

Abstract
The aim of this study was to develop a new methodology that is suitable for DNA methylation diagnostics and to demonstrate its clinical applicability. We developed a new anion‐exchange column for high‐performance liquid chromatography (HPLC) with electrostatic and hydrophobic properties. Both cytosine and thymine, corresponding to methylated and unmethylated cytosine after bisulfite modification, respectively, are captured by electrostatic interaction and then discriminated from each other by their hydrophobic interactions. The DNA methylation levels of synthetic DNA were quantified accurately and reproducibly within 10 minutes without time‐consuming pretreatment of PCR products, and the measured values were unaffected by the distribution of methylated CpG within the synthetic DNA fragments. When the DNA methylation status of the FAM150A gene, a marker of the CpG island methylator phenotype specific to clear cell renal cell carcinoma (ccRCC), was examined in 98 patients with ccRCC, bulk specimens of tumorous tissue including cancer cells showing DNA methylation of the FAM150A gene were easily identifiable by simply viewing the differentiated chromatograms, even when the cancer cell content was low. Sixteen ccRCC showing DNA methylation more frequently exhibited clinicopathological parameters reflecting tumor aggressiveness (ie, a larger diameter, higher histological grade, vascular involvement, renal vein tumor thrombi, infiltrating growth, tumor necrosis, renal pelvis invasion and higher pathological TNM stage), and had significantly lower recurrence‐free and overall survival rates. These data indicate that HPLC analysis using this newly developed anion‐exchange column could be a powerful tool for DNA methylation diagnostics, including prognostication of patients with cancers, in a clinical setting.
Keywords: anion‐exchange column for high‐performance liquid chromatography, clear cell renal cell carcinoma, CpG island methylator phenotype, DNA methylation diagnostics, prognostication
1. INTRODUCTION
DNA methylation alterations are known to occur during multistage human carcinogenesis,1, 2 even from the early and precancerous stage. Such alterations at the precancerous stage are inherited by the cancers themselves and frequently associated with clinicopathological aggressiveness and outcome.3, 4 Once DNA methylation alterations have occurred during carcinogenesis, they are stably preserved on DNA double strands by maintenance methylation involving DNA (cytosine‐5)‐methyltransferase 1 (DNMT1).5, 6 Therefore, DNA methylation status in specific regions can be an excellent biomarker for carcinogenetic risk estimation, early diagnosis and prognostication of cancers derived from various organs.7
In general, clear cell renal cell carcinomas (ccRCC) are curable by nephrectomy at the early stage.8 However, some ccRCC relapse and metastasize to distant organs, even if resection is considered complete.9 The effectiveness of any therapy using novel targeting agents is restricted unless relapsed or metastasized tumors are diagnosed early by close follow‐up.10 Therefore, reliable prognostic criteria need to be established. Recently, our group categorized CpG island methylator phenotype (CIMP)‐positive ccRCC characterized by accumulation of DNA hypermethylation of CpG islands, clinicopathological aggressiveness and poor outcome, based on genome‐wide DNA methylation analysis.11 We also identified 17 genes as ccRCC‐specific CIMP marker genes.11 Furthermore, we identified therapeutic targets for more aggressive CIMP‐positive ccRCC using multilayer/integrated omics analysis.12 Therefore, evaluation of the DNA methylation status of ccRCC‐specific CIMP marker genes could be a potentially powerful tool for prognostication and companion diagnosis when considering molecular targeted therapy for patients with ccRCC.
Several methods for analysis of DNA methylation (eg, quantitative methylation‐specific PCR [MSP],13 MassARRAY14 and pyrosequencing)15 have often been used for quantitative analyses, especially in research.16, 17 Existing methods generally require long analysis times, complex procedures and expensive, large‐scale equipment. Therefore, to facilitate dissemination of DNA methylation diagnostics, it is crucial to develop an analytical system capable of delivering data readily, quickly and precisely, especially for diseased cells with DNA methylation abnormalities that need to be discriminated from contaminating cells.
In order to devise a new analytical procedure that satisfies these requirements, we have developed an anion‐exchange high‐performance liquid chromatography (HPLC) column for detection of methylated DNA. We examined the clinical applicability of this newly developed HPLC column by evaluating the DNA methylation status of the FAM150A CIMP marker gene using 98 samples of ccRCC tissue.
2. MATERIALS AND METHODS
2.1. Preparation of the column packing material
A reactor equipped with a stirrer was filled with 2000 mL of a 3% w/w aqueous solution of polyvinyl alcohol (Nippon Synthetic Chemical Industry, Osaka, Japan), and a mixture of 200 g tetraethylene glycol dimethacrylate (Shin Nakamura Chemical, Wakayama, Japan), 100 g triethylene glycol dimethacrylate (Shin Nakamura Chemical), 100 g glycidyl methacrylate (Wako Pure Chemical Industries, Osaka, Japan) and 1 g benzoyl peroxide (Kishida Chemical, Tokyo, Japan) was added to the solution. The contents were heated to 80°C with stirring and polymerized at that temperature for 1 hour in a nitrogen atmosphere. In this initial step, polymer particles with hydrophobic properties were formed.
Subsequently, a monomer solution of 100 g ethyl methacrylate trimethyl ammonium chloride (Wako Pure Chemical Industries) dissolved in 100 mL ion‐exchange water was added to the reactor, and the contents were polymerized at 80°C for 2 hour in a nitrogen atmosphere to prepare a polymerization composite. This composite was rinsed with ion‐exchange water and then with acetone. In this second step, polymer chains with quaternary ammonium groups were introduced onto the surface of the polymer particles.
Finally, 10 g of polymer particles were dispersed in 100 mL ion‐exchange water to prepare an unreacted slurry. Next, 10 mL N,N‐dimethylamino propyl amine (Wako Pure Chemical Industries) was added to the slurry with stirring, and the contents were allowed to react at 70°C for 4 hour. After completion of the reaction, the supernatant was removed using a centrifugal separator, Himac CR20G (Hitachi Koki, Tokyo, Japan), and the remaining contents were rinsed with ion‐exchange water. After rinsing, an additional supernatant was removed using a centrifugal separator. Rinsing with ion‐exchange water was then performed an additional 4 times. In this final step, tertiary amino groups were introduced onto the surface of the polymer particles. Thus, as shown in Figure 1, newly designed anion‐exchange packing materials with electrostatic and hydrophobic properties were prepared. The average particle size was 10 μm, as measured using AccuSizer 780 (Particle Sizing Systems, Florida, USA), an instrument for determination of particle size distribution.
Figure 1.
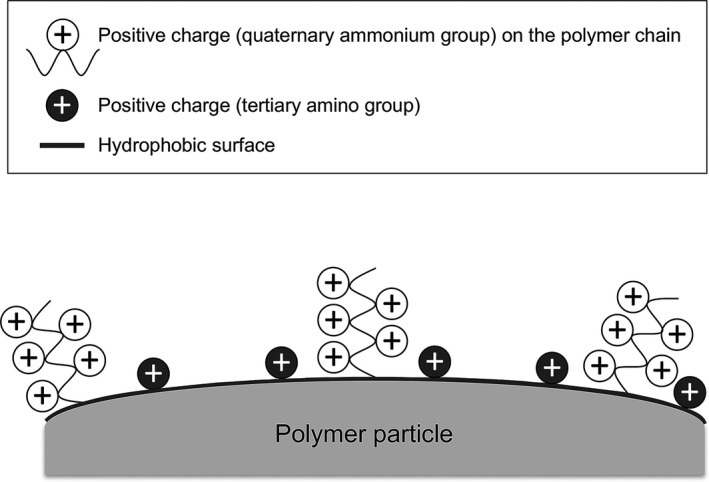
Schematic view of the newly developed packing material with electrostatic and hydrophobic properties for the high‐performance liquid chromatography column
2.2. Chromatographic analysis
High‐performance liquid chromatography was performed on an LC‐20A system (Shimadzu Corporation, Kyoto, Japan) equipped with a stainless steel column (20 × 4.6 mm I.D.) filled with the new packing material possessing anion‐exchange groups, an oven, an auto‐injector, a photo diode array detector, a degassing module and a data analysis system. Eluent buffer A was 25 mmol/L Tris‐HCl (pH 7.5) and eluent buffer B was the same buffer containing 1 mol/L ammonium sulfate. PCR products were separated on a gradient of 40%‐100% buffer B for 10 minutes at a flow rate of 1.0 mL/min. The separated PCR products were detected at 260 nm. HPLC analysis was performed at a column temperature of 70°C.
2.3. Synthetic DNA fragments
Synthetic DNA fragments of 384 base pairs (bp) containing 39 CpG sites (Eurofins Genomics, Tokyo, Japan) were designed based on a sequence encompassing the promoter region of a ccRCC‐specific CIMP marker, the FAM150A gene (Table 1a). It was expected that if 0 (0%), 8 (20%), 10 (25%), 13 (30%), 20 (50%), 30 (75%) and 39 (100%) CpG sites in the promoter region were methylated, then Fragments 1‐10 in Table 1b would be obtained after bisulfite modification. To verify the quantitative accuracy and reproducibility of the newly developed HPLC column, the 10 384‐bp synthetic DNA fragments, including Fragments 5‐8 that include 20 (50%) methylated CpG dinucleotides scattered in various positions (Table 1b), were applied to the column.
Table 1.
The 384‐bp synthetic DNA fragments designed based on a sequence encompassing the promoter region of the FAM150A gene
| Sequence | ||
|---|---|---|
| (a) Original sequence of the promoter region of the FAM150A gene | ||
| GGGAGGACCC AGTAGGGTAA CTGCCGCGTC GCCCCGGCGG TTCTCCCTGG GCTCTGTCTC CCGCCGCCTC CACCCCCCGA GCCTCGGGGT CCGTCACGGC TTCCCCTGGC TGGCGGGGTC AGTAGAACCC GCGGCGCCTA GGTCCGGACG GAAAAAAGCA GGGCCGGGGT GCGGCCTGGA TGAGCGGAGA TCTCCGCGCC TTGGGCTCAA AGGTGCGGGG TGCGCTCTGC TGCCGAGCCC CTGCTCGCTC AGGAACACTG GCCACGCCGT CACGCCAGCC GCCCCTGCCC CAGGTCTGGA GGCCCGACCT GCTCTCCTAG GCGCAGCACC GCGTTCTCTT CCGCGTGGGG GAGCGGCGGG CGGAAGAGGT CTGGGGCTGG GCAC | ||
| Synthetic DNA | DNA methylation levels (%) | Sequence |
|---|---|---|
| (b) Sequence of synthetic DNA fragments designed based on the status after bisulfite modification | ||
| Fragment 1 | 0 | GGGAGGATTTAGTAGGGTAATTGT TGTGTTGTTTTGGTGGTTTTTTTTGGGTTTTGTTTTTTGTTGTTTTTATTTTTTGAGTTTTGGGGTTTGTTATGGTTTTT TTTGGTTGGTGGGGTTAGTAGAATTTGTGGTGTTTAGGTTTGGATGGAAAAAAGTAGGGTTGGGGTGTGGTTTGGATGAGTGGAGATTTTTGTGTTTTGGGTTTAAAGGTGTGGGGTGTGTTTTGTTGTTGAGTTTTTGTTTGTTTAGGAATATTGGTTATGTTGTTATGTTAGTTGTTTTTGTTTTAGGTTTGGAG GTTTGATTTGTTTTTTTAGGTGTAGTATTGTGTTTTTTTTTGTGTGGGGGAGTGGTGGGTG GAAGAGGTTTGGGGTTGGGTAT |
| Fragment 2 | 20 | GGGAGGATTTAGTAGGGTAATTGT TGTGTTGTTTCGGTGGTTTTTTTTGGGTTTTGTTTTTTGTTGTTTTTATTTTTTGAGTTTCGGGGTTTGTTATGGTTTTTTTTGGTTGGTGGGGTTAGTAGAATTTG CGGTGTTTAGGTTTGGATGGAAAAAAGTAGGGTTGGGGTGCGGTTTGGATGAGTGGAGATTTTTGTGTTTTGGGTTTA AAGGTGTGGGGTGCGTTTTGTTGTTGAGTTTTTGTTTGTTTAGGAATATTGGTTATGTTGTTACGTTAGTTGTTTTTGTTTTAGGTTTGGAGGTTTGATTT GTTTTTTTAGGTGTAGTATTG CGTTTTTTTTTGTGTGGGGGAGTGGTGGGCG GAAGAGGTTTGGGGTTGGGTAT |
| Fragment 3 | 25 | GGGAGGATTTAGTAGGGTAATTGT TGTGTCGTTTTGGTGGTTTTTTTTGGGTTTTGTTTTTTGTCGTTTTTATTTTTTGAGTTTTGGGGTTTGTTACGGTTTTTT TTGGTTGGTGGGGTTAGTAGAATTTGTGGCGTTTAGGTTTGGATGGAAAAAAGTAGGGTTGGGGTGCGGTTTGGATGAGTGGAGATTTTTGTGTTTTGGGTT TAAAGGTGCGGGGTGTGTTTTGTTGTTGAGTTTTTGTTTGTTTAGGAATATTGGTTACGTTGTTATGTTAGTTGTTTTTGTTTTAGGTTTGGAGGTTCGATTTGT TTTTTTAGGTGTAGTATTGTGTTTTTTTTCG TGTGGGGGAGTGGTGGGCG GAAGAGGTTTGGGGTTGGGTAT |
| Fragment 4 | 30 | GGGAGGATTTAGTAGGGTAATTGT TGTGTCGTTTTGGTGGTTTTTTTTGGGTTTTGTTTTTCGTTGTTTTTATTTTTTGAGTTTCGGGGTTTGTTATGGTTTTTTTTGGTTGGCGGGGTTAGTAGAATTTGTGGCGTTTAGGTTTGGATGGAAAAAAGTAGGGTCGGGGTGTGGTTTGGATGAGTGGAGATTTTCG TGTTTTGGGTTTAAA GGTGTGGGGTGCGTTTTGTTGTTGAGTTTTTGTTTGTTTAGGAATATTGGTTACGTTGTTATGTTAGTCGTTTTTGTTTTAGGTTTGGAGGTTTGATTTGTTT TTTTAGGTGTAGTATCG TGTTTTTTTTTG CGTGGGGGAGTGGTGGGCG GAAGAGGTTTGGGGTTGGGTAT |
| Fragment 5 | 50 | GGGAGGATTTAGTAGGGTAATTGT CG TGTCGTTTTGGCGGTTTTTTTTGGGTTTTGTTTTTTGTCGTTTTTATTTTTTGAGTTTCGGGGTTTGTTACGGTTTTT TTTGGTTGGTGGGGTTAGTAGAATTCG TGGCGTTTAGGTTTGGACGGAAAAAAGTAGGGTTGGGGTGCGGTTTGGATGAGTGGAGATTTTCG TGTTTTG GGTTTAAAGGTGCGGGGTGTGTTTTGTTGTCGAGTTTTTGTTTGTTTAGGAATATTGGTTACGTTGTTACGTTAGTTGTTTTTGTT TTAGGTTTGGAGGTTCGATTTGTTTTTTTAGGTGTAGTATCG TGTTTTTTTTCG TGTGGGGGAGCGGTGGGCG GAAGAGGTTTGGGGTTGGGTAT |
| Fragment 6 | 50 | GGGAGGATTTAGTAGGGTAATTGT CGCGTCGTTTCGGCGGTTTTTTTTGGGTTTTGTTTTTCGTCGTTTTTATTTTTCGAGTTTCGGGGTTCGTTACGGTTTTTTT TGGTTGGCGGGGTTAGTAGAATTCGCGGCGTTTAGGTTCGGACGGAAAAAAGTAGGGTCGGGGTGCGGTTTGGATGAGCGGAGATTTTTGTGTTTTGGGT TTAAAGGTGTGGGGTGTGTTTTGTTGTTGAGTTTTTGTTTGTTTAGGAATATTGGTTATGTTGTTATGTTAGTTGTTTTTGTTTTAGGTTTGGAGGTTTGATTTGT TTTTTTAGGTGTAGTATTGTGTTTTTTTTTGTGTGGGGGAGTGGTGGGTG GAAGAGGTTTGGGGTTGGGTAT |
| Fragment 7 | 50 | GGGAGGATTTAGTAGGGTAATTGT TGTGTTGTTTTGGTGGTTTTTTTTGGGTTTTGTTTTTTGTTGTTTTTATTTTTTGAGTTTTGGGGTTTGTTATGGTTTTTTT TGGTTGGTGGGGTTAGTAGAATTTGTGGTGTTTAGGTTTGGATGGAAAAAAGTAGGGTTGGGGTGTGGTTTGGATGAGCGGAGATTTTCGCG TTTTGGGTTTAAAGGTGCGGGGTGCGTTTTGTTGTCGAGTTTTTGTTCGTTTAGGAATATTGGTTACGTCGTTACGTTAGTCG TTTTTGTTTTAGGTTTGGAGGTTCGATTTGTTTTTTTAGGCGTAGTATCGCGTTTTTTTTCGCGTGGGGGAGCGGCGGGCG GAAGAGGTTTGGGGTTGGGTAT |
| Fragment 8 | 50 | GGGAGGATTTAGTAGGGTAATTGT TGTGTTGTTTTGGTGGTTTTTTTTGGGTTTTGTTTTTTGTTGTTTTTATTTTTTGAGTTTTGGGGTTTGTTACGGTTTTT TTTGGTTGGCGGGGTTAGTAGAATTCGCGGCGTTTAGGTTCGGACGGAAAAAAGTAGGGTCGGGGTGCGGTTTGGATGAGCGGAGATTTTCGCG TTTTGGGTTTAAAGGTGCGGGGTGCGTTTTGTTGTCGAGTTTTTGTTCGTTTAGGAATATTGGTTACGTCGTTACGTTAGTCG TTTTTGTTTTAGGTTTGGAGGTTTGATTTGTTTTTTTAGGTGTAGTATTGTGTTTTTTTTTGTGTGGGGGAGTGGTGGGTG GAAGAGGTTTGGGGTTGGGTAT |
| Fragment 9 | 75 | GGGAGGATTTAGTAGGGTAATTGT CGCGTCGTTTTGGCGGTTTTTTTTGGGTTTTGTTTTTCGTCGTTTTTATTTTTTGAGTTTCGGGGTTCGTTACG GTTTTTTTTGGTTGGTGGGGTTAGTAGAATTCGCGGCGTTTAGGTTTGGACGGAAAAAAGTAGGGTCGGGGTGCGGTTTGGATGAGTGGAGATTTTCGCG TTTTGGGTTTAAAGGTGCGGGGTGTGTTTTGTTGTCGAGTTTTTGTTCGTTTAGGAATATTGGTTACGTTGTTACGTTAGTCGTTTTTGTTTTAGGTTTGGAGGTT CGATTTGTTTTTTTAGGTGTAGTATCGCGTTTTTTTTCG TGTGGGGGAGCGGCGGGCG GAAGAGGTTTGGGGTTGGGTAT |
| Fragment 10 | 100 | GGGAGGATTTAGTAGGGTAATTGT CGCGTCGTTTCGGCGGTTTTTTTTGGGTTTTGTTTTTCGTCGTTTTTATTTTTCGAGTTTCGGGGTTCGTTACGGTTTT TTTTGGTTGGCGGGGTTAGTAGAATTCGCGGCGTTTAGGTTCGGACGGAAAAAAGTAGGGTCGGGGTGCGGTTTGGATGAGCGGAGATTTTCGCG TTTTGGGTTTAAAGGTGCGGGGTGCGTTTTGTTGTCGAGTTTTTGTTCGTTTAGGAATATTGGTTACGTCGTTACG TTAGTCGTTTTTGTTTTAGGTTTGGAGGTTCGATTTGTTTTTTTAGGCGTAGTATCGCGTTTTTTTTCGCGTGGGGGAGCGGCGGGCG GAAGAGGTTTGGGGTTGGGTAT |
Dinucleotides left as CpG even after bisulfite treatment due to methylation are shown in bold and unmethylated and modified dinucleotides (TpG) are underlined. PCR primer positions are double‐underlined.
2.4. Patients and tissue samples
Ninety‐eight samples of cancerous tissue obtained from specimens surgically resected from 98 patients with primary ccRCC were subjected to the present analysis. These patients did not receive preoperative treatment and underwent nephrectomy at the National Cancer Center Hospital, Tokyo, Japan. There were 69 men and 29 women with a mean (± SD) age of 62.82 ± 10.56 years. Histological diagnosis was made in accordance with the World Health Organization classification.18 All of the tumors were classified according to the pathological TNM classification.19 The criteria for macroscopic configuration of RCC20 followed those established for hepatocellular carcinoma (HCC): type 3 (contiguous multinodular type) HCC show poorer histological differentiation and a higher incidence of intrahepatic metastasis than type 1 (single nodular type) and type 2 (single nodular type with extranodular growth) HCC.21 The presence or absence of vascular involvement was examined microscopically on slides stained with H&E and Elastica van Gieson. The presence or absence of tumor thrombi in the main trunk of the renal vein was examined macroscopically. The clinicopathological characteristics of the ccRCC in the present study are summarized in Table S1. All 98 ccRCC had been included in our previous studies,11, 16 and 84 and 14 of them were diagnosed as CIMP‐negative and CIMP‐positive, respectively (4 of the ccRCC in the previous study were not included in the present study due to a shortage of genomic DNA samples).
This study was approved by the Ethics Committees of the National Cancer Center, Tokyo and Keio University School of Medicine, and was performed in accordance with the Declaration of Helsinki. All patients provided written informed consent prior to inclusion in the study.
2.5. Bisulfite modification and PCR
Tissue specimens were taken and frozen immediately after surgical removal and then stored in liquid nitrogen until DNA extraction. High‐molecular‐weight DNA was extracted from the fresh‐frozen tissue samples using phenol‐chloroform followed by dialysis. One microgram of genomic DNA was subjected to bisulfite treatment using an EpiTect Bisulfite Kit (QIAGEN GmbH, Hilden, Germany), in accordance with the manufacturer's protocol. The primer positions for PCR encompassing the promoter region of the FAM150A gene are indicated by double underlining in Table 1. A portion of each PCR product was subjected to 3% agarose gel electrophoresis and visualized by staining with ethidium bromide to confirm the amplification, after which the PCR products were subjected to chromatographic analysis.
2.6. Statistics
The linearity of retention times and cytosine contents was examined by correlation analysis. Differences in retention times were evaluated by t test. Correlations between DNA methylation status and clinicopathological parameters were examined by Wilcoxon rank sum test and Fisher's exact test. Survival curves of patients were generated using the Kaplan‐Meier method, and the differences were compared by log‐rank test. Statistical analyses were performed using programming language R and IBM SPSS Statistics for Windows, version 24.0 (IBM, Armonk, NY, USA). Differences at P‐value of <.05 were considered significant.
3. RESULTS
3.1. Determination of quantitativeness and reproducibility of the newly developed high‐performance liquid chromatography column using synthetic DNA fragments
Synthetic DNA fragments 1, 3, 5, 6, 7, 8, 9 and 10 corresponding to the sequences after bisulfite modification of 0%, 25% 50%, 75% and 100% methylated promoter regions (Table 1) were subjected to PCR amplification and applied to the newly developed HPLC column. The chromatograms obtained are shown in Figure 2A,B. The targeted peaks were completely separated from peaks derived from impurities in the PCR mixture (Figure 2A), indicating that the PCR products can be applied immediately to the newly developed HPLC column without any pretreatment. The peaks derived from synthetic DNA fragments corresponding to 0%‐100% methylation were detected between 7 and 8 minutes (Figure 2B), indicating that the assay using the newly developed HPLC column can be completed within a short period of time. The linear decrease of the retention time associated with the increase in methylation levels from 0% to 100% (R = −0.993, P = 3.19 × 10−8) confirmed the quantitativeness of the newly developed HPLC column (Figure 2C). The peaks obtained for the synthetic DNA fragments 5, 6, 7 and 8, which contained 20 (50%) methylated CpG dinucleotides scattered in various positions (Table 1), completely overlapped each other, indicating that the retention time was not affected by the positions of methylated CpG (Figure 2A,B).
Figure 2.
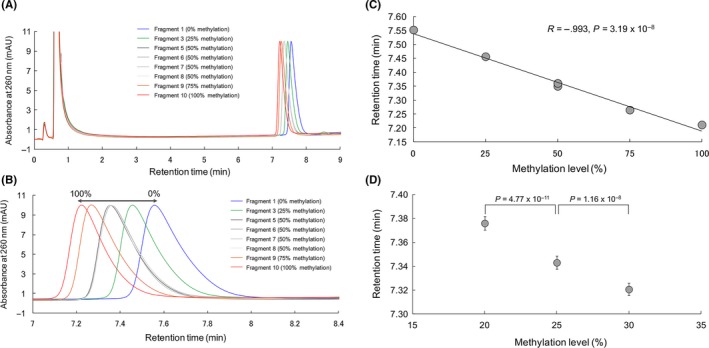
Separation of PCR products amplified from the synthetic DNA fragments by the newly developed anion‐exchange high‐performance liquid chromatography method. A, Chromatograms of synthetic DNA fragments 1, 3, 5, 6, 7, 8, 9 and 10 corresponding to the sequences after bisulfite modification of 0%, 25% 50%, 75% and 100% methylated promoter regions of the FAM150A gene (Table 1). Flow rate, 1.0 mL/min; buffer A, 25 mmol/L Tris‐HCl, pH 7.5; buffer B, 1 mol/L NH 4(SO 4)2 in buffer A; linear gradient from 40%‐100% buffer B in 10 minutes; column temperature, 70°C; injection volume, 5 μL. B, Enlarged view of the chromatograms of (A); retention time: 7‐8.4 minutes. C, Linearity of the decrease in the retention time associated with an increase in the methylation level from 0% to 100%. D, Within‐run reproducibility test using PCR products obtained from Fragments 2, 3 and 4 corresponding to the sequences after bisulfite modification of 20%, 25% and 30% methylated promoter regions of the FAM150A gene (Table 1). Error bar: SD
To confirm whether this HPLC method is reproducible, we carried out a within‐run reproducibility test using PCR products obtained from Fragments 2, 3 and 4 corresponding to DNA methylation levels of 20%, 25% and 30%, respectively (Table 1), which were around the cut‐off value (27.2%) of the DNA methylation level of the FAM150A gene for discrimination of more aggressive CIMP‐positive ccRCC from CIMP‐negative ccRCC in our previous study using specimens of tumorous tissue and MassARRAY analysis.16 After 10 measurements were taken for each fragment, excellent concordance of the retention time for each fragment was revealed (Figure 2D), indicating the reproducibility of HPLC analysis using the newly developed column. In addition, it was revealed that a difference in methylation level of 5% (ie, 20% vs 25% [P = 4.77 × 10−11] and 25% vs 30% [P = 1.16 × 10−8]) would be statistically distinguishable (Figure 2D) in a clinically feasible setting.
3.2. High‐performance liquid chromatography analysis of clear cell renal cell carcinoma tissue specimens
The FAM150A gene is a CIMP marker gene showing a high area under the curve value (0.968) in receiver operating characteristic analysis for discrimination of CIMP‐positive ccRRC from CIMP‐negative ccRCC.16 In the present study, 384‐bp PCR products encompassing the promoter region of the FAM150A gene from the 98 ccRCC were subjected to HPLC analysis. PCR products from synthetic DNA fragments 1 and 10 with methylation levels of 0% and 100%, respectively (Table 1), were measured simultaneously as control samples. Representative chromatograms are shown in Figure 3A. Eighty‐two ccRCC showed a single peak that had retention times similar to the unmethylated DNA control (eg, Case 3 in Figure 3A). In 7 ccRCC, a single peak with a shoulder was observed, as Cases 84 and 89 in Figure 3A. In 9 ccRCC, a bimodal peak pattern consisting of 2 peaks with retention times similar to the unmethylated and methylated DNA was observed (eg, Case 91 in Figure 3A). Chromatograms of all 98 ccRCC examined are shown in Figure S1.
Figure 3.
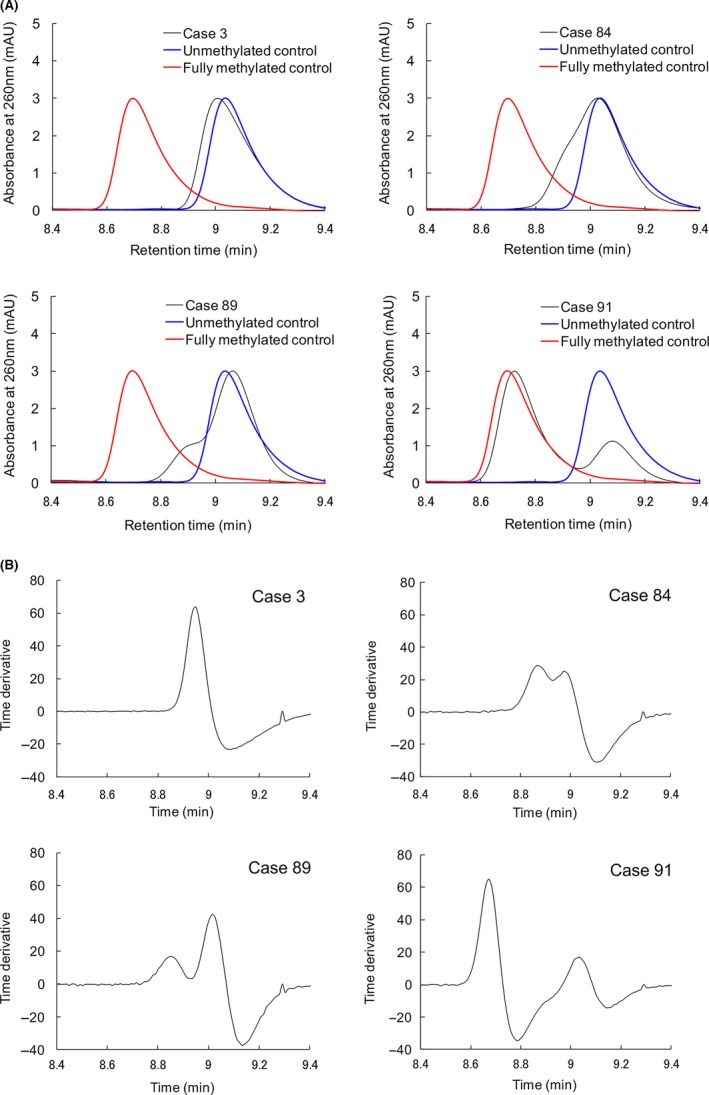
High‐performance liquid chromatography (HPLC) analysis using the newly developed anion‐exchange column for tissue specimens of clear cell renal cell carcinoma (ccRCC). A, Representative chromatograms. PCR products of 384 bp encompassing the promoter region of the FAM150A gene from the 98 ccRCC were subjected to HPLC analysis. Case 3 ccRCC showed a single peak with retention times similar to the unmethylated DNA control. In Cases 84 and 89 ccRCC, a single peak with a shoulder was observed. In Case 91 ccRCC, a bimodal peak pattern consisting of 2 peaks which had retention times similar to the unmethylated and methylated DNA was observed. Chromatograms of all of the 98 ccRCC examined are shown in Figure S1. B, Differential processing of the chromatograms. The differential patterns of chromatograms in (A) are shown in this panel. In Case 3, a chromatogram showing a “single peak” pattern was converted to a differential curve with 1 upward convex portion. In Cases 84 and 89, chromatograms showing a “single peak with a shoulder” pattern were converted to differential curves with 2 upward convex portions. In Case 91, a “bimodal peak” pattern chromatogram was converted to a differential curve with 2 upward convex portions
The differential patterns of Figure 3A are shown in Figure 3B. All 82 chromatograms showing “a single peak” pattern were converted to differential curves with a single upward convex shape as in Case 3 in Figure 3B. In contrast, chromatograms with “a bimodal peak” pattern were converted to differential curves with 2 upward convex portions as in Case 91 in Figure 3B. Furthermore, all 7 chromatograms showing “a single peak with a shoulder” pattern were converted to differential curves with 2 upward convex portions without exception (Cases 84 and 89 in Figure 3B). Thus, we were able to clearly stratify ccRCC into 2 groups, those showing the “one upward convex pattern” and those showing the “two upward convex pattern.” by differential analysis of the chromatograms.
3.3. Clinicopathological impact of DNA methylation status of clear cell renal cell carcinoma assessed by high‐performance liquid chromatography analysis using the newly developed column
Sixteen ccRCC showing the “two upward convex pattern” showed significant correlations with clinicopathological parameters reflecting tumor aggressiveness (Table 2): ccRCC showing a larger diameter, a type 3 macroscopic configuration, higher histological grade, vascular involvement, renal vein tumor thrombi, infiltrating growth, tumor necrosis, renal pelvis invasion and higher pathological TNM stage were observed more frequently in the “two upward convex pattern” group than in the “one upward convex pattern” group.
Table 2.
Correlation between DNA methylation status (“one upward convex pattern” vs “two upward convex pattern”) and clinicopathological parameters of clear cell renal cell carcinomas
| Clinicopathological parameters | One upward convex pattern (n = 82) | Two upward convex pattern (n = 16) | P |
|---|---|---|---|
| Age | 61.93 ± 10.33 | 67.38 ± 10.89 | 7.93 × 10−2 b |
| Sex | |||
| Male | 56 | 13 | 3.80 × 10−1 c |
| Female | 26 | 3 | |
| Tumor diameter (cm) | 5.17 ± 3.24 | 8.69 ± 2.75 | 1.36 × 10−4 b , a |
| Macroscopic configuration | |||
| Type 1 | 32 | 2 | 2.51 × 10−3 c , a |
| Type 2 | 27 | 2 | |
| Type 3 | 23 | 12 | |
| Predominant histological gradesd | |||
| G1 | 42 | 2 | 1.12 × 10−4 c , a |
| G2 | 32 | 5 | |
| G3 | 7 | 7 | |
| G4 | 1 | 2 | |
| Highest histological gradese | |||
| G1 | 7 | 0 | 4.91 × 10−3 c , a |
| G2 | 39 | 2 | |
| G3 | 21 | 5 | |
| G4 | 15 | 9 | |
| Vascular involvement | |||
| Negative | 48 | 2 | 8.03 × 10−4 c , a |
| Positive | 34 | 14 | |
| Renal vein tumor thrombi | |||
| Negative | 64 | 5 | 4.62 × 10−4 c , a |
| Positive | 18 | 11 | |
| Predominant growth patternd | |||
| Expansive | 76 | 9 | 8.15 × 10−4 c , a |
| Infiltrative | 6 | 7 | |
| Most aggressive growth patterne | |||
| Expansive | 54 | 5 | 2.39 × 10−3 c , a |
| Infiltrative | 28 | 11 | |
| Tumor necrosis | |||
| Negative | 63 | 4 | 1.25 × 10−4 c , a |
| Positive | 19 | 12 | |
| Invasion to renal pelvis | |||
| Negative | 76 | 12 | 5.51 × 10−2 c |
| Positive | 6 | 4 | |
| Pathological Tumor‐Node‐Metastasis stage | |||
| Stage I | 46 | 0 | 1.09 × 10−5 c , a |
| Stage II | 2 | 2 | |
| Stage III | 19 | 5 | |
| Stage IV | 15 | 9 | |
P‐values of <.05.
Wilcoxon rank sum test.
Fisher's exact test.
If the tumor showed heterogeneity, findings in the predominant area were described.
If the tumor showed heterogeneity, the most aggressive features of the tumor were described.
Figure 4A shows the Kaplan‐Meier survival curves for recurrence‐free survival of patients with ccRCC showing the “one upward convex” and “two upward convex” patterns, and for whom curative resection of both the primary and metastatic lesions (if present) had been performed (n = 84). Figure 4B presents the Kaplan‐Meier curves for overall survival of patients with ccRCC, showing the “one‐upward convex” and “two upward convex” patterns (n = 98). The period covered ranged from 34 to 5719 days (mean, 2638 days). The recurrence‐free and overall survival rates of patients in the “two upward convex pattern” group were significantly lower than those of patients in the “one upward convex pattern” group (Figure 4).
Figure 4.
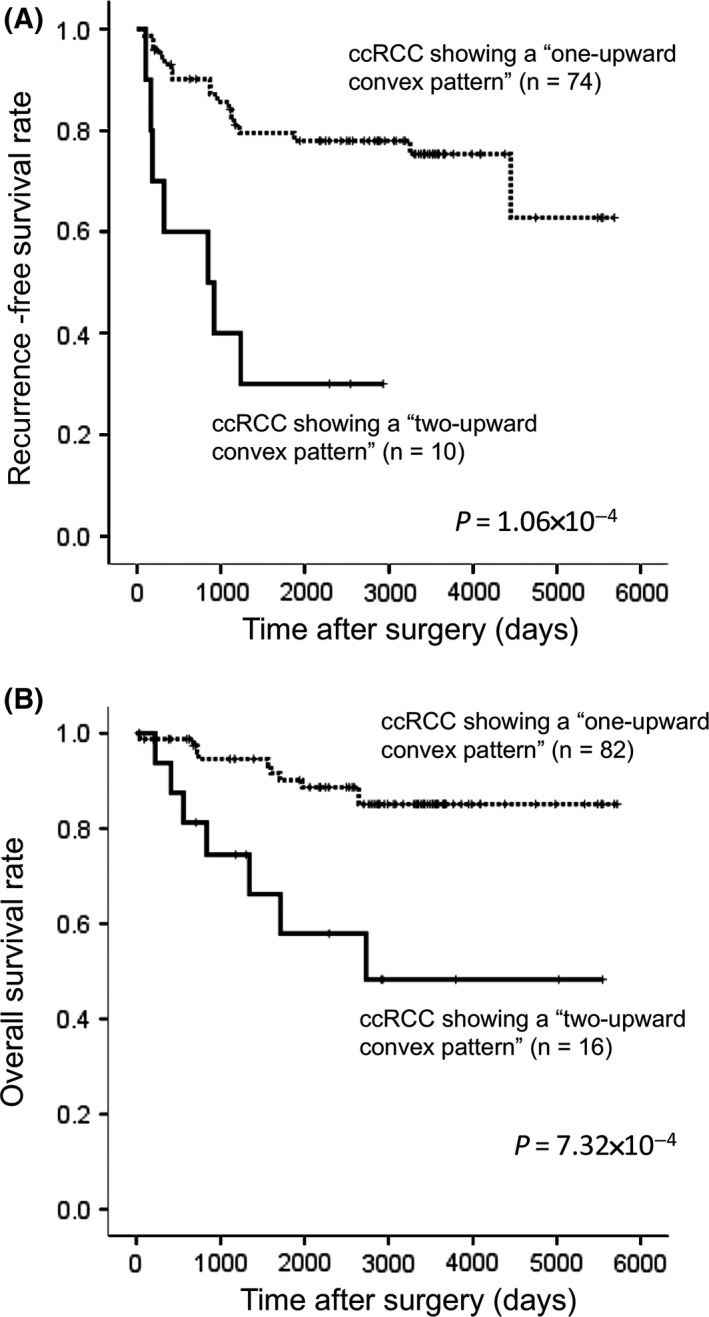
Kaplan‐Meier survival curves of patients with clear cell renal cell carcinoma (ccRCC) showing the “one upward convex pattern” and the “two upward convex pattern.” A, Recurrence‐free survival was examined in patients for whom curative resection of both primary and metastatic lesions (if present) had been performed (n = 84). B, Overall survival was examined for all patients (n = 98)
4. DISCUSSION
Ion‐exchange HPLC is widely used for simple and quick separation analysis of biopolymers such as nucleic acids, proteins and polysaccharides.22, 23, 24, 25 However, only a few reports have described analysis of DNA methylation using ion‐exchange HPLC, as it is not easy to separate cytosine and thymine corresponding to methylated and unmethylated cytosine after bisulfite modification, respectively, using an ordinary ion‐exchange column which relies on electrostatic properties alone. To facilitate analysis of DNA methylation by ion‐exchange HPLC, we developed a novel anion‐exchange packing material having both electrostatic properties based on ion‐exchange groups (quaternary ammonium and tertiary amino groups), and hydrophobic properties based on the polymer particles made up of hydrophobic monomers. Quaternary ammonium groups introduced by polymerization and tertiary amino groups introduced by an open‐ring reaction at the glycidyl group are arranged three‐dimensionally on the polymer particles (Figure 1). Thus, bisulfite‐PCR products passing through the column interact first with the quaternary ammonium groups and are then captured by the tertiary amino groups on the surface of the polymer particles. Thereafter, the hydrophobicity of the polymer particles plays a role in discrimination between cytosine derived from methylated CpG and thymine derived from unmethylated CpG within the PCR products. Although the amounts of negative charge from the phosphate groups in cytosine and thymine are the same, hydrophobic interaction between the polymer particles and the PCR products containing thymine is stronger due to the methyl group at the 5′ position of the pyrimidine ring in comparison with that between the polymer particles and the PCR products containing cytosine.
It was found that the novel HPLC column has excellent quantitativeness (Figure 2C). Synthetic DNA fragments 5, 6, 7 and 8, all showing 50% methylation (Table 1), showed the same retention times regardless of differences in the positions of methylated CpG dinucleotides within the target sequence (Figure 2A,B). Even though methylated CpGs are distributed unevenly, it is likely that the interaction between the packing materials and PCR products are averaged out by isotropic behavior while passing through the column. Therefore, the new HPLC column can be useful for DNA methylation analysis regardless of the sequences of the target genes. Subsequently, in Figure 2D, 10 measurements in a within‐run reproducibility test revealed excellent reproducibility, and differences in methylation level of only 5% were clearly distinguishable. In addition, we have confirmed that the DNA methylation levels of the FAM150A gene quantified using the new HPLC column are in excellent accordance with those evaluated by the conventional MassARRAY method (Figure S2).
Another HPLC method, denaturing HPLC (DHPLC), is an ion‐pair reverse‐phase HPLC technique that can detect DNA methylation at a specific partial denaturation temperature.26, 27 Some cancer researchers have reported that the DHPLC method is able to detect DNA methylation of the promoter region of tumor suppressor genes.28, 29, 30 However, because heteroduplex formation is necessary during analysis, the total analysis time is prolonged. In addition, the shapes of the peaks and retention time changed appreciably with a difference in column temperature of only 1°C (Figure S3A). Furthermore, even if the total number of methylated CpG in the target sequence is the same (20 CpG [50% methylation], Table 1), the retention time of Fragments 5‐8 is changed considerably due to differences in the positions of the methylated CpG (Figure S3B), resulting in inaccurate measurement of the DNA methylation level when the DHPLC method is used. In contrast, use of our anion‐exchange HPLC method makes it possible to directly apply PCR products without any time‐consuming preanalytic procedures, and the analysis can be completed within 10 minutes (Figure 2A,B). The newly developed column can yield reproducible data using a conventional HPLC device without strict temperature control.
Finally, we applied our newly developed HPLC method to analysis of DNA methylation status using tissue samples. Three types of peak pattern were observed: a “single peak,” a “single peak with a shoulder” and a “bimodal peak” (Figure 3A). Because ccRCC are hypervascular tumors, as shown in Figure S4, specimens of cancerous tissue may contain considerable numbers of vascular endothelial cells. A “single peak with a shoulder” and a “bimodal peak” may be formed by a mixture of methylated DNA from cancer cells and unmethylated DNA derived from non‐cancerous cells such as endothelial cells. In the “bimodal peak” pattern where the DNA methylation level of the target region in cancer cells is high, as shown in Case 91 in Figure 3A (close to the fully methylated control DNA), we can identify the presence of cancer cells having methylated DNA even if the content of such cells is low. In contrast, when the DNA methylation level of the target region in cancer cells is not so high, and when methylated and unmethylated peaks show a high degree of overlap, discrimination of a “single peak with a shoulder” from a “single peak” may be unclear, as shown in Case 84 in Figure 3A. Therefore, we attempted to differentiate the chromatogram data and establish criteria for stratification of cancers. A “single peak” pattern was converted to a peak pattern with one upward convex portion, and a “single peak with a shoulder” and a “bimodal peak” were converted to a peak pattern with 2 upward convex portions without exception (Figure 3B). Simple viewing of the differential curves (Figure 3B) made it possible to identify tissue samples including cancer cells having methylated DNA and showing the “two upward convex pattern,” allowing discrimination from tissue samples lacking methylated DNA and showing the “one upward convex pattern,” even if the content of cancer cells showing DNA methylation and the DNA methylation level of the target region were low.
As shown in Figure S5, it was confirmed that this HPLC method was able to detect at least a 5% mixture of fully methylated DNA by simply viewing the differential curve. Although the 1% mixture could not be detected in this setting, the discriminability may be improved sufficiently for detection of a 1% content of fully methylated DNA by introduction of fluorescence detection for PCR products tagged with fluorescent substances. In comparison to ordinary methods, such as MassARRAY and pyrosequencing, for which the reliability of measured values of less than 10% is restricted, simple and reliable detection of only a small number of cancer cells having methylated DNA using our HPLC system would be advantageous for analysis of clinical specimens.
The present study using our newly developed HPLC column revealed a “two upward convex pattern” of the differential curve in 16 ccRCC. These 16 ccRCC including cancer cells showing various degree of DNA methylation of the FAM150A gene showed a significantly higher incidence of clinicopathological features reflecting tumor aggressiveness (Table 2) and shorter recurrence‐free and overall survival times than 82 ccRCC showing a “one upward convex pattern” (Figure 4). In our previous study, 14 ccRCC included in the present study were found to be CIMP‐positive using genome‐wide DNA methylation analysis by Infinium assay11 and DNA methylation quantification of the 7 ccRCC‐specific CIMP marker genes using the MassARRAY system.16 Although the present stratification of 16 ccRCC showing 2 upward convex portions showed appreciable overlap with the previous stratification of CIMP‐positive ccRCC, the present “two upward convex pattern” can be identified by simple and quick analysis of only one gene, FAM150A. Taken together, the present data suggest that our HPLC column provides an excellent prognostication system for patients with ccRCC. Moreover, this HPLC method is applicable for not only prognostication of patients with ccRCC but also for evaluation of other marker genes, which have been identified from biopsy and surgically resected tissue samples,31, 32, 33 regardless of the target sequences.
However, methods for efficient capture of circulating tumor cells (CTC) from blood samples have been recently developed for liquid biopsy.34, 35, 36 Adams et al34 report that pathologically definable, apoptotic and epithelial‐mesenchymal transition‐like CTC accounted for 17%, 16% and 58% of cells isolated by one such capture method, respectively. Because our HPLC method is able to detect 5% DNA methylation (Figure S5), 16%‐58% of cancer cells in liquid biopsy samples would be detectable using our system. In addition, unlike ordinary methods that tend to quantify the different DNA methylation levels in cancer cells and contaminating blood cells as a whole, our HPLC system that quantifies the various levels of DNA methylation in individual cell lineages would be more suitable for liquid biopsy samples containing CTC and contaminating blood cells. Moreover, pharmacoepigenetic studies have recently revealed that drug response can be predicted based on the DNA methylation profiles of blood cells, because germline DNA methylation status regulates the expression levels of drug‐metabolizing enzymes and drug transporters.37, 38 Therefore, quantification of DNA methylation using our HPLC system would be applicable to the prediction of anti‐cancer drug response.
Unlike clinical sequencing and RNA sequencing, our HPLC method has been developed for quantification of DNA methylation. DNA methylation alterations accumulate upon exposure to carcinogenetic factors, even in precancerous conditions, and generally precede genetic alterations revealed by clinical sequencing. DNA methylation profiles generally reflect each of the steps of multistage carcinogenesis and are frequently correlated with the clinicopathological features of individual cancers. In addition, unlike alterations of mRNA and/or protein expression and metabolomic features, which can be easily affected by the microenvironment of cancer cells, DNA methylation alterations are stably preserved on DNA double strands by covalent bonds. Therefore, DNA methylation levels at appropriate marker CpG sites, which can be quantified using our HPLC method, would appear to be more optimal biomarkers. Moreover, our HPLC method has excellent cost‐effectiveness. These characteristics make our method outstanding in a clinical setting.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Supporting information
Yotani T, Yamada Y, Arai E, et al. Novel method for DNA methylation analysis using high‐performance liquid chromatography and its clinical application. Cancer Sci. 2018;109:1690–1700. https://doi.org/10.1111/cas.13566
Funding information
The Program for Promotion of Fundamental Studies in Health Sciences (10‐42) from The National Institute of Biomedical Innovation (NiBio), The Project for Utilizing Glycans in the Development of Innovative Drug Discovery Technologies (17ae0101020h0002) from The Japan Agency for Medical Research and Development (AMED) and KAKENHI (16H02472 and 16K08720) from The Japan Society for the Promotion of Science.
REFERENCES
- 1. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17:630‐641. [DOI] [PubMed] [Google Scholar]
- 2. Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 2016;8:a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arai E, Kanai Y. DNA methylation profiles in precancerous tissue and cancers: carcinogenetic risk estimation and prognostication based on DNA methylation status. Epigenomics. 2010;2:467‐481. [DOI] [PubMed] [Google Scholar]
- 4. Kanai Y. Genome‐wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci. 2010;101:36‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garvilles RG, Hasegawa T, Kimura H, et al. Dual functions of the RFTS domain of Dnmt1 in replication‐coupled DNA methylation and in protection of the genome from aberrant methylation. PLoS ONE. 2015;10:e0137509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J, Gao Q, Li P, et al. S phase‐dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 2011;21:1723‐1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kanai Y, Arai E. Multilayer‐omics analyses of human cancers: exploration of biomarkers and drug targets based on the activities of the International Human Epigenome Consortium. Front Genet. 2014;5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ljungberg B, Campbell SC, Cho HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615‐621. [DOI] [PubMed] [Google Scholar]
- 9. Donat SM, Diaz M, Bishoff JT, et al. Follow‐up for clinically localized renal neoplasms: AUA guideline. J Urol. 2013;190:407‐416. [DOI] [PubMed] [Google Scholar]
- 10. Ko JJ, Xie W, Kroeger N, et al. The International Metastatic Renal Cell Carcinoma Database Consortium model as a prognostic tool in patients with metastatic renal cell carcinoma previously treated with first‐line targeted therapy: a population‐based study. Lancet Oncol. 2015;16:293‐300. [DOI] [PubMed] [Google Scholar]
- 11. Arai E, Chiku S, Mori T, et al. Single‐CpG‐resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenisis. 2012;33:1487‐1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arai E, Gotoh M, Tian Y, et al. Alterations of the spindle checkpoint pathway in clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Int J Cancer. 2015;137:2589‐2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation‐specific PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821‐9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ehrich M, Nelson MR, Stanssens P, et al. Quantitative high‐throughput analysis of DNA methylation patterns by base‐specific cleavage and mass spectrometry. Proc Natl Acad Sci USA. 2005;102:15785‐15790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shen L, Guo Y, Chen X, Ahmed S, Issa JP. Optimizing annealing temperature overcomes bias in bisulfite PCR methylation analysis. Biotechniques. 2007;42:48‐58. [DOI] [PubMed] [Google Scholar]
- 16. Tian Y, Arai E, Gotoh M, Komiyama M, Fujimoto H, Kanai Y. Prognostication of patiants with clear cell renal cell carcinomas based on quantification of DNA methylation levels of CpG island methylator phenotype marker genes. BMC Cancer. 2014;14:772‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagashio R, Arai E, Ojima H, Kosuge T, Kondo Y, Kanai Y. Carcinogenetic risk eatimation based on quantification of DNA methylation levels in liver tissue at the precancerous stage. Int J Cancer. 2011;129:1170‐1179. [DOI] [PubMed] [Google Scholar]
- 18. Moch H, Humphrey PA, Ulbright TM, Reuter VE, eds. World Health Organization Classification of Tumours of the Urinary System and Male Genital Organs. Lyon: IARC Press, 2016;11‐44. [Google Scholar]
- 19. Sobin LH, Gospodarowicz MK, Wittekind C, eds. International Union Against Cancer: TNM Classification of Malignant Tumors, 7th edn Chichester: Wiley; 2009. [Google Scholar]
- 20. Arai E, Kanai Y, Ushijima S, Fujimoto H, Mukai K, Hirohashi S. Regional DNA hypermethylation and DNA methyltransferase (DNMT) 1 protein overexpression in both renal tumors and corresponding nontumorous renal tissues. Int J Cancer. 2006;119:288‐296. [DOI] [PubMed] [Google Scholar]
- 21. Kanai T, Hirohashi S, Upton MP. Pathology of small hepatocellular carcinoma. A proposal for a new gross classification. Cancer. 1987;60:810‐819. [DOI] [PubMed] [Google Scholar]
- 22. Mauri PL, Pietta PG, Pace M. Analysis and purification of DNA restriction fragments by high‐performance liquid chromatography. J Chromatogr. 1991;548:281‐287. [DOI] [PubMed] [Google Scholar]
- 23. Strege MA, Lagu A. Anion‐exchange chromatography of DNA restriction fragments. J Chromatogr. 1991;555:109‐124. [DOI] [PubMed] [Google Scholar]
- 24. Kasai K. Size‐dependent chromatographic separation of nucleic acids. J Chromatogr. 1993;618:203‐221. [DOI] [PubMed] [Google Scholar]
- 25. Gustavasson PE, Lemmens R, Nyhammar T, Busson P, Larsson PO. Purification of plasmid DNA with a new type of anion‐exchange beads having a non‐charged surface. J Chromatogr A. 2004;1038:131‐140. [DOI] [PubMed] [Google Scholar]
- 26. Huber CG, Oefner PJ, Preuss E, Bonn GK. High‐resolution liquid chromatography of DNA fragments on non‐porous poly (stylene‐divinylbenzene) particles. Nucl Acids Res. 1993;21:1061‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xiao W, Oefner PJ. Denaturing high perfomance liquid chromatography: a review. Hum Mutat. 2001;17:439‐474. [DOI] [PubMed] [Google Scholar]
- 28. Deng D, Deng G, Smith MF, et al. Simultanious detection of CpG methylation and single nucleotide polymorphism by denaturing high perfomance liquid chromatography. Nucl Acids Res. 2002;30:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Omaruddin RA, Chaudhry MA. Detection of genomic DNA methylation with denaturing high performance liquid chromatography. Hum Cell. 2010;23:41‐49. [DOI] [PubMed] [Google Scholar]
- 30. Couvert P, Poirier K, Carrié A. DHPLC‐based method for DNA methylation analysis of differential methylated regions from imprinted genes. Biotechniques. 2003;34:356‐362. [DOI] [PubMed] [Google Scholar]
- 31. Wang M, Li Y, Gao J, et al. p16 Methylation is associated with chemosensitivity to fluorouracil in patients with advanced gastric cancer. Med Oncol. 2014;31:988. [DOI] [PubMed] [Google Scholar]
- 32. Lv L, Deng H, Li Y, et al. The DNA methylation‐regulated miR‐193a‐3p dictates the multi‐chemoresistance of bladder cancer via repression of SRSF2/PLAU/HIC2 expression. Cell Death Dis. 2014;5:e1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watanabe Y, Maeda I, Oikawa R, et al. Aberrant DNA methylation status of DNA repair genes in breast cancer treated with neoadjuvant chemotherapy. Genes Cells. 2013;18:1120‐1130. [DOI] [PubMed] [Google Scholar]
- 34. Adams DL, Stefansson S, Haudenschild C, et al. Cytometric characterization of circulating tumor cells captured by microfiltration and their correlation to the Cell Search® CTC test. Cytometry A. 2015;87:137‐144. [DOI] [PubMed] [Google Scholar]
- 35. Schreier S, Sawaisorn P, Udomsangpetch R, Triampo W. Advances in rare cell isolation: an optimization and evaluation study. J Transl Med. 2017;15:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murlidhar V, Zeinali M, Grabauskiene S, et al. A radial flow microfluidic device for ultra‐high‐throughput affinity‐based isolation of circulating tumor cells. Small. 2014;10:4895‐4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nasr R, Sleiman F, Awada Z, Zgheib NK. The pharmacoepigenetics of drug metabolism and transport in breast cancer: review of the literature and in silico analysis. Pharmacogenomics. 2016;17:1573‐1585. [DOI] [PubMed] [Google Scholar]
- 38. Candelaria M, de la Cruz‐Hernández E, Pérez‐Cárdenas E, Trejo‐Becerril C, Gutiérrez‐Hernández O, Dueñas‐González A. Pharmacogenetics and pharmacoepigenetics of gemcitabine. Med Oncol. 2010;27:1133‐1143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials