

Abstract
Background and Purpose
Patients with irritable bowel syndrome suffer from chronic visceral pain (CVP) and limited analgesic therapeutic options are currently available. We have shown that α‐conotoxin Vc1.1 induced activation of GABAB receptors on the peripheral endings of colonic afferents and reduced nociceptive signalling from the viscera. However, the analgesic efficacy of more stable, cyclized versions of Vc1.1 on CVP remains to be determined.
Experimental Approach
Using ex vivo colonic afferent preparations from mice, we determined the inhibitory actions of cyclized Vc1.1 (cVc1.1) and two cVc1.1 analogues on mouse colonic nociceptors in healthy and chronic visceral hypersensitivity (CVH) states. Using whole‐cell patch clamp recordings, we also assessed the inhibitory actions of these peptides on the neuronal excitability of colonic innervating dorsal root ganglion neurons. In vivo, the analgesic efficacy of these analogues was assessed by determining the visceromotor response to colorectal distension in healthy and CVH mice.
Key Results
cVc1.1 and the cVc1.1 analogues, [C2H,C8F]cVc1.1 and [N9W]cVc1.1, all caused concentration‐dependent inhibition of colonic nociceptors from healthy mice. Inhibition by these peptides was greater than those evoked by linear Vc1.1 and was substantially greater in colonic nociceptors from CVH mice. cVc1.1 also reduced excitability of colonic dorsal root ganglion neurons, with greater effect in CVH neurons. CVH mice treated with cVc1.1 intra‐colonically displayed reduced pain responses to noxious colorectal distension compared with vehicle‐treated CVH mice.
Conclusions and Implications
Cyclic versions of Vc1.1 evoked significant anti‐nociceptive actions in CVH states, suggesting that they could be novel candidates for treatment of CVP.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Abbreviations
- cVc1.1
cyclized Vc1.1
- CVH
chronic visceral hypersensitivity
- CVP
chronic visceral pain
- DRG
dorsal root ganglion
- EMG
electromyography
- IBS
irritable bowel syndrome
- TL
thoracolumbar
- TNBS
trinitrobenzene sulphonic acid
- Vc1.1
‘linear’ Vc1.1
- vfh
von Frey hair
- VGCCs
voltage‐gated calcium channels
- VMR
visceromotor response
Introduction
Irritable bowel syndrome (IBS) is a prevalent, chronic gastrointestinal disorder that detracts from the quality of life for ~11% of the global population (Chey et al., 2015; Enck et al., 2016). IBS is characterized by abdominal pain, discomfort, bloating and altered bowel habits (Chey et al., 2015; Enck et al., 2016). Although the pathophysiology of IBS is not completely understood, it is becoming clear that changes to peripheral cellular and sensory mechanisms play key roles in the associated pain (Brierley and Linden, 2014; Enck et al., 2016; Bellono et al., 2017). In particular, chronic visceral hypersensitivity (CVH) of colonic afferents is implicated in the development and maintenance of chronic visceral pain (CVP) in IBS patients (Brierley and Linden, 2014). Characteristic features of CVH include nociceptor hypersensitivity, increased signalling of noxious colorectal distension (CRD) within the spinal cord and allodynia and hyperalgesia to colorectal distension (Castro et al., 2013; de Araujo et al., 2014; Castro et al., 2017). Recent evidence suggests sensory afferents display up‐regulation of various membrane receptors and ion channels in animal models of CVH (Brierley, 2016), making them targets for analgesic treatment.
The α‐conotoxin family of venom‐derived peptides from marine cone snails provides a rich source of novel disulfide‐bonded peptides that target a wide variety of membrane receptors and ion channels (Lewis et al., 2000; Schroeder and Craik, 2012; Vetter and Lewis, 2012; Adams and Berecki, 2013). In particular, α‐conotoxin Vc1.1, a ‘linear’ 16‐amino acid synthetic version of a peptide derived from Conus victoriae, has anti‐nociceptive actions in vitro and anti‐hyperalgesic actions in numerous in vivo models of neuropathic pain (Satkunanathan et al., 2005; Clark et al., 2010; Klimis et al., 2011). We recently showed that Vc1.1 inhibits sensory afferent pathways within the splanchnic and pelvic innervation of the colon and that these inhibitory actions of Vc1.1 are enhanced in an animal model of CVH (Castro et al., 2017). Notably, we demonstrated that this inhibitory effect occurs via Vc1.1 activation of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=242, which is expressed by colonic afferents, and the subsequent down‐stream inhibition of the voltage‐gated calcium channels (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=80), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=533 (N‐type) and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=534 (R‐type) (Castro et al., 2017). We also demonstrated that Vc1.1 reduces the excitability of human dorsal root ganglion (DRG) neurons via a GABAB receptor‐mediated mechanism (Castro et al., 2017). These studies confirmed recent recombinant cell line studies demonstrating the human GABAB receptor is the primary, and high affinity target for Vc1.1 (Callaghan and Adams, 2010; Clark et al., 2010), rather than the originally proposed target, α9α10 http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76 (Vincler et al., 2006; Vincler & McIntosh, 2007; McIntosh et al., 2009). Furthermore, these studies show that GABAB receptor activation by Vc1.1 causes down‐stream inhibition of the VGCCs CaV2.2 (N‐type) and CaV2.3 (R‐type) but not http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=532 (P/Q‐type) (Adams et al., 2012; Berecki et al., 2014).
These findings are promising for the development of Vc1.1 as a therapeutic treatment for CVP, particularly as Vc1.1 (also called ACV1) has been used in Phase I and Phase IIA clinical trials for the treatment of neuropathic pain (ASX, 2006a; ASX, 2006b; ASX, 2007). In these studies, ACV1 was reported to be safe and well tolerated, with a clean safety and side effect profile. In spite of such promise, peptides as therapeutics can be limited by their susceptibility to degradation within the body. However, the use of synthetic cyclization, whereby the N and C termini are joined by a linker sequence of residues, can stabilize the structure of the peptide and reduce the susceptibility to proteolysis, thus potentially enhancing therapeutic potential (Clark et al., 2010). We have previously shown that cyclization of Vc1.1 improves its stability and is orally active (Clark et al., 2010). Intriguingly, this cyclized Vc1.1 (called cVc1.1) has two additional serendipitous benefits, which are likely to be a result of its extra rigidity. First, it is a more potent inhibitor of the GABAB receptor‐modulated CaV2.2 channel, the proposed target for analgesia, than the native ‘linear’ Vc1.1. Second, it is also more selective for the inhibition of VGCC currents over the α9α10 nAChR subtype when compared to ‘linear’ Vc1.1 (Clark et al., 2010). Therefore, it has increased potency for GABAB receptor‐mediated inhibition of VGCCs and a reduced activity at the α9α10 nAChR compared to linear Vc1.1 (Clark et al., 2010; Yu et al., 2015). Accordingly, cVc1.1 has been used as an orally active peptide, which displays analgesic activity in rat models of neuropathic pain (Clark et al., 2010; Yu et al., 2015). Therefore, in the present study, we assessed the anti‐nociceptive activity of cVc1.1 and two modified cyclic analogues of Vc1.1 in colonic afferents and colonic DRG neurons from healthy and CVH mice. One of the cyclic analogues of Vc1.1, [C2H,C8F]cVc1.1, was designed to be a simplified version of cVc1.1 in terms of its production, whereas the second, [N9W]cVc1.1, was designed to have increased potency at the α9α10 nAChR. We also compared the most potent cVc1.1 analogue with Vc1.1, using an in vivo model assessing visceral sensitivity in both healthy and CVH mice.
Methods
Animals
All animal care and experimental procedures were approved by the Animal Ethics Committees of the South Australian Health and Medical Research Institute (SAHMRI), The University of Adelaide and Flinders University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). A total of 116 male C57BL/6J mice aged 13–17 weeks of age were used in the reported experiments. We have previously demonstrated that mice provide an appropriate model for investigating CVH relevant to clinical conditions such as IBS (Castro et al., 2013; Brierley and Linden, 2014). Therefore, we chose mice as the species for the current study.
Mice were acquired from an in‐house C57BL/6J breeding programme (from Jax strain #000664 originally purchased from The Jackson Laboratory; Bar Harbor, ME) within SAHMRI's specific and opportunistic pathogen‐free animal care facility. Mice were group housed (five mice per cage) within individual ventilated cages (IVC), which were filled with chip coarse dust‐free aspen bedding (Cat# – ASPJMAEB‐CA; PuraBed, Niederglatt, Switzerland). These cages were stored on IVC racks in specific housing rooms within a temperature‐controlled environment of 22°C and a 12 h light/12 h dark cycle. Mice had free access to JL Rat and Mouse/Auto6F chow (Cat# – 5K52; LabDiet, St. Louis, MO) and autoclaved reverse osmosis purified water (EcoPure. UK). Mice had an average weight of 29 g on the experimental day.
Mice were randomly assigned to healthy control or trinitrobenzene sulphonic acid (TNBS) treatment groups. Following TNBS administration, mice were individually housed in IVCs to allow for accurate clinical monitoring until the experimental day in question. Mice were randomly assigned to study sub‐groups and the order of treatment was also randomized. Where possible, investigators were blinded to either the drugs being administered or the treatment group during analysis.
Model of chronic visceral hypersensitivity (CVH)
Colitis was induced by administration of TNBS as described previously (Hughes et al., 2009b; Castro et al., 2013; de Araujo et al., 2014; Hughes et al., 2014; Osteen et al., 2016; Castro et al., 2017). Briefly, 13‐week‐old mice, anaesthetized with isofluorane, were administered an intracolonic enema of 0.1 mL TNBS (135 μL·mL−1 of 1 M solution in 35% ethanol), via a polyethylene catheter inserted 3 cm from the anus. Mice were then individually housed and monitored up to three times daily for clinical assessment for changes in body weight, physical appearance and behaviour. Our previous studies using this model show mucosal architecture, cellular infiltrate, crypt abscesses and goblet cell depletion confirming that TNBS induces significant damage of the colonic mucosa by day 3 post treatment. This damage largely spontaneously recovers by day 7 and is fully resolved by day 28. At the 28 day time point, the high‐threshold nociceptors in these mice display significant mechanical hypersensitivity and lower mechanical activation thresholds (Hughes et al., 2009b). Mice from this model also display increased neuronal activation in the dorsal horn of the spinal cord in response to noxious colorectal distension, as well as sprouting of colonic afferent terminals within the dorsal horn (Harrington et al., 2012). This model also induces hyperalgesia and allodynia to colorectal distension (Adam et al., 2006) and is therefore termed ‘Chronic Visceral Hypersensitivity’ (Hughes et al., 2009b; Castro et al., 2013; de Araujo et al., 2014; Hughes et al., 2014; Osteen et al., 2016; Castro et al., 2017).
Ex vivo single fibre colonic splanchnic afferent recording preparation
Mice were humanely killed, by CO2 inhalation at days 0 (healthy) and 28 (CVH) after TNBS administration. The colon and rectum (5–6 cm) and attached splanchnic nerves were removed, and afferent recordings from splanchnic nerves were performed as described previously (Brierley et al., 2004; Brierley et al., 2005b; Hughes et al., 2009b). Briefly, colons were removed, dissected open and pinned flat, mucosal side up, in a specialized organ bath. The colonic compartment was superfused with a modified Krebs solution [in mM: 117.9 NaCl, 4.7 KCl, 25 NaHCO3, 1.3 NaH2PO4, 1.2 MgSO4, 2.5 CaCl2, 11.1 d‐glucose], bubbled with carbogen (95% O2, 5% CO2) at a temperature of 34°C. All solutions contained the L‐type calcium channel antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2514 (1 μM) to suppress smooth muscle activity and the prostaglandin synthesis inhibitor http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1909 (3 μM) to block endogenous prostaglandin production. The nerve bundle was extended into a paraffin‐filled recording compartment in which finely dissected strands were laid onto a mirror, and single fibres placed on the platinum recording electrode. Action potentials, generated by mechanical stimuli to the colon's receptive field, were recorded by a differential amplifier, filtered and sampled (20 kHz) using a 1401 interface (Cambridge Electronic Design, Cambridge, UK) and stored on a PC for offline analysis.
Colonic afferent classification and selection
Receptive fields were identified by systematically stroking the mucosal surface of the colon with a stiff brush to activate all subtypes of mechanoreceptors. Categorization of afferents properties was in accordance with our previously published classification system (Brierley et al., 2004; Brierley et al., 2005b). Once identified, receptive fields were tested with three distinct mechanical stimuli to enable classification: static probing with calibrated von Frey hairs (vfh) (2 g force; applied three times for a period of 3 s), mucosal stroking with calibrated vfh (10 mg force; applied 10 times) or circular stretch (5 g; applied for a period of 1 min). We tested the effect of cVc1.1 and the cVc1.1 analogues, [C2H,C8F]cVc1.1 and [N9W]cVc1.1, on serosal afferents, also termed vascular afferents (Brookes et al., 2013), recorded from the splanchnic pathway. These colonic afferents have high‐mechanical activation thresholds and respond to noxious distension (40 mmHg), stretch (≥7 g) or vfh filaments (2 g) but not to fine mucosal stroking (10 mg vfh) (Brierley et al., 2004; Hughes et al., 2013; de Araujo et al., 2014; Osteen et al., 2016). The algesic ion channels and receptors, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=578 (Osteen et al., 2016), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=507 (Brierley et al., 2005b), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=485 (Hughes et al., 2009a; Brierley et al., 2011), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=510 (Brierley et al., 2008), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=480 (Brierley et al., 2005b), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=41 (Brierley et al., 2005a), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 (Beyak, 2010) and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1870 (Hughes et al., 2013), are highly expressed in these afferents. In addition, serosal afferents become mechanically hypersensitive in models of CVP (Hughes et al., 2009b) and have a nociceptor phenotype (Brierley et al., 2004; Castro et al., 2013; de Araujo et al., 2014; Carstens et al., 2016; Osteen et al., 2016; Castro et al., 2017). In the present study, they are therefore referred to as colonic ‘nociceptors’.
Peptide application to colonic afferents
The peptides cVc1.1, [C2H,C8F]cVc1.1 and [N9W]cVc1.1 were prepared from stock solutions, diluted to appropriate final concentrations (1, 10, 100 or 1000 nM) in Krebs solution. These concentrations of peptide were applied sequentially to the same individual afferent receptive field. There was no washout duration per se with the next drug added within ~30 s of completing mechanical testing following the previous drug addition. This process involved determining baseline splanchnic colonic nociceptor mechanosensitivity in response to application of 3 × 3 s 2 g vfh probes to the afferent receptive field. A small chamber was then applied to the mucosal surface of the colon, which surrounded the afferent receptive field. Residual Krebs solution within the chamber was aspirated and 1 nM of the respective peptide applied for 5 min. Mechanical sensitivity was then re‐tested in response to application of 3 × 3 s 2 g vfh probes to the afferent receptive field. This process was then repeated for 10, 100 and 1000 nM of the respective peptide and mechanical sensitivity re‐tested after each concentration (Castro et al., 2013; de Araujo et al., 2014; Carstens et al., 2016; Osteen et al., 2016; Castro et al., 2017).
Statistical analysis of afferent recording data
The data and statistical analyses in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Action potentials were analysed offline using the Spike 2 (version 5.21) software (Cambridge Electronic Design, Cambridge, UK) and discriminated as single units based on distinguishable waveforms, amplitudes and durations. Data are expressed as mean ± SEM. n = the number of afferents recorded. N = the number of animals used for those specific experiments. In some instances, data are presented as ‘change from baseline’. This is calculated by determining the change in mechanosensitivity of individual afferents between the normal ‘baseline’ response in healthy or CVH conditions compared to the respective mechanical responses following peptide addition. This difference is then averaged across all afferents with a cohort to obtain a final mean ± SEM of ‘change in response from baseline’. Data were statistically compared using Prism 7 software (GraphPad Software, San Diego, CA, USA) and, where appropriate, were analysed using a one or two‐way ANOVA with Bonferroni post hoc tests. Differences were considered significant at a level of P < 0.05.
Retrograde tracing to identify colonic thoracolumbar (TL) DRG neurons
Cholera toxin subunit B conjugated to AlexaFluor 488 (Invitrogen, Carlsbad, CA, USA) was injected at three sites sub‐serosally within the wall of the distal colon of healthy control or CVH mice (Brierley et al., 2009; Harrington et al., 2012; de Araujo et al., 2014; Castro et al., 2017). After 4 days, animals were humanely killed by CO2 inhalation for subsequent TL (T10‐L1) DRG removal and dissociation.
Cell culture of colonic DRG neurons
DRGs (thoracic 9 to lumbar 1) were digested with 4 mg·mL−1 collagenase II (GIBCO, Invitrogen) and 4 mg·mL−1 dispase (GIBCO) for 30 min at 37°C, followed by 4 mg·mL−1 collagenase II for 10 min at 37°C. A single‐cell suspension was achieved via trituration of DRG's through fire‐polished Pasteur pipettes of descending diameter. Neurons were resuspended in DMEM (GIBCO) containing 10% FCS (Invitrogen), 2 mM L‐glutamine (GIBCO), 100 μM MEM non‐essential amino acids (GIBCO) and 100 mg·mL−1 penicillin/streptomycin (Invitrogen). Neurons were spot‐plated on clean 13 mm coverslips cut in half and coated with laminin (20 μg·mL−1) and poly‐D‐lysine (800 μg·mL−1) and maintained in an incubator at 37°C in 5% CO2.
Patch clamp recordings of colonic DRG neurons
Whole‐cell patch clamp recordings were made from fluorescently labelled colonic TL DRG neurons (from N = 5 mice per group) 24–48 h after plating, using fire‐polished glass electrodes with a resistance of 2–5 MΩ. Inclusion criteria for cells included: (i) being retrogradely traced; (ii) having small diameter (maximum soma diameter <20 μm); (iii) having a resting membrane potential more negative than −40 mV; (iv) a series resistance of <10 MΩ; and (v) a capacitance of ≤30 pF. Resting membrane potential was −49.19 ± 1.0 mV for healthy colonic innervating DRG neurons and −47.64 ± 1.3 mV for CVH colonic innervating DRG neurons. For all colonic DRG neurons, the membrane potential was held at −70 mV. In current clamp mode, a series of depolarizing pulses (10 pA current step, 500 ms duration) were applied from the holding potential and the amount of current required to elicit an action potential (rheobase) determined in normal external bath solution and following either the addition of Vc1.1 (10 nM) or cVc1.1 (10 nM). Control solutions and peptides were applied with a gravity‐driven multi‐barrel perfusion system positioned within 1 mm of the neuron under investigation, as used previously (Brierley et al., 2011). This involved an initial baseline rheobase recording being made during continuous perfusion of normal external bath solution. This was followed by a 2 min continuous perfusion with either Vc1.1 (10 nM) or cVc1.1 (10 nM) diluted in external bath solution, after which a second recording was made to determine rheobase in the presence of the individual peptides. Pipettes were filled with intracellular solutions containing (in mM): 135 KCl; 2 MgCl2; 2 MgATP; 5 EGTA‐Na; 10 HEPES‐Na; adjusted to pH 7.4. Extracellular bath solutions contained (in mM) the following: 140 NaCl; 4 KCl; 2 MgCl2; 2 CaCl2; 10 HEPES‐Na; 5 glucose; adjusted to pH 7.4.
Statistical analysis of patch clamp data
All data were analysed using Prism 7 software (GraphPad Software, San Diego, CA, USA) using paired t‐tests or one‐way ANOVAs, to compare before and after effects of Vc1.1 or cVc1.1 on rheobase. Data are expressed as mean ± SEM. A neuron inhibited by Vc1.1 or cVc1.1 was defined as exhibiting a ≥10% change in rheobase from baseline control, as described previously (Osteen et al., 2016; Castro et al., 2017). An unpaired t‐test was used to determine differences in the rheobase of colonic DRG neurons from healthy and CVH mice. Differences between specific drug and baseline responses were considered significant at a level of P < 0.05. n = the number of colonic innervating DRG neurons recorded. N = the number of animals used for those specific experiments.
Visceromotor responses (VMR) to colorectal distension
Noxious distension of the colorectum triggers the VMR, a nociceptive brainstem reflex consisting of the contraction of the abdominal muscles (Ness and Gebhart, 1988). Using abdominal electromyography (EMG), this technique allows assessment of visceral sensitivity in vivo in fully awake animals (Christianson and Gebhart, 2007; Deiteren et al., 2014). Under isoflurane anaesthesia, the bare endings of two Teflon‐coated stainless steel wires (Advent Research Materials Ltd, Oxford, UK) were sutured into the right abdominal muscle and tunnelled subcutaneously to be exteriorized at the base of the neck for future access. At the end of the surgery, mice received prophylactic antibiotic (Baytril®; 5 mg·kg−1 s.c.) and analgesic (buprenorphine; 0.09 mg kg−1 s.c.), were housed individually and allowed to recover for at least 3 days before assessment of VMR. On the day of VMR assessment, mice were briefly anaesthetized using isoflurane and received a 100 μL enema of 1 μM Vc1.1, 1 μM cVc1.1 or vehicle (sterile H2O). A lubricated balloon (2.5 cm length) was gently introduced through the anus and inserted into the colorectum up to 0.25 cm past the anal verge. The balloon catheter was secured to the base of the tail and connected to a barostat (Isobar 3, G&J Electronics, Willowdale, Canada) for graded and pressure‐controlled balloon distension. Mice were allowed to recover from anaesthesia in a restrainer with dorsal access for 15 min prior to initiation of the distension sequence. Distensions were applied at 20–40–60–80 mmHg (20 s duration) at a 4 min interval so that the last distension was performed 30 min after intra colonic treatment. Following the final distension, mice were humanely killed by cervical dislocation. The EMG electrodes were relayed to a data acquisition system, and the signal was recorded (NL100AK headstage), amplified (NL104), filtered (NL 125/126, Neurolog, Digitimer Ltd, bandpass 50–5000 Hz) and digitized (CED 1401, Cambridge Electronic Design, Cambridge, UK) to a PC for offline analysis using Spike2 (Cambridge Electronic Design). The analogue EMG signal was rectified and integrated. To quantify the magnitude of the VMR at each distension pressure, the AUC during the distension (20 s) was corrected for the baseline activity (AUC pre‐distension, 20 s).
Colonic compliance
Colonic compliance was assessed by applying graded volumes (40–200 μL, 20 s duration) to the balloon in the colorectum of fully awake mice, during the recording of the corresponding colorectal pressure, as described previously (Deiteren et al., 2014).
Statistical analysis of VMR data
Data are presented as means ± SEM, where N represents the number of animals. Data were statistically analysed by generalized estimating equations followed by the LSD post hoc test when appropriate, using SPSS 23.0. Analysis and figures were prepared in GraphPad (Prism 7 Software, San Diego, CA, USA).
Design of cVc1.1 and cVc1.1 analogue variants
Vc1.1 was synthesized via solid‐phase peptide chemistry as described previously (Clark et al., 2006). Cyclic Vc1.1 (cVc1.1) was engineered by joining the N‐ and C‐termini of the peptide without affecting the three‐dimensional structure or biological activity (Clark et al., 2010). cVc1.1 was designed because a major obstacle generally impeding the use of bioactive peptides as drugs is their susceptibility to enzymic degradation and lack of oral bioavailability. The cyclic variant proved to be stable and orally active in the rat chronic constriction injury model of neuropathic pain (Clark et al., 2010).
Two mutants of the cyclic peptide were also studied (Figure 1). [N9W]cVc1.1 was designed based on the increased potency at α9α10 nAChR of ‘linear’ Vc1.1 resulting from the substitution N9W (Yu et al., 2013). This variant was instrumental in determining that Vc1.1 preferentially blocks the α10(+)α9(−) orthosteric binding site (Yu et al., 2013). [N9W]Vc1.1 is 30‐fold more active than the parent peptide at the human α9α10 nAChR, with an IC50 of 33 nM compared with the rat α9α10 nAChR of 975 nM. A one‐disulfide variant of cVc1.1, namely, [C2H,C8F]cVc1.1, was also designed to reduce the possibility of disulfide bond shuffling, which can limit the bioavailability of disulfide‐rich peptides (Yu et al., 2015). The disulfide bond between the first and third Cys was replaced by two hydrophobic residues, which we showed by NMR spectroscopy analysis to form a small hydrophobic core to the peptide. The cost of this simplifying mutation is that the inhibitory activity of [C2H,C8F]cVc1.1 falls by threefold compared to the native peptide at the human α9α10 (IC50 of 13 μM) and at VGCCs via activation of GABAB receptors (IC50 of 900 pM) (Yu et al., 2015).
Figure 1.
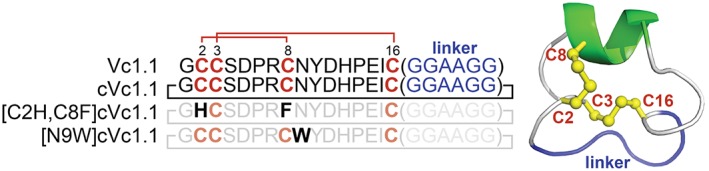
Solution structure of cVc1.1 and sequences of cVc1.1 wild‐type and variants [C2H,C8F]cVc1.1 and [N9W]cVc1.1 used in this study. cVc1.1 is an engineered peptide in which a cyclizing linker (blue) was added to confer stability and oral activity to the analgesic peptide Vc1.1. This peptide comprises two disulfide bonds, which are shown in red. The conserved positions of the cVc1.1 variants are shown using lighter colour fonts to highlight the substituted positions.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/2018 (Alexander et al., 2017a,b,c).
Results
Cyclized Vc1.1 (cVc1.1) inhibits colonic nociceptor mechanosensitivity
We have previously shown that Vc1.1 inhibits colonic nociceptors from both healthy and CVH mice. However, we observed that Vc1.1 evoked a larger inhibitory effect in colonic afferents from a mouse model of CVH (Castro et al., 2017). Given that cVc1.1 has improved stability and increased potency and efficacy for GABAB receptor‐mediated inhibition of VGCC currents than Vc1.1 (Clark et al., 2010; Yu et al., 2015), we hypothesized that cVc1.1 should evoke greater inhibition of colonic nociceptors. To test this hypothesis, we performed ex vivo single fibre afferent recordings of colonic nociceptors from both healthy and CVH mice (Castro et al., 2013; de Araujo et al., 2014; Osteen et al., 2016; Castro et al., 2017). We assessed colonic nociceptor mechanosensitivity before and after increasing doses of cVc1.1 (1, 10, 100 nM and 1 μM) and observed that cVc1.1 concentration‐dependently inhibited colonic nociceptor mechanosensitivity from both healthy (IC50: 12.2 nM, Figure 2A) and CVH (IC50: 3.5 nM, Figure 2B) mice. Notably, at each of the cVc1.1 concentrations tested, CVH colonic nociceptors displayed lower mechanosensory responses than in healthy nociceptors, with greatest inhibition observed at a concentration of 1 μM (Figure 2C, D, E). A direct comparison of the inhibitory actions evoked by ‘linear’ Vc1.1 and cyclic cVc1.1 demonstrated, at each concentration tested, that cVc1.1 caused greater inhibition than Vc1.1. of colonic nociceptors from both healthy (IC50 values, Vc1.1: 23.4 nM; cVc1.1: 12.2 nM, Figure 2F) and CVH (IC50 values, Vc1.1: 10.2 nM; cVc1.1: 3.5 nM, Figure 2G) mice. Overall, these data show that cVc1.1 causes potent inhibition of colonic nociceptors, particularly those from CVH mice.
Figure 2.
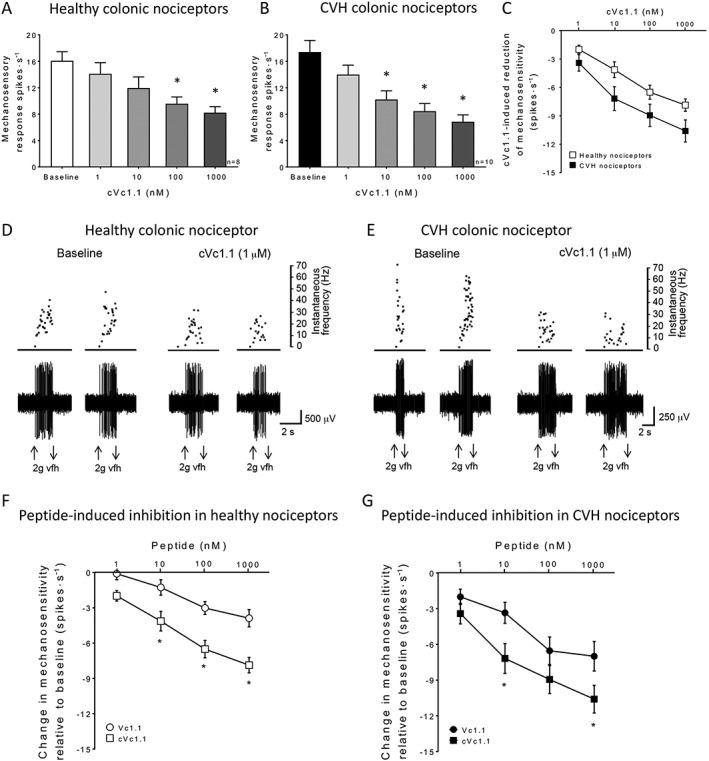
Cyclized Vc1.1 (cVc1.1) inhibits colonic nociceptors from healthy and CVH mice. (A) Healthy colonic nociceptor mechanosensitivity is significantly reduced following increasing concentrations of cVc1.1, applied to the mucosal epithelium, for 5 min at each concentration. cVc1.1 at 100 nM and 1 μM significantly reduced colonic nociceptor mechanosensitivity compared with baseline responses. n = 8 afferents from N = 5 healthy mice. *P < 0.05, one‐way ANOVA, with Bonferroni post hoc test. (B) In a model of CVH, colonic nociceptors display increased mechanosensitivity at baseline but are potently and concentration‐dependently inhibited by cVc1.1. Concentrations of cVc1.1 at 10, 100 nM and 1 μM significantly reduced the mechanical response of colonic nociceptors compared with baseline responses. n = 10 afferents from N = 5 CVH mice. *P < 0.05 one‐way ANOVA, with Bonferroni post hoc test. (C) Change in mechanosensitivity induced by cVc1.1 in healthy and CVH nociceptors compared to their respective baseline responses. cVc1.1 caused more inhibition, at every concentration assessed, in CVH nociceptors compared with healthy nociceptors. n = 8 afferents from N = 5 healthy mice, n = 10 CVH from N = 5 CVH mice). *P < 0.05, two‐way ANOVA. (D) Original recordings of a colonic nociceptor from a healthy control mouse, showing action potential firing in response to a 2 g vfh probe at baseline and in the presence of cVc1.1 (1 μM). (E) Original recordings of a colonic nociceptor from a CVH mouse, showing action potential firing in response to a 2 g vfh probe at baseline and reduced action potential firing in the presence of cVc1.1 (1 μM). (F) Comparative inhibition of healthy colonic nociceptors by cVc1.1 versus Vc1.1 expressed as change in nociceptor mechanosensitivity with respect to baseline response. Overall, cVc1.1 (n = 8 afferents, N = 5 healthy mice) caused greater inhibition of healthy colonic nociceptors relative to Vc1.1 (n = 10 afferents, N = 5 healthy mice). *P < 0.05, significantly different from Vc1.1; two‐way ANOVA, with Bonferroni post hoc test. Vc1.1 data are re‐analysed from Castro et al. (2017). Data for cVc1.1 effects on healthy nociceptors are also shown in (C). (G) Inhibition of CVH colonic nociceptors by cVc1.1 compared with that by Vc1.1, expressed as percentage inhibition of baseline response. Overall, cVc1.1 (n = 10 afferents, N = 5 mice) caused greater inhibition of CVH colonic nociceptors relative to Vc1.1 (n = 10 afferents, N = 5 mice). *P < 0.05 two‐way ANOVA, with Bonferroni post hoc test. Vc1.1 data are re‐analysed from Castro et al. (2017). Data for cVc1.1's effects on CVH nociceptors are also shown in (C).
cVc1.1 inhibits colonic DRG neurons with greater efficacy in CVH mice
We have previously shown that Vc1.1 reduces the excitability of a sub‐population of human DRG neurons (Castro et al., 2017). We therefore wanted to determine if both Vc1.1 and cVc1.1 inhibited colonic DRG neurons from healthy mice and if their inhibitory actions were enhanced in CVH mice. To assess the excitability of colonic DRG neurons, we performed whole‐cell patch clamp recordings in current clamp mode. Accordingly, we determined the effects of Vc1.1 and cVc1.1 on the rheobase, the amount of injected current required to fire an action potential, in colonic DRG neurons from healthy and CVH mice. ‘Linear’ Vc1.1 (10 nM) had a modest but significant effect on reducing the neuronal excitability of large population of colonic DRG neurons from healthy mice (8.5 ± 1.8% inhibition relative to baseline, Figure 3A, panels i–iii). Furthermore, cVc1.1 (10 nM) also caused inhibition of a subpopulation of colon‐innervating DRG neurons from healthy mice (22.8 ± 5.1% inhibition relative to baseline Figure 3B, panels i–iii), an effect which was greater than that observed with Vc1.1 (Figure 3A, panel iii).
Figure 3.

α‐Conotoxins Vc1.1 and cVc1.1 inhibit excitability in colon‐innervating DRG neurons from healthy and CVH mice. (A, panel i) Vc1.1 (10 nM) caused a modest but significant inhibition of a subpopulation (8 of 13) colonic DRG neurons from healthy mice, as determined by an increase in rheobase. n = 8 neurons from N = 5 healthy mice. *P < 0.05, significantly different from baseline; paired t‐test). (A, panel ii) Vc1.1 (10 nM) did not affect a small subpopulation of colon‐innervating DRG neurons (5 of 13 neurons tested). (A, panel iii) Current clamp recordings of colon‐innervating DRG neurons from healthy mice in the absence and presence of Vc1.1 (10 nM). Representative whole‐cell current clamp recording of a retrogradely traced colon‐innervating DRG neuron in response to 500 ms 10 pA step current injection at rheobase. (B, panel i) cVc1.1 (10 nM) increased the rheobase of a subpopulation of healthy colonic DRG neurons indicating that cVc1.1 also inhibits neuroexcitability. n = 10 neurons of 19 tested from N = 5 healthy mice. *P < 0.05, significantly different from baseline, paired t‐test). (B, panel ii) cVc1.1 (10 nM) did not affect a subpopulation of colon‐innervating DRG neurons (9 of 19 neurons tested) from healthy mice. (B, panel iii) Current clamp recordings of colon‐innervating DRG neurons from healthy mice in the presence and absence of cVc1.1 (10 nM). (C, panel i) In colon‐innervating DRG neurons from CVH mice, Vc1.1 caused a significant increase in rheobase, indicative of a reduction in neuroexcitability. n = 9 of 15 neurons tested from N = 5 CVH mice. *P < 0.05; significantly different from baseline, paired t‐test). (C, panel ii) Vc1.1 (10 nM) did not affect a small subpopulation of colon‐innervating DRG neurons (6 of 15 neurons tested) from CVH mice. (C, panel iii) Current clamp recordings of colon‐innervating DRG neurons from CVH mice in the absence and presence of Vc1.1 (10 nM). (D, panel i) cVc1.1 increased the rheobase of a subpopulation of CVH colonic DRG neurons (15 of 18 neurons tested from N = 5 CVH mice), indicative of a reduction in neuroexcitability. *P < 0.05; significantly different from baseline, paired t‐test). (D, panel ii) cVc1.1 (10 nM) did not affect a subpopulation of colon‐innervating DRG neurons (3 of 18 neurons tested) from CVH mice. (D, panel iii) Current clamp recordings of colon‐innervating DRG neurons from CVH mice in the presence and absence of cVc1.1 (10 nM).
Colonic DRG neurons from CVH mice displayed pronounced hyper‐excitability compared with their healthy control counterparts, as reflected in their rheobase (healthy: 222.8 ± 20.29 pA; n = 32 neurons, vs. CVH: 145.8 ± 8.49 pA; n = 33 neurons, *P < 0.05, unpaired t‐test; Figure 3A–D). Vc1.1 caused significant inhibition of a subpopulation of colon‐innervating DRG neurons from CVH (10.5 ± 1.6% inhibition relative to CVH baseline, Figure 3C, panels i, ii). Furthermore, cVc1.1 also caused significant inhibition of neuroexcitability in a large sub‐population of CVH colonic DRG neurons (26.7 ± 6.8% inhibition relative to CVH baseline, Figure 3D panels i, ii). These data indicated that cVc1.1 has increased efficacy in reducing neuronal excitability compared to Vc1.1 at the same concentration in CVH mice. Overall, these findings are consistent with our ex vivo afferent recording studies, which demonstrate that cVc1.1 causes greater inhibition of colonic nociceptors from CVH mice.
Effects of cVc1.1 analogues on colonic nociceptor mechanosensitivity
Given that cVc1.1 has potent inhibitory effects on colonic nociceptors, we tested several analogues of cVc1.1, which have modifications to improve synthesis and production, for their effects on colonic nociceptors. The cVc1.1 analogue, [C2H,C8F]cVc1.1, has a similar three‐dimensional structure and activity to Vc1.1. However, because it has only one possible disulfide isomer, the cost of peptide synthesis and purification is reduced compared to the parent peptide (Yu et al., 2015). Application of [C2H,C8F]cVc1.1 caused inhibition of healthy colonic nociceptors (IC50: 8.6 nM, Figure 4A, D) and inhibition in CVH colonic nociceptors (IC50: 7.0 nM, Figure 4B, C, E). The other cyclic mutant, [N9W]cVc1.1, also inhibited colonic nociceptors from both healthy (IC50: 6.3 nM, Figure 5A, D) and CVH mice (IC50: 23.1 nM, Figure 5B, C, E).
Figure 4.
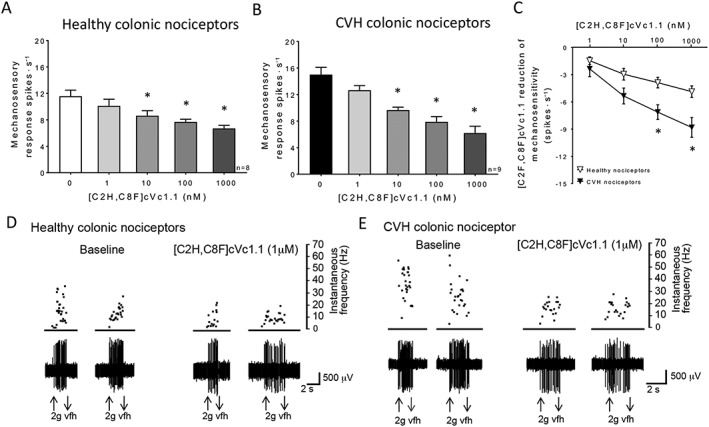
The cVc1.1 analogue, [C2H,C8F]cVc1.1, inhibits the mechanosensitivity of colonic nociceptors from healthy and CVH mice. (A) The mechanosensitivity of colonic nociceptors from healthy mice is reduced following increasing concentrations of [C2H,C8F]cVc1.1 applied to the mucosal epithelium, for 5 min at each concentration. Significant reductions in healthy colonic nociceptor mechanosensitivity relative to baseline responses were observed at 10, 100 nM and 1 μM. n = 8 afferents from N = 5 healthy mice. *P < 0.05; significantly different from baseline; one‐way ANOVA, with Bonferroni post hoc test. (B) [C2H,C8F]cVc1.1 potently and concentration‐dependently inhibited CVH nociceptors at 10, 100 nM and 1 μM. n = 9 afferents from N = 5 CVH mice. *P < 0.05; one‐way ANOVA, with Bonferroni post hoc test. (C) [C2H,C8F]cVc1.1 caused significantly more inhibition of CVH colonic nociceptors at 100 nM and 1 μM, compared with responses in healthy nociceptors. n = 8 afferents from N = 5 healthy mice, n = 9 afferents from N = 5 CVH mice. *P < 0.05; significantly different from helathy mice; two‐way ANOVA, with Bonferroni post hoc test. (D) Original recordings of a healthy colonic nociceptor, showing action potential firing in response to a 2 g vfh probe at baseline and in the presence of [C2H,C8F]cVc1.1 (1 μM). (E) Original recordings of a CVH colonic nociceptor showing action potential firing in response to a 2 g vfh probe at baseline (control) and in the presence of [C2H,C8F]cVc1.1 (1 μM).
Figure 5.
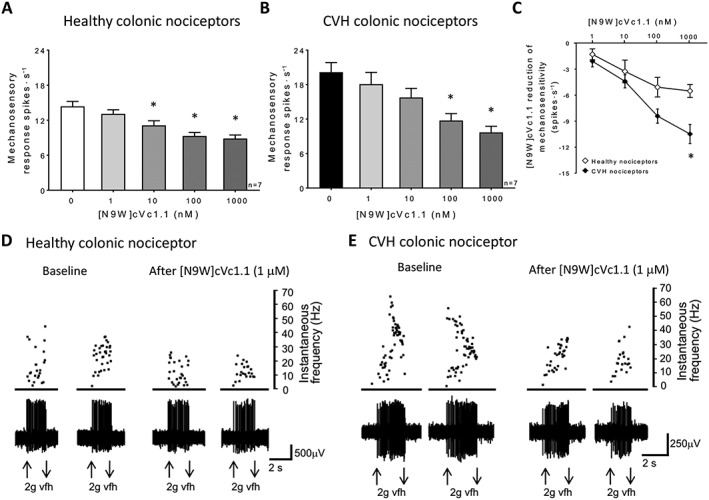
[N9W]cVc1.1 inhibits the mechanosensitivity of colonic nociceptors from healthy and CVH mice. (A) Colonic nociceptor mechanosensitivity in healthy mice is significantly reduced following application of [N9W]cVc1.1, at concentrations of 10, 100 nM and 1 μM. n = 7 afferents from N = 5 healthy mice. *P < 0.05; significantly different from control; one‐way ANOVA, with Bonferroni post hoc test. (B) [N9W]cVc1.1 inhibited CVH colonic nociceptors at concentrations of 100 nM (*P < 0.05) and 1 μM. n = 7 afferents from N = 5 CVH mice. *P < 0.05, significantly different from control; one‐way ANOVA, with Bonferroni post hoc test. (C) [N9W]cVc1.1 caused significantly more inhibition at 1 μM in CVH colonic nociceptors compared with healthy colonic nociceptors (n = 7 afferents from N = 5 healthy mice, n = 7 afferents from N = 5 CVH mice. *P < 0.05, significantly different from healthy mice; one‐way ANOVA, with Bonferroni post hoc test. (D) Healthy colonic nociceptor recording, showing action potential firing in response to a 2 g vfh probe at baseline and in the presence of [N9W]cVc1.1 (1 μM). (E) Recording of a CVH colonic nociceptor showing action potential firing in response to a 2 g vfh probe at baseline and in the presence of [N9W]cVc1.1 (1 μM).
A direct comparison of anti‐nociceptive efficacy of the analogues shows that ‘linear’ Vc1.1 caused the least amount of inhibition in both healthy (Figure 6A) and CVH (Figure 6B) states. Conversely, cVc1.1 evoked the greatest degree of inhibition in healthy and CVH colonic nociceptors (Figure 6A, B).
Figure 6.
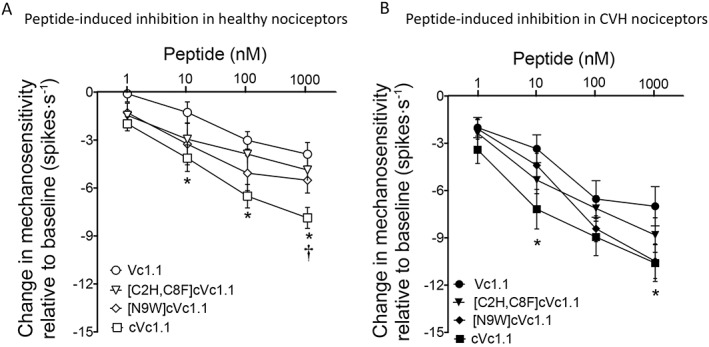
Direct comparison of the inhibitory effects of the various Vc1.1 analogues on colonic nociceptors from healthy and CVH mice. (A) Overall cVc1.1 evoked the largest inhibitory effect on healthy colonic nociceptors, with Vc1.1 evoking the least amount of inhibition. The cVc1.1 analogues evoked inhibition between that of Vc1.1 and cVc1.1. Analogues inhibited colonic nociceptors from healthy mice in the following rank order cVc1.1 > [N9W]cVc1.1 > [C2H,C8F]cVc1.1 > Vc1.1. Vc1.1: n = 10 afferents from N = 5 mice; cVc1.1: n = 8 afferents from N = 5 healthy mice; [C2H,C8F]cVc1.1: n = 8 afferents from N = 5 healthy mice, [N9W]cVC1.1: n = 7 afferents from N = 5 healthy mice. Analysis represents a total of 33 afferents from N = 20 healthy mice. *P < 0.05, cVc1.1 significantly different from Vc1.1; † P < 0.05, cVc1.1 significantly different from [C2H,C8F]cVc1.1; two‐way ANOVA with Bonferroni post hoc test. (B) Colonic nociceptors from CVH mice displayed the greatest degree of inhibition in the presence of cVc12.1, with Vc1.1 causing the least amount of inhibition. Analogues inhibited colonic nociceptors from CVH mice in the following rank order cVc1.1 > [N9W]cVc1.1 > [C2H,C8F]cVc1.1 > Vc1.1. Vc1.1: n = 10 afferents from N = 5 CVH mice; cVc1.1: n = 10 afferents from N = 5 CVH mice; [C2H,C8F]cVc1.1: n = 9 afferents from N = 5 CVH mice, [N9W]cVC1.1: n = 7 afferents from N = 5 CVH mice. Analysis represents a total of 36 afferents from N = 20 CVH mice. *P < 0.05, cVc1.1 significantly different from Vc1.1; two‐way ANOVA with Bonferroni post hoc test.
Intra‐colonic administration of Vc1.1 or cVc1.1 inhibits in vivo colonic pain responses in mice with CVH
To translate our ex vivo and in vitro findings to an in vivo model, we recorded the VMR, a nociceptive brainstem reflex consisting of the contraction of the abdominal muscles, in response to CRD (Ness and Gebhart, 1988). By using abdominal EMG, we were able to assess the effect of intra‐colonically administered Vc1.1 or cVc1.1 on visceral sensitivity in vivo in fully awake healthy or CVH mice (Christianson and Gebhart, 2007; Deiteren et al., 2014; Carstens et al., 2016). We chose Vc1.1 and cVc1.1 as they had the least and highest anti‐nociceptive actions in our colonic nociceptor recordings respectively. Intra‐colonic administration of linear Vc1.1 (1 μM) had no significant effect on the VMR of healthy mice compared with the VMRs of vehicle‐treated mice (Figure 7A). In contrast, linear Vc1.1 significantly reduced the VMR of CVH mice to CRD, particularly at noxious distension pressures of 40 and 60 mmHg (Figure 7B). Intra‐colonic administration of cVc1.1 did not affect VMRs in healthy mice (Figure 7C, E). However, cVc1.1 did significantly reduce enhanced VMRs to CRD in CVH mice at noxious distension pressures of 40, 60 and 80 mmHg (Figure 7D, E). Notably, colonic compliance was not affected by intra‐colonic administration of cVc1.1 in either healthy or CVH mice (Figure 7F), suggesting that the anti‐nociceptive actions of cVc1.1 involve the inhibition of colonic nociceptors, rather than relaxation of the colonic smooth muscle.
Figure 7.
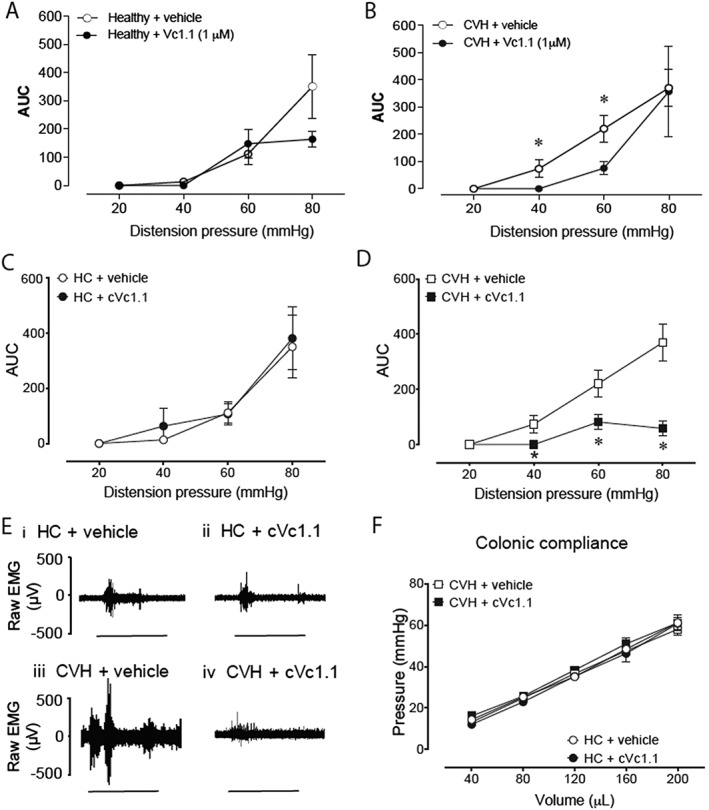
Effects of Vc1.1 and cVc1.1 on pseudo‐related pain responses to colorectal distension in healthy and CVH mice. (A) In healthy mice, VMR to colorectal distension (CRD) were not significantly changed by intra‐colonic administration of linear Vc1.1 (1 μM), relative to administration of vehicle. Vehicle: N = 10 mice; Vc1.1: N = 6 mice. Data shown are AUC of the corresponding EMG signal (means ± SEM). (B) In CVH mice, intra‐colonic administration of linear Vc1.1 (1 μM) significantly inhibited the VMR to CRD, particularly at distension pressures of 40 mmHg and 60 mmHg. Vehicle: N = 10 mice, Vc1.1: N = 7 mice. *P < 0.05; significantly different from vehicle; generalized estimating equations test followed by LSD post hoc test. (C) Intra‐colonic administration of cVc1.1 (1 μM) did not affect VMRs to CRD in healthy mice relative to vehicle treated mice. Vehicle: N = 8 mice, cVc1.1: N = 7 mice. (D) In CVH mice, intra‐colonic administration of cVc1.1 significantly reduced VMRs to colorectal distension, particularly at distension pressures of 40 mmHg, 60 mmHg and 80 mmHg. Vehicle: N = 10 mice, cVc1.1: N = 8 mice. *P < 0.05, significantly different from vehicle; generalized estimating equations test followed by LSD post hoc test. (E) Representative tracing of the raw abdominal EMG signals assessing the VMR to 60 mmHg (20 s duration, indicated by horizontal bar) of CRD in healthy control (HC) mice after (E, panel i) vehicle or (E, panel ii) cVc1.1 and in mice with CVH after (E, panel iii) vehicle or (E, panel iv) cVc1.1. Length of recording shown 45 s. (F) Colonic compliance in both healthy and CVH mice was not altered by intra‐colonic cVc1.1 (1 μM) administration, compared with intra‐colonic administration of vehicle. Data were statistically analysed by generalized estimating equations followed by the LSD post hoc test.
Discussion
This study provided evidence that cyclized analogues of the α‐conotoxin Vc1.1 inhibited colonic nociception and that these inhibitory effects were enhanced in a mouse model of CVH. Notably, the cyclized Vc1.1 analogues, cVc1.1 and [C2H,C8F]cVc1.1, evoked greater anti‐nociceptive effects than Vc1.1, the ‘linear’ native version of the peptide. We also demonstrated that peripheral administration of the cVc1.1 peptide inhibited visceral pain in a mouse model of CVH. These findings highlighted the potential therapeutic value of cyclized versions of Vc1.1 in the treatment of CVP.
Comparing the anti‐nociceptive and analgesic effects of Vc1.1 relative to cVc1.1
We recently showed that ‘linear’ Vc1.1 inhibits mouse colonic nociceptors and low‐threshold distension‐sensitive colonic afferents (Castro et al., 2017). Although Vc1.1 can act upon both the GABABR and α9α10 nAChR with IC50 values in the nM range (Adams et al., 2012; Mohammadi & Christie, 2015), we have demonstrated that ‘linear’ Vc1.1 evokes inhibition of colonic afferents via activation of GABAB receptors, as the inhibitory effects of Vc1.1 on colonic afferents were blocked by a selective antagonist of these receptors. Conversely, the inhibitory actions of Vc1.1 can be mimicked by the GABAB receptor agonist baclofen and reproduced by using inhibitors of the VGCCs CaV2.2 and CaV2.3, which are known downstream targets of Vc1.1‐induced activation of GABAB receptors (Berecki et al., 2014; Castro et al., 2017). Importantly for translation to humans, ‘linear’ Vc1.1 also reduces human DRG neuroexcitability, via a mechanism mediated by activation of GABAB receptors. Correspondingly, both mouse colonic DRG neurons and human DRG neurons co‐express the activation of GABAB receptor subunits R1 and R2, plus CaV2.2 and/or CaV2.3 channels, which combined are the required molecular components for Vc1.1‐induced inhibition (Castro et al., 2017). This anti‐nociceptive action of Vc1.1 also translates in vivo, as intra‐colonic administration of Vc1.1 to mice inhibited the signalling of noxious information from the colon into the spinal cord (Castro et al., 2017).
In the present study, we took these observations further and showed that both ‘linear’ Vc1.1 and cyclized cVc1.1 cause a greater reduction in the excitability of colonic DRG neurons from mice with CVH. Moreover, both Vc1.1 and cVc1.1 administration reduced colonic pain in CVH mice. One possible explanation for the increased efficacy of both Vc1.1 and cVc1.1 during CVH is our observation that colonic DRG neurons from CVH mice display up‐regulation of CaV2.2 exon‐37a (Castro et al., 2017), which is a known nociceptive variant of CaV2.2 (Altier et al., 2007). In this scenario, the increase in CaV2.2 exon‐37a expression may contribute, in part, to the hyper‐excitability of colonic DRG neurons from CVH mice. Correspondingly, we have observed that the selective inhibitor of CaV2.2 channels, ω‐conotoxin CVID, causes greater inhibition of colonic nociceptors from CVH mice, relative to healthy mice (Castro et al., 2017). Consequently, activation of GABAB receptors, induced by Vc1.1 or cVc1.1, would result in a greater net inhibition of VGCC currents in CVH neurons, relative to healthy colonic DRG neurons.
What is becoming apparent from our work and those of others is that the downstream effector channels of GABAB receptors appear to be linked to the agonist, the manner in which the GABAB receptor is activated and the ion channels expressed by the cell in question. Recent studies have shown that baclofen‐induced activation of GABAB receptors results in the downstream blockade of CaV2.1, CaV2.2 and CaV2.3 channels. However, Vc1.1‐induced activation of GABAB receptors results in the downstream inhibition of CaV2.2 and CaV2.3, but not CaV2.1 channels (Berecki et al., 2014). Furthermore, although baclofen‐induced activation of GABAB receptors may couple to http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=74 http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=74 channels in some cell types (Takeda et al., 2015), previous studies have shown that neither Vc1.1 nor a related α‐conopeptide, Rg1A, target GIRK channels via GABAB receptors (McIntosh et al., 2009; Huynh et al., 2015). These findings suggest that activation of GABAB receptors by Vc1.1 and cVc1.1 results in a more biased, less promiscuous set of downstream inhibitory targets than baclofen. The reason for these actions are not clear at present but may involve GPCR‐biased signalling and recruitment of selective second messenger systems.
The increased efficacy of cVc1.1 relative to ‘linear’ Vc1.1 is probably a result of the improved stability of cVc1.1 and increased affinity of the peptide for the GABAB receptor, which results in greater inhibition of VGCC currents compared with ‘linear’ Vc1.1 (Clark et al., 2010; Yu et al., 2015). This interpretation is consistent with studies using Vc1.1 and cVc1.1, in rat models of neuropathic pain. Although Vc1.1 has clear anti‐nociceptive actions in vitro and anti‐hyperalgesic actions in vivo (Satkunanathan et al., 2005; Clark et al., 2010; Klimis et al., 2011), it is not orally active. By contrast, cVc1.1 is orally active and displays significant analgesic activity in rat chronic constriction injury models of neuropathic pain (Clark et al., 2010; Yu et al., 2015). Accordingly, our collective findings indicate that cVc1.1 has greater analgesic properties for both visceral pain and neuropathic pain than linear Vc1.1.
Cyclic analogues of cVc1.1
Although cVc1.1 has clear anti‐nociceptive and analgesic properties, potentially, it can be improved further. One approach was to focus on the disulfide bond network. Because there are four cysteine residues in the peptide primary sequence of cVc1.1, it can form three disulfide‐bond isomers, with only one of them being active (Yu et al., 2015). The formation of multiple isomers can complicate synthesis procedures and significantly increase the cost of peptide production, especially on the scale of therapeutic production. However, recent studies have shown for other disulfide‐rich conotoxins that only certain disulfide bonds are crucial for activity and stability of the peptide (Barnham et al., 1998; Flinn et al., 1999; Pennington et al., 1999; Carrega et al., 2005; Khoo et al., 2009; Yu et al., 2015). Therefore, we used in silico modelling to design disulfide‐deleted variants and found that removing one disulfide bond of cVc1.1 to produce [C2H,C8F]cVc1.1 leads to a well‐folded peptide (Yu et al., 2015). This peptide has a similar three‐dimensional structure and activity to Vc1.1 but has a larger hydrophobic core than cVc1.1 and, potentially, additional surface salt bridge interactions. The advantage of [C2H,C8F]cVc1.1 is that it has only one possible disulfide isomer, reducing the cost of peptide synthesis and purification compared to the parent peptide (Yu et al., 2015). Specifically, cVc1.1 folds into two isomers in a 72:28 ratio (Clark et al., 2010), whereas [C2H,C8F]cVc1.1 forms only one isomer, therefore gaining an immediate improvement of 28% in folding yield (Yu et al., 2015). Here, we found that [C2H,C8F]cVc1.1 inhibited the mechanosensitivity of colonic nociceptors from healthy mice and that this inhibitory effect was greater in CVH colonic nociceptors. In both cases, the inhibitory effect of [C2H,C8F]cVc1.1 was greater than that observed with Vc1.1 but less than that of cVc1.1. These observations are consistent with their respective IC50 values determined for inhibition of rat DRG neuron CaV2.2 channels and human CaV2.3 channels (Callaghan et al., 2008; Clark et al., 2010; Berecki et al., 2014; Yu et al., 2015).
Another mutant of the cyclic peptide, [N9W]cVc1.1, designed in an earlier study to preferentially target human over rat nAChRs, but also to act via GABAB receptors, inhibited colonic nociceptors from both healthy and CVH mice. However, [N9W]cVc1.1 evoked less inhibition than cVc1.1, which has a greater efficacy on GABAB receptors and a reduced affinity at nAChRs. Taken together, these data again suggest that Vc1.1 inhibits colonic nociception via a mechanism mediated by GABAB receptors, rather than a nAChR‐mechanism, as we have demonstrated previously (Castro et al., 2017).
The lack of in vivo analgesic effect observed in healthy mice with intra‐colonic administration of either ‘linear’ Vc1.1 or cVc1.1 contrasts with our previous findings with a truncated form of Vc1.1, an 8‐amino acid peptide called Vc1.1(1–8). In these earlier studies, we found that the same intra‐colonic delivery of this shorter peptide caused significant reductions in colonic pain response in healthy animals (Carstens et al., 2016). The reasons for these differences may relate to the size of the peptide and the time it takes to reach efficacious concentrations at the afferent ending of the nociceptor within the colonic wall. In CVH mice, we did observe analgesic actions of both linear Vc1.1 and cVc1.1 following intra‐colonic administration. This key difference between the healthy and CVH states may relate to increased permeability of the colonic epithelium in this post‐inflammatory CVH model. Such increases in mucosal permeability have also been reported in patients with IBS (Brierley and Linden, 2014; Enck et al., 2016). Notably, the inhibitory effect of Vc1.1 at higher distension pressures (80 mmHg) in CVH mice was more variable than that observed with cVc1.1. As these recordings were performed at the furthest time point after administration, this variability may relate to reduced stability of Vc1.1 relative to cVc1.1, which increases the rationale for designing and developing cyclized analogues of Vc1.1.
In conclusion, our findings demonstrated an anti‐nociceptive action for cVc1.1 and cVc1.1 analogues in colonic nociceptors. This anti‐nociceptive action was greater in a model of CVH than that observed in healthy mice. The use of cyclized Vc1.1 analogues increased both the anti‐nociceptive and analgesic effects relative to ‘linear’ Vc1.1. Because altered visceral sensory function is a hallmark of IBS, cVc1.1 represents a potential novel therapy to reduce nociceptive stimuli from the colon and rectum to the CNS. These findings highlight the potential therapeutic value of cyclized Vc1.1 analogues in the treatment of CVP.
Author contributions
S.M.B., D.J.A. and D.J.C. conceived the study. S.M.B., D.J.A., J.C., L.G., A.D., A.M.H., T.O.D., J.M., S.G.‐C., G.Y.R. and J.M. designed, conducted and analysed experiments. D.J.C., Q.K. and R.Y. synthesized Vc1.1, cVc1.1 and the associated cVc1.1 analogues and assisted with critical revision of the manuscript for important intellectual content. S.M.B. wrote the paper and all authors contributed to revising the manuscript.
Conflict of interests
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was funded by the National Health and Medical Research Council (NHMRC) of Australia Project Grant #1049928 awarded to D.J.A, S.M.B and D.J.C. A.M.H received funding via the Australian Research Council (ARC) Discovery Early Career Research Award. D.J.C is an ARC Australian Laureate Fellow (FL150100146). S.M.B is an NHMRC R.D Wright Biomedical Research Fellow (APP1126378).
Castro, J. , Grundy, L. , Deiteren, A. , Harrington, A. M. , O'Donnell, T. , Maddern, J. , Moore, J. , Garcia‐Caraballo, S. , Rychkov, G. Y. , Yu, R. , Kaas, Q. , Craik, D. J. , Adams, D. J. , and Brierley, S. M. (2018) Cyclic analogues of α‐conotoxin Vc1.1 inhibit colonic nociceptors and provide analgesia in a mouse model of chronic abdominal pain. British Journal of Pharmacology, 175: 2384–2398. doi: 10.1111/bph.14115.
References
- Adam B, Liebregts T, Gschossmann JM, Krippner C, Scholl F, Ruwe M et al (2006). Severity of mucosal inflammation as a predictor for alterations of visceral sensory function in a rat model. Pain 123: 179–186. [DOI] [PubMed] [Google Scholar]
- Adams DJ, Berecki G (2013). Mechanisms of conotoxin inhibition of N‐type (CaV2.2) calcium channels. Biochim Biophys Acta 1828: 1619–1628. [DOI] [PubMed] [Google Scholar]
- Adams DJ, Callaghan B, Berecki G (2012). Analgesic contoxins: block and G‐protein‐coupled receptor modulation of the N‐type (CaV2.2) calcium channels. Br J Pharmacol. 166: 486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altier C, Dale CS, Kisilevsky AE, Chapman K, Castiglioni AJ, Matthews EA et al (2007). Differential role of N‐type calcium channel splice isoforms in pain. J Neurosci 27: 6363–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASX (2006a). Metabolic Pharmaceuticals: metabolics neuropathic pain drug ACV1‐additional preclinical studies reveal greater potential. http://wwwasxcomau/asxpdf/20061123/pdf/3zqm91n1jhpffpdf.
- ASX (2006b). Metabolic Pharmaceuticals: metabolics neuropathic pain drug, ACV1‐ Clinical trials update. http://wwwasxcomau/asxpdf/20061129/pdf/3zv2c96tyh1nxpdf.
- ASX (2007). Metabolic Pharmaceuticals: metabolic discontinues clinical trial programme for neuropathic pain drug, ACV1. http://wwwasxcomau/asxpdf/20070814/pdf/313yjgpf7jl4lgpdf.
- Barnham KJ, Torres AM, Alewood D, Alewood PF, Domagala T, Nice EC et al (1998). Role of the 6‐20 disulfide bridge in the structure and activity of epidermal growth factor. Protein Sci 7: 1738–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellono NW, Bayrer JR, Leitch DB, Castro J, Zhang C, O'Donnell TA et al (2017). Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell 29: 170: 185–198.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berecki G, McArthur JR, Cuny H, Clark RJ, Adams DJ (2014). Differential CaV2.1 and CaV2.3 channel inhibition by baclofen and α‐conotoxin Vc1.1 via GABABR activation. J Gen Physiol 143: 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyak MJ (2010). Visceral afferents ‐ Determinants and modulation of excitability. Auton Neurosci 153: 69–78. [DOI] [PubMed] [Google Scholar]
- Brierley SM (2016). Altered ion channel/receptor expression and function in extrinsic sensory neurons: the cause of and solution to chronic visceral pain? Adv Exp Med Biol 891: 75–90. [DOI] [PubMed] [Google Scholar]
- Brierley SM, Linden DR (2014). Neuroplasticity and dysfunction after gastrointestinal inflammation. Nature Rev Gastroenterol Hepatol Oct 11: 611–627. [DOI] [PubMed] [Google Scholar]
- Brierley SM, Jones RCW III, Gebhart GF, Blackshaw LA (2004). Splanchnic and pelvic mechanosensory afferents signal different qualities of colonic stimuli in mice. Gastroenterology 127: 166–178. [DOI] [PubMed] [Google Scholar]
- Brierley SM, Jones RCW III, Xu L, Gebhart GF, Blackshaw LA (2005a). Activation of splanchnic and pelvic colonic afferents by bradykinin in mice. Neurogastro & Motil 17: 854–862. [DOI] [PubMed] [Google Scholar]
- Brierley SM, Jones RCW III, Xu L, Robinson DR, Hicks GA, Gebhart GF et al (2005b). Differential chemosensory function and receptor expression of splanchnic and pelvic colonic afferents in mice. J Physiol (Lond) 567: 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley SM, Page AJ, Hughes PA, Adam B, Liebregts T, Cooper NJ et al (2008). Selective role for TRPV4 ion channels in visceral sensory pathways. Gastroenterology 134: 2059–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley SM, Hughes PA, Page AJ, Kwan KY, Martin CM, O'Donnell TA et al (2009). The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology 137: 2084–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley SM, Castro J, Harrington AM, Hughes PA, Page AJ, Rychkov GY et al (2011). TRPA1 contributes to specific mechanically activated currents and sensory neuron mechanical hypersensitivity. J Physiol 15: 3575–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes SJ, Spencer NJ, Costa M, Zagorodnyuk VP (2013). Extrinsic primary afferent signalling in the gut. Nature Rev Gastroenterol Hepatol 10: 286–296. [DOI] [PubMed] [Google Scholar]
- Callaghan B, Adams DJ (2010). Analgesic α‐conotoxins Vc1.1 and RgIA inhibit N‐type calcium channels in sensory neurons of α9 nicotinic receptor knockout mice. Channels (Austin) 4: 51–54. [DOI] [PubMed] [Google Scholar]
- Callaghan B, Haythornthwaite A, Berecki G, Clark RJ, Craik DJ, Adams DJ (2008). Analgesic α‐conotoxins Vc1.1 and Rg1A inhibit N‐type calcium channels in rat sensory neurons via GABABR activation. J Neurosci 28: 10943–10951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrega L, Mosbah A, Ferrat G, Beeton C, Andreotti N, Mansuelle P et al (2005). The impact of the fourth disulfide bridge in scorpion toxins of the α‐KTx6 subfamily. Proteins 61: 1010–1023. [DOI] [PubMed] [Google Scholar]
- Carstens BB, Berecki G, Daniel JT, Lee HS, Jackson KA, Tae HS et al (2016). Structure‐activity studies of cysteine‐rich α‐conotoxins that inhibit high voltage‐activated calcium channels via gabab receptor activation reveal a minimal functional motif. Angew Chem Int Ed Engl 55: 4692–4696. [DOI] [PubMed] [Google Scholar]
- Castro J, Harrington AM, Hughes PA, Martin CM, Ge P, Shea CM et al (2013). Linaclotide inhibits colonic nociceptors and relieves abdominal pain via guanylate cyclase‐C and extracellular cyclic guanosine 3′,5′‐monophosphate. Gastroenterology 145 : 1334‐1346 e1331‐1311: 1334–1346.e11. [DOI] [PubMed] [Google Scholar]
- Castro J, Harrington AM, Garcia‐Caraballo S, Maddern J, Grundy L, Zhang J et al (2017). α‐Conotoxin Vc1.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABAB receptors. Gut 66: 1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chey WD, Kurlander J, Eswaran S (2015). Irritable bowel syndrome: a clinical review. JAMA 313: 949–958. [DOI] [PubMed] [Google Scholar]
- Christianson JA, Gebhart GF (2007). Assessment of colon sensitivity by luminal distension in mice. Nat Protoc 2: 2624–2631. [DOI] [PubMed] [Google Scholar]
- Clark RJ, Fischer H, Nevin ST, Adams DJ, Craik DJ (2006). The synthesis, structural characterization, and receptor specificity of the α‐conotoxin Vc1.1. J Biol Chem 281: 23254–23263. [DOI] [PubMed] [Google Scholar]
- Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ (2010). The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew Chem Int Ed Engl 49: 6545–6548. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo AD, Mobli M, Castro J, Harrington AM, Vetter I, Dekan Z et al (2014). Selenoether oxytocin analogues have analgesic properties in a mouse model of chronic abdominal pain. Nat Commun 5: 3165. [DOI] [PubMed] [Google Scholar]
- Deiteren A, De Man JG, Ruyssers NE, Moreels TG, Pelckmans PA, De Winter BY (2014). Histamine H4 and H1 receptors contribute to postinflammatory visceral hypersensitivity. Gut 63: 1873–1882. [DOI] [PubMed] [Google Scholar]
- Enck P, Aziz Q, Barbara G, Farmer AD, Fukudo S, Mayer EA et al (2016). Irritable bowel syndrome. Nat Rev Dis Primers 2: 16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinn JP, Pallaghy PK, Lew MJ, Murphy R, Angus JA, Norton RS (1999). Role of disulfide bridges in the folding, structure and biological activity of ω‐conotoxin GVIA. Biochim Biophys Acta 1434: 177–190. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AM, Brierley SM, Isaacs N, Hughes PA, Castro J, Blackshaw LA (2012). Sprouting of colonic afferent central terminals and increased spinal mitogen‐activated protein kinase expression in a mouse model of chronic visceral hypersensitivity. J Comp Neurol 520: 2241–2255. [DOI] [PubMed] [Google Scholar]
- Hughes PA, Brierley SM, Blackshaw LA (2009a). Post‐inflammatory modification of colonic afferent mechanosensitivity. Clin Exp Pharmacol Physiol 36: 1034–1040. [DOI] [PubMed] [Google Scholar]
- Hughes PA, Brierley SM, Martin CM, Brookes SJ, Linden DR, Blackshaw LA (2009b). Post‐inflammatory colonic afferent sensitisation: different subtypes, different pathways and different time courses. Gut 58: 1333–1341. [DOI] [PubMed] [Google Scholar]
- Hughes PA, Harrington AM, Castro J, Liebregts T, Adam B, Grasby DJ et al (2013). Sensory neuro‐immune interactions differ between irritable bowel syndrome subtypes. Gut 62: 1456–1465. [DOI] [PubMed] [Google Scholar]
- Hughes PA, Castro J, Harrington AM, Isaacs N, Moretta M, Hicks GA et al (2014). Increased kappa‐opioid receptor expression and function during chronic visceral hypersensitivity. Gut 63: 1199–1200. [DOI] [PubMed] [Google Scholar]
- Huynh TG, Cuny H, Slesinger PA, Adams DJ (2015). Novel mechanism of voltage‐gated N‐type (CaV2.2) calcium channel inhibition revealed through α‐conotoxin Vc1.1 activation of the GABAB receptor. Mol Pharmacol 87: 240–250. [DOI] [PubMed] [Google Scholar]
- Khoo KK, Feng ZP, Smith BJ, Zhang MM, Yoshikami D, Olivera BM et al (2009). Structure of the analgesic μ‐conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry 48: 1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimis H, Adams DJ, Callaghan B, Nevin S, Alewood PF, Vaughan CW et al (2011). A novel mechanism of inhibition of high‐voltage activated calcium channels by α‐conotoxins contributes to relief of nerve injury‐induced neuropathic pain. Pain 152: 259–266. [DOI] [PubMed] [Google Scholar]
- Lewis RJ, Nielsen KJ, Craik DJ, Loughnan ML, Adams DA, Sharpe IA et al (2000). Novel ω‐conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J Biol Chem 275: 35335–35344. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Brit J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M (2009). Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem Pharmacol 78: 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi SA, Christie MJ (2015). Conotoxin interactions with a9a10‐nAChRs: Is the a9a10‐nicotinic acetylcholine receptor an important therapeutic target for pain management? Toxins (Basel) 7: 3916–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness TJ, Gebhart GF (1988). Colorectal distension as a noxious visceral stimulus: physiologic and pharmacologic characterization of pseudaffective reflexes in the rat. Brain Res 450: 153–169. [DOI] [PubMed] [Google Scholar]
- Osteen JD, Herzig V, Gilchrist J, Emrick JJ, Zhang C, Wang X et al (2016). Selective spider toxins reveal a role for the NaV1.1 channel in mechanical pain. Nature 534: 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington MW, Lanigan MD, Kalman K, Mahnir VM, Rauer H, McVaugh CT et al (1999). Role of disulfide bonds in the structure and potassium channel blocking activity of ShK toxin. Biochemistry 38: 14549–14558. [DOI] [PubMed] [Google Scholar]
- Satkunanathan N, Livett B, Gayler K, Sandall D, Down J, Khalil Z (2005). Alpha‐conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res 1059: 149–158. [DOI] [PubMed] [Google Scholar]
- Schroeder CI, Craik DJ (2012). Therapeutic potential of conopeptides. Fut Med Chem 4: 1243–1255. [DOI] [PubMed] [Google Scholar]
- Takeda M, Nasu M, Kanazawa T, Shimazu Y (2015). Activation of GABA(B) receptors potentiates inward rectifying potassium currents in satellite glial cells from rat trigeminal ganglia: in vivo patch‐clamp analysis. Neuroscience 288: 51–58. [DOI] [PubMed] [Google Scholar]
- Vetter I, Lewis RJ (2012). Therapeutic potential of cone snail venom peptides (conopeptides). Curr Topics Med Chem 12: 1546–1552. [DOI] [PubMed] [Google Scholar]
- Vincler M, McIntosh JM (2007). Targeting the a9a10 nicotinic acetylcholine receptor to treat severe pain. Expert Opin Ther Targets 11: 891–897. [DOI] [PubMed] [Google Scholar]
- Vincler M, Wittenauer S, Parker R, Ellison M, Olivera BM, McIntosh JM (2006). Molecular mechanism for analgesia involving specific antagonism of a9a10‐nicotinic acetylcholine receptors. Proc Natl Acad Sci USA 103: 17880–17884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Kompella SN, Adams DJ, Craik DJ, Kaas Q (2013). Determination of the α‐conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J Med Chem 56: 3557–3567. [DOI] [PubMed] [Google Scholar]
- Yu R, Seymour VA, Berecki G, Jia X, Akcan M, Adams DJ et al (2015). Less is more: design of a highly stable disulfide‐deleted mutant of analgesic cyclic α‐conotoxin Vc1.1. Sci Rep 5: 13264. [DOI] [PMC free article] [PubMed] [Google Scholar]