

Abstract
Perhexiline, a chiral drug, is a potent antiischemic agent whose clinical utility is limited by hepatic and neural toxicities. It inhibits mitochondrial carnitine palmitoyltransferase‐1, however, excessive inhibition predisposes toward tissue steatosis. This pilot study investigated the distribution of the two enantiomers and their toxicological potential. Dark Agouti rats (n = 4 per group) were administered vehicle or 200 mg/kg daily of racemic, (+)− or (−)‐perhexiline maleate orally for 8 weeks. Plasma biochemical liver function tests and Von Frey assessments of peripheral neural function were performed. Hepatic and neuronal histology, including lipid and glycogen content, was assessed using electron microscopy. Concentrations of the perhexiline enantiomers and metabolites were quantified in plasma, liver and heart. Plasma perhexiline concentrations following administration of racemate, (+)− or (‐)‐enantiomer were within the mid‐upper clinical therapeutic range. There was extensive uptake of both enantiomers into liver and heart, with 2.5‐ to 4.5‐fold greater net uptake of (+)‐ compared to (−)‐perhexiline (P < .05) when administered as pure enantiomers, but not when administered as racemate. There was no biochemical or gross histological evidence of hepatotoxicity. However, livers of animals administered (+)‐perhexiline had higher lipid (P < .01) and lower glycogen (P < .05) content, compared to those administered (−)‐perhexiline. Animals administered racemic perhexiline had reduced peripheral neural function (P < .05) compared to controls or animals administered (−)‐perhexiline. For the same plasma concentrations, differences in tissue distribution may contribute to disparities in the effects of (+)‐ and (−)‐perhexiline on hepatic histology and neural function.
Keywords: enantiomer, hepatic histology, neural function, perhexiline, tissue uptake
Abbreviation
- ALP
alkaline phosphatase
- ALT
alanine transaminase
- AST
aspartate transaminase
- GGT
gamma glutamyl transpeptidase
- LDH
lactate dehydrogenase
1. INTRODUCTION
In Australia, perhexiline is approved for treating patients with refractory angina pectoris or patients with angina in whom other therapies are contraindicated.1 However, it also has other clinically beneficial effects, including enhancing myocardial energetics, in heart failure2 and hypertrophic cardiomyopathy,3 and improving New York Heart Association functional class in inoperable aortic stenosis.4 Perhexiline inhibits carnitine palmitoyltransferase 1 (CPT‐1),5 the enzyme that initiates mitochondrial translocation of free fatty acids, the rate‐limiting step for their β‐oxidation. Inhibition of CPT‐1 (and β‐oxidation), facilitates the utilization of carbohydrates,6 thus maintaining myocardial production of ATP with lower oxygen consumption.7
Although deficient myocardial energetics are increasingly recognized as an underlying feature of cardiovascular disease,7 the potential for perhexiline to cause hepatotoxicity and peripheral neuropathy has limited its clinical use for extended exposures.8, 9 Both adverse effects have been associated with altered lipid storage, including steatosis and lysosomal phospholipidosis, which may reflect excessive CPT‐1 inhibition, as previously described with other drugs.10 Although current clinical practice incorporating therapeutic monitoring of perhexiline in plasma has practically abolished the risk of hepatic and neural toxicity,11, 12 it remains a major impediment to wider clinical use.
Perhexiline, a chiral compound, is on the market as a racemic mixture, with no data on the pharmacology of the individual enantiomers, and only pharmacokinetic investigations of the enantiomers, particularly their extensive metabolism by cytochrome P450 2D6 (CYP2D6).13, 14, 15 The female Dark Agouti (DA) rat, traditionally an animal model for the human CYP2D6 poor metabolizer phenotype, has previously been utilized to investigate the hepatic and neural toxicity of racemic perhexiline.16, 17 This pilot study was designed to investigate and compare the distribution of the two enantiomers and the racemate, and their toxicological potential during long‐term exposures, as we have previously demonstrated that DA rats display metabolism of the racemate similar to human CYP2D6 extensive/intermediate metabolizers at clinically relevant plasma perhexiline concentrations.17 With this model, we now demonstrate significant stereoselectivity with respect to perhexiline's tissue distribution, as well as its effects on hepatic histology and peripheral neural function.
2. MATERIALS AND METHODS
2.1. Chemicals
(+)‐ and (−)‐Perhexiline maleate were prepared as previously described,18 and Sigma Pharmaceuticals (Rowville, Victoria, Australia) donated the racemate, (±)‐perhexiline maleate. Prenylamine lactate and (R)‐(−)‐1‐(1‐naphthyl) ethyl isocyanate were purchased from Sigma Aldrich (Castle Hill, NSW, Australia). Sodium hydroxide, n‐hexane, ethyl acetate, acetone, and methanol were obtained from ThermoFisher (Scoresby, Australia); All other solvents and reagents were of HPLC or analytical grade, respectively. DA rats were obtained from the animal facilities of the University of Adelaide.
2.2. Animal study
2.2.1. In vivo procedures
This study was approved by the University of Adelaide (M‐079‐2007) and the SA Pathology (120/07) animal ethics committees. All experiments were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Based on pilot experiments conducted with racemic perhexiline,17 vehicle or (±)‐, (+)‐ or (−)‐perhexiline maleate (200 mg/kg daily) were administered to four groups of female DA rats (four per group) for 8 weeks. To mimic a “sustained‐release” and avoid gastrointestinal irritation observed following oral gavage, the test drugs were prepared as a peanut paste mixture, coated onto the food pellets, and left for the rats to consume overnight, with complete consumption verified each morning.17 The animals’ average daily food pellet consumption had been calculated over a 5‐day period prior to commencement of the dosing study. At the end of week 4, blood was collected from the tail vein, and plasma was separated and stored at −20°C. On the day of the final week 8 dose, Von Frey filament testing was used to assess peripheral neural function.19 The next morning, blood was collected under anesthesia, via a cardiac puncture, and plasma was separated and stored at −20°C. After being euthanized, the liver, heart and neuronal tissues were harvested. Tissues were divided, with part snap frozen immediately in liquid nitrogen, some placed into a solution of fixative for subsequent electron microscopy and the remainder paraffin embedded for hematoxylin and eosin (H&E) staining.
Due to the small quantity of (+)‐perhexiline maleate available, one of the four animals was treated for only 4 weeks. The plasma concentration data for this animal were still used for pharmacokinetic analyses, assuming pharmacokinetic steady state had been attained. However, we could not assume that pharmacodynamic effects had reached steady state and it was omitted from week 8 analyses of histology, neuronal function or plasma biochemistry (see below).
2.2.2. Hepatic and neuronal morphology
Gross hepatic morphology was assessed, using formalin‐fixed, paraffin‐embedded 6 μm H&E stained sections. Hepatic and neuronal tissues (approximately 0.5 mm3) were also processed for analysis by transmission electron microscopy as previously described.17 For liver electron micrographs, lipid vesicles, and glycogen were identified20 and total content measured as a percentage of the field of view using Image J software (http://imagej.nih.gov/ij/). The average content was calculated from examining two to three de‐identifed sections of each tissue. To confirm histological assessment of glycogen content, biochemical analysis was also performed using stored frozen hepatic tissue, as described by Unger et al.21 Neuronal sections were assessed for the presence of Mallory bodies.16
2.2.3. Plasma biochemistry
A Beckman Coulter instrument (Gladesville, NSW, Australia) in the Clinical Chemistry Unit of SA Pathology (Adelaide, Australia) was used to measure the plasma concentrations of glucose, lactate, triglycerides, total bilirubin, albumin, lactate dehydrogenase (LDH), alanine transaminase (ALT), aspartate transaminase (AST), gamma glutamyl transpeptidase (GGT) and alkaline phosphatase (ALP). For tests of liver function, the 4‐ and 8‐week values for control animals were combined to establish a normal range (n = 8), and hepatotoxicity was defined as values >1.5‐times above the upper limit of normal, or values below the lower limit of normal for albumin.
2.3. Quantitation of perhexiline and its OH‐metabolites
Heart and liver homogenates were prepared, using 0.15 mol/L phosphate buffer at pH 7.4 (2 mL per g of tissue, on ice) and stored (−80°C) pending analysis. The concentrations of (+)‐ and (−)‐perhexiline in plasma and homogenates of heart or liver were measured by high performance liquid chromatography,18 as were the concentrations of cis‐, trans1‐ and trans2‐OH‐perhexiline in plasma and liver homogenate.22 The lower limit of quantification for parent enantiomers and the metabolites was 0.01 mg/L.
2.4. Statistical analyses
Concentrations of (+)− and (−)‐perhexiline in plasma and tissue were normalized to a daily dose of 100 mg/kg, to allow for the two‐fold difference in enantiomeric dose between administration of racemate and individual enantiomers. Between‐group comparisons were performed using Residual Maximum Likelihood (REML), incorporating rats as random and treatments as fixed to account for pairing of the (+)‐ and (−)‐ perhexiline concentrations arising from administering (±)‐ perhexiline, and the unpaired nature of between‐group comparisons (GenStat 13th edition, VSN International Ltd, UK). GraphPad Prism (V4.02 for Windows, GraphPad, San Diego CA) was used to perform other statistical analyses. Between‐group comparisons of hepatic lipid vesicle and glycogen contents, and neuronal function were carried out by Kruskal Wallis analysis, with Dunn's multiple comparisons post‐test. P < .05 was considered statistically significant. Nonparametric Spearman correlation was used to investigate possible relationships between hepatic concentrations of (+)‐ and (−)‐perhexiline and changes in glycogen or lipid contents.
3. RESULTS
3.1. Perhexiline concentrations
Concentrations of the enantiomers and their metabolites in plasma and tissue are presented in Table 1. The ratio of (+)‐ to (−)‐enantiomers in plasma was approximately 2:1 regardless of whether animals were administered the individual enantiomers or racemate. Figure 1 shows the plasma and tissue concentrations of (+)‐ and (−)‐perhexiline normalized to an enantiomeric dose of 100 mg/kg per day. No difference was observed in the dose‐adjusted concentrations of (+)‐perhexiline in plasma between enantiomeric and racemic administrations. Similarly, (−)‐perhexiline also displayed dose‐independent plasma pharmacokinetics.
Table 1.
Mean (SD) steady‐state concentrations of (+)‐ and (−)‐perhexiline, and the OH‐metabolites, in DA rats administered 200 mg/kg per day of (±)‐, (+)‐ or (−)‐perhexiline maleate for 8 weeks in rat chow pellets provided each day at 1700 hour
| Dosage form | |||
|---|---|---|---|
| (±) | (+) | (−) | |
| Plasma (mg/L) | |||
| (+)‐Perhexiline | 0.38 (0.29) | 0.67 (0.13) | n.d. |
| (−)‐Perhexiline | 0.19 (0.12) | n.d. | 0.30 (0.09) |
| cis‐OH‐perhexiline | 0.42 (0.14) | 0.21 (0.06) | 0.60 (0.06) |
| trans1‐OH‐perhexiline | 0.03 (0.01) | 0.05 (0.03) | <LLOQ |
| trans2‐OH‐perhexiline | 0.03 (0.02) | n.d. | <LLOQ |
| Liver (μg/g) | |||
| (+)‐Perhexiline | 19.4 (14.0) | 104.9 (36.8) | n.d. |
| (−)‐Perhexiline | 8.9 (6.0) | n.d. | 17.1 (2.8) |
| cis‐OH‐perhexiline | 11.8 (3.2) | 13.9 (6.6) | 15.5 (6.6) |
| trans1‐OH‐perhexiline | 1.1 (0.4) | 4.0 (1.7) | 0.6 (0.2) |
| trans2‐OH‐perhexiline | 1.1 (0.5) | 0.8 (0.7) | 1.9 (1.2) |
| Heart (μg/g) | |||
| (+)‐Perhexiline | 15.6 (6.8) | 65.1 (39.1) | |
| (−)‐Perhexiline | 6.3 (2.4) | 6.5 (3.3) | |
See Figure 1 for statistical analysis of dose‐corrected (+)‐ and (−)‐perhexiline concentrations. Samples were taken in the morning after the last dose (n.d. = not detectable; <LLOQ, below lower limit of quantification, 0.01 mg/L or μg/g).
Figure 1.
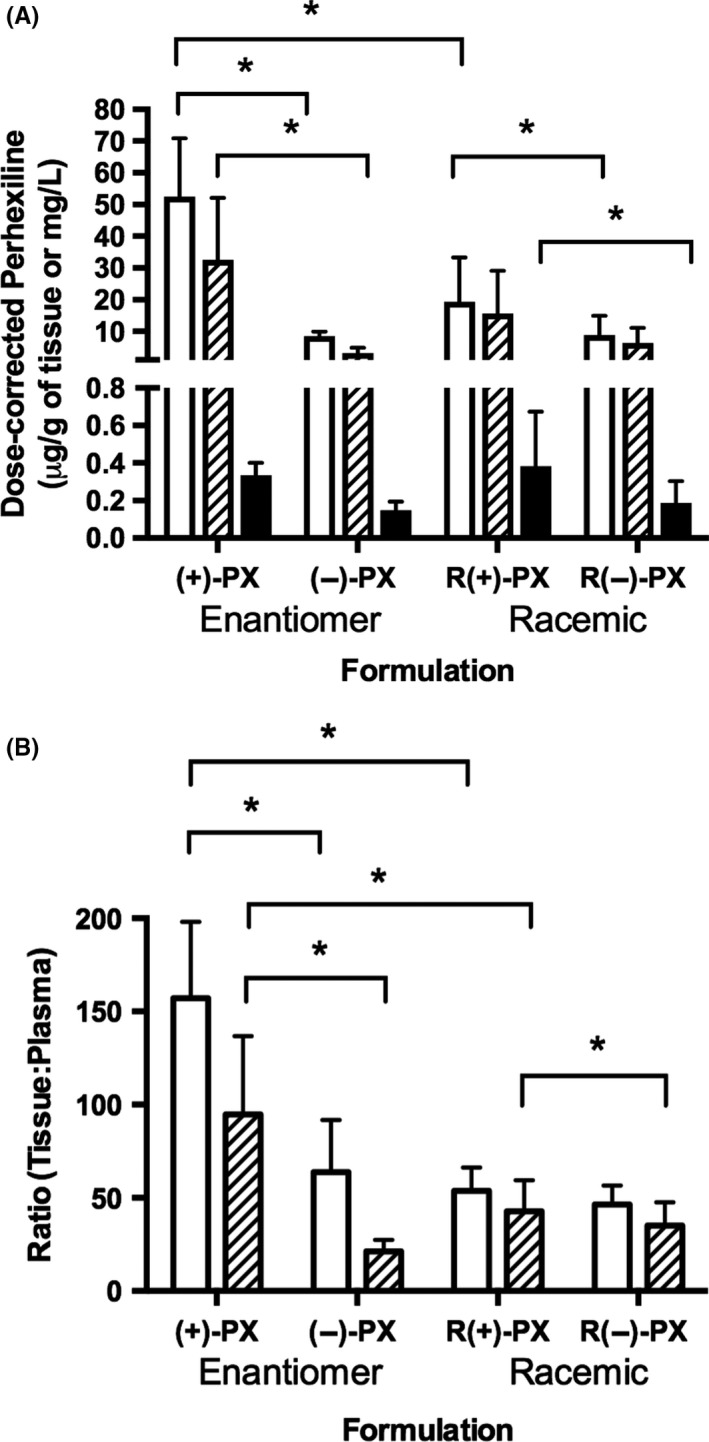
(A) Mean (SD) dose‐corrected perhexiline (PX) concentrations in liver (white), myocardium (hatched) and plasma (black), and (B) tissue:plasma perhexiline concentration ratios for liver (white) and myocardium (hatched) in DA rats administered (+)‐, (‐)‐ or (±)‐perhexiline for 8 weeks (*P < .05 using Residual Maximum Likelihood, n = 4/group)
For both enantiomers, the concentrations in tissue (heart or liver) were at least 20‐times greater than the corresponding concentrations in plasma (Figure 1B) and, similar to plasma, tissue (+)‐perhexiline concentrations were generally higher than those of (−)‐perhexiline (Figure 1A). However, when pure enantiomers were administered, the concentrations of (+)‐perhexiline in the two tissues were 6 to 10 times higher than (−)‐perhexiline (P < .05), and the tissue (heart or liver):plasma concentration ratios for (+)‐perhexiline were also significantly higher than (−)‐perhexiline (P < .05) (Figure 1B). In contrast, following administration of racemate, (+)‐ and (−)‐perhexiline displayed similar ratios of the concentrations of tissue:plasma with respect to both heart and the liver. For (−)‐perhexiline, tissue accumulation was similar regardless of the dosage form administered (i.e., pure enantiomer or racemic mixture). However, there was a significantly lower (P < .05) accumulation of (+)−perhexiline in the heart and liver after administering the racemate compared to the pure enantiomer (Figure 1B).
3.2. OH‐metabolite concentrations
The formation of cis− and trans‐mono‐hydroxy metabolites is shown in Table 1. Cis‐OH‐perhexiline was the most abundant metabolite formed after individual administration of either (+)‐ or (−)‐perhexiline. For (−)‐perhexiline, the ratio of total metabolites: parent was significantly higher than observed for (+)‐perhexiline, in both plasma (mean SD values of 2.34 ± 0.96 vs. 0.39 ± 0.13, P < .05) and liver (1.11 ± 0.60 vs. 0.18 ± 0.06, P < .05). Following administration of racemate, it was not analytically possible to distinguish between the metabolites of (+)− and (−)‐perhexiline. However, total cis‐OH‐(±)‐perhexiline was also the most abundant metabolite in plasma and tissue from liver.
3.3. Plasma biochemistry
Normal ranges were established for each biochemical parameter by combining the control group data for weeks 4 and 8. Only two drug‐treated animals had liver function test results >1.5‐times the upper limit of normal. At week 8, one animal treated with (−)‐perhexiline developed elevated AST (278 U/L, 1.6‐times the upper limit) and LDH (1091 U/L, 2.7‐times the upper limit). At week 4, one animal treated with (+)‐perhexiline developed elevated AST (375 U/L, 2.2‐times the upper limit) and LDH (703 U/L, 1.8‐times the upper limit). Unfortunately, dosing of this animal ceased after week 4 due to a lack of pure (+)‐enantiomer. No other biochemistry abnormalities were observed.
3.4. Hepatic histology
Microscopic examination of formalin‐fixed, paraffin‐embedded H&E sections of livers revealed no significant morphological changes in treated or control groups. There was no evidence of hepatocellular degeneration in the form of hydropic degeneration or fatty change (lipidosis, steatosis) and no evidence of necrosis or apoptosis of hepatocytes.
At the ultrastructural level, there were very mild changes in hepatic lipid vacuole and glycogen content, as shown in Figures 2 and 3. When measured as a percentage of the overall field under view, median hepatic lipid vacuole content was significantly higher in (+)‐ compared to (−)‐perhexiline treated rats (Figure 3A, P < .01). When the control, racemic‐ and (+)‐perhexiline groups were combined, there was a significant correlation between hepatic (+)‐perhexiline concentrations and lipid contents (r s 2 = .46, P = .02). Median (range) hepatic glycogen content was significantly higher in (−)‐ compared to (+)‐perhexiline treated rats (Figure 3B, P < .05). When the control, racemic‐ and (−)‐perhexiline groups were combined, there was a significant correlation between hepatic (−)‐perhexiline concentrations and glycogen contents (r s 2 = .44, P = .02). Although there was sufficient frozen tissue to biochemically assess hepatic glycogen concentrations in only eleven animals, there was a significant association between histological and biochemical assessments, using linear regression analysis (r 2 = .89, P < .0001). Similar to the histological results, median hepatic glycogen concentrations in animals administered (−)‐perhexiline were significantly higher than those in animals administered (+)‐perhexiline (0.83 (0.56‐1.33) vs. 0.22 (0.17‐0.39) μmol/g tissue, respectively P < .05).
Figure 2.
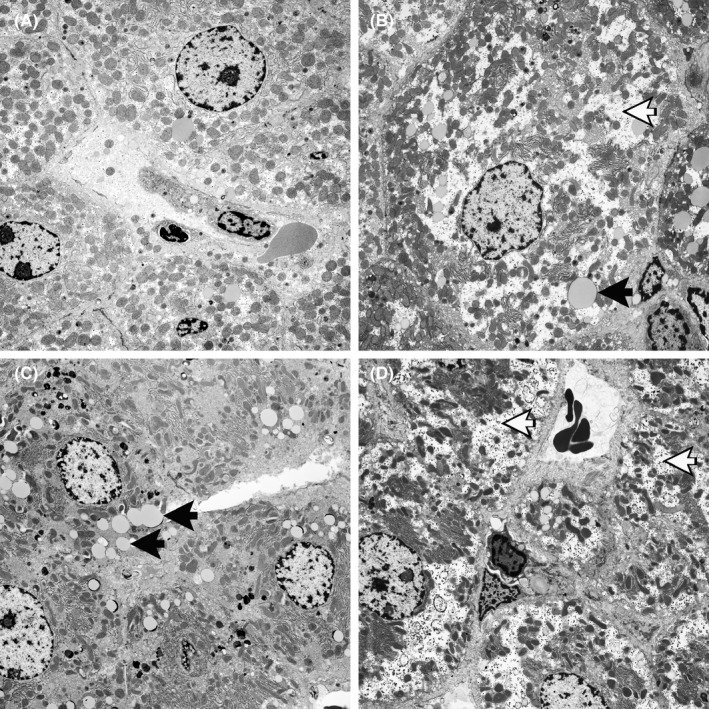
Transmission electron micrographs of livers from DA rats treated for 8 weeks with (A) vehicle, (B) (±)‐perhexiline, (C) (+)‐perhexiline and (D) (−)‐perhexiline. White arrows indicate glycogen “granules” and black arrows indicate lipid vesicles
Figure 3.
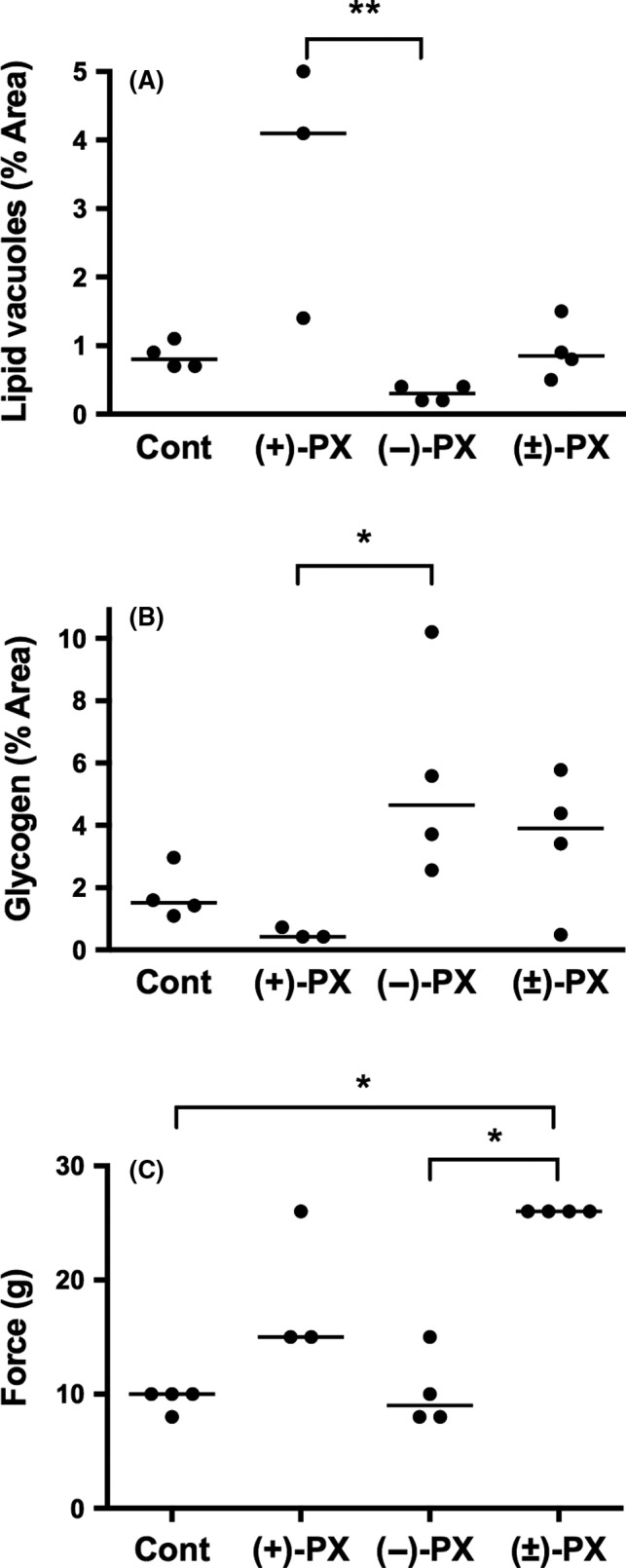
(A) hepatic lipid vacuole content, (B) hepatic glycogen content and C) Von Frey paw withdrawal thresholds in DA rats treated for 8 weeks with vehicle (Cont), (+)‐, (−)‐ or (±)‐perhexiline (PX) (*P < .05, **P < .01, following Kruskal Wallis analysis; line indicates median)
3.5. Neural function and histology
Peripheral neural function assessed as paw withdrawal threshold in the Von Frey test is shown in Figure 3C. All of the animals treated with racemic or (+)‐perhexiline had thresholds above the range of the control animals, but the difference was significant (P < .05) only for racemic perhexiline. In contrast, rats treated with (−)‐perhexiline had significantly lower paw withdrawal thresholds compared to those administered racemic perhexiline, and only one of the four had a threshold above the range of the controls. In rats treated with (+)‐perhexiline, electron micrographs of dorsal root ganglia showed the presence of Mallory bodies in approximately 1% of neurones. However, Mallory bodies were not identified in any of the other treatment groups.
4. DISCUSSION
It is increasingly apparent that perhexiline's inhibition of CPT‐1 may not sufficiently account for its improvement of myocardial function.21 Other mechanisms may be important also, such as sensitizing the response to nitric oxide and its anti‐inflammatory properties.21, 23, 24 Perhexiline's mechanisms of action have remained poorly defined, partly due to the lack of investigation of the individual enantiomers’ pharmacodynamic/toxicological properties. This pilot study in vivo is the first one investigating distribution of the enantiomers in tissue and their effects on neuronal and hepatic function, at clinically relevant concentrations.
This study confirmed that the female DA rat displays perhexiline pharmacokinetics similar to those in humans, with the chosen dosage regimen attaining plasma perhexiline concentrations that approximate the mid to high range of therapeutic concentrations (0.15‐0.60 mg/L). Similar to humans,13, 14, 25 we report enantioselective plasma pharmacokinetics of perhexiline in female DA rats, consistent with a greater apparent oral clearance of the (−)‐enantiomer. Furthermore, similar to human CYP2D6 extensive / intermediate metabolisers,22 the dominant metabolite in plasma following administration of the racemate was cis‐OH‐perhexiline. The majority of cis‐, trans‐1‐ and trans‐2‐OH‐metabolites in plasma were derived from (−)‐, (+)‐ and (−)‐perhexiline, respectively, generally consistent with in vitro metabolism in human liver microsomes.25
Both enantiomers of perhexiline distributed extensively into the liver and heart, which may have been facilitated by uptake transporters and/or tissue binding, as suggested by the high tissue:plasma concentration ratios. Similar tissue accumulation has previously been reported for (±)‐perhexiline in animals16 and humans.26 Indeed, in this study, both total and individual enantiomer concentrations in plasma and heart following administration of the racemate to rats were similar to those observed clinically.26, 27 Although there was no evidence of substantial enantioselectivity in the in vivo uptake of perhexiline into human myocardium following clinical administration of racemate,27 this current study has revealed significant enantioselectivity in the hepatic and myocardial distribution of (+)‐ and (−)‐perhexiline, but it was evident only following administration of the pure enantiomers.
Since the apparent oral clearance of each enantiomer in DA rats was unaffected by the dosage form administered (enantiomer or racemate), and the plasma protein binding of perhexiline is not enantioselective,14 the steady‐state tissue:plasma concentration ratios should reflect the net tissue uptake of each enantiomer (i.e., the balance between uptake into and elimination from tissue). Therefore, our observations suggest enantioselectivity in the net‐uptake (or tissue binding) of the pure enantiomers, favouring (+)‐perhexiline (2.5‐ to 4.5‐fold greater). The loss of this enantioselectivity following administration of the racemate, suggests that (−)‐perhexiline and/or its metabolites may inhibit the net uptake (or tissue binding) of (+)‐perhexiline. However, this will need to be confirmed experimentally in a larger study. Enantiomer‐enantiomer interactions for uptake transporters have previously been reported for the antihistamine fexofenadine, leading to significant differences in the uptake of (+)‐ and (−)‐fexofenadine by Caco‐2 cells depending on whether they are studied individually or as a racemic mixture.28
In this study there was no evidence of hepatotoxicity, with plasma biochemistry for liver specific enzymes (ALT, GGT) showing no significant differences between treated or control groups, and no significant morphological changes in H&E sections. However, differences in the hepatic distribution of (+)‐ and (−)‐perhexiline following administration as pure enantiomers was associated with very mild changes in ultrastructural hepatic histology. Inhibition of hepatic CPT‐1 is associated with lipid accumulation10 and there are reports of hepatic steatosis with clinical use of racemic perhexiline.29 Racemic perhexiline inhibits rat myocardial and hepatic CPT‐1 (IC50 of 77 and 148 μmol/L, respectively5), with no apparent difference in potency between the enantiomers (Chong et al., manuscript in preparation). If one assumes that the tissue density is 1 g/mL, administration of the individual enantiomers achieved mean hepatic (+)‐perhexiline concentrations (375 μmol/L) 2.5‐fold higher than its IC50 for inhibiting hepatic CPT‐1, and six‐fold higher than the corresponding hepatic concentrations of (−)‐perhexiline (62 μmol/L). Thus, the ultrastructural differences in hepatic lipid contents between (+)‐ and (−)‐perhexiline may reflect greater inhibition of hepatic CPT‐1 in animals treated with (+)‐perhexiline, and may be the first signs of very mild microvesicular steatosis.
In contrast, animals treated with (−)‐perhexiline showed a significantly greater hepatic content of glycogen compared to (+)‐perhexiline. Again, these observations are consistent with inhibition of CPT‐1 by (+)‐perhexiline, leading to increased carbohydrate utilization and diminished glycogen stores. However, primary human hepatocyte cultures exposed to supra‐therapeutic concentrations of racemic perhexiline accumulate glycogen,30 and clinical reports have linked the use of racemic perhexiline to hepatic glycogen accumulation, with features similar to type‐1 glycogen storage disease.31 Perhexiline may have insulin‐sensitizing effects,32, 33 which would result in enhanced hepatic glycogen storage, and may be beneficial, also leading to enhanced myocardial glucose utilization and function.34 Perhexiline increases cardiac efficiency in isolated rat hearts without altering fatty acid oxidation, consistent with CPT‐1 independent actions,21 which could include a direct effect on carbohydrate (or glycogen) utilization.
Racemic perhexiline has been reported to decrease peripheral neuronal function in DA rats and induce, within dorsal root ganglia, an accumulation of Mallory bodies.16 In our study, Mallory and lamellar bodies were only observed in animals treated with (+)‐perhexiline: although these were present in less than 1% of cells. The association of (+)‐perhexiline with neurotoxicity was further supported by our Von Frey testing of peripheral neural function, as all animals administered (+)‐ or (±)‐perhexiline had thresholds for paw withdrawal that were above the range for the controls. It is possible that inhibition of neuronal CPT‐1 contributes to the development of peripheral neuropathy with long‐term use of perhexiline. However, a similar effect of the (−)‐enantiomer cannot be ruled out, given the significantly lower systemic exposure in rats administered (−)‐perhexiline.
In conclusion, these pilot data suggest that, although the two enantiomers of perhexiline are physicochemically identical, they display significantly different patterns of tissue distribution, which were not explained by enantioselectivity in their plasma clearance. Our study suggests that the extensive tissue exposure to (+)‐perhexiline and consequent inhibition of CPT‐1, may be associated with greater hepatic lipid vacuole content and peripheral neural dysfunction, as observed in cases of clinical toxicity from perhexiline. Importantly, for the same plasma concentrations, differences in tissue distribution may contribute to disparities in the effects of (+)‐ and (−)‐perhexiline on hepatic histology and neural function, when administered individually. However, further studies of the individual enantiomers and their effects on hepatic and myocardial energy metabolism and function are necessary to confirm these early data, particularly to determine concentration‐ and time‐dependence of our pilot observations, and whether a single enantiomer may have an improved therapeutic index compared to the current racemic dosage form.
DISCLOSURE
The authors are inventors on a patent application for the use of perhexiline enantiomers.
ACKNOWLEDGMENTS
We thank JK Brealey (Electron Microscope Unit, The Queen Elizabeth Hospital), Dr J Pierides (Surgical Pathology and Cytopathology, SA Pathology) and Dr John Finnie (Senior Veterinary Pathologist, SA Pathology) for their assistance with the histological procedures, and Dr J Field (University of Adelaide) for assistance with statistical analyses. Dr G Licari was supported by a postgraduate scholarship from the Queen Elizabeth Hospital Research Foundation and Adelaide University.
Licari G, Milne RW, Somogyi AA, Sallustio BC. Enantioselectivity in the tissue distribution of perhexiline contributes to different effects on hepatic histology and peripheral neural function in rats. Pharmacol Res Perspect. 2018;e00406 https://doi.org/10.1002/prp2.406
REFERENCES
- 1. Rossi S, ed. Australian medicines handbook. Adelaide: Australian Medicines Handbook Pty Ltd; 2017. [Google Scholar]
- 2. Lee L, Campbell R, Scheuermann‐Freestone M, Taylor R, Gunaruwan P, Williams L, et al. Metabolic modulation with perhexiline in chronic heart failure. A randomized, controlled trial of short‐term use of a novel treatment. Circulation. 2005;112:3280‐3288. [DOI] [PubMed] [Google Scholar]
- 3. Abozguia K, Elliot P, McKenna W, Phan TT, Nallur Shivu G, Ahmed I, et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in syptomatic hypertrophic cardiomyopathy. Circulation. 2010;122:1562‐1569. [DOI] [PubMed] [Google Scholar]
- 4. Unger SA, Robinson MA, Horowitz JD. Perhexiline improves symptomatic status in elderly patients with severe aortic stenosis. Aust NZ J Med. 1997;27:24‐28. [DOI] [PubMed] [Google Scholar]
- 5. Kennedy JA, Unger SA, Horowitz JD. Inhibition of carnitine palmitoyltransferase‐1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol. 1996;52:273‐280. [DOI] [PubMed] [Google Scholar]
- 6. Hue L, Taegtmeyer H. The Randle cycle revisited: A new head for an old hat. Am J Physiol Endocrinol Metab. 2009;297:E578‐E591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horowitz JD, Chirkov YY, Kennedy JA, Sverdlov AL. Modulation of myocardial metabolism: An emerging therapeutic principle. Curr Opin Cardiol. 2010;25:329‐334. [DOI] [PubMed] [Google Scholar]
- 8. Singlas E, Goujet MA, Simon P. Pharmacokinetics of perhexiline maleate in anginal patients with and without peripheral neuropathy. Eur J Clin Pharmacol. 1978;14:195‐201. [DOI] [PubMed] [Google Scholar]
- 9. Morgan MY, Reshef R, Shah RR, Oates NS, Smith RL, Sherlock S. Impaired oxidation of debrisoquine in patients with perhexiline liver injury. Gut. 1984;25:1057‐1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fromenty B, Pessayre D. Inhibition of mitochondrial beta‐oxidation as a mechanism of hepatotoxicity. Pharmac Ther. 1995;67:101‐154. [DOI] [PubMed] [Google Scholar]
- 11. Horowitz JD, Sia STB, Macdonald PS, Goble AJ, Louis WJ. Perhexiline maleate treatment for severe angina pectoris ‐ correlations with pharmacokinetics. Int J Cardiol. 1986;13:219‐229. [DOI] [PubMed] [Google Scholar]
- 12. Cole PL, Beamer AD, McGowan N, Cantillon CO, Benfell K, Kelly RA, et al. Efficacy and safety of perhexiline maleate in refractory angina. A double‐blind placebo‐controlled clinical trial of a novel antianginal agent. Circulation. 1990;81:1260‐1270. [DOI] [PubMed] [Google Scholar]
- 13. Gould BJ, Amoah AGB, Parke DV. Stereoselective pharmacokinetics of perhexiline. Xenobiotica. 1986;16:491‐502. [DOI] [PubMed] [Google Scholar]
- 14. Inglis SC, Herbert MK, Davies BJ, Coller JK, James HM, Horowitz JD, et al. Effect of CYP2D6 metabolizer status on the disposition of the (+) and (‐) enantiomers of perhexiline in patients with myocardial ischaemia. Pharmacogenet Genomics. 2007;17:305‐312. [DOI] [PubMed] [Google Scholar]
- 15. Davies BJ, Herbert MK, Coller JK, Somogyi AA, Milne RW, Sallustio BC. Steady‐state pharmacokinetics of the enantiomers of perhexiline in CYP2D6 poor and extensive metabolisers administered rac‐perhexiline. Br J Clin Pharmacol. 2008;65:347‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meier C, Wahllaender A, Hess CW, Preisig R. Perhexiline‐induced lipidosis in the dark agouti (DA) rat: An animal model of genetically determined neurotoxicity. Brain. 1986;109:649‐660. [DOI] [PubMed] [Google Scholar]
- 17. Licari G, Somogyi AA, Milne RW, Sallustio BC. Comparison of CYP2D metabolism and hepatotoxicity of the myocardial metabolic agent perhexiline in Sprague Dawley and Dark Agouti rats. Xenobiotica. 2015;45:3‐9. [DOI] [PubMed] [Google Scholar]
- 18. Davies BJ, Herbert MK, Culbert JA, Pyke SM, Coller JK, Somogyi AA, et al. Enantioselective assay for the determination of perhexiline enantiomers in human plasma by liquid chromatography. J Chromatogr B. 2006;832:114‐120. [DOI] [PubMed] [Google Scholar]
- 19. Chaplan SR, Bach FW, Pogrel W, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55‐63. [DOI] [PubMed] [Google Scholar]
- 20. Singh A, Gilroy C, Chu I, Villeneuve DC. Toxicity of PCB 105 in the rat liver: An ultrastructural and biochemical study. Ultrastruct Pathol. 1997;21:143‐151. [DOI] [PubMed] [Google Scholar]
- 21. Unger SA, Kennedy JA, McFadden‐Lewis K, Minerds K, Murphy GA, Horowitz JD. Dissociation between metabolic and efficiency effects of perhexiline in normoxic rat myocardium. J Cardiovasc Pharmacol. 2005;46:849‐855. [DOI] [PubMed] [Google Scholar]
- 22. Davies BJ, Herbert MK, Coller JK, Somogyi AA, Milne RW, Sallustio BC. Determination of the 4‐monohydroxy metabolites of perhexiline in human plasma, urine and liver microsomes by liquid chromatography. J Chromatogr B. 2006;843:302‐309. [DOI] [PubMed] [Google Scholar]
- 23. Willoughby SR, Stewart S, Chirkov YY, Kennedy JA, Holmes AS, Horowitz JD. Beneficial clinical effects of perhexiline in patients with stable angina pectoris and acute coronary syndromes are associated with potentiation of platelet responsiveness to nitric oxide. Eur Heart J. 2002;23:1946‐1954. [DOI] [PubMed] [Google Scholar]
- 24. Kennedy JA, Beck‐Oldach K, McFadden‐Lewis K, Murphy GA, Wong YW, Zhang Y, et al. Effect of the anti‐anginal agent, perhexiline, on neutrophil, valvular and vascular superoxide formation. Eur J Pharmacol. 2006;531:13‐19. [DOI] [PubMed] [Google Scholar]
- 25. Davies BJ, Coller JK, Somogyi AA, Milne RW, Sallustio BC. CYP2B6, CYP2D6, and CYP3A4 catalyze the primary oxidative metabolism of perhexiline enantiomers by human liver microsomes. Drug Metab Dispos. 2007;35:128‐138. [DOI] [PubMed] [Google Scholar]
- 26. Drury NE, Licari G, Chong C‐R, Howell NJ, Frenneaux MP, Horowitz JD, et al. Relationship between plasma, atrial and ventricular perhexiline concentrations in humans: Insights into factors affecting myocardial uptake. Br J Clin Pharmacol. 2014;77:789‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chong C‐R, Drury NE, Licari G, et al. Stereoselective handling of perhexiline: implications regarding accumulation within the human myocardium. Eur J Clin Pharmacol. 2015;71:1485‐1491. [DOI] [PubMed] [Google Scholar]
- 28. Togami K, Tosaki Y, Chono S, Morimoto K, Hayasaka M, Tada H. Enantioselective uptake of fexofenadine by Caco‐2 cells as model intestinal epithelial cell. J Pharm Pharmacol. 2013;65:22‐29. [DOI] [PubMed] [Google Scholar]
- 29. Lewis D, Wainwright HC, Kew MC, Zwi S, Isaacson C. Liver damage associated with perhexiline maleate. Gut. 1979;20:186‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lageron A, Scotto J, Gautier M. Effects of Pexid on liver cell cultures. Ultrastructural and histoenzymological studies. Eur J Clin Pharmacol. 1981;19:417‐421. [DOI] [PubMed] [Google Scholar]
- 31. Lageron A, Poupon R, de Saint‐Maur PP, Levy VG. Liver ganglioside storage after perhexiline maleate. Lancet. 1977;309:483. [DOI] [PubMed] [Google Scholar]
- 32. Stewart S, Voss DW, Northey DL, Horowitz JD. Relationship between plasma perhexiline concentration and symptomatic status during short‐term perhexiline therapy. Ther Drug Monit. 1996;18:635‐639. [DOI] [PubMed] [Google Scholar]
- 33. Nobuhara M, Saotome M, Watanabe T, Urushida T, Katoh H, Sato H, et al. Mitochondrial dysfunction caused by saturated fatty acid loading induces myocardial insulin‐resistance in differentiated H9c2 myocytes: A novel ex vivo myocardial insulin‐resistance model. Exp Cell Res. 2013;319:955‐966. [DOI] [PubMed] [Google Scholar]
- 34. Fath‐Ordoubadi F, Beatt KJ. Glucose‐insulin‐potassium therapy for treatment of acute myocardial infarction: An overview of randomized placebo‐controlled trials. Circulation. 1997;96:1152‐1156. [DOI] [PubMed] [Google Scholar]