of these cells in neuroinflammatory diseases to be examined.
Similar to the M1/M2 polarization of macrophages and microglia, subpopulations of astrocytes have been reported that produce pro‐inflammmatory mediators (A1) and immunoregulatory mediators (A2). The A1 astrocytes that secrete Il‐1a, tumour necrosis factor alpha (TNFα) and C1q are considered to be neuroinflammatory, and damage neurons and oligodendrocytes in vitro as well as inducing apoptosis, suppressing T helper cell activation, proliferation and function of activated T‐cells. In contrast, A2 astrocytes are neuroprotective, promoting neuronal growth, survival and synaptic repair.52 Astrocytes respond to a plethora of insults and are frequently observed as hypertrophic in many neurodegenerative diseases, including stroke, TBI, MS, ALS and viral infections and other inflammatory conditions.52 A1 reactive astrocytes have been suggested as having toxic effects in ALS, AD, MS, PD, HD, schizophrenia and ageing,52, 53 whilst synapse‐promoting A2 astrocytes may be responsible for unwanted synapses in epilepsy and neuropathic pain.54 As well as the classical innate immune cells, i.e. microglia and astrocytes, oligodendrocytes also contribute to innate immune reactions, expressing receptors and producing immunomodulatory cytokines and chemokines. During CNS insults and disease, oligodendrocytes can aid protective and regenerative processes, but can also contribute to neurodegeneration through poor production or repair of myelin. Cross‐talk between oligodendrocytes and microglia is a key area of interest in many CNS diseases.55 NK cells have long been considered as lymphocytes that kill tumour cells and virally infected cells. However, recent studies have identified regulatory roles in T‐cell responses and homeostasis. Dysfunction of these regulatory roles has been linked to MS,56 and reduced levels of NK cells have been found in depression.57 Mast cells mediate BBB permeability and recruitment of immune cells into the brain, sustaining CNS inflammation in a potentially detrimental manner.58 Recent studies show that mast cells interact with the gut microbiota and gut permeability, and may therefore influence many diseases in which the gut microbiota is of importance, including MS, AD, ALS, PD and epilepsy.59
The complement system is an ancient part of the immune system, which protects from microbes, removes debris and promotes cell survival. Recent studies have highlighted further roles of the complement system, including control of cellular reprogramming and intracellular metabolic programming.60 These newly discovered functions are still under investigation, and their meaning could have important relevance for therapeutic targeting as the complement system has been implicated in many diseases, including AD, ALS, MS, PD, epilepsy, TBI, schizophrenia and depression, predominantly through driving inflammation.61 It is now emerging that the complement system may form a link between innate and adaptive immunity, aiding in stimulation and regulation of lymphocytes, and antigen presentation.62
The classical image of innate immunity being strictly non‐specific has been confounded in recent times, with emerging evidence of innate immunity having some memory capacity, mostly through epigenetic changes. Monocytes, macrophages and NK cells have all been shown to have enhanced responses to previously‐encountered insults, although less specific than adaptive immune responses.63
Adaptive immunity
The role of adaptive immunity in neurodegenerative disorders is supported by alterations in T‐ and B‐cell subsets and (auto)antibody levels in the blood, cerebrospinal fluid (CSF) and brain tissues during disease (Fig. 2). Whether the cell subsets have a detrimental role (i.e. Th1 or Th17), or anti‐inflammatory role [i.e. Th 2 or regulatory cells (Tregs)] is based, for example, on cytokines and chemokines profiles. The role of the adaptive immune responses in neurological diseases is best illustrated by the spectrum of autoimmune encephalopathy syndromes, including the paraneoplastic neurological disorders (PNND).64, 65 In many of these disorders the use of IVIG or removal of the tumour expressing the aberrant antigen in PNND is frequently sufficient to treat these disorders. Traditionally, MS has been characterized by the invasion of the CNS by adaptive immune cells, i.e. T and B lymphocytes; however, the role of the adaptive immunity in PD and ALS is gradually gaining interest, although in AD the inflammation is primarily by CNS‐resident microglia. For example, in early PD, increased numbers of Th17 cells are observed in the blood, some of which recognize α‐synuclein,66 although whether these T‐cells are pathogenic is unclear. T‐ and B‐cells are also present in the CNS during X‐ALD, and may represent a secondary phenomenon as immunosuppressive therapies have little impact on the course of the disease. In mouse models of ALS, lower numbers of Tregs are concomitant with motor neuron death and shorter survival times, while transfer of Tregs suppresses neuroinflammation and prolongs survival. In line with these findings, Tregs have been reported to be dysfunctional in people with ALS,67 although the relevance for the disease progression has not been examined. In an experimental model of AD T‐cells aid clearance of plaques in transgenic mice yet also drive the pathology and cognitive impairments that can be rectified using anti‐CD3 or IL‐2 treatment.68, 69 That T‐cells are more likely to be beneficial in AD is supported by studies showing that transplantation of splenocytes from young mice improved spatial learning and memory in amyloid precursor protein (APP)swe/PSENldE9 transgenic mice.70 Whether aged T‐cells in AD are pathogenic is unknown; however, such protection in early life may be due to haematopoietic stem cell proliferation known to reduce with age,71 leading to a decrease in naïve and memory B‐cells, impaired antibody levels, number and function72 of T‐cells characterized by an inverted CD4+/CD8+ ratio, and an accumulation of CD8+/CD28− cells.73
Figure 2.
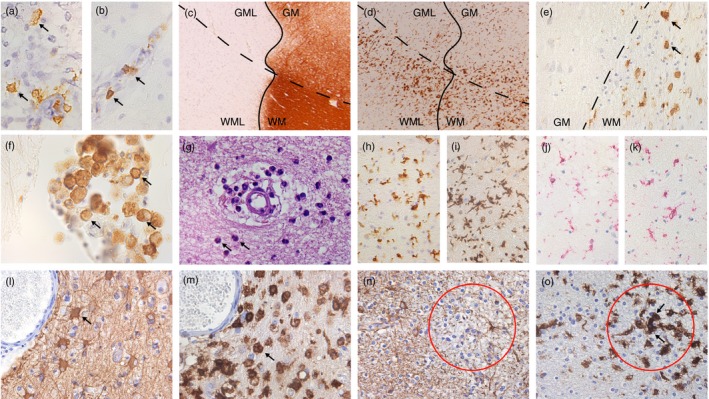
Immune responses in human and experimental inflammatory neurodegenerative disorders. B‐cells (arrows) are observed in white (a) and grey matter lesions (b) in multiple sclerosis (MS). (c) and (d) depict an MS leucocortical lesion. The white matter (WML) is associated with HLA + microglia (d, WML) in contrast to the lack of HLA + microglia in the grey matter (d, GML). A similar pattern of HLA + cells is seen in the white and grey matter in an X‐ALD case (e) and where peripheral macrophages infiltrate the white matter (f). Granulocytes (arrow) in suspected vasculitis cases (g). Ageing influences the activity of microglia in a mouse model of MS: microglia in the central nervous system (CNS) of young mice (h; Iba1 staining) are less active than in aged mice (i). In MS cases microglia in normal appearing white matter express P2Y12 (j) and TMEM119 (k). In progressive multifocal leucoencephalopathy astrocytes (l, arrow) and activated microglia/macrophages (m, arrow) are highly reactive in an area of demyelination. The paucity of astrocytic glial fibrillary acidic protein expression (red circle, n) is associated with an area of microglial activation (red circle, o) in acute haemorrhagic leucoencephalitis.
That the adaptive immune response plays a major role in early MS is evidenced by the effectivity of anti‐inflammatory therapies that modify the natural evolution of disease. Although widely‐considered to be a T‐cell‐mediated disease, CD20 therapy is surprisingly effective, indicating a key role of B‐cells in the disease. This is further supported by recent data indicating memory B‐cells are major targets for effective immunotherapy in relapsing–remitting MS.74 However, approaches targeting the adaptive immune response are less effective when administered to patients in the progressive phase of the disease, indicating a less important role for B‐cells in disease progression.
In addition to the cellular involvement, the presence of immunoglobulins, i.e. oligoclonal IgG in the CSF, is a diagnostic marker in MS; however, it is still unclear whether these antibodies are pathogenic, or merely arise due to aberrant intrathecal B‐cells activated by Epstein–Barr virus infection. This issue remains controversial and deserves further study. In contrast to the as yet unknown role of antibodies in MS, the closely related disorder NMO, once classified within the MS spectrum, is now considered a separate entity as the identification of the target antigen of the antibody was identified as the water channel AQP4. That antibodies to AQP4 are pathogenic has been demonstrated in animal models and also in vitro, resulting in astrocyte damage.75
Microbiome
Emerging evidence indicates that the microbiome influences CNS function and that disturbances in the microbiota–gut–brain axis may play a key role in susceptibility to, as well as augmenting, neuroinflammatory disorders (Fig. 3). Systemic infections contribute to neurodegenerative disorders76 by ‘priming’ innate immune cells in the CNS, implying that priming may also occur following release of microbiome bacteria, viruses, fungi and protozoa, and their toxins and metabolic products. The composition of the gut microbiome is largely dictated by early‐life occurrences, such as caesarean section,77 breastfeeding and the early use of antibiotics. During infancy to adulthood, the microbiome is relatively stable, although changes may occur in later life when neurodegenerative diseases arise.78 That recent studies have linked functional alterations in the gut microbiota to several neurodegenerative diseases, for example, ALS, AD, MS, PD, autism, bipolar disorder, depression and schizophrenia,79, 80, 81 suggests that such alterations may contribute to disease. In AD and PD, exposure to gut bacterial infections is associated with disease,82, 83 and in AD and MS, fewer anti‐inflammatory gut bacteria and an increase in pro‐inflammatory gut bacteria are associated with disease.79 Furthermore, the gut microbiome from people with MS were shown to induce experimental neurological disease in mice,84 supporting the notion that some components of the microbiome activate a pathogenic inflammatory response. Conversely, some gut bacteria regulate immune responses, raising the possibility that probiotic biotherapies or faecal microbiota transplants may be therapeutic for a range of inflammatory diseases, including neurodegenerative diseases.85
Figure 3.
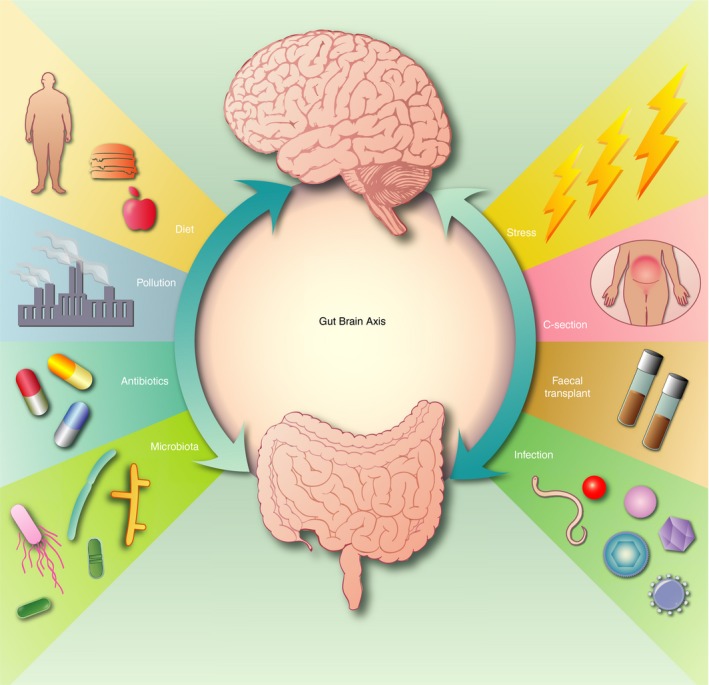
Proposed factors that influence the gut–brain axis in neuroinflammmatory disorders. Cross‐talk (arrows) between the gut and brain indicates that lifestyle and the environment influence brain function that feeds back to the gut brain axis. Altered gut microbiota composition as a result of lifestyle, for example, poor diet, stress, infection and other environmental factors, enhances the risk of neuroinflammatory disorders. During development maternal inflammation and caesarean section may influence brain development and microbiome of the fetus. Therapeutic approaches using faecal transplants, controlled and restricted diet and probiotics may help establish a healthy microbiome and therefore improve brain health.
Environmental triggers and lifestyle risk factors
Several neurodegenerative diseases are clearly genetic, for example, spinal muscular atrophy (SMA), genetic white matter disorders, HD, spinocerebellar ataxia and familial forms of ALS. However, the age of onset, progression and severity of disease are often influenced by environmental factors and lifestyle risk factors, such as smoking.86 For example, family members of people with ALS share the same ‘causal’ genetic mutation, yet may develop disease onset at considerably different ages, some with, and some without, cognitive impairment.87 A recent study showed an increased risk for ALS in areas with higher concentrations of airborne pollutants,88 and many other environmental factors have also been implicated. Such variation is also seen in MS, of which where the incidence is influenced by latitude and vitamin D.89 Identification of environmental risk factors is highly reliant on large epidemiological studies, which usually report only weak associations between risk factors and disease. However, many environmental and lifestyle factors increase the risk of several neurodegenerative and neuroinflammatory diseases (Table 2).
Table 2.
Environmental and lifestyle risk factors in neuroinflammmatory diseases
| Risk factor | Potential mechanisms | Disease | References |
|---|---|---|---|
| Viral infections | ↑pro‐inflammatory cytokines, chemokines, ↑macrophages, ↑NK cells | ↑ALS, ↑MS, ↑stroke, ↑autisma, ↑schizophreniaa, ↑bipolar disordera | 91, 93, 94, 95, 96, 98, 99 |
| Bacterial infections | ↑neutrophils, ↑complement, ↑pro‐inflammatory cytokines | ↑MS, ↑stroke, ↑schizophreniaa, ↑bipolar disordera | 94, 95, 98, 99 |
| Fungal infections | ↑neutrophils, ↑pro‐inflammatory cytokines, chemokines, ↑macrophages | ↑MS, ↑ALS, ↑AD, ↑stroke | 87, 92, 94, 95 |
| Pollution | ↑ROS, microglial activation, BBB changes, ↑pro‐inflammatory cytokines, infiltrating monocytes, astrogliosis | ↑stroke, ↑AD, ↑PD, ↑MS, ↑ALS, ↑autisma | 90, 91, 96 |
| Metals exposure | Neurotoxicity and metal aggregates, ↑ROS | ↑ALS, ↑PD, ↑autisma | 91, 92, 96 |
| Pesticides | Neurotoxicity, ↑ROS, BBB changes, UPS inhibition, defective autophagy, ER stress, mitochondrial dysfunction | ↑ALS, ↑PD, ↑AD | 91, 92 |
| Moderate alcohol consumption | ↑pro‐inflammatory cytokines, ↑ROS, ↑chemokines, astrogliosis | ↓ALS, ↓AD, ↓PD, ↓MS, ↑stroke, ↑depression, ↑bipolar disorder | 91, 92, 93, 95, 97, 99 |
| Smoking | ↑ROS, neurotoxicity, ↑pro‐inflammatory cytokines | ↑ALS, ↓PD, ↑AD, ↑MS, ↑stroke, ↑autisma (debated), ↑depression, ↑bipolar disordera | 88, 90, 91, 93, 94, 95, 97 |
| Regular exercise | ↑monocytes, ↑neutrophils, ↑NK cells | ↑ALS, ↓AD, ↓PD, ↓depression | 91, 92, 97 |
| Obesity | ↑macrophages, ↑pro‐inflammatory cytokines, ER stress | ↑ALS, ↑PD, ↑AD, ↑MS, ↑stroke, ↑depression | 91, 92, 93, 95, 97 |
| Head injury | ↑neutrophils, ↑complement, ↑pro‐inflammatory cytokines and chemokines, T‐cell migration | ↑ALS, ↑PD, ↑AD, ↑MS, ↑bipolar disorder | 91, 92, 93, 99 |
Prenatal exposure.
An emerging threat that is spreading rapidly in nearly all countries on the American continents is the flavivirus Zika (ZIKV).100 The tropism of ZIKV has been reported to be directed to neurons and neural stem cells, increasing its risk in CNS disease development. Despite the asymptomatic nature of ZIKV infection in adults, neurological complications have been reported in children, including Guillain–Barré syndrome, myelitis, seizures and meningoencephalitis.100 Another recent, yet more deadly, virus is Ebola (EVD), with a mortality rate ranging up to 90%.101 The recent outbreak in 2014 has given insight into its pathological mechanisms as it can cross the BBB and affect the CNS during infection. Subsequently, EVD has been reported to cause neurological symptoms such as seizures and delirium, while on a neuropathological level glial nodules, (chorio)meningoencephalitis, perivascular cuffing and cerebral haemorrhages are found.101
Ageing
Ageing, a major risk factor in neurodegenerative diseases, has a predominantly negative effect on both innate and adaptive immune responses, reducing the efficacy of vaccinations, and increasing susceptibility to infectious, chronic, autoimmune and neurodegenerative diseases.72 Ageing has been associated with a low‐grade sterile inflammatory status of the immune system, frequently termed inflammaging,102 in which pro‐inflammatory cytokines (e.g. IL‐6, TNF, IL‐1β) are key players in unhealthy ageing. Inflammaging might be the most important aetiological factor in age‐related neurodegenerative diseases, as ‘neuro‐inflammaging’ is associated with significantly decreased numbers of neurons, neuronal arborization, spines and cortical volume.103 With ageing, both macrophages and microglia display impaired and prolonged activation to insults, reduced motility and impaired phagocytosis.104 This over‐activation induces reactive oxygen species (ROS) production and attracts peripheral leucocytes, which affects the metabolic and trophic support glial cells provide their environment.105 Impaired phagocytosis results in increased toxic protein accumulation, which is associated with progressive pathology of Aβ in AD and α‐synuclein in PD.106 Furthermore, the self‐renewing capacity of glial cells drives telomere shortening, which was found to contribute towards AD pathology.107 While not yet fully investigated in other neurodegenerative diseases, this may be an important factor that drives progressive neuropathology with ageing. Ageing also affects one of the most important regenerative processes in the brain, viz: remyelination. The efficiency of this process progressively declines during ageing as a result of reduced signalling from macrophages to regulate differentiation of oligodendrocyte precursor cells.108
Monitoring neuroinflammation
Although biomarkers of neuroinflammation are considered essential for monitoring disease diagnosis, progression and response to therapy, there is a lack of accurate and reliable biomarkers for many neurological diseases.109 Many biomarkers present in blood or CSF are a consequence of the CNS pathology, for example, cytokines and chemokines,109, 110 loss of BBB integrity105 or indicators of neuronal damage, such as the increased levels of neurofilament111 or decreased N‐acetyl‐aspartate levels on MRS.109 One such pathological marker, extracellular vesicles (EVs), are released by neurons, oligodendrocytes, astrocytes, microglia and epithelial cells, as well as by immune cells that enter the brain during inflammation.112 In neurodegeneration, EVs are widely considered to act as vehicles for the spread of aggregated pathogenic proteins. The composition of EVs closely reflects the cell from which they are derived. Upon entering the CSF or blood they should therefore be considered as potential biomarkers of disease progression.
While blood and CSF are commonly used to monitor biomarkers of neuroinflammation, in vivo imaging of the CNS during disease has become a more widely accepted approach due to its non‐ or minimally invasive nature. Such techniques include magnetic resonance imaging (MRI), positron emission tomography (PET), single‐photon emission computed tomography (SPECT) and optical imaging.113 In this way, neuroinflammation can be studied by: (i) monitoring activation of resident CNS immune cells, for example, microglia activation; (ii) BBB permeability, for example, upregulation of adhesion molecules; (iii) CNS infiltration of immune cells; and (iv) pathology as a result of neuroinflammation, for example, demyelination and cell death (Table 3). For example, imaging of resident immune cells is frequently performed using PET to examine the translocator protein 18 kDa (TSPO) as an indicator of neuroinflammation in stroke,119 AD,120 MS121 and epilepsy.122
Table 3.
Biomarkers and imaging of neuroinflammatory diseases
| Target type | Target | Marker | Methods | References |
|---|---|---|---|---|
| Resident CNS cells | Translocator protein | Innate immune activation | PET, SPECT | 109 |
| Monoamine oxidase‐b | Reactive astrocytes | PET | 109 | |
| Cyclooxygenase 1 | Activated microglia and astrocytes | PET | 109 | |
| Myeloperoxidase | Inflammatory mediator found in leucocytes | MRI, PET | 114 | |
| Adenosine receptors | Cell injury | PET | 115 | |
| a4b2 nicotinic acetylcholine receptors | Activated microglia and astrocytes | PET | 109 | |
| Myo‐inositol | Astrocyte hypertrophy | MRS | 109 | |
| N‐acetyl‐aspartate | Neuronal integrity | MRS | 109 | |
| Iron accumulation | Free radical formation, mitochondrial or neuronal dysfunction | MRI | 116 | |
| Myelin | Demyelination and loss of myelin integrity in white matter disorders | PET | 109 | |
| BBB integrity | Vascular cell adhesion molecule 1 | Activation BBB | Molecular imaging | 109 |
| P‐glycoprotein | Alterations of expression in relation to BBB activity | PET, optical imaging | 109 | |
| Immune markers | Cytokines | Pro‐ or anti‐inflammatory signals | CSF | 110 |
| Chemokines | Pro‐ or anti‐inflammatory signals | CSF | 110 | |
| Superparamagnetic particles of iron oxide (SPIO) | SPIO‐labelled phagocytic cells | MRI | 117 | |
| Antibodies | Oligoclonal bands | IgG of unknown specificity | CSF | 111 |
| Anti‐aquaporin 4 antibodies | Antibodies to aquaporin 4 (water channel protein) | Blood | 111 | |
| Anti‐NF antibodies | Neuronal damage | Blood | 111 | |
| Free proteins | Neurofilaments | Neuronal damage | CSF | 111 |
| MicroRNAs | Circulating microRNAs involved in inflammation | Blood | 111 | |
| β‐amyloid | Proteins involved in disease pathology | Blood | 118 | |
| Tau | Proteins involved in disease pathology | Blood | 112 | |
| Annexin V | Apoptosis | PET, SPECT, blood | 109 | |
| Exosomes | A potential mechanism by which pathology is spread and/or toxic proteins are transported | CSF/blood | 112 |
In addition, BBB permeability, regarded as the hallmark of neuroinflammation, is imaged by leakage of gadolinium using MRI, or by nuclear imaging of P‐glycoprotein and vascular cell adhesion molecule (VCAM‐1), which are differentially expressed in MS,123 stroke,124 AD and vascular dementia.125
Indicators of leucocyte function include markers of oxidative stress, such as pro‐inflammatory and oxidative enzymes secreted by activated monocytes and neutrophils. One such product is myeloperoxidase (MPO) that can be detected by gadolinium (Gd) (MPO‐Gd) to track the oxidative activity of MPO non‐invasively. Thus, MPO has been used as a potential biomarker of neuroinflammation in experimental models of MS, namely experimental autoimmune encephalomyelitis126 and experimental stroke.127
Cell‐labelling approaches include radiolabelled antibodies and radiolabelled cytokines, which are imaged using SPECT, PET or optical imaging. Radiolabelling of anti‐CD3, anti‐CD4, IL‐1 and IL‐2 have all been used to visualize T‐lymphocytes in MS128 and rheumatoid arthritis.129 As well as ongoing neuroinflammation, several approaches image the resultant pathology. As an example, PET ligands have been used to visualize myelin damage in MS,130, 131 while many approaches are used to visualize cell death, for example, neuronal loss, such as annexin‐V, caspases and ML‐10.113 Imaging of neuro‐inflammatory biomarkers is an expanding topic with the potential to expedite diagnosis, and improve disease and therapeutic monitoring. Unfortunately, while many approaches are examined in preclinical models, fewer are available for studies in humans.109
Immune therapies
The accumulating evidence supporting the notion that inflammation plays a key role in neurodegenerative diseases of the CNS has stimulated an increasing number of immunotherapeutic strategies to modulate neuroinflammatory diseases (Fig. 4; Table 4). Many of these approaches that have been examined in MS may also be effective in other neurodegenerative diseases, as shown with Gilenya (FTY720, S1P‐R agonist) for experimental PD.165 Some approaches, such as gene silencing,166, 167 target a specific aggregated protein, while immune‐based therapies including plasmapheresis or IVIG are specific for antibody‐mediated disorders. However, other approaches, including antioxidant compounds, ion channel blocks, and approaches promoting neuroprotection and regeneration, such as haematopoietic stem cell transplantation and modified stem cells, are now being exploited in neurodegenerative disorders such as cerebral adrenoleucodystrophy.168 Other approaches for modulating aberrant innate and adaptive immune cells are also under investigation, including the use of exogenous HSPs. As an example, HSPB5 exerts neuroprotective effects in several models of neurodegeneration as well in MS.169
Figure 4.
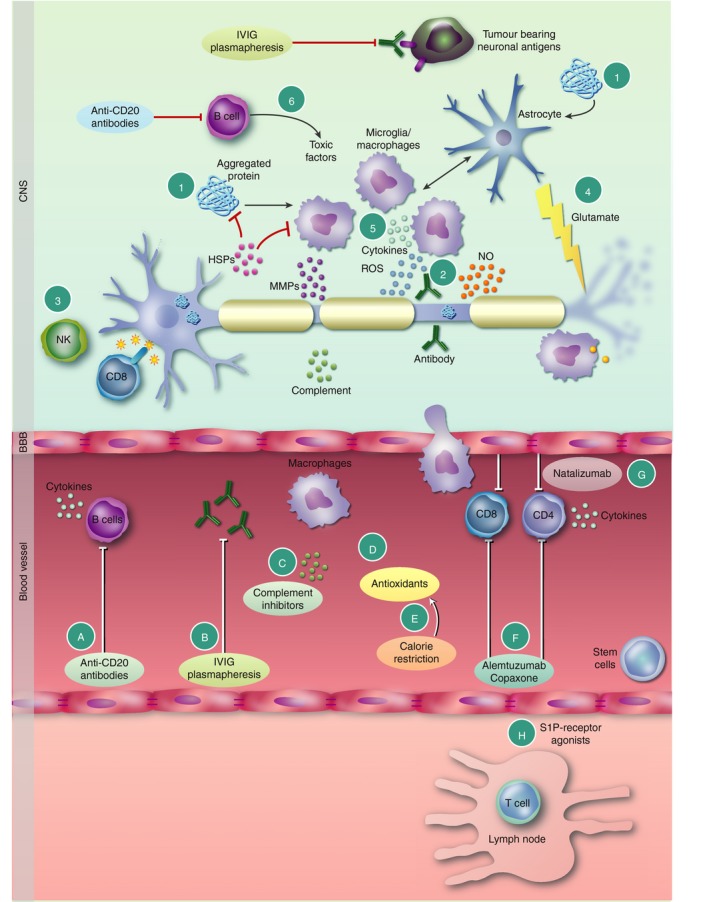
Mechanisms of damage and therapeutic control of inflammation in neurodegenerative diseases. (1) In the central nervous system (CNS), damage to neurons or genetic mutations leads to accumulation of misfolded and aggregated proteins characteristic of many neurodegenerative diseases. Such damage‐associated molecular patterns (DAMPs) and pathogen‐associated molecular patterns (PAMPs) activate microglia and astrocytes to release pro‐inflammatory factors. Likewise, stressed neurons and glia release heat‐shock proteins (HSPs) in an effort to counter the formation of aggregated proteins. Some HSPs, for example, HSPB5, induce regulatory microglia and astrocyte phenotypes. Neuronal specific antibodies (2) activate the complement system or induce Fc receptor (FcR)‐mediated damage. (3) Natural killer (NK) and T‐cells damage neurons via MHC class‐I, CD8+ T‐cells or non‐classical MHC molecules. (4) Excessive production of glutamate together with reduced glutamate uptake by astrocytes leads to excitotoxic damage of neurons. (5) Macrophage/microglia activation triggers reactive oxygen (ROS) and nitrogen (NOS) species, MMPs, chemokines and cytokines known to damage axons and neurons. (6) B‐cells secrete pathogenic antibodies and toxic factors that damage axons and oligodendrocytes. Therapeutic approaches to control neuroinflammation include (A) anti‐CD20 antibodies to deplete B‐cells that play multiple roles in immune‐mediated neurodegeneration (see text for details). (B) IVIG and plasmapheresis block pathogenic antibodies, including those triggered by tumours as in paraneoplastic disorders. (C) Complement inhibitors control activity of complement, while (D) antioxidants and (E) calorie restriction reduce ROS and NO levels that contribute to neurodegeneration. (F) In multiple sclerosis (MS), inhibition of T‐ and B‐cells entry across the blood–brain barrier (BBB) into the CNS, for example, (G) Natalizumab (Tysabri®), or immune therapies that deplete T‐ and B‐cells [Alemtuzumab (Lemtrada®), or alter their function Glatiramer acetate (Copaxone®)] in the periphery, or (H) block immune cell trafficking from the lymph nodes (FTY720, S1PR‐agonists) controls neuroinflammation in the CNS.
Table 4.
Immune therapies in neuroinflammatory disorders
| Therapeutic intervention | Proposed mode of action | Disease | References |
|---|---|---|---|
| Antibodies to abnormal protein aggregations e.g. a‐synuclein, PrP | Clearance of aggregates | AD, prion diseases, HD, ALS, stroke, PND | 132, 133, 134, 135 |
| Plasmapheresis (+tumour removal) | Removal of pathogenic antibody | Paraneoplastic disorders | 136 |
| Stem cell therapy | Creating non‐pathogenic and reparative cell populations | MS, SMA, ALS, TBI, AD, PD, prion diseases, stroke, HD | 137, 138, 139, 140, 141, 142, 143, 144 |
| Antibiotics e.g. minocycline | Inhibition of inflammation and anti‐apoptotic activity | AD, ischaemia, PD, HD, MS | 145, 146 |
| Cannabinoids | Attenuates excitotoxic glutamatergic neurotransmission, modulation of microglia and astrocytes | AD, PD, HD, ALS, epilepsy, MS, autism | 133, 147, 148, 149 |
| Non‐steroidal anti‐inflammatory | Inhibition of COX‐1 and ‐2 | AD | 132 |
| Diet and calorie restriction | Antioxidant functions, inhibits COX‐2 and iNOS, reduction in free radicals and oxidative stress | Epilepsy, ALS, AD, PD | 150, 151, 152, 153 |
| Anti‐lymphocyte therapy |
B‐cell depletion (CD20) Plasma cells depletion (CD19) Blocking T‐cell responses Modulating T‐cell phenotypes |
MS, stroke, depression | 135, 137, 154 |
| Innate immunity based therapy | Macrophage apoptosis and macrophage suppression. inhibition NLRP inflammasome inhibiting/modulating microglia phenotypes. | MS, depression, ischaemia, TBI | 137, 141, 154 |
| Targeting ionotropic and metabotropic receptors | Agonist/antagonist antibodies or ligands e.g. NMDA, glutamate receptor antagonist | AD, PD, HD, MS, SMA, ALS, prion disease, epilepsy, bipolar disorder, TBI, depression, schizophrenia | 132, 133, 134, 137, 138, 141, 154, 155, 156, 157, 158 |
| Antioxidants | Reducing ROS, upregulating antioxidant genes. Targeting Nrf2 pathway (e.g. BG12). HSPs. |
MS, HD, ALS, stroke, TBI, schizophrenia, depression Friedreich's ataxia |
134, 135, 137, 141, 154, 158, 159 |
| RAGE antagonists | Reduction of formation or activation of innate immune responses by blocking/inhibiting advanced glycation end‐products (AGEs) | AD | 160 |
| Complement inhibition | Blocking complement mediated neuronal damage | Stroke, TBI, epilepsy | 161, 162, 163 |
| Potassium and sodium channel targets | Neuroprotection e.g. lamotrigine, fampridine acid‐sensing ion channel 1 (ASIC1) | MS | 137 |
| Chemokine/cytokine modulation |
Promoting regenerative microenvironment e.g. IL‐4, CD28, amplification of Tregs. CXCR3 |
AD, TBI, epilepsy, depression, schizophrenia, bipolar disorder | 132, 141, 154, 157, 158, 164 |
Conclusions
That the nervous and immune systems are inextricably interlinked is reinforced by recent studies revealing meningeal vessels that directly link the brain with the lymphatic system. In many neurodegenerative diseases the innate immune response in the CNS plays a key role in the onset and progression of disease, but is equally important for resolution of inflammation. During development microglia aid in synaptic pruning and neurophagy,53 which is important for neuronal development. Even at this early stage, environmental factors including maternal infections or alcohol intake influence microglial responses in later life. Similarly, adaptive and innate immune cells that enter the CNS trigger damage but are crucial for immune regulation. As well as the well‐recognized roles of microglia, astrocytes and neurons, oligodendrocytes also contribute to immune surveillance and regulation in the CNS.
Thus, despite beneficial roles of immune responses, such responses must remain under tight control to prevent CNS damage. During ageing and repeated activation, immune cells undergo senescence, implying that the CNS is not fully protected. The factors that drive chronic inflammation include misfolded and aggregated proteins, HSPs and other DAMPs that trigger local innate responses. Infectious agents also play a role, including those arising from the gut microbiome and toxic compounds in the environment. Gene silencing to prevent protein aggregation is effective in experimental settings, and it is encouraging that studies in humans are now underway. Thus, development of novel therapeutic approaches to target the pathogenic mechanisms leading to detrimental inflammation, as well as harnessing endogenous protective pathways, will be key to controlling neuroinflammatory diseases.
Disclosures
The authors have no conflict of interests to disclose.
Acknowledgements
The authors gratefully acknowledge the financial support of the Multiple Sclerosis Society, and the Stichting MS Research for their work referenced in this review. Dr JM van Noort is gratefully acknowledged for reviewing the paper and for his helpful comments. The authors also thank Alison Schroeer for her expertise in producing Figs 1, 3 and 4.
References
- 1. Wimo A, Guerchet M, Ali GC, Wu YT, Prina AM, Winblad B et al The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimers Dement 2017; 13:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cova I, Markova A, Campini I, Grande G, Mariani C, Pomati S. Worldwide trends in the prevalence of dementia. J Neurol Sci 2017; 379:259. [DOI] [PubMed] [Google Scholar]
- 3. Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun 2004; 18:407–13. [DOI] [PubMed] [Google Scholar]
- 4. Ngugi AK, Kariuki SM, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Incidence of epilepsy A systematic review and meta‐analysis. Neurology 2011; 77:1005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh A, Trevick S. The epidemiology of global epilepsy. Neurol Clin 2016; 34:837–47. [DOI] [PubMed] [Google Scholar]
- 6. Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D et al Inflammation in neurodegenerative diseases–an update. Immunology 2014; 142:151–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mackenzie IS, Morant SV, Bloomfield GA, MacDonald TM, O'riordan J. Incidence and prevalence of multiple sclerosis in the UK 1990–2010: a descriptive study in the General Practice Research Database. J Neurol Neurosurg Psychiatry 2014; 85:76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Myall DJ, Pitcher TL, Pearson JF, Dalrymple‐Alford JC, Anderson TJ, MacAskill MR. Parkinson's in the oldest old: impact on estimates of future disease burden. Parkinsonism Relat Disord 2017; 42:78–84. [DOI] [PubMed] [Google Scholar]
- 9. Tysnes OB, Storstein A. Epidemiology of Parkinson's disease. J Neural Transm 2017; 124:901–5. [DOI] [PubMed] [Google Scholar]
- 10. Rawlins MD, Wexler NS, Wexler AR, Tabrizi SJ, Douglas I, Evans SJ et al The prevalence of Huntington's disease. Neuroepidemiology 2016; 46:144–53. [DOI] [PubMed] [Google Scholar]
- 11. Robertson A, Aartsma‐Rus A, Jones CC, Lochmüller H, Wilson IJ, Verhaart IE et al Prevalence, incidence and carrier frequency of 5q‐linked spinal muscular atrophy–a literature review. Orphanet J Rare Dis 2017; 12:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chio A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA et al Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 2013; 41:118–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arthur KC, Calvo A, Price TR, Geiger JT, Chio A, Traynor BJ. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun 2016; 7:12 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mohan KM, Wolfe CD, Rudd AG, Heuschmann PU, Kolominsky‐Rabas PL, Grieve AP. Risk and cumulative risk of stroke recurrence. Stroke 2011; 42:1489–94. [DOI] [PubMed] [Google Scholar]
- 15. Stevens EGV, Emmet ES, Wang Y, McKevitt CJ, Wolfe CDA. The burden of stroke in Europe report King's college London. In Stroke Alliance for Europe. 2017. ISBN 0661.651.450, p 108.
- 16. Nguyen R, Fiest KM, McChesney J, Kwon CS, Jette N, Frolkis AD et al The international incidence of traumatic brain injury: a systematic review and meta‐analysis. Can J Neurol Sci 2016; 43:774–85. [DOI] [PubMed] [Google Scholar]
- 17. UNAIDS (2017) Fact sheet 2017. URL http://www.unaids.org/en/resources/fact-sheet [accessed on 05 December 2017]
- 18. Harrison LH, Trotter CL, Ramsay ME. Global epidemiology of meningococcal disease. Vaccine 2009; 27:B51–63. [DOI] [PubMed] [Google Scholar]
- 19. Scott JG, Duhig M, Hamlyn J, Norman RE. Environmental contributions to autism: explaining the rise in incidence of autistic spectrum disorders. J Environ Immunol Toxicol 2014; 1:75–9. [Google Scholar]
- 20. Baxter AJ, Brugha TS, Erskine HE, Scheurer RW, Vos T, Scott JG. The epidemiology and global burden of autism spectrum disorders. Psychol Med 2015; 45:601–13. [DOI] [PubMed] [Google Scholar]
- 21. Ferrari AJ, Somerville AJ, Baxter AJ, Norman R, Patten SB, Vos T et al Global variation in the prevalence and incidence of major depressive disorder: a systematic review of the epidemiological literature. Psychol Med 2013; 43:471–81. [DOI] [PubMed] [Google Scholar]
- 22. Hidaka BH. Depression as a disease of modernity: explanations for increasing prevalence. J Affect Disord 2012; 140:205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burns JK, Tomita A, Kapadia AS. Income inequality and schizophrenia: increased schizophrenia incidence in countries with high levels of income inequality. Int J Soc Psychiatry 2014; 60:185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Merikangas KR, Jin R, He JP, Kessler RC, Lee S, Sampson NA et al Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch Gen Psychiatry 2011; 68:241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yutzy SH, Woofter CR, Abbott CC, Melhem IM, Parish BS. The increasing frequency of mania and bipolar disorder: causes and potential negative impacts. J Nerv Ment Dis 2012; 200:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wong CH, Jenne CN, Tam PP, Léger C, Venegas A, Ryckborst K et al Prolonged activation of invariant natural killer T cells and TH2‐skewed immunity in stroke patients. Front Neurol 2017; 8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zheng YH, Xiao SL, He B, He Y, Zhou HY, Chen Z et al The role of IL‐27 and its receptor in the pathogenesis of HIV/AIDS and anti‐viral immune response. Curr HIV Res 2017; 15:279–84. [DOI] [PubMed] [Google Scholar]
- 28. Deeks SG. HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med 2011; 62:141–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ribes S, Nessler S, Heide EC, Malzahn D, Perske C, Brück W et al The early adaptive immune response in the pathophysiology of pneumococcal meningitis. J Infect Dis 2017; 215:150–8. [DOI] [PubMed] [Google Scholar]
- 30. Careaga M, Rogers S, Hansen RL, Amaral DG, Van de Water J, Ashwood P. Immune endophenotypes in children with autism spectrum disorder. Biol Psychiat 2017; 81:434–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hughes MM, Connor TJ, Harkin A. Stress‐related immune markers in depression: implications for treatment. Int J Neuropsychopharmacol 2016; 19:pii: pyw001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karpiński P, Frydecka D, Sąsiadek MM, Misiak B. Reduced number of peripheral natural killer cells in schizophrenia but not in bipolar disorder. Brain Behav Immun 2016; 54:194–200. [DOI] [PubMed] [Google Scholar]
- 33. Kéri S, Szabó C, Kelemen O. Antipsychotics influence Toll‐like receptor (TLR) expression and its relationship with cognitive functions in schizophrenia. Brain Behav Immun 2017; 62:256–64. [DOI] [PubMed] [Google Scholar]
- 34. Ascoli BM, Géa LP, Colombo R, Barbé‐Tuana FM, Kapczinski F, Rosa AR. The role of macrophage polarization on bipolar disorder: identifying new therapeutic targets. Aust N Z J Psychiatry 2016; 50:618–30. [DOI] [PubMed] [Google Scholar]
- 35. Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol 2008; 67:1113–21. [DOI] [PubMed] [Google Scholar]
- 36. Wilhelm I, Nyúl‐Tóth Á, Suciu M, Hermenean A, Krizbai IA. Heterogeneity of the blood–brain barrier. Tissue Barriers 2016; 4:e1143544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iliff JJ, Nedergaard M. Is there a cerebral lymphatic system? Stroke 2013; 44(6 Suppl 1):S93–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M et al A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015; 212:991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD et al Structural and functional features of central nervous system lymphatic vessels. Nature 2015; 523:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis 2010; 37:13–25. [DOI] [PubMed] [Google Scholar]
- 41. Gorlé N, Van Cauwenberghe C, Libert C, Vandenbroucke RE. The effect of aging on brain barriers and the consequences for Alzheimer's disease development. Mamm Genome 2016; 27:407–20. [DOI] [PubMed] [Google Scholar]
- 42. Bertram L. The role of TREM2 R47H as a risk factor for Alzheimer's disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson's disease. Alzheimers Dement 2015; 1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 122:787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ruckh JM, Zhao JW, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ et al Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 2012; 10:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wes PD, Holtman IR, Boddeke EW, Möller T, Eggen BJ. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia 2016; 64:197–213. [DOI] [PubMed] [Google Scholar]
- 46. Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton‐Jones M et al Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid‐β pathology. Brain 2016; 139:1265–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schaafsma W, Basterra LB, Jacobs S, Brouwer N, Meerlo P, Schaafsma A et al Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol Dis 2017; 106:291–300. [DOI] [PubMed] [Google Scholar]
- 48. Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP et al Microglial brain region‐dependent diversity and selective regional sensitivities to aging. Nat Neurosci 2016; 19:504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Erny D, de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E et al Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 2015; 18:965–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL et al Selective chemokine receptor usage by central nervous system myeloid cells in CCR2‐red fluorescent protein knock‐in mice. PLoS ONE 2010; 5:e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017; 140:1900–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity 2017; 46:957–67. [DOI] [PubMed] [Google Scholar]
- 53. Vilalta A, Brown GC. Neurophagy–the phagocytosis of live neurons and synapses by glia–contributes to brain development and disease. FEBS J 2017; https://doi.org/10.1111/febs.14323. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 54. Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature 2010; 468:223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peferoen L, Kipp M, Valk P, Noort JM, Amor S. Oligodendrocyte‐microglia cross‐talk in the central nervous system. Immunology 2014; 141:302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gross CC, Schulte‐Mecklenbeck A, Wiendl H, Marcenaro E, de Rosbo NK, Uccelli A et al Regulatory functions of natural killer cells in multiple sclerosis. Front Immunol 2016; 7:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Suzuki H, Savitz J, Teague TK, Gandhapudi SK, Tan C, Misaki M et al Altered populations of natural killer cells, cytotoxic T lymphocytes, and regulatory T cells in major depressive disorder: association with sleep disturbance. Brain Behav Immun 2017; 66:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Skaper SD, Facci L, Zusso M, Giusti P. Neuroinflammation, mast cells, and glia: dangerous liaisons. Neuroscientist 2017; 23:478–98. [DOI] [PubMed] [Google Scholar]
- 59. Girolamo F, Coppola C, Ribatti D. Immunoregulatory effect of mast cells influenced by microbes in neurodegenerative diseases. Brain Behav Immun 2017; 65:68–89. [DOI] [PubMed] [Google Scholar]
- 60. Elvington M, Liszewski MK, Atkinson JP. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol Rev 2016; 274:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McGeer PL, Lee M, McGeer EG. A review of human diseases caused or exacerbated by aberrant complement activation. Neurobiol Aging 2017; 52:12–22. [DOI] [PubMed] [Google Scholar]
- 62. Bennett KM, Rooijakkers SH, Gorham RD Jr. Let's tie the knot: marriage of complement and adaptive immunity in pathogen evasion, for better or worse. Front Microbiol 2017; 8:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG et al Trained immunity: a program of innate immune memory in health and disease. Science 2016; 352:aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kalman B. Autoimmune encephalitides: a broadening field of treatable conditions. Neurologist 2017; 22:1–3. [DOI] [PubMed] [Google Scholar]
- 65. Balint B, Vincent A, Meinck HM, Irani SR, Bhatia KP. Movement disorders with neuronal antibodies: syndromic approach, genetic parallels and pathophysiology. Brain 2018; 141:13–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin‐Liebes J et al T cells from patients with Parkinson's disease recognize α‐synuclein peptides. Nature 2017; 546:656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Beers DR, Zhao W, Wang J, Zhang X, Wen S, Neal D et al ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight 2017; 2:e89530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alves S, Churlaud G, Audrain M, Michaelsen‐Preusse K, Fol R, Souchet B et al Interleukin‐2 improves amyloid pathology, synaptic failure and memory in Alzheimer's disease mice. Brain 2017; 140:826–42. [DOI] [PubMed] [Google Scholar]
- 69. Laurent C, Dorothée G, Hunot S, Martin E, Monnet Y, Duchamp M et al Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 2016; 140:184–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang F, Shen X, Li S, Chen L, Wang Y, Qin J et al Splenocytes derived from young WT mice prevent AD progression in APPswe/PSENldE9 transgenic mice. Oncotarget 2015; 6:20 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D et al Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci USA 2010; 107:5465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Castelo‐Branco C, Soveral I. The immune system and aging: a review. Gynecol Endocrinol 2014; 30:16–22. [DOI] [PubMed] [Google Scholar]
- 73. Olsson J, Wikby A, Johansson B, Löfgren S, Nilsson BO, Ferguson FG. Age‐related change in peripheral blood T‐lymphocyte subpopulations and cytomegalovirus infection in the very old: the Swedish longitudinal OCTO immune study. Mech Ageing Dev 2001; 121:187–201. [DOI] [PubMed] [Google Scholar]
- 74. Baker D, Marta M, Pryce G, Giovannoni G, Schmierer K. Memory B cells are major targets for effective immunotherapy in relapsing multiple sclerosis. EBioMedicine 2017; 16:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vaknin‐Dembinsky A, Karussis D, Avichzer J, Abramsky O. NMO spectrum of disorders: a paradigm for astrocyte‐targeting autoimmunity and its implications for MS and other CNS inflammatory diseases. J Autoimmun 2014; 54:93–9. [DOI] [PubMed] [Google Scholar]
- 76. Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 2007; 7:161–7. [DOI] [PubMed] [Google Scholar]
- 77. Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H et al Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med 2016; 8:343ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dinan TG, Cryan JF. Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. J Physiol 2017; 595:489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Quigley EM. Microbiota‐brain‐gut axis and neurodegenerative diseases. Curr Neurol Neurosci Rep 2017; 17:94. [DOI] [PubMed] [Google Scholar]
- 80. Chen J, Chia N, Kalari KR, Yao JZ, Novotna M, Soldan MM et al Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep 2016; 6:28 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mangiola F, Ianiro G, Franceschi F, Fagiuoli S, Gasbarrini G, Gasbarrini A. Gut microbiota in autism and mood disorders. World J Gastroenterol 2016; 22:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Andreadou E, Pantazaki AA, Daniilidou M, Tsolaki M. Rhamnolipids, microbial virulence factors, in Alzheimer's disease. J Alzheimers Dis 2017; 59:209–22. [DOI] [PubMed] [Google Scholar]
- 83. Goldman SM, Kamel F, Ross GW, Jewell SA, Marras C, Hoppin JA et al Peptidoglycan recognition protein genes and risk of Parkinson's disease. Mov Disord 2014; 29:1171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z et al Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA 2017; 114:10 719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cohen NA, Maharshak N. Novel indications for fecal microbial transplantation: update and review of the literature. Dig Dis Sci 2017; 62:131–1145. [DOI] [PubMed] [Google Scholar]
- 86. Malek AM, Barchowsky A, Bowser R, Heiman‐Patterson T, Lacomis D, Rana S et al Environmental and occupational risk factors for amyotrophic lateral sclerosis: a case–control study. Neurodegener Dis 2014; 14:31–8. [DOI] [PubMed] [Google Scholar]
- 87. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B et al TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008; 319:1668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Seelen M, Campos RA, Veldink JH, Visser AE, Hoek G, Brunekreef B et al Long‐term air pollution exposure and amyotrophic lateral sclerosis in Netherlands: a population‐based case–control study. Environ Health Perspect 2017; 125:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jelinek GA, Marck CH, Weiland TJ, Pereira N, van der Meer DM, Hadgkiss EJ. Latitude, sun exposure and vitamin D supplementation: associations with quality of life and disease outcomes in a large international cohort of people with multiple sclerosis. BMC Neurol 2015; 15:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Block ML, Calderón‐Garcidueñas L. Air pollution: mechanisms of neuroinflammation and CNS disease. Trends Neurosci 2009; 32:506–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol 2015; 7:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Campdelacreu J. Parkinson's disease and Alzheimer disease: environmental risk factors. Neurología 2014; 29:541–9. [DOI] [PubMed] [Google Scholar]
- 93. Belbasis L, Bellou V, Evangelou E, Ioannidis JP, Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta‐analyses. Lancet Neurol 2015; 14:263–73. [DOI] [PubMed] [Google Scholar]
- 94. Libbey JE, Cusick MF, Fujinami RS. Role of pathogens in multiple sclerosis. Int Rev Immunol 2014; 33:266–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mettananda C, Rothwell P, Li L, Mehta Z, Gutnikov S. Comparison of risk factors for stroke subtypes versus acute coronary syndrome: a population‐based study. In proceedings of the 17th Conference on Postgraduate Research, International Postgraduate Research Conference 2016, Faculty of Graduate Studies, University of Kelaniya, Sri Lanka. p 128.
- 96. Lyall K, Schmidt RJ, Hertz‐Picciotto I. Maternal lifestyle and environmental risk factors for autism spectrum disorders. Int J Epidemiol 2014; 43:443–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chang SC, Pan A, Kawachi I, Okereke OI. Risk factors for late‐life depression: a prospective cohort study among older women. Prev Med 2016; 91:144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. van Os J, Kenis G, Rutten BP. The environment and schizophrenia. Nature 2010; 468:203–12. [DOI] [PubMed] [Google Scholar]
- 99. Marangoni C, Hernandez M, Faedda GL. The role of environmental exposures as risk factors for bipolar disorder: a systematic review of longitudinal studies. J Affect Disord 2016; 193:165–74. [DOI] [PubMed] [Google Scholar]
- 100. Duca LM, Beckham JD, Tyler KL, Pastula DM. Zika virus disease and associated neurologic complications. Curr Infect Dis Rep 2017; 19:4. [DOI] [PubMed] [Google Scholar]
- 101. Billioux BJ. Neurological complications and sequelae of Ebola virus disease. Curr Infect Dis Rep 2017; 19:19. [DOI] [PubMed] [Google Scholar]
- 102. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E et al Inflamm‐aging: an evolutionary perspective on immunosenescence. Ann N Y Acad Sci 2000; 908:244–54. [DOI] [PubMed] [Google Scholar]
- 103. Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathmechdis Mech Dis 2008; 3:41–66. [DOI] [PubMed] [Google Scholar]
- 104. Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW. Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 2016; 139:653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chinta SJ, Woods G, Rane A, Demaria M, Campisi J, Andersen JK. Cellular senescence and the aging brain. Exp Gerontol 2015; 68:3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Spittau B. Aging microglia–phenotypes, functions and implications for age‐related neurodegenerative diseases. Front Aging Neurosci 2017; 9:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 2007; 10:61–74. [DOI] [PubMed] [Google Scholar]
- 108. Franklin RJ, Zhao C, Sim FJ. Ageing and CNS remyelination. NeuroReport 2002; 13:923–8. [DOI] [PubMed] [Google Scholar]
- 109. Albrecht DS, Granziera C, Hooker JM, Loggia ML. In vivo imaging of human neuroinflammation. ACS Chem Neurosci 2016; 7:470–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kothur K, Wienholt L, Brilot F, Dale RC. CSF cytokines/chemokines as biomarkers in neuroinflammatory CNS disorders: a systematic review. Cytokine 2016; 77:227–37. [DOI] [PubMed] [Google Scholar]
- 111. El Ayoubi NK, Khoury SJ. Blood biomarkers as outcome measures in inflammatory neurologic diseases. Neurotherapeutics 2017; 14:135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Croese T, Furlan R. Extracellular vesicles in neurodegenerative diseases. Mol Aspects Med 2017; 60:52–61. [DOI] [PubMed] [Google Scholar]
- 113. Pulli B, Chen JW. Imaging neuroinflammation–from bench to bedside. J Clin Cell Immunol 2014; 5:pii: 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhang Y, Seeburg DP, Pulli B, Wojtkiewicz GR, Bure L, Atkinson W et al Myeloperoxidase nuclear imaging for epileptogenesis. Radiology 2015; 278:822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Petroni D, Giacomelli C, Taliani S, Barresi E, Robello M, Daniele S et al Toward PET imaging of A 2B adenosine receptors: a carbon‐11 labeled triazinobenzimidazole tracer: synthesis and imaging of a new A2B PET tracer. Nucl Med Biol 2016; 43:309–17. [DOI] [PubMed] [Google Scholar]
- 116. Walsh AJ, Blevins G, Lebel RM, Seres P, Emery DJ, Wilman AH. Longitudinal MR imaging of iron in multiple sclerosis: an imaging marker of disease. Radiology 2014; 270:186–96. [DOI] [PubMed] [Google Scholar]
- 117. Hussain SM, Krestin GP. Superparamagnetic iron oxide contrast agents: physicochemical characteristics and application in MR imaging. Eur J Radiol 2001; 11:2319–31. [DOI] [PubMed] [Google Scholar]
- 118. Lue LF, Guerra A, Walker DG. Amyloid beta and tau as Alzheimer's disease blood biomarkers: promise from new technologies. Neurol Ther 2017; 6:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Heiss WD, Graf R, Fujita T, Ohta K, Bauer B, Löttgen J et al Early detection of irreversibly damaged ischemic tissue by flumazenil positron emission tomography in cats. Stroke 1997; 28:2045–52. [DOI] [PubMed] [Google Scholar]
- 120. Pascual B, Prieto E, Arbizu J, Marti‐Climent JM, Peñuelas I, Quincoces G et al Decreased carbon‐11‐flumazenil binding in early Alzheimer's disease. Brain 2012; 135:2817–25. [DOI] [PubMed] [Google Scholar]
- 121. Freeman L, Garcia‐Lorenzo D, Bottin L, Leroy C, Louapre C, Bodini B et al The neuronal component of gray matter damage in multiple sclerosis: a [11C] flumazenil positron emission tomography study. Ann Neurol 2015; 78:554–67. [DOI] [PubMed] [Google Scholar]
- 122. Juhasz C, Nagy F, Watson C, Da Silva EA, Muzik O, Chugani DC et al Glucose and [11C] flumazenil positron emission tomography abnormalities of thalamic nuclei in temporal lobe epilepsy. Neurology 1999; 53:2037–45. [DOI] [PubMed] [Google Scholar]
- 123. Serres S, Mardiguian S, Campbell SJ, McAteer MA, Akhtar A, Krapitchev A et al VCAM‐1‐targeted magnetic resonance imaging reveals subclinical disease in a mouse model of multiple sclerosis. FASEB J 2011; 25:4415–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gauberti M, Montagne A, Marcos‐Contreras OA, Le Béhot A, Maubert E, Vivien D. Ultra‐sensitive molecular MRI of vascular cell adhesion molecule‐1 reveals a dynamic inflammatory penumbra after strokes. Stroke 2013; 44:1988–96. [DOI] [PubMed] [Google Scholar]
- 125. Montagne A, Gauberti M, Macrez R, Jullienne A, Briens A, Raynaud JS et al Ultra‐sensitive molecular MRI of cerebrovascular cell activation enables early detection of chronic central nervous system disorders. NeuroImage 2012; 63:760–70. [DOI] [PubMed] [Google Scholar]
- 126. Chen JW, Breckwoldt MO, Aikawa E, Chiang G, Weissleder R. Myeloperoxidase‐targeted imaging of active inflammatory lesions in murine experimental autoimmune encephalomyelitis. Brain 2008; 131:1123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Breckwoldt MO, Chen JW, Stangenberg L, Aikawa E, Rodriguez E, Qiu S et al Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci USA 2008; 105:18 584–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Costa GL, Sandora MR, Nakajima A, Nguyen EV, Taylor‐Edwards C, Slavin AJ et al Adoptive immunotherapy of experimental autoimmune encephalomyelitis via T cell delivery of the IL‐12 p40 subunit. J Immunol 2001; 167:2379–87. [DOI] [PubMed] [Google Scholar]
- 129. Barrera P, Van der Laken CJ, Boerman OC, Oyen WJ, Van de Ven MT, Van Lent PL et al Radiolabelled interleukin‐1 receptor antagonist for detection of synovitis in patients with rheumatoid arthritis. Rheumatology 2000; 39:870–4. [DOI] [PubMed] [Google Scholar]
- 130. Wang C, Wu C, Popescu DC, Zhu J, Macklin WB, Miller RH et al Longitudinal near‐infrared imaging of myelination. J Neurosci 2011; 31:2382–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Frullano L, Wang C, Miller RH, Wang Y. A myelin‐specific contrast agent for magnetic resonance imaging of myelination. J Am Chem Soc 2011; 133:1611–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. O'Brien JT, Holmes C, Jones M, Jones R, Livingston G, McKeith I et al Clinical practice with anti‐dementia drugs: a revised (third) consensus statement from the British Association for Psychopharmacology. J Psychopharmacol 2017; 31:147–68. [DOI] [PubMed] [Google Scholar]
- 133. Brudek T, Winge K, Folke J, Christensen S, Fog K, Pakkenberg B et al Autoimmune antibody decline in Parkinson's disease and Multiple System Atrophy; a step towards immunotherapeutic strategies. Mol Neurodegener 2017; 12:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wyant KJ, Ridder AJ, Dayalu P. Huntington's disease–update on treatments. Curr Neurol Neurosci Rep 2017; 17:33. [DOI] [PubMed] [Google Scholar]
- 135. Siniscalchi A, Iannacchero R, Anticoli S, Romana Pezzella F, De Sarro G, Gallelli L. Anti‐inflammatory strategies in stroke: a potential therapeutic target. Curr Vasc Pharmacol 2016; 14:98–105. [DOI] [PubMed] [Google Scholar]
- 136. Cortese I, Cornblath DR. Therapeutic plasma exchange in neurology: 2012. J Clin Apheresis 2013; 28:16–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Torkildsen Ø, Myhr KM, Bø L. Disease‐modifying treatments for multiple sclerosis–a review of approved medications. Eur J Neurol 2016; 23(S1):18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wirth B, Barkats M, Martinat C, Sendtner M, Gillingwater TH. Moving towards treatments for spinal muscular atrophy: hopes and limits. Expert Opin Emerg Drugs 2015; 20:353–6. [DOI] [PubMed] [Google Scholar]
- 139. Czarzasta J, Habich A, Siwek T, Czapliński A, Maksymowicz W, Wojtkiewicz J. Stem cells for ALS: an overview of possible therapeutic approaches. Int J Dev Neurosci 2017; 57:46–55. [DOI] [PubMed] [Google Scholar]
- 140. Gros‐Louis F, Soucy G, Larivière R, Julien JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem 2010; 113:1188–99. [DOI] [PubMed] [Google Scholar]
- 141. Pearn ML, Niesman IR, Egawa J, Sawada A, Almenar‐Queralt A, Shah SB et al Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell Mol Neurobiol 2017; 37:571–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Fong H, Tong LM, Huang Y. Stem cell therapy for Alzheimer's disease and related disorders: current status and future perspectives. Exp Mol Med 2015; 47:e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Shan Z, Hirai Y, Nakayama M, Hayashi R, Yamasaki T, Hasebe R et al Therapeutic effect of autologous compact bone‐derived mesenchymal stem cell transplantation on prion disease. J Gen Virol 2017; 98:2615–27. [DOI] [PubMed] [Google Scholar]
- 144. Bhasin A, Srivastava MP, Mohanty S, Bhatia R, Kumaran SS, Bose S. Stem cell therapy: a clinical trial of stroke. Clin Neurol Neurosurg 2013; 115:1003–8. [DOI] [PubMed] [Google Scholar]
- 145. Budni JL, Garcez M, Medeiros JD, Cassaro E, Bellettini‐Santos T, Mina F et al The anti‐inflammatory role of minocycline in Alzheimer′s disease. Curr Alzheimer Res 2016; 13:1319–29. [DOI] [PubMed] [Google Scholar]
- 146. Orsucci D, Calsolaro V, Mancuso M, Siciliano G. Neuroprotective effects of tetracyclines: molecular targets, animal models and human disease. CNS Neurol Disord Drug Targets 2009; 8:222–31. [DOI] [PubMed] [Google Scholar]
- 147. Fattore L, ed. Cannabinoids in Neurologic and Mental Disease. United States of Amserica: Academic Press,2015. [Google Scholar]
- 148. Pryce G, Riddall DR, Selwood DL, Giovannoni G, Baker D. Neuroprotection in experimental autoimmune encephalomyelitis and progressive multiple sclerosis by cannabis‐based cannabinoids. J Neuroimmune Pharmacol 2015; 10:281–92. [DOI] [PubMed] [Google Scholar]
- 149. Habib SS, Al‐Regaiey K, Bashir S, Iqbal M. Role of endocannabinoids on neuroinflammation in autism spectrum disorder prevention. J Clin Diagn Res 2017; 11:CE01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Martin K, Jackson CF, Levy RG, Cooper PN. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst Rev 2016; 2:CD001903. [DOI] [PubMed] [Google Scholar]
- 151. Ngo ST, Steyn FJ, McCombe PA. Body mass index and dietary intervention: implications for prognosis of amyotrophic lateral sclerosis. J Neurol Sci 2014; 340:5–12. [DOI] [PubMed] [Google Scholar]
- 152. Otaegui‐Arrazola A, Amiano P, Elbusto A, Urdaneta E, Martínez‐Lage P. Diet, cognition, and Alzheimer's disease: food for thought. Eur J Nutr 2014; 53:1–23. [DOI] [PubMed] [Google Scholar]
- 153. Perez‐Pardo P, Dodiya HB, Broersen LM, Douna H, van Wijk N, Lopes da Silva S et al Gut–brain and brain–gut axis in Parkinson's disease models: effects of a uridine and fish oil diet. Nutr Neurosci 2017; https://doi.org/10.1080/1028415X.2017.1294555. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 154. Komori T. The significance of proinflammatory cytokines and Th1/Th2 balance in depression and action of antidepressants. Neuropsychiatry 2017; 7:57–60. [Google Scholar]
- 155. Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM et al Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion‐infected mice. Sci Transl Med 2013; 5:206ra138. [DOI] [PubMed] [Google Scholar]
- 156. Berkovic SF. Epilepsy research in 2016: new treatment directions. Lancet Neurol 2017; 16:7. [DOI] [PubMed] [Google Scholar]
- 157. Yang AC, Tsai SJ. New targets for schizophrenia treatment beyond the dopamine hypothesis. Int J Mol Sci 2017; 18:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Rosenblat JD, Kakar R, Berk M, Kessing LV, Vinberg M, Baune BT et al Anti‐inflammatory agents in the treatment of bipolar depression: a systematic review and meta‐analysis. Bipolar Disord 2016; 18:89–101. [DOI] [PubMed] [Google Scholar]
- 159. Williams UE, Philip‐Ephraim EE, Oparah SK. Multidisciplinary interventions in motor neuron disease. J Neurodegener Dis 2014; 2014:435 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Cai Z, Liu N, Wang C, Qin B, Zhou Y, Xiao M et al Role of RAGE in Alzheimer's disease. Cell Mol Neurobiol 2016; 36:483–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Alawieh A, Tomlinson S. Injury site‐specific targeting of complement inhibitors for treating stroke. Immunol Rev 2016; 274:270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Fluiter K, Opperhuizen AL, Morgan BP, Baas F, Ramaglia V. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J Immunol 2014; 192:2339–48. [DOI] [PubMed] [Google Scholar]
- 163. Liguori C, Romigi A, Izzi F, Placidi F, Nuccetelli M, Cordella A et al Complement system dysregulation in patients affected by idiopathic generalized epilepsy and the effect of antiepileptic treatment. Epilepsy Res 2017; 137:107–11. [DOI] [PubMed] [Google Scholar]
- 164. Dey A, Kang X, Qiu J, Du Y, Jiang J. Anti‐inflammatory small molecules to treat seizures and epilepsy: from bench to bedside. Trends Pharmacol Sci 2016; 37:463–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Zhao P, Yang X, Yang L, Li M, Wood K, Liu Q et al Neuroprotective effects of fingolimod in mouse models of Parkinson's disease. FASEB J 2017; 31:172–9. [DOI] [PubMed] [Google Scholar]
- 166. Lundberg C, Bjorklund T, Carlsson T, Jakobsson J, Hantraye P, Déglon N et al Applications of lentiviral vectors for biology and gene therapy of neurological disorders. Curr Gene Ther 2008; 8:461–73. [DOI] [PubMed] [Google Scholar]
- 167. Safety T. Pharmacokinetics and pharmacodynamics of IONIS‐HTTRx in patients with early manifest Huntington's disease. NCT02519036. 2015.
- 168. Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ et al Hematopoietic stem‐cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med 2017; 377:1630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. van Noort JM, Bsibsi M, Nacken PJ, Verbeek R, Venneker EH. Therapeutic intervention in multiple sclerosis with alpha B‐crystallin: a randomized controlled phase IIa trial. PLoS ONE 2015; 10:e0143366. [DOI] [PMC free article] [PubMed] [Google Scholar]