

Abstract
Neuropathic pain is a complex and debilitating syndrome for which there are few effective pharmacological treatments. Opioid‐based medications are initially effective for acute pain, but tolerance to their analgesic effects quickly develops, and long‐term use often leads to physical dependence and addiction. Furthermore, neuropathic pain is generally resistant to non‐steroidal anti‐inflammatory drugs. Other classes of medications including antidepressants, antiepileptics and voltage‐gated calcium channel inhibitors are only partially effective in most patients, may be associated with significant side effects and have few disease‐modifying effects on the underlying pathology. Medications that act through new mechanisms of action, and particularly ones that have disease‐modifying properties, would be highly desirable. In the last decade, a potential new target for the treatment of neuropathic pain has emerged: the α9‐containing nicotinic acetylcholine receptor (nAChR). Recent studies indicate that antagonists of α9‐containing nAChRs are analgesic in animal models of neuropathic pain. These nerve injury models include chronic constriction injury, partial sciatic nerve ligation, streptozotocin‐induced diabetic neuropathy and chemotherapeutic‐induced neuropathy. This review details the history and state of the field regarding the role that α9‐containing nAChRs may play in neuropathic pain. An alternative hypothesis that α‐conotoxins exert their therapeutic effect through blocking N‐type calcium channels via activation of GABAB receptors is also reviewed. Understanding how antagonists of α9‐containing nAChRs exert their therapeutic effects may ultimately result in the development of medications that not only treat but also prevent the development of neuropathic pain states.
Linked Articles
This article is part of a themed section on Nicotinic Acetylcholine Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.11/issuetoc
Abbreviations
- CCI
chronic constriction injury
- DRG
dorsal root ganglia
- KO
knockout
- nAChR
nicotinic ACh receptor
- PSNL
partial sciatic nerve ligation
- VGCC
voltage‐gated calcium channel
- WT
wild type
- α‐Ctx
α‐conotoxin
Introduction
Compounds that target http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76 have been investigated for some time as analgesics, and almost all have been agonists or positive allosteric modulators of α4β2 or α7 nAChRs (Umana et al., 2013; Uteshev, 2014; Dineley et al., 2015). Although ligands that activate http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=468&familyId=76&familyType=IC are hypothesized to be analgesic and anti‐inflammatory (de Jonge and Ulloa, 2007; Medhurst et al., 2008; Feuerbach et al., 2009), positive findings with α7 agonists have not always been observed (Wang et al., 2005; Freitas et al., 2015). The α7‐ and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=469&familyId=76&familyType=IC share a similar pharmacological profile, and therefore, one potential complication is that agonists of α7 may not be selective but instead may also activate α9α10 nAChRs. For example, the prototypical agonist of α7 nAChRs, choline, also activates α9‐containing subtypes (Verbitsky et al., 2000; Sgard et al., 2002). Intriguingly, new data indicate that nAChR antagonists may also be analgesic, including those that target α9α10 nAChRs (Vincler et al., 2006; Vincler and McIntosh, 2007; Del Bufalo et al., 2014). Accordingly, stimulation of α7 or inhibition of α9α10 nAChRs may produce analgesia and thus agonists that do not discriminate between α7 and α9α10 may have mutually counteracting effects. Studies using such agonists may underestimate potential beneficial effects. Therefore, the functional activity and subtype selectivity of candidate therapeutic compounds may be critical.
nAChRs containing the α9 subunit are a recently identified subtype (Elgoyhen et al., 1994). The first demonstration of a physiological role for α9‐containing nAChRs was described in studies of the cochlea where they are expressed in hair cells and are functionally coupled to calcium‐activated potassium channels (Vetter et al., 1999; Glowatzki and Fuchs, 2000; Nie et al., 2004). Shortly after the discovery of α9, an additional nicotinic subunit was identified that is now known as α10 (Elgoyhen et al., 2001; Lustig et al., 2001; Sgard et al., 2002). These two nicotinic subunits share a high sequence homology, and their expression patterns overlap considerably. However, in contrast to the α9 subunit, mammalian α10 subunits have not been shown to form functional receptors by themselves but do assemble with α9 subunits to form α9α10 heteromers (Elgoyhen et al., 2001; Sgard et al., 2002).
The presence of α9α10 nAChRs in immune cells has led to the hypothesis that this subtype may be involved in immunological responses. Evidence for such a role has been documented in studies using experimental models of the autoimmune diseases Pemphigus vulgaris (Nguyen et al., 2000) and multiple sclerosis (Simard et al., 2013). Curiously, α9α10 nAChRs in immune cells do not appear to function as ionotropic receptors (Peng et al., 2004; Hecker et al., 2015; Richter et al., 2016) in contrast to their known functions in cochlear hair cells. Additionally, some experiments have reported the presence of transcripts for α9 and α10 subunits in rat DRG neurons, and it has been suggested that the expression of α9α10 nAChRs may be a universal feature of all DRG neurons (Lips et al., 2002). However, as in studies of immune cells, patch clamp electrophysiology experiments using cultured DRG neurons have failed to detect currents that could be attributed to α9α10 nAChRs (Rau et al., 2005; Hone et al., 2012). Transcripts for http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=470 subunits are consistently reported in DRG neurons, but detection of transcripts for α9 subunits is highly variable and, in some cases, absent (Lips et al., 2002; Haberberger et al., 2004; Lips et al., 2006; Callaghan and Adams, 2010; Hone et al., 2012). The significance of α10 subunit expression in the absence of α9 subunits is currently unknown, and therefore, whether or not DRG neurons express α9α10 nAChRs remains unclear. However, one possibility is that a receptor composed of α7 and α10 subunits may be present as reported in sympathetic neurons (Lips et al., 2006).
Discovery of the analgesic α‐conotoxin Vc1.1
A PCR screen of mRNAs present in the venom duct of the marine snail Conus victoriae identified a sequence encoding a 17 amino acid peptide belonging to a conopeptide subclass called α‐conotoxins (α‐Ctxs) (Sandall et al., 2003). This α‐Ctx, Vc1.1, was shown to be an antagonist of native α3β4*AChRs (asterisk denotes the possible presence of additional subunits) expressed in bovine adrenal chromaffin cells and most potently inhibited heterologously expressed rat http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=464, relative to other subtypes tested (Clark et al., 2006). Groundbreaking functional studies in rat models of neuropathic pain demonstrated that Vc1.1 was capable of reversing allodynia and accelerating the recovery of sensory nerves (Satkunanathan et al., 2005). Vc1.1 was also shown to reduce the excitability of human sural nerves via antagonism of α3β4* nAChRs (Lang et al., 2005). It is notable, however, that the concentrations required for inhibition of α3β4 nAChRs in these and other studies were in the μM range (Sandall et al., 2003; Clark et al., 2006), concentrations that are unlikely to be sustained during extended in vivo animal studies at the dosages used. This suggested that a molecular target other than α3‐containing nAChRs was responsible for the effects observed in rodent models of neuropathic pain. Subsequent studies examining the nAChR subtype selectivity of Vc1.1 revealed that rat α9α10 nAChRs heterologously expressed in oocytes were inhibited at concentrations >2000‐fold lower than the α3β4 subtype (Vincler et al., 2006). However, although the higher potency of Vc1.1 for inhibition of α9α10 nAChRs suggested that it could be a molecular target involved in the analgesic effects of Vc1.1, the lack of high nAChR subtype specificity prevented unequivocal assignment of the analgesic effects to the α9α10 subtype.
α‐Conotoxins RgIA and Vc1.1 are analgesic in rodent models of neuropathic pain
Advances in molecular cloning strategies rapidly accelerated the discovery of novel peptides from cone snails, and in 2006, a sequence isolated from a genomic DNA library from Conus regius was described that encoded an α‐Ctx now called http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4008 (Ellison et al., 2006). Importantly, RgIA was shown to be >1000‐fold more potent at inhibiting α9α10 than other nAChR subtypes, and its discovery offered the possibility of selectively studying the role of α9α10 nAChRs in neuropathic pain.
The discovery of RgIA led to the possibility of testing the hypothesis that antagonism of α9α10 nAChRs produced analgesic effects. In a side‐by‐side comparison of the activities of RgIA and Vc1.1 in rat, both peptides ameliorated neuropathic pain (Vincler et al., 2006). Daily administration of RgIA and Vc1.1 in rats subjected to chronic constriction injury (CCI) dose‐dependently inhibited mechanical hypersensitivity. Remarkably, these peptides also reduced the accumulation of immune cells at the site of injury. These studies confirmed and extended previous findings that Vc1.1 possessed analgesic properties (Satkunanathan et al., 2005) and were the first to suggest α9α10 nAChRs as a molecular target for treatment of neuropathic pain. RgIA was subsequently shown to prevent sciatic nerve damage when given to rats following CCI (Di Cesare Mannelli et al., 2014).
α‐Conotoxin RgIA is analgesic in oxaliplatin‐induced peripheral neuropathy
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7433 is an anticancer drug used in chemotherapy treatments of certain colorectal, ovarian and pancreatic cancers. A severe complication accompanying the use of this drug is the development of chemotherapy‐induced peripheral neuropathy. Currently, there are no effective treatments for the prevention of this type of neuropathic pain. Oxaliplatin and other chemotherapeutics are used in animal models of peripheral neuropathy (Flatters and Bennett, 2004; Xiao et al., 2012). Rats administered oxaliplatin show evidence of peripheral neuropathy indicated by hypersensitivity to mechanical stimuli and a decreased tolerance to non‐noxious cold. Oxaliplatin‐based drugs have also been shown to produce morphological changes in DRG neurons similar to those observed in the DRG of rats subjected to CCI. In this model, daily intramuscular administration of 3.1 or 15.7 μg of RgIA in rats increased the pain threshold back to control levels and significantly reduced oxaliplatin‐induced sensitivity to non‐noxious stimuli (Pacini et al., 2016). Additionally, pathological changes in the morphological characteristics of DRG neurons were reduced by administration of RgIA.
Development of an RgIA analogue with high potency at human α9α10 nAChRs
Although RgIA is effective in rodent models of neuropathic pain, its utility in humans is likely to be limited due to the fact that it is orders of magnitude less potent on human compared to rat α9α10 nAChRs (Azam and McIntosh, 2012). However, one advantage of using α‐Ctx peptides as a platform for ligand development is that analogues can be generated with increased potency and specificity for particular receptor subtypes of interest. This can be accomplished through various techniques including systematically mutating residues, replacing cysteine residues with non‐standard amino acids such as selenocysteine, replacing the sulfur bridges between cysteines with dicarba bridges and by backbone cyclization (McIntosh et al., 2004; Halai et al., 2009; 2011; Clark et al., 2010; Carstens et al., 2011; 2016a,b; Daly et al., 2011; Hone et al., 2013; Chhabra et al., 2014; Yu et al., 2015). A structure–activity relationship study was conducted with RgIA to generate analogues with increased potency for human α9α10 nAChRs. These experiments generated a number of analogues that displayed ~10 000‐fold increase in potency for the human receptor (Romero et al., 2017). One of these analogues, RgIA4, was further tested for activity on a panel of receptors, ion channels and transporters and was found to have relatively very low or no activity on non‐α9α10 nAChR targets. RgIA4 was also demonstrated to be active in animal models of neuropathic pain. In vivo testing in the oxaliplatin model of peripheral neuropathy showed that daily injections of RgIA4 prevented the expression of cold allodynia and mechanical hyperalgesia in rats. RgIA4 also prevented the expression of oxaliplatin‐induced cold allodynia in wild‐type (WT) (CBA/CaJ) mice but did not attenuate neuropathic pain behaviours in CHRNA9 gene knockout (α9 KO) mice, indicating that α9‐containing nAChRs were necessary for the therapeutic effects of RgIA4.
CHRNA9 KO mice show attenuated pain expression in traumatic, inflammatory and chemotherapy‐induced pain models
In addition to pharmacological studies demonstrating that antagonism of α9α10 nAChRs reduces symptoms of neuropathic pain and neuropathy induced by physical or chemical nerve injury, studies in α9 KO mice have implicated α9‐containing nAChRs in the involvement of neuropathic pain. In a recent study by Mohammadi and Christie (2014), WT (129Sv/Ae) and α9 KO mice were subjected to CCI to model neuropathic pain or injected in the hind paw with complete Freund's adjuvant (CFA) to model inflammatory pain. α9 KO mice displayed normal responses to noxious mechanical and thermal stimuli. Chronic cold mechanical allodynia also developed normally in both models. However, α9 KO mice showed a distinct phenotype in the development of mechanical hyperalgesia. WT mice subjected to CCI continued to display elevated mechanical hyperalgesia 21 days after injury, whereas α9 KO mice showed substantially reduced hyperalgesia by day 14 following CCI surgery. Furthermore, the overall magnitude of hyperalgesia was significantly blunted in α9 KO mice in both CCI and CFA models (Mohammadi and Christie, 2014). Overall, however, the phenotype exhibited by the α9 KO mice significantly differed from the analgesic effects observed after injection of Vc1.1 or RgIA calling into question the mechanistic importance of the α9 subunit (Mohammadi and Christie, 2015).
A recent report utilizing this same mouse strain (129Sv/Ev) also showed behavioural differences in α9 subunit null mice. These alterations included differences in stress‐induced arousal, measures of anhedonia‐ and anxiety‐like behaviour, circadian patterns of activity and corticosterone responses (Mohammadi et al., 2017). Such changes could affect pain‐related behaviour. Functional α9α10 nAChRs have been demonstrated in rat adrenal medulla, and transcripts have been reported in the pituitary gland (Elgoyhen et al., 1994; Colomer et al., 2010). Therefore, differences in the hypothalamic–pituitary–adrenal axis may underlie such effects. Behavioural changes were not reported in human clinical trials of the α9α10 nAChR antagonist Vc1.1 (AdisInsight, 2016). However, decreased potency of Vc1.1 for the human α9α10 nAChR could have obscured potential findings. Separate studies, in a different strain of α9 KO mice (CBA/CaJ), have been conducted in the oxaliplatin model of peripheral neuropathy. Oxaliplatin produced robust cold allodynia in WT mice, but the expression of cold‐allodynia in α9 KO mice was attenuated in this strain in both magnitude and symptom duration (Romero et al., 2017). By 72 h post oxaliplatin injection, WT mice, but not α9 KO mice, exhibited pronounced cold allodynia.
Together, these studies suggest that germline deletion of α9‐containing nAChRs per se can attenuate the development and progression of some aspects of neuropathic pain. However, in studies of germline‐deleted genes, developmental compensation often alters aspects of the mutant mouse phenotype compared with acute pharmacological silencing of function in adult WT animals. Therefore, a close phenotypic correlation between KO mice and acute pharmacological treatment in WT animals may or may not occur.
Potential disease‐modifying effects
A particularly interesting aspect of the compounds discussed in this review is their capacity to produce long‐lasting effects. Pivotal work by Khalil and colleagues demonstrated that administration of Vc1.1 not only produced analgesia but also produced long‐lasting effects (Satkunanathan et al., 2005). Vc1.1 attenuated mechanical hyperalgesia after CCI or partial sciatic nerve ligation (PSNL), and these analgesic effects persisted for 1 week (last time point measured) following cessation of treatment. In addition, functional recovery of the sciatic nerve has been demonstrated (normalization of vascular response to substance P). Long‐lasting reductions in pain responses produced by α‐Ctxs have been measured in subsequent studies. Preservation of nerve morphology after CCI was observed and is consistent with disease‐modifying effects (Di Cesare Mannelli et al., 2014). Remarkably, the apparent disease‐modifying effects of α9α10 antagonists are not limited to traumatic nerve injury. Vc1.1 has also been shown to attenuate established neuropathic pain in streptozotocin‐induced diabetic rats. In these studies, Vc1.1 produced a progressive improvement in tactile allodynia and mechanical hyperalgesia that persisted for 10 days (last time points measured) after cessation of drug treatment (Metabolic, 2006; McIntosh et al., 2009). In addition, α9α10 antagonists have been shown to prevent chemotherapy‐induced neuropathic pain (Wala et al., 2012; Pacini et al., 2016; Romero et al., 2017). The effects of RgIA4 lasted 72 h post injection (last time point examined), a notable result given that RgIA4 is a peptide (Romero et al., 2017). A common drawback of peptides as drug candidates is their susceptibility to degradation by proteolytic enzymes. Peptide pharmacokinetics are typified by short bloodstream half‐lives, often in the order of min, leading to relatively brief biological effects (Diao and Meibohm, 2013). Vc1.1 and RgIA4 have plasma half‐lives of 0.5–3 h and <20 min respectively (Metabolic, 2006; Mercado et al., 2014). Thus, the pharmacokinetic profiles of Vc1.1 and RgIA are consistent with the time course of the acute pharmacodynamic effect observed but do not fit with the extended pharmacodynamic effects observed in animal studies, which suggest potential restorative properties. The long‐term effects on neuropathic pain produced by Vc1.1 and RgIA might be influenced by acute modulation of immune cell infiltration into the site of injury and the release of inflammatory substances by these cells. Evidence for the acute modulatory effects of α‐Ctxs has been observed in the CFA‐induced inflammatory pain model where a single dose of Vc1.1 attenuated mechanical hyperalgesia albeit only at a high dose of 2.4 mg·kg−1 (Metabolic, 2006). These results suggest that Vc1.1 may be less effective in altering CFA‐induced cutaneous inflammation than in modifying nerve injury‐induced immune responses. The respective immune mediated pathways for inflammatory and neuropathic pain remain active areas of investigation (Pavlov et al., 2003; Ren and Dubner, 2010; McMahon et al., 2015) but probably differ in key elements that might explain the differences in α‐Ctx potency in the different pain models.
Rat sciatic nerves subjected to CCI show a number of morphological changes that indicate Wallerian degeneration including oedema, axon demyelination and degeneration, and also show increased infiltration of immune cells into the site of injury (Basbaum et al., 1991; Nuytten et al., 1992; Sommer et al., 1995). Additionally, the cell bodies of these axons, located in the DRG, show signs of damage including oedema and eccentrically located nucleoli. Animals subjected to CCI and treated with RgIA showed largely preserved nerve morphology including a higher number of nerve fibres, increased myelin thickness and axon diameter, and a general reduction in oedema compared with control animals receiving injections of vehicle alone (Di Cesare Mannelli et al., 2014). Furthermore, a significant reduction in the number of activated macrophages (CD86+) was observed in the nerve. Both Vc1.1 and RgIA have been shown to reduce immune cell infiltration into CCI‐injured nerves (Vincler et al., 2006). In the oxaliplatin model of peripheral neuropathy, RgIA reduced oxaliplatin‐induced morphological changes in nucleoli of DRG neurons. CNS glial cells have been shown to modulate pain. RgIA reduced the number of microglial cells (Iba+) present in the dorsal horn of the spinal column of oxaliplatin‐treated rats; RgIA alone increased microglia in brain (Pacini et al., 2016). Additional studies are needed to further elucidate the mechanism of how Vc1.1 and RgIA produce these disease‐modifying effects.
Expression of α9α10 nAChRs in immune cells
Accumulating evidence indicates that immune cells play an important role in the development of neuropathic pain following injury to nerves (Grace et al., 2014; Ji et al., 2016; Peng et al., 2016). α9 and α10 subunits have consistently been reported to be expressed by a variety of immune cells (Peng et al., 2004; Biallas et al., 2007; Chernyavsky et al., 2010; Koval et al., 2011; Hecker et al., 2015; Richter et al., 2016). In nerve injury models of neuropathic pain, immune cells infiltrate the damaged nerve where they release a variety of cytokines, chemokines and growth factors which may promote inflammation and sensitize nerves to nociceptive stimuli (Leskovar et al., 2000; George et al., 2004; Hendriks et al., 2005). A reduction in immune cell infiltrate into nerves is associated with attenuated inflammation as well as disease‐modifying effects including delayed or reduced Wallerian degeneration (Liu et al., 2000). Additionally, athymic nude rats that lack the ability to produce mature T‐lymphocytes show significantly attenuated neuropathic pain symptoms following CCI injury (Moalem et al., 2004). Vc1.1 and RgIA have been demonstrated to reduce the number of immune cells infiltrating into sciatic nerves and DRG that have been subjected to CCI or PSNL injuries (Vincler et al., 2006; Di Cesare Mannelli et al., 2014). These cell types include ED1+ and CD86+ macrophages, CD2+ lymphocytes and cells positive for choline acetyltransferase (Vincler et al., 2006). Collectively, these observations support the possibility that antagonism of α9α10 nAChRs expressed by immune cells may be a mechanism through which α9α10 nAChR antagonists exert their analgesic and anti‐inflammatory properties. However, the function of α9α10 nAChRs in immune cells has been difficult to demonstrate. Patch clamp studies have failed to detect nAChR‐mediated currents in these cells (Peng et al., 2004; Hecker et al., 2015). Recently, however, function for immune cell α9α10* nAChRs has been convincingly demonstrated. Stimulation of purine http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=77 in the human monocyte cell line U937 leads to the release of the cytokine http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974, and nAChR agonists inhibit this release (Hecker et al., 2015; Richter et al., 2016). In these studies, a combination of tools including siRNA, the α9α10 antagonist RgIA4, and α9 or α10 nAChR subunit KO mice indicated that an α9α10* nAChR modulates P2X receptor function and cytokine release in these cells. Interestingly, the pharmacology and biophysical properties of these monocyte‐expressed α9α10 nAChRs is distinct from that of heterologously expressed α9α10 nAChRs or those present in cochlear hair cells. In U937 cells, stimulation with nicotinic agonists does not evoke ionic current. Furthermore, nicotine appears to behave as an agonist of α9α10* nAChRs in contrast to the known activity of this ligand in other expression systems (Elgoyhen et al., 1994; Elgoyhen et al., 2001). It is currently not known whether these features of monocyte‐expressed α9α10 nAChRs are generalizable to other immune cells. Additional studies examining the functional role of α9α10 nAChRs in other immune cells and in particular those that invade damaged neural tissue during injury are needed to better understand the role of immune cell‐expressed α9α10 nAChRs in neuropathic pain.
An alternative hypothesis for the mechanism of α‐contoxins and their analogues
Several studies of a subset of α‐Ctxs have postulated the analgesic mechanism of action to be dependent on activation of G‐protein coupled http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=26 (Callaghan et al., 2008; Callaghan and Adams, 2010; Klimis et al., 2011; Cuny et al., 2012; van Lierop et al., 2013; Berecki et al., 2014; Huynh et al., 2015; Castro et al., 2017). Studies examining the activity profile of Vc1.1 and its analogues raised questions as to whether α9α10 nAChRs were the actual target that mediated the analgesic effects of Vc1.1. Vc1.1 is a synthetic version of the natural peptide found in Conus victoriae. The natural peptide (vc1a, also known as Vc1.1 ptm) contains two post‐translational modifications not present in synthetic Vc1.1 (hydroxyproline at position 6 and γ‐carboxyglutamate at position 14). When tested on rat α9α10 nAChRs expressed in oocytes, Vc1.1, the native peptide vc1a and the two Vc1.1 analogues Vc1.1(P6O) and Vc1.1(E14γ) each inhibited ACh‐induced responses with similar potencies (Nevin et al., 2007). However, when tested at high doses (60 μg per rat) for their ability to produce analgesia in the PSNL model of neuropathic pain in rats, only Vc1.1 showed effects on mechanical allodynia. These results suggested that activity at α9α10 nAChRs was not necessary for the analgesic effects of Vc1.1 and are consistent with those of Livett et al., who also reported decreased or a lack of effects of vc1a on mechanical thresholds in CCI rats after low‐dose (0.37 μg per rat) administration. In contrast, low doses of both Vc1.1 (0.36 μg per animal) and vc1a (0.37 μg per animal) accelerated the functional recovery of injured sensory nerves (Livett et al., 2008). This was demonstrated using an assay that measured the ability of injured sciatic nerve terminals to produce an inflammatory vascular response following exposure to substance P. Vc1.1 showed an 83% recovery compared with controls, while vc1a showed an 85% recovery (Satkunanathan et al., 2005; Livett et al., 2008). Thus, in this vascular response assay, activity at GABAB receptors was not necessary for effect since vc1a lacks GABAB receptor agonist activity (Callaghan et al., 2008).
http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=80 are present in sensory neurons of DRG and are functionally coupled to GABAB receptors. Activation of GABAB receptors by the agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1084 has been shown to inhibit the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=533 VGCC subtype via G‐protein coupled mechanisms. This inhibition in turn decreases neurotransmitter release and synaptic transmission between DRG neurons and second‐order neurons in the spinal column and thus impairs the transmission of nociceptive information to the brain. Evidence supporting the agonist activity of α‐Ctxs on GABAB receptors comes from studies examining their effects on VGCC currents in cultured DRG neurons. In an initial study by Callaghan et al., Vc1.1 and RgIA were shown to be capable of inhibiting VGCC currents in DRG neurons, an effect that was prevented by competitive GABAB receptor antagonists (Callaghan et al., 2008). These inhibitory effects on VGCC currents were not observed when vc1a, Vc1.1(P6O), or Vc1.1(E14γ) were tested in the same assays. Inhibition of Cav2.2 channel currents by Vc1.1 could also be prevented by inclusion of a tyrosine kinase inhibitor in the patch pipette or by pertussis toxin, suggesting a G‐protein coupled mechanism of VGCC inhibition. Vc1.1 also lacked direct inhibitory activity on VGCCs heterologously expressed in oocytes. Thus, it is possible that acute analgesic effects of some α‐Ctxs are mediated by activation of GABAB receptors. In further support of this mechanism, three α‐Ctxs (Vc1.1, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3973 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3970) with dissimilar pharmacological profiles were compared. α‐Ctx Vc1.1 has 1.5 nM potency for partial inhibition of VGCC currents in DRG and 19–64 nM potency for α9α10 nAChRs (see Klimis et al., 2011, and the references therein). Vc1.1 potently reversed mechanical allodynia (rat PSNL model), an effect that was reversed by the GABAB receptor antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1075. AuIB displayed 1.5 nM potency for partial inhibition of VGCC currents in DRG but did not block α9α10 nAChRs expressed in oocytes. AuIB potently reversed mechanical allodynia with an IC50 of 1.88 (0.04–8.75) μg, indicating that activity at α9α10 nAChRs was not necessary for this analgesic effect (Klimis et al., 2011). AuIB also inhibits α3β4 (IC50 750 nM) and α6β4 (IC50 7.3 μM) nAChRs, subtypes known to be expressed by DRG neurons (Genzen et al., 2001; Rau et al., 2005; Hone et al., 2012; Smith et al., 2013). However, it seems unlikely that the high nM to low μM concentrations required for inhibition of α3β4 or α6β4 subtypes would be sustained in order to produce analgesia in vivo. Surprisingly, MII, which blocks α3β2 and α6‐containing nAChRs but does not block α9α10 nAChRs or activate GABAB receptors, was also analgesic, though the IC50, 9.2 (2.7–30.5) μg, was lower (but note overlapping confidence intervals with IC50 for AuIB) and the effects were not significant beyond 4 h. These results indicate that for MII, a mechanism other than activity at α9α10 nAChRs or GABAB receptors may also produce analgesic effects.
Multiple structure–function studies have been undertaken to further understand the selectivity of α‐Ctxs for GABAB receptors versus α9α10 nAChRs and derive significantly improved ligands (Halai et al., 2009; Carstens et al., 2011; 2016b Daly et al., 2011; Halai et al., 2011; Yu et al., 2015). For instance, dicarba analogues of Vc1.1 show different activity for these receptors (van Lierop et al., 2013). Synthetic cyclization was used to produce the analogue cVc1.1 that retains high potency for inhibiting VGCC currents and is orally active in the CCI model (Clark et al., 2010). In a separate structure–function study, a core peptide motif for inhibiting VGCC currents via GABAB receptors was identified that enabled development of the Vc1.1 analogue [Ser3]Vc1.1(1–8) (Carstens et al., 2016b). [Ser3]Vc1.1(1–8) inhibited VGCC currents in rat and mouse DRG neurons, an effect prevented by the GABAB receptor antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1088. This analogue failed to inhibit human α9α10 nAChRs at 1 μM yet fully blocked human α7 nAChRs at this same concentration. In a mouse model of visceral pain, intracolonic [Ser3]Vc1.1(1–8) reduced the visceromotor response to colorectal distension (Carstens et al., 2016a), further supporting the idea that GABAB receptors rather than α9α10 nAChRs produce an analgesic effect. As the parent peptide Vc1.1 is known to be much more potent on rat versus human α9α10 nAChRs, it would be of interest to determine the activity of [Ser3]Vc1.1(1–8) on rat α9α10 nAChRs.
Separately, studies with conditional KO mice call into question the importance of peripheral versus CNS GABAB receptors in neuropathic pain. The http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=240 subunit is required for GABAB receptor function. Conditional KO mice that specifically lack the GABAB1 subunit in sensory nociceptors in DRG but retain GABAB1 in spinal cord and brain were generated. Notably, neuropathic pain (spared nerve injury model) developed normally in these animals (Gangadharan et al., 2009), and thus, the pain phenotype seems to differ from that seen after administration of putative α‐Ctx GABAB receptor agonists in WT mice. Therefore, it is unclear whether α‐Ctxs agonism of the GABAB receptors expressed by nociceptive DRG neurons would alone be sufficient for the attenuation of neuropathic pain.
Lastly, although some antagonists of GABAB receptors prevent α‐Ctx inhibition of VGCCs, competition binding studies with Vc1.1 and RgIA failed to demonstrate displacement of the competitive GABAB receptor antagonist [3H]‐CGP54626 (McIntosh et al., 2009). Subsequently, it was proposed that Vc1.1 acts at an allosteric site on the GABAB receptors to produce agonist activity (Adams et al., 2012), and receptor mutation studies are consistent with this mechanism (Huynh et al., 2015).
A separate difficulty with the GABAB receptor hypothesis is that the functional effects of the clinically available GABAB receptor agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1084 do not closely resemble the effects of α‐Ctxs. Baclofen has shown limited efficacy in the treatment of neuropathic pain (Loubser and Akman, 1996). In addition, the effects that are observed are thought to be centrally mediated (Bowery, 2006; Goudet et al., 2009) (necessitating intrathecal administration in humans). In contrast, α‐Ctxs are effective following peripheral administration; if they were acting on GABAB receptors, the tissue site would probably be peripheral, since they are unlikely to efficiently cross the blood‐brain‐barrier. Furthermore, tolerance to baclofen's therapeutic effects is problematic. In contrast, tolerance to the therapeutic effects of α‐Ctxs has not been observed.
Variable agonist effects of α‐conotoxins on GABAB receptors
α‐Ctxs Vc1.1 and RgIA have been reported to produce inhibition of VGCCs in rat DRG sensory neurons (Figure 1A, B) (Callaghan et al., 2008; Callaghan and Adams, 2010; Klimis et al., 2011). However, for no obvious reasons, agonist activity at GABAB receptors has not been observed in a variety of other experimental conditions. In the work by Wright et al. (2015), RgIA failed to inhibit VGCC currents in DRG neurons (Wright et al., 2015). Vc1.1, however, produced a variable (on average, small) and reversible inhibition of VGCC currents in a minority of DRG neurons. The magnitude of VGCC current inhibition by Vc1.1 did not correlate with the inhibition produced by baclofen, making it unlikely that the observed effect was mediated by GABAB receptors. Furthermore, Vc1.1 has also been shown to lack effects on excitatory post‐synaptic currents (EPSCs) in spinal cord neurons (Napier et al., 2012). Dorsal horn neurons are excited by glutamate released from DRG afferents that express GABAB receptors. Activation of presynaptic GABAB receptors by baclofen inhibits EPSCs and prevents the release of glutamate and, consequently, transmission of nociceptive information from the periphery to the brain. In contrast to the results obtained with baclofen, Vc1.1 failed to inhibit these EPSCs, suggesting a lack of activity on GABAB receptors (Figure 1C, D). It is difficult to reconcile these findings with the hypothesis that α‐Ctxs produce analgesia via inhibition of DRG neuron‐expressed VGCCs when synaptic transmission at this synapse is unaffected by Vc1.1.
Figure 1.
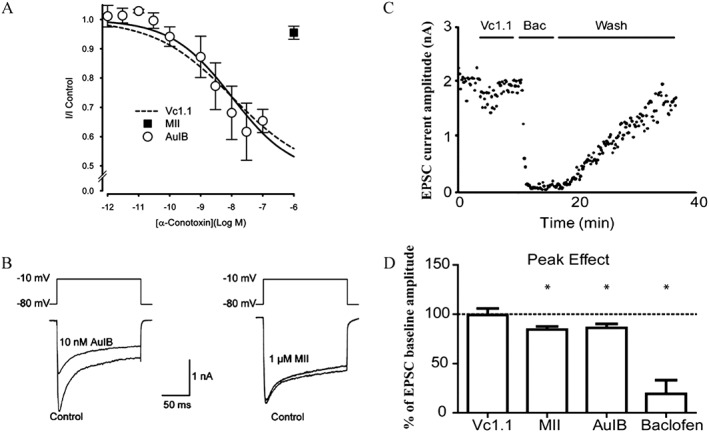
Effects of analgesic α‐Ctxs on VGCC currents in DRG neurons and EPSCs in spinal cord neurons. An alternative hypothesis is that α‐Ctxs exert their analgesic effects by inhibition of VGCCs via activation of GABAB receptors rather than by inhibition of α9α10 nAChRs. (A and B) Examples of experiments that demonstrate α‐Ctx activity on GABAB receptors, whereas (C) and (D) show experiments demonstrating a lack of α‐Ctx activity on GABAB receptors. (A) Concentration–response relationship for inhibition of VGCC currents by Vc1.1, AuIB and MII in cultured rat DRG neurons (figures used with permission of Klimis et al., 2011). Similar activity has been reported for analogues of Vc1.1 and for RgIA (see text for full discussion and references). One study did not replicate key findings for Vc1.1 or RgIA in DRG neurons (Wright et al., 2015) (data not shown). (B) AuIB, but not MII, inhibits VGCC currents in DRG neurons. These findings support the hypothesis that the analgesic effects of some α‐Ctxs are mediated through activation of GABAB receptors. However, Vc1.1 failed to inhibit EPSCs in spinal cord neurons via activation of presynaptic GABAB receptors on DRG afferents. (C) Electrophysiological recordings from rat spinal cord neurons in a slice preparation showing the effect of Vc1.1 and baclofen (Bac) on EPSC amplitudes. (D) Histogram of EPSCs in the presence of α‐Ctxs or baclofen compared with baseline controls. Note lack of effect of Vc1.1 and small inhibitory effects of AuIB and MII (the latter attributed to block of α3‐containing nAChRs). The GABAB receptor agonist baclofen is shown as a positive control (figures used with permission, Napier et al., 2012).
Agonism of GABAB receptors is known to produce activation of http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=74 via G protein‐coupled mechanisms (White et al., 1998). However, GABA, but neither Vc1.1 nor RgIA, activated heterologously expressed GABAB receptors coupled to Kir channels (McIntosh et al., 2009; Huynh et al., 2015) (Figure 2A, B). Furthermore, in two functional assays of GABAB receptors, RgIA4 failed to activate GABAB receptors (Figure 2C, D) (Romero et al., 2017). We also observed that Vc1.1, AuIB or RgIA had no agonist effects on GABAB receptors expressed in cell lines (unpublished observations).
Figure 2.
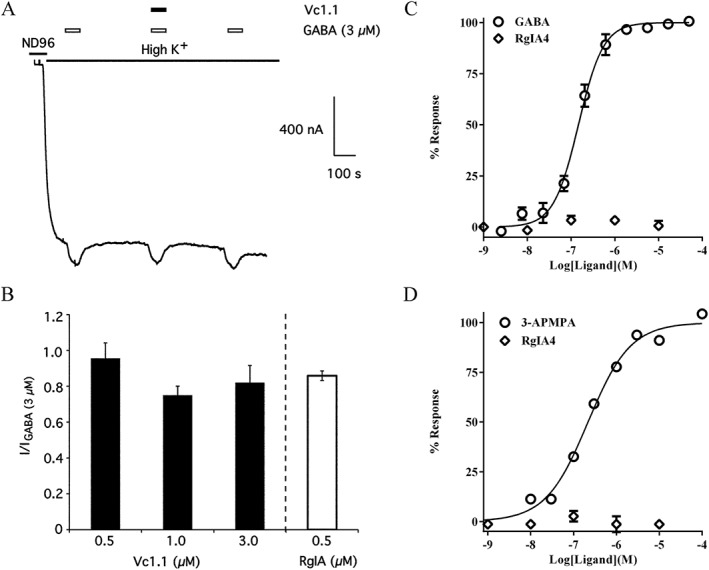
Analgesic α‐Ctxs Vc1.1, RgIA and RgIA4 fail to activate heterologously expressed GABAB receptors. α‐Ctxs have not shown agonist activity in a variety of functional assays designed to measure GABAB receptor activity. (A) Vc1.1 failed to activate human GABAB receptors functionally coupled to Kir (GIRK) channels expressed in Xenopus oocytes. The oocytes were stimulated with 45 mM K+ and then pulsed with GABA or GABA + Vc1.1 to stimulate GABAB receptor activation of Kir channels. Note that GABA, but not Vc1.1, increased K+ currents. Similar results were obtained with RgIA in the same assay. (B) Histogram showing the lack of effect of Vc1.1 and RgIA on Kir channel activation. (figures in A and B were used with permission of McIntosh et al., 2009). (C) Gi‐coupled cAMP modulation was assessed in CHO cells expressing GABAB1a/B2 receptors with Gi/o coupling. GABA produced an increase in cAMP response in a concentration‐dependent manner, whereas RgIA4 had no effect at any of the concentrations tested. (D) Cellular dielectric spectroscopy was used to assay changes in electrical impedance using CHO cells expressing human GABAB1a/B2 receptors during stimulation with the GABAB receptor agonist 3‐aminopropyl(methyl)phosphonic acid (3‐AMPA). 3‐AMPA produced changes in cellular impedance in a concentration‐dependent manner, whereas RgIA4 had no effect. (Figures in C and D were used with the permission of Romero et al., 2017).
Finally, extended duration of action and disease modification of several compounds should be considered in the context of the GABAB receptor hypothesis. The mechanism for producing the extended pharmacodynamics effects is unknown. N‐type calcium channel blockade is caused by agonist action on GABAB receptors (Zamponi and Currie, 2013) and has been proposed as the therapeutic mechanism for Vc1.1, Vc1.1 analogues, RgIA and AuIB (Klimis et al., 2011; Adams et al., 2012; Huynh et al., 2015). In this regard, it is noteworthy that in side‐by‐side trials with Vc1.1, peripheral administration of the N‐type calcium blocker http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2536 did not alter short‐term or long‐term mechanical pain thresholds in CCI rats and did not accelerate functional recovery of neurons (Livett et al., 2008). To our knowledge, in humans, neither the FDA‐approved MVIIA (ziconotide) nor the FDA‐approved GABAB receptor agonist baclofen has been associated with disease‐modifying effects in neuropathic pain conditions.
As reviewed above, a striking characteristic of ligands that inhibit α9α10 nAChRs is the capacity to prevent or modify the development of chronic neuropathic pain (Sandall et al., 2003; Satkunanathan et al., 2005; Livett et al., 2006; Vincler et al., 2006; Vincler and McIntosh, 2007; McIntosh et al., 2009; Di Cesare Mannelli et al., 2014; Luo et al., 2015; Li et al., 2016; Pacini et al., 2016; Romero et al., 2017). α‐Ctx derivatives that lack GABAB receptor activity show effects consistent with the modification of disease progression (Romero et al., 2017). In addition, small‐molecule antagonists of α9α10 nAChRs reverse or prevent the development of neuropathic pain (Holtman et al., 2011; Zheng et al., 2011; Wala et al., 2012). Furthermore, a new set of αO family Ctxs that inhibit α9α10 nAChRs was recently discovered (Luo et al., 2015). These peptides are structurally unrelated to α‐Ctxs and lack GABAB receptor activity yet are analgesic. Furthermore, the analgesia produced by the αO‐Ctx lasts at least 2 weeks after cessation of drug, consistent with possible disease‐modifying effects (Li et al., 2016). In contrast, known GABAB receptor agonists have not been shown to have similar properties. Thus, the acute analgesic action of some α‐Ctxs may be mediated by activity at GABAB receptors, although the lack of reproducibility across laboratories and/or systems renders this interpretation unclear. Multiple pharmacological families of compounds that inhibit α9α10 nAChRs have been shown to favourably alter the expression and/or maintenance of chronic pain in a variety of experimental paradigms. Evidence from α9 subunit null mice also supports a role for α9α10 nAChRs in attenuating the expression of neuropathic pain (Mohammadi and Christie, 2014; Romero et al., 2017).
Non‐α‐conotoxin antagonists of α9α10 nAChRs are analgesic
Studies demonstrating that Vc1.1 and RgIA produce analgesia in rodent models of neuropathic pain provided initial evidence for α9α10 nAChRs as an important target in the treatment of neuropathic pain syndromes. Since the initial studies with α‐Ctxs, small molecules and other selective antagonists of α9α10 nAChRs (tetrakis‐, tris‐ and bis‐azaaromatic quaternary ammonium salts) have been discovered and tested for their ability to produce effects similar to those observed with α‐Ctxs (Holtman et al., 2011; Zheng et al., 2011; Wala et al., 2012). One such molecule, ZZ‐204G has been shown to be effective at decreasing nociceptive behaviour and reducing mechanical hyperalgesia (Holtman et al., 2011). Another small molecule ZZ1‐61C is effective in reducing symptoms of peripheral neuropathy induced by the chemotherapeutic agent vincristine in rodents (Wala et al., 2012). More recently, a novel conopeptide belonging to the O1‐Ctx superfamily was discovered by a PCR screen of the venom duct from Conus generalis (Luo et al., 2015). A well‐known member of this superfamily is the VGCC antagonist ω‐Ctx MVIIA (FDA approved as ziconotide). However, in contrast to the potent inhibition of VGCCs by MVIIA, GeXIVA lacks inhibitory activity on rat DRG neuron‐expressed VGCCs. Instead, GeXIVA was observed to be a potent inhibitor of rat α9α10 nAChRs expressed in Xenopus oocytes. A closer examination of the activity of GeXIVA on α9α10 nAChRs revealed that the peptide binds to a site distinct from that of RgIA. Nevertheless, when tested in the rat CCI model of neuropathic pain, GeXIVA significantly reduced signs of mechanical hyperalgesia induced by CCI injury (Luo et al., 2015; Li et al., 2016). Taken together, evidence obtained from studies using α‐Ctxs, small molecule compounds and GeXIVA as receptor antagonists are consistent with an important role for α9α10 nAChRs in neuropathic pain. Table 1 summarizes the effects of these compounds in the various models of neuropathic pain.
Table 1.
Therapeutic effects of small molecule antagonists of α9α10 nAChRs, α‐Ctxs, and α‐Ctx analogues in neuropathic pain
| Ligand | Pain model | Therapeutic effects |
|---|---|---|
| RgIA | CCI | Reduced mechanical allodynia and mechanical hyperalgesia; disease‐modifying effectsa , b |
| Oxaliplatin‐induced neuropathy | Reduced mechanical allodynia and mechanical hyperalgesia; reduced cold allodynia; disease‐modifying effectsc | |
| RgIA4 | Oxaliplatin‐induced neuropathy | Reduced mechanical hyperalgesia; reduced cold allodynia; prolonged effectsd |
| Vc1.1 | CCI | Reduced mechanical allodynia and mechanical hyperalgesia; disease‐modifying effectsb , e |
| PSNL | Reduced mechanical allodynia and mechanical hyperalgesiae , f , g | |
| Streptozotocin‐induced neuropathy | Reduced mechanical allodyniah , i | |
| CVH | Reduced excitability of nociceptorsj | |
| vc1a | CCI | No effect on mechanical allodyniag; accelerated recovery of sensory nervesh |
| cVc1.1 | CCI | Reduced mechanical allodyniak |
| [Ser3]Vc1.1(1–8) | VMR | Reduced visceromotor responsel |
| AuIB | PSNL | Reduced mechanical allodyniaf , g |
| αO‐GeXIVA | CCI | Reduced mechanical allodyniam , n |
| ZZ‐204G | CCI | Reduced mechanical hyperalgesiao |
| Formalin test | Reduced inflammatory paino | |
| Tail flick | No effecto | |
| ZZ1‐61c | Vincristine‐induced neuropathy | Reduced mechanical allodynia and mechanical hyperalgesiap |
CVP, chronic visceral hypersensitivity; VMR, visceromotor response.
Mechanical allodynia measured with the Von Frey test; mechanical hyperalgesia measured with the paw pressure test.
Di Cesare Mannelli et al., 2014.
Vincler et al., 2006.
Pacini et al., 2016.
Romero et al., 2017.
Satkunanathan et al., 2005.
Napier et al., 2012.
Klimis et al., 2011.
Nevin et al., 2007.
Livett et al., 2008.
McIntosh et al., 2009.
Castro et al., 2017.
Clark et al., 2010.
Carstens et al., 2016a.
Luo et al., 2015.
Li et al., 2016.
Holtman et al., 2011.
Wala et al., 2012.
Summary and conclusions
In summary, three separate structural families of molecules that share the common feature of being selective antagonists of α9α10 nAChRs (α‐Ctxs, an O1‐superfamily of Ctxs and quaternary ammonium salts) have been identified. Members from each of these families have been shown to not only reverse pain but also prevent pain. As disease modification is not a common feature of analgesics, these effects support a role for α9* nAChRs. While we cannot rule out the possibility that each of these molecules also acts on some unidentified, yet therapeutic, target, the parsimonious explanation is that their common characteristic, inhibition of α9* nAChRs, is mechanistically important. Evidence from α9 subunit null mice further supports a role for α9* nAChRs; however, since these are germline deletions, rather than conditional KO mice, developmental compensation may affect both pain phenotype expression and response to compounds, particularly if complex neuroimmuno‐modulatory processes are involved. In contrast, multiple compelling studies indicate that one of these families, the α‐Ctxs, have GABAB receptor agonist activity and are analgesic. One member, AuIB, lacks activity at the α9α10 nAChR yet is analgesic, calling into question the necessity of activity at α9α10 nAChRs. Furthermore, analogues of α‐Ctxs that have α9α10 nAChR activity but lack GABAB receptor activity have diminished or lack analgesic activity. In the midst of these otherwise convincing data, the very activity of these compounds on GABAB receptors is inconsistent or absent across assays and laboratories somewhat diminishing the confidence in this explanation. In addition, the therapeutic profile of the one FDA‐approved GABAB receptor agonists, baclofen, does not closely resemble that of the α‐Ctxs with GABAB receptor activity. While there is clearly much yet to learn, as a group, these α9α10 antagonists not only produce analgesia, but perhaps more importantly, may also alter the course of disease progression, thereby averting sequela normally associated with nerve injury. These findings raise the possibility of developing therapeutics that may be given prophylactically to protect against the development of nerve injury‐induced neuropathic pain.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Conflict of interest
Conotoxins, including some of those referenced in this paper, and quaternary ammonium salts have been patented by the University of Utah, Hainan University or the University of Kentucky, or with J.M.M. listed as an inventor.
Acknowledgements
Some of the work summarized in this review were supported by NIH grants P01 GM48677 and R01 GM103801, U.S. Department of Defense grant W81XWH‐15‐2‐0057 and a sponsored research agreement from Kineta Inc. to J.M.M, and the European Research Council FP7 to D.S.
Hone, A. J. , Servent, D. , and McIntosh, J. M. (2018) α9‐containing nicotinic acetylcholine receptors and the modulation of pain. British Journal of Pharmacology, 175: 1915–1927. doi: 10.1111/bph.13931.
References
- Adams DJ, Callaghan B, Berecki G (2012). Analgesic conotoxins: block and G protein‐coupled receptor modulation of N‐type (Ca(V) 2.2) calcium channels. Br J Pharmacol 166: 486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AdisInsight (2016). ACV1. Available at: http://adisinsight.springer.com/drugs/800020787 (accessed 04/17/2017).
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, McIntosh JM (2012). Molecular basis for the differential sensitivity of rat and human alpha9alpha10 nAChRs to alpha‐conotoxin RgIA. J Neurochem 122: 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Gautron M, Jazat F, Mayes M, Guilbaud G (1991). The spectrum of fiber loss in a model of neuropathic pain in the rat: an electron microscopic study. Pain 47: 359–367. [DOI] [PubMed] [Google Scholar]
- Berecki G, McArthur JR, Cuny H, Clark RJ, Adams DJ (2014). Differential Cav2.1 and Cav2.3 channel inhibition by baclofen and alpha‐conotoxin Vc1.1 via GABAB receptor activation. J Gen Physiol 143: 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biallas S, Wilker S, Lips KS, Kummer W, Grando SA, Padberg W et al. (2007). Immunohistochemical detection of nicotinic acetylcholine receptor subunits alpha9 and alpha10 in rat lung isografts and allografts. Life Sci 80: 2286–2289. [DOI] [PubMed] [Google Scholar]
- Bowery NG (2006). GABAB receptor: a site of therapeutic benefit. Curr Opin Pharmacol 6: 37–43. [DOI] [PubMed] [Google Scholar]
- Callaghan B, Adams DJ (2010). Analgesic alpha‐conotoxins Vc1.1 and RgIA inhibit N‐type calcium channels in sensory neurons of alpha9 nicotinic receptor knockout mice. Channels (Austin) 4: 51–54. [DOI] [PubMed] [Google Scholar]
- Callaghan B, Haythornthwaite A, Berecki G, Clark RJ, Craik DJ, Adams DJ (2008). Analgesic alpha‐conotoxins Vc1.1 and Rg1A inhibit N‐type calcium channels in rat sensory neurons via GABAB receptor activation. J Neurosci 28: 10943–10951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens BB, Berecki G, Daniel JT, Lee HS, Jackson KA, Tae HS et al. (2016a). Structure‐activity studies of cysteine‐rich alpha‐conotoxins that inhibit high‐voltage‐activated calcium channels via GABA(B) receptor activation reveal a minimal functional motif. Angew Chem Int Ed Engl 55: 4692–4696. [DOI] [PubMed] [Google Scholar]
- Carstens BB, Clark RJ, Daly NL, Harvey PJ, Kaas Q, Craik DJ (2011). Engineering of conotoxins for the treatment of pain. Curr Pharm Des 17: 4242–4253. [DOI] [PubMed] [Google Scholar]
- Carstens BB, Swedberg J, Berecki G, Adams DJ, Craik DJ, Clark RJ (2016b). Effects of linker sequence modifications on the structure, stability, and biological activity of a cyclic alpha‐conotoxin. Biopolymers 106: 864–875. [DOI] [PubMed] [Google Scholar]
- Castro J, Harrington AM, Garcia‐Caraballo S, Maddern J, Grundy L, Zhang J et al. (2017). alpha‐Conotoxin Vc1.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABAB receptors. Gut 66: 1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyavsky AI, Arredondo J, Skok M, Grando SA (2010). Auto/paracrine control of inflammatory cytokines by acetylcholine in macrophage‐like U937 cells through nicotinic receptors. Int Immunopharmacol 10: 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra S, Belgi A, Bartels P, van Lierop BJ, Robinson SD, Kompella SN et al. (2014). Dicarba analogues of alpha‐conotoxin RgIA. Structure, stability, and activity at potential pain targets. J Med Chem 57: 9933–9944. [DOI] [PubMed] [Google Scholar]
- Clark RJ, Fischer H, Nevin ST, Adams DJ, Craik DJ (2006). The synthesis, structural characterization, and receptor specificity of the alpha‐conotoxin Vc1.1. J Biol Chem 281: 23254–23263. [DOI] [PubMed] [Google Scholar]
- Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ (2010). The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew Chem Int Ed Engl 49: 6545–6548. [DOI] [PubMed] [Google Scholar]
- Colomer C, Olivos‐Ore LA, Vincent A, McIntosh JM, Artalejo AR, Guerineau NC (2010). Functional characterization of alpha9‐containing cholinergic nicotinic receptors in the rat adrenal medulla: implication in stress‐induced functional plasticity. J Neurosci 30: 6732–6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuny H, de Faoite A, Huynh TG, Yasuda T, Berecki G, Adams DJ (2012). gamma‐Aminobutyric acid type B (GABAB) receptor expression is needed for inhibition of N‐type (Cav2.2) calcium channels by analgesic alpha‐conotoxins. J Biol Chem 287: 23948–23957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly NL, Callaghan B, Clark RJ, Nevin ST, Adams DJ, Craik DJ (2011). Structure and activity of alpha‐conotoxin PeIA at nicotinic acetylcholine receptor subtypes and GABA(B) receptor‐coupled N‐type calcium channels. J Biol Chem 286: 10233–10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bufalo A, Cesario A, Salinaro G, Fini M, Russo P (2014). Alpha9 alpha10 nicotinic acetylcholine receptors as target for the treatment of chronic pain. Curr Pharm Des 20: 6042–6047. [DOI] [PubMed] [Google Scholar]
- Di Cesare Mannelli L, Cinci L, Micheli L, Zanardelli M, Pacini A, McIntosh JM et al. (2014). alpha‐Conotoxin RgIA protects against the development of nerve injury‐induced chronic pain and prevents both neuronal and glial derangement. Pain 155: 1986–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao L, Meibohm B (2013). Pharmacokinetics and pharmacokinetic‐pharmacodynamic correlations of therapeutic peptides. Clin Pharmacokinet 52: 855–868. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Pandya AA, Yakel JL (2015). Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci 36: 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S (1994). Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79: 705–715. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J (2001). alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A 98: 3501–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison M, Haberlandt C, Gomez‐Casati ME, Watkins M, Elgoyhen AB, McIntosh JM et al. (2006). Alpha‐RgIA: a novel conotoxin that specifically and potently blocks the alpha9alpha10 nAChR. Biochemistry 45: 1511–1517. [DOI] [PubMed] [Google Scholar]
- Feuerbach D, Lingenhoehl K, Olpe HR, Vassout A, Gentsch C, Chaperon F et al. (2009). The selective nicotinic acetylcholine receptor alpha7 agonist JN403 is active in animal models of cognition, sensory gating, epilepsy and pain. Neuropharmacology 56: 254–263. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ (2004). Ethosuximide reverses paclitaxel‐ and vincristine‐induced painful peripheral neuropathy. Pain 109: 150–161. [DOI] [PubMed] [Google Scholar]
- Freitas KC, Carroll FI, Negus SS (2015). Effects of nicotinic acetylcholine receptor agonists in assays of acute pain‐stimulated and pain‐depressed behaviors in rats. J Pharmacol Exp Ther 355: 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangadharan V, Agarwal N, Brugger S, Tegeder I, Bettler B, Kuner R et al. (2009). Conditional gene deletion reveals functional redundancy of GABAB receptors in peripheral nociceptors in vivo. Mol Pain 5: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genzen JR, Van Cleve W, McGehee DS (2001). Dorsal root ganglion neurons express multiple nicotinic acetylcholine receptor subtypes. J Neurophysiol 86: 1773–1782. [DOI] [PubMed] [Google Scholar]
- George A, Buehl A, Sommer C (2004). Wallerian degeneration after crush injury of rat sciatic nerve increases endo‐ and epineurial tumor necrosis factor‐alpha protein. Neurosci Lett 372: 215–219. [DOI] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA (2000). Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science 288: 2366–2368. [DOI] [PubMed] [Google Scholar]
- Goudet C, Magnaghi V, Landry M, Nagy F, Gereau RW, Pin JP (2009). Metabotropic receptors for glutamate and GABA in pain. Brain Res Rev 60: 43–56. [DOI] [PubMed] [Google Scholar]
- Grace PM, Hutchinson MR, Maier SF, Watkins LR (2014). Pathological pain and the neuroimmune interface. Nat Rev Immunol 14: 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberberger RV, Bernardini N, Kress M, Hartmann P, Lips KS, Kummer W (2004). Nicotinic acetylcholine receptor subtypes in nociceptive dorsal root ganglion neurons of the adult rat. Auton Neurosci 113: 32–42. [DOI] [PubMed] [Google Scholar]
- Halai R, Callaghan B, Daly NL, Clark RJ, Adams DJ, Craik DJ (2011). Effects of cyclization on stability, structure, and activity of alpha‐conotoxin RgIA at the alpha9alpha10 nicotinic acetylcholine receptor and GABA(B) receptor. J Med Chem 54: 6984–6992. [DOI] [PubMed] [Google Scholar]
- Halai R, Clark RJ, Nevin ST, Jensen JE, Adams DJ, Craik DJ (2009). Scanning mutagenesis of alpha‐conotoxin Vc1.1 reveals residues crucial for activity at the alpha9alpha10 nicotinic acetylcholine receptor. J Biol Chem 284: 20275–20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker A, Kullmar M, Wilker S, Richter K, Zakrzewicz A, Atanasova S et al. (2015). Phosphocholine‐modified macromolecules and canonical nicotinic agonists inhibit ATP‐induced IL‐1beta release. J Immunol 195: 2325–2334. [DOI] [PubMed] [Google Scholar]
- Hendriks JJ, Teunissen CE, de Vries HE, Dijkstra CD (2005). Macrophages and neurodegeneration. Brain Res Brain Res Rev 48: 185–195. [DOI] [PubMed] [Google Scholar]
- Holtman JR, Dwoskin LP, Dowell C, Wala EP, Zhang Z, Crooks PA et al. (2011). The novel small molecule alpha9alpha10 nicotinic acetylcholine receptor antagonist ZZ‐204G is analgesic. Eur J Pharmacol 670: 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hone AJ, Meyer EL, McIntyre M, McIntosh JM (2012). Nicotinic acetylcholine receptors in dorsal root ganglion neurons include the alpha6beta4* subtype. FASEB J 26: 917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hone AJ, Ruiz M, Scadden M, Christensen S, Gajewiak J, Azam L et al. (2013). Positional scanning mutagenesis of alpha‐conotoxin PeIA identifies critical residues that confer potency and selectivity for alpha6/alpha3beta2beta3 and alpha3beta2 nicotinic acetylcholine receptors. J Biol Chem 288: 25428–25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh TG, Cuny H, Slesinger PA, Adams DJ (2015). Novel mechanism of voltage‐gated N‐type (Cav2.2) calcium channel inhibition revealed through alpha‐conotoxin Vc1.1 activation of the GABA(B) receptor. Mol Pharmacol 87: 240–250. [DOI] [PubMed] [Google Scholar]
- Ji RR, Chamessian A, Zhang YQ (2016). Pain regulation by non‐neuronal cells and inflammation. Science 354: 572–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, Ulloa L (2007). The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151: 915–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimis H, Adams DJ, Callaghan B, Nevin S, Alewood PF, Vaughan CW et al. (2011). A novel mechanism of inhibition of high‐voltage activated calcium channels by alpha‐conotoxins contributes to relief of nerve injury‐induced neuropathic pain. Pain 152: 259–266. [DOI] [PubMed] [Google Scholar]
- Koval L, Lykhmus O, Zhmak M, Khruschov A, Tsetlin V, Magrini E et al. (2011). Differential involvement of alpha4beta2, alpha7 and alpha9alpha10 nicotinic acetylcholine receptors in B lymphocyte activation in vitro. Int J Biochem Cell Biol 43: 516–524. [DOI] [PubMed] [Google Scholar]
- Lang PM, Burgstahler R, Haberberger RV, Sippel W, Grafe P (2005). A conus peptide blocks nicotinic receptors of unmyelinated axons in human nerves. Neuroreport 16: 479–483. [DOI] [PubMed] [Google Scholar]
- Leskovar A, Moriarty LJ, Turek JJ, Schoenlein IA, Borgens RB (2000). The macrophage in acute neural injury: changes in cell numbers over time and levels of cytokine production in mammalian central and peripheral nervous systems. J Exp Biol 203: 1783–1795. [DOI] [PubMed] [Google Scholar]
- Li X, Hu Y, Wu Y, Huang Y, Yu S, Ding Q et al. (2016). Anti‐hypersensitive effect of intramuscular administration of alphaO‐conotoxin GeXIVA[1,2] and GeXIVA[1,4] in rats of neuropathic pain. Prog Neuropsychopharmacol Biol Psychiatry 66: 112–119. [DOI] [PubMed] [Google Scholar]
- van Lierop BJ, Robinson SD, Kompella SN, Belgi A, McArthur JR, Hung A et al. (2013). Dicarba alpha‐conotoxin Vc1.1 analogues with differential selectivity for nicotinic acetylcholine and GABAB receptors. ACS Chem Biol 8: 1815–1821. [DOI] [PubMed] [Google Scholar]
- Lips KS, Konig P, Schatzle K, Pfeil U, Krasteva G, Spies M et al. (2006). Coexpression and spatial association of nicotinic acetylcholine receptor subunits alpha7 and alpha10 in rat sympathetic neurons. J Mol Neurosci 30: 15–16. [DOI] [PubMed] [Google Scholar]
- Lips KS, Pfeil U, Kummer W (2002). Coexpression of alpha 9 and alpha 10 nicotinic acetylcholine receptors in rat dorsal root ganglion neurons. Neuroscience 115: 1–5. [DOI] [PubMed] [Google Scholar]
- Liu T, van Rooijen N, Tracey DJ (2000). Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain 86: 25–32. [DOI] [PubMed] [Google Scholar]
- Livett BG, Khalil Z, Down J, Sandall DW, Keays D (2008). α‐Contoxoin peptides with analgesic properties. US Patent 7,348,400 B2.
- Livett BG, Sandall DW, Keays D, Down J, Gayler KR, Satkunanathan N et al. (2006). Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon 48: 810–829. [DOI] [PubMed] [Google Scholar]
- Loubser PG, Akman NM (1996). Effects of intrathecal baclofen on chronic spinal cord injury pain. J Pain Symptom Manage 12: 241–247. [DOI] [PubMed] [Google Scholar]
- Luo S, Zhangsun D, Harvey PJ, Kaas Q, Wu Y, Zhu X et al. (2015). Cloning, synthesis, and characterization of alphaO‐conotoxin GeXIVA, a potent alpha9alpha10 nicotinic acetylcholine receptor antagonist. Proc Natl Acad Sci U S A 112: E4026–E4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig LR, Peng H, Hiel H, Yamamoto T, Fuchs PA (2001). Molecular cloning and mapping of the human nicotinic acetylcholine receptor alpha10 (CHRNA10). Genomics 73: 272–283. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M (2009). Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem Pharmacol 78: 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A et al. (2004). Analogs of alpha‐conotoxin MII are selective for alpha6‐containing nicotinic acetylcholine receptors. Mol Pharmacol 65: 944–952. [DOI] [PubMed] [Google Scholar]
- McMahon SB, La Russa F, Bennett DL (2015). Crosstalk between the nociceptive and immune systems in host defence and disease. Nat Rev Neurosci 16: 389–402. [DOI] [PubMed] [Google Scholar]
- Medhurst SJ, Hatcher JP, Hille CJ, Bingham S, Clayton NM, Billinton A et al. (2008). Activation of the alpha7‐nicotinic acetylcholine receptor reverses complete Freund adjuvant‐induced mechanical hyperalgesia in the rat via a central site of action. J Pain 9: 580–587. [DOI] [PubMed] [Google Scholar]
- Mercado J, Peckham DW, JP, KN, Tarcha E, Bartron J, et al. (2014). Preclinical development of novel cone snail peptide‐based analgesics that selectively inhibit the alpha9alpha10 nicotinic acetylcholine receptor. International Association for the Study of Pain, 15th World Congress on Pain, Meeting Abstract #2026.
- Metabolic PL (2006). ACVI: a novel therapeutic for neuropathic pain. Technical Summary of Preclinical and Phase I Data. Available at: http://wwwmetaboliccomau/_files/QTO7TA4EWA/ACV_NonConfidentialPackage_February2007pdf (accessed 03/01/2007)
- Moalem G, Xu K, Yu L (2004). T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience 129: 767–777. [DOI] [PubMed] [Google Scholar]
- Mohammadi S, Christie MJ (2014). alpha9‐nicotinic acetylcholine receptors contribute to the maintenance of chronic mechanical hyperalgesia, but not thermal or mechanical allodynia. Mol Pain 10: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi SA, Burton TJ, Christie MJ (2017). alpha9‐nAChR knockout mice exhibit dysregulation of stress responses, affect and reward‐related behaviour. Behav Brain Res 328: 105–114. [DOI] [PubMed] [Google Scholar]
- Mohammadi SA, Christie MJ (2015). Conotoxin interactions with alpha9alpha10‐nAChRs: is the alpha9alpha10‐nicotinic acetylcholine receptor an important therapeutic target for pain management? Toxins (Basel) 7: 3916–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier IA, Klimis H, Rycroft BK, Jin AH, Alewood PF, Motin L et al. (2012). Intrathecal alpha‐conotoxins Vc1.1, AuIB and MII acting on distinct nicotinic receptor subtypes reverse signs of neuropathic pain. Neuropharmacology 62: 2202–2207. [DOI] [PubMed] [Google Scholar]
- Nevin ST, Clark RJ, Klimis H, Christie MJ, Craik DJ, Adams DJ (2007). Are alpha9alpha10 nicotinic acetylcholine receptors a pain target for alpha‐conotoxins? Mol Pharmacol 72: 1406–1410. [DOI] [PubMed] [Google Scholar]
- Nguyen VT, Ndoye A, Grando SA (2000). Novel human alpha9 acetylcholine receptor regulating keratinocyte adhesion is targeted by Pemphigus vulgaris autoimmunity. Am J Pathol 157: 1377–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie L, Song H, Chen MF, Chiamvimonvat N, Beisel KW, Yamoah EN et al. (2004). Cloning and expression of a small‐conductance Ca(2+)‐activated K+ channel from the mouse cochlea: coexpression with alpha9/alpha10 acetylcholine receptors. J Neurophysiol 91: 1536–1544. [DOI] [PubMed] [Google Scholar]
- Nuytten D, Kupers R, Lammens M, Dom R, Van Hees J, Gybels J (1992). Further evidence for myelinated as well as unmyelinated fibre damage in a rat model of neuropathic pain. Exp Brain Res 91: 73–78. [DOI] [PubMed] [Google Scholar]
- Pacini A, Micheli L, Maresca M, Branca JJ, McIntosh JM, Ghelardini C et al. (2016). The alpha9alpha10 nicotinic receptor antagonist alpha‐conotoxin RgIA prevents neuropathic pain induced by oxaliplatin treatment. Exp Neurol 282: 37–48. [DOI] [PubMed] [Google Scholar]
- Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ (2003). The cholinergic anti‐inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med 9: 125–134. [PMC free article] [PubMed] [Google Scholar]
- Peng H, Ferris RL, Matthews T, Hiel H, Lopez‐Albaitero A, Lustig LR (2004). Characterization of the human nicotinic acetylcholine receptor subunit alpha (alpha) 9 (CHRNA9) and alpha (alpha) 10 (CHRNA10) in lymphocytes. Life Sci 76: 263–280. [DOI] [PubMed] [Google Scholar]
- Peng J, Gu N, Zhou L, B Eyo U, Murugan M, Gan WB et al. (2016). Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun 7: 12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau KK, Johnson RD, Cooper BY (2005). Nicotinic AChR in subclassified capsaicin‐sensitive and ‐insensitive nociceptors of the rat DRG. J Neurophysiol 93: 1358–1371. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R (2010). Interactions between the immune and nervous systems in pain. Nat Med 16: 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter K, Mathes V, Fronius M, Althaus M, Hecker A, Krasteva‐Christ G et al. (2016). Phosphocholine – an agonist of metabotropic but not of ionotropic functions of alpha9‐containing nicotinic acetylcholine receptors. Sci Rep 6: 28660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero HK, Christensen S, Di Cesare Mannelli L, Gajewiak J, Ramachandra R, Elmslie KS et al. (2017). Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy‐induced neuropathic pain. Proc Natl Acad Sci U S A 114: 1825–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandall DW, Satkunanathan N, Keays DA, Polidano MA, Liping X, Pham V et al. (2003). A novel alpha‐conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry 42: 6904–6911. [DOI] [PubMed] [Google Scholar]
- Satkunanathan N, Livett B, Gayler K, Sandall D, Down J, Khalil Z (2005). Alpha‐conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res 1059: 149–158. [DOI] [PubMed] [Google Scholar]
- Sgard F, Charpantier E, Bertrand S, Walker N, Caput D, Graham D et al. (2002). A novel human nicotinic receptor subunit, alpha10, that confers functionality to the alpha9‐subunit. Mol Pharmacol 61: 150–159. [DOI] [PubMed] [Google Scholar]
- Simard AR, Gan Y, St‐Pierre S, Kousari A, Patel V, Whiteaker P et al. (2013). Differential modulation of EAE by alpha9*‐ and beta2*‐nicotinic acetylcholine receptors. Immunol Cell Biol 91: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, Hone AJ, Memon T, Bossi S, Smith TE, McIntosh JM et al. (2013). Comparative functional expression of nAChR subtypes in rodent DRG neurons. Front Cell Neurosci 7: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C, Lalonde A, Heckman HM, Rodriguez M, Myers RR (1995). Quantitative neuropathology of a focal nerve injury causing hyperalgesia. J Neuropathol Exp Neurol 54: 635–643. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umana IC, Daniele CA, McGehee DS (2013). Neuronal nicotinic receptors as analgesic targets: it's a winding road. Biochem Pharmacol 86: 1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev VV (2014). The therapeutic promise of positive allosteric modulation of nicotinic receptors. Eur J Pharmacol 727: 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbitsky M, Rothlin CV, Katz E, Elgoyhen AB (2000). Mixed nicotinic–muscarinic properties of the alpha9 nicotinic cholinergic receptor. Neuropharmacology 39: 2515–2524. [DOI] [PubMed] [Google Scholar]
- Vetter DE, Liberman MC, Mann J, Barhanin J, Boulter J, Brown MC et al. (1999). Role of alpha9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron 23: 93–103. [DOI] [PubMed] [Google Scholar]
- Vincler M, McIntosh JM (2007). Targeting the alpha9alpha10 nicotinic acetylcholine receptor to treat severe pain. Expert Opin Ther Targets 11: 891–897. [DOI] [PubMed] [Google Scholar]
- Vincler M, Wittenauer S, Parker R, Ellison M, Olivera BM, McIntosh JM (2006). Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors. Proc Natl Acad Sci U S A 103: 17880–17884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wala EP, Crooks PA, McIntosh JM, Holtman JR Jr (2012). Novel small molecule alpha9alpha10 nicotinic receptor antagonist prevents and reverses chemotherapy‐evoked neuropathic pain in rats. Anesth Analg 115: 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Su DM, Wang RH, Liu Y, Wang H (2005). Antinociceptive effects of choline against acute and inflammatory pain. Neuroscience 132: 49–56. [DOI] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH et al. (1998). Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396: 679–682. [DOI] [PubMed] [Google Scholar]
- Wright AB, Norimatsu Y, McIntosh JM (2015). & Elmslie KS (2015). Limited efficacy of alpha‐conopeptides, Vc1.1 and RgIA, to inhibit sensory neuron Ca current. eNeuro 2: e0057–e0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao WH, Zheng H, Bennett GJ (2012). Characterization of oxaliplatin‐induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience 203: 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Seymour VA, Berecki G, Jia X, Akcan M, Adams DJ et al. (2015). Less is more: design of a highly stable disulfide‐deleted mutant of analgesic cyclic alpha‐conotoxin Vc1.1. Sci Rep 5: 13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Currie KP (2013). Regulation of Ca(V)2 calcium channels by G protein coupled receptors. Biochim Biophys Acta 1828: 1629–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Zhang Z, Dowell C, Wala E, Dwoskin LP, Holtman JR et al. (2011). Discovery of non‐peptide, small molecule antagonists of alpha9alpha10 nicotinic acetylcholine receptors as novel analgesics for the treatment of neuropathic and tonic inflammatory pain. Bioorg Med Chem Lett 21: 2476–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]