

Abstract
Understanding the mechanism of lymph node metastasis, a poor prognostic sign for prostate cancer, and the further dissemination of the disease is important to develop novel treatment strategies. Recent studies have reported that C‐C chemokine receptor 7 (CCR7), whose ligand is CCL21, is abundantly expressed in lymph node metastasis and promotes cancer progression. Tumor necrosis factor‐α (TNF‐α) is chronically produced at low levels within the tumor microenvironment. The aim of this study was to determine whether TNF‐α promotes prostate cancer dissemination from metastatic lymph nodes through activation of the CCL21/CCR7 axis. First, human prostate cancer cells were determined to express both TNF‐α and CCR7. Second, low concentrations of TNF‐α were confirmed to induce CCR7 in prostate cancer cells through phosphorylation of ERK. Finally, CCL21 was found to promote the migration of prostate cancer cells through phosphorylation of the protein kinase p38. Our results suggest that TNF‐α leads to the induction of CCR7 expression and that the CCL21/CCR7 axis might increase the metastatic potential of prostate cancer cells in lymph node metastasis.
Keywords: C‐C chemokine receptor type 7, lymph node, metastasis, prostate cancer, tumor necrosis factor‐α
1. INTRODUCTION
Prostate cancer is one of the most frequently diagnosed malignancies in men worldwide.1 Approximately 70%‐80% of patients with prostate cancer develop bone metastases,2 and regional lymph node metastasis is observed in 5%‐12% of patients with prostate cancer with clinically organ‐confined cancer.3 The detection of lymph node metastasis in prostate cancer, which remains incurable, indicates a poor prognosis for patients.4, 5, 6 Therefore, understanding the mechanism of lymph node metastasis and the further dissemination of the disease is important to develop novel treatment strategies.
Tumor necrosis factor‐α (TNF‐α), which has pluripotent effects on tumorigenesis and tumor progression, is produced by cancer cells as well as the tumor microenvironment.7, 8 Increased C‐C chemokine receptor 7 (CCR7) expression in tumor cells has been reported to be significantly associated with lymph node metastasis in several cancer types.9, 10, 11 Interestingly, CCR7 has been found to mediate TNF‐α‐induced lymphatic metastasis of gallbladder cancer.12, 13 C‐C chemokine receptor 7 is a receptor for the chemokine CCL21 and is expressed in various immune cells that migrate to and within lymphoid tissues. Chemokine CCL21 is abundantly expressed on fibroblastic reticular cells in lymph node metastasis.14 Although several chemokine receptors contribute to the increased migration ability of cancer cells in specific organs,15, 16, 17 whether TNF‐α and CCR7 deteriorate prostate cancer cells within the microenvironment of lymph node metastasis remains unclear.
In the present study, we found that low TNF‐α concentrations increase CCR7 expression through the phosphorylation of ERK in prostate cancer cells and that the CCL21/CCR7 axis promotes prostate cancer cell migration through phosphorylation of p38.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
Recombinant human TNF‐α (210‐TA) and CCL21 (366‐6C) were purchased from R&D Systems (Minneapolis, MN, USA). The following antibodies were used in Western blot analyses and immunocytochemistry: rabbit anti‐TNF‐α (ab6671), goat anti‐rabbit FITC‐conjugated IgG H&L (ab6717), and rabbit anti‐CCR7 (ab103404) from Abcam (Cambridge, MA, USA); rabbit anti‐p38 (8690), rabbit anti‐phospho‐p38 (Thr180/Tyr182; 4511), rabbit anti‐ERK (9102), rabbit anti‐phospho‐ERK (Thr202/Tyr204; 9101), and HRP‐conjugated anti‐rabbit IgG (7074) antibodies from Cell Signaling Technology (Danvers, MA, USA); and mouse anti‐GAPDH (NB300‐221) antibody from Novus Biologicals (Littleton, CO, USA). The ERK inhibitor U0126 (9903) and the p38 inhibitor SB203580 (5633) were purchased from Cell Signaling Technology.
2.2. Cell culture
The following human prostate cancer cell lines were purchased from ATCC (Manassas, VA, USA): PC‐3, DU145, and LNCaP. Prostate cancer cells were maintained in a growth medium containing RPMI‐1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 5% FBS and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA). All experiments were carried out using cells grown in a humidified incubator containing 5% CO2 at 37°C. The androgen‐independent LNCaP cell line LNCaP‐SF was established in our laboratory by the long‐term subculture of parental LNCaP cells in RPMI‐1640 supplemented with 5% charcoal‐stripped FBS (Hyclone, Logan, UT, USA) and 1% penicillin/streptomycin.
To investigate CCR7 upregulation in prostate cancer cells, cells were seeded in 6‐well plates and allowed to reach 70%‐80% confluence, followed by treatment with recombinant human TNF‐α (10 ng/mL) for 6 hour in serum‐free medium.
To evaluate the ERK signaling pathway in prostate cancer cells, cells were seeded in 6‐well plates and allowed to reach 60%‐70% confluence. The cells were synchronized by starvation in serum‐free RPMI‐1640 medium for 12 hour, followed by incubation with U0126 (10 μmol/L) in serum‐free RPMI‐1640 for 1 hour. Next, the cells were incubated with recombinant human TNF‐α (10 ng/mL) for 20 minutes in serum‐free medium. The following procedures were the same as above.
2.3. Reverse transcription‐PCR
Total RNA was isolated from cultured cells using the RNeasy Mini kit (Qiagen, Germantown, MD, USA), following the manufacturer's instructions. RNA concentrations were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific). First‐strand cDNA was prepared from RNA templates (500 ng) using the iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA, USA). Reverse transcription was carried out using the following conditions: 45°C for 5 minutes, 42°C for 30 minutes, and 85°C for 5 minutes. Conditions for amplification by PCR for GAPDH, CCR7, and TNF‐α were as follows: GAPDH, 98°C for 10 seconds, 60°C for 30 seconds, 72°C for 60 seconds, 26 cycles; CCR7, 98°C for 10 seconds, 60°C for 30 seconds, 72°C for 60 seconds, 35 cycles; and TNF‐α, 98°C for 10 seconds, 68°C for 30 seconds, 72°C for 60 seconds, 30 cycles. Polymerase chain reaction was carried out using template cDNA and Takara Ex Taq Hot Start Version PCR kit (Takara Bio, Kusatsu, Japan). The PCR products were separated by electrophoresis on 1.5% agarose gels and stained with ethidium bromide. Primer sequences were as follows: GAPDH forward, 5′‐TCC ACC ACC CTG TTG GTG TA‐3′; GAPDH reverse, 5′‐GAC CAC AGT CCA TGC CAT CA‐3′; CCR7 forward, 5′‐TCC TTC TCA TCA GCA AGC TGT‐3′; CCR7 reverse, 5′‐GAG GCA GCC CAG GTC CTT GAA G‐3′; TNF‐α forward, 5′‐AGT GAC AAG CCT GTA GCC C‐3′; and TNF‐α reverse, 5′‐GCA ATG ATC CCA AAG TAG ACC‐3′.
2.4. Western blot analysis
Cell lysates were prepared with the M‐PER Mammalian Protein Extraction reagent (Thermo Fisher Scientific). Soluble lysates (25 μg) were mixed with the lithium dodecyl sulfate sample buffer and a sample reducing agent, both from Thermo Fisher Scientific, and were resolved by SDS‐PAGE. Proteins were transferred to nitrocellulose membranes, which were blocked with 1% gelatin and 0.05% Tween in Tris‐buffered saline for 1 hour at room temperature. The membranes were then incubated overnight at 4°C with primary anti‐CCR7, anti‐TNF‐α, anti‐p38, anti‐phospho‐p38, anti‐ERK, or anti‐phospho‐ERK primary antibodies following the manufacturers’ instructions. Membranes were washed three times before incubation with HRP‐conjugated anti‐rabbit or anti‐mouse secondary antibodies for 1 hour at room temperature. Protein loading was determined with an anti‐GAPDH antibody. Protein bands were detected using Super Signal West Femto Maximum Sensitivity substrate (Thermo Fisher Scientific).
2.5. Immunocytochemistry
Prostate cancer cells were seeded at a concentration of 1.0 × 105 cells/well into 6‐well plates and cultured until they reached 60%‐70% confluence for immunocytochemistry. The cells were washed with PBS and fixed in 4% paraformaldehyde in PBS for 15 minutes at room temperature. After washing three times with ice‐cold PBS, the cells were permeabilized with incubation in PBS containing 0.25% Triton X‐100 for 15 minutes at room temperature. The cells were washed three times with PBS and blocked with polybutylene succinate‐co‐butylene terephthalate containing 10% serum from the species of the secondary antibody for 30 minutes at room temperature. Next, the cells were incubated with the indicated primary antibodies overnight at 4°C, as described by the manufacturer. Tumor necrosis factor‐α was detected by incubating cells for 1 hour with an FITC‐conjugated TNF‐α antibody (1:1000). The cells were counterstained with 4',6‐Diamidino‐2‐phenylindole dihydrochloride (Sigma‐Aldrich, St. Louis, MO, USA), and images were captured using an Olympus FSX100 (Olympus, Tokyo, Japan).
2.6. Cell migration assay
The cell migration assay was carried out using Transwell inserts placed in 24‐well plates, which had a pore size of 8.0 μm (Greiner Bio‐One, Monroe, NC, USA). PC‐3 and DU145 prostate cancer cell lines were grown to 80% confluence in growth medium, followed by synchronization by starvation in serum‐free RPMI‐1640 containing 0.5% BSA for 16 hour in a humidified incubator containing 5% CO2 at 37°C. To determine migration in LNCaP cells, the cells were seeded in 6‐well plates and allowed to reach 70%‐80% confluence, followed by incubation with or without recombinant human TNF‐α (10 ng/mL) in serum‐free medium for 6 hour. Approximately 2.5 × 104 cells (PC‐3 and DU145) or 5.0 × 104 cells (LNCaP) in 200 μL RPMI‐1640 culture medium supplemented with 0.1% or 1% FBS were placed in the upper compartment, and the lower compartment was filled with 800 μL RPMI‐1640 containing 2% or 10% FBS with or without different concentrations of CCL21. PC‐3 and DU145 cells were incubated for 24 hour at 37°C in 5% CO2, whereas LNCaP cells were incubated for 40 hour under the same conditions, followed by fixation with 4% paraformaldehyde in PBS for 10 minutes. The cells on the upper surface of the filter were removed carefully with a cotton swab, and the cells on the lower surface of the filter were stained with 0.1% crystal violet for 15 minutes. The stained filters were photographed. The crystal violet dye retained on the filters was extracted into 33% acetic acid, and cell migration was measured by reading the absorbance of crystal violet at 595 nm, followed by correction at 450 nm on a microplate reader. Statistical analysis was undertaken using Student's t‐test. A P‐value <.05 was considered statistically significant.
2.7. Wound‐healing assay
PC‐3 and DU145 (2.5 × 105 cells/well) and LNCaP cells (5.0 × 105 cells/well) were seeded into 6‐well plates. After reaching 100% confluence, the cell monolayers were scratched with a 100‐μL sterile pipette tip to create a vertical wound. To avoid any influence from the rate of cell growth on wound healing, the tumor cell culture medium was changed from RPMI‐1640 with 5% FBS to serum‐free RPMI‐1640. The cells were incubated with 10 μmol/L SB203580 at 37°C for 1 hour, followed by incubation with or without CCL21 (50 ng/mL) for 24 or 48 hour. Phase‐contrast images were captured at 0 and 24 hour with a 10× objective using an Olympus FSX100 microscope. Migration activity was expressed using the transferred distance method determined using ImageJ software (NIH, Bethesda, MD, USA). Three separate visual fields were measured in each experiment. Statistical analysis was carried out using Student's t‐test. A P‐value <.05 was considered statistically significant.
2.8. Proliferation assay
PC‐3 cells were seeded at a density of 2.0 × 103 cells/well in 96‐well plates in media containing RPMI‐1640 and 5% FBS. Twelve hours after seeding, the medium was changed to serum‐free RPMI‐1640 with or without TNF‐α at different concentrations. Cell growth was determined at 0, 24, 48, and 72 hour using the WST‐1 assay (Takara Bio). Briefly, WST‐1 (10 μL/well) was added to each well and incubated for 105 minutes at 37°C. Absorbance was measured at 595 nm and corrected at 450 nm on a microplate reader.
2.9. Statistical analysis
Student's t‐test was used to determine the statistical significance of differences in the results of the wound healing assay between two groups. For multiple comparisons, one‐way anova, followed by Bonferroni's test, was used to determine the statistical significance of differences in the proliferation and Transwell migration assay results. P < .05 was considered statistically significant.
3. RESULTS
3.1. Tumor necrosis factor‐α and CCR7 expression and TNF‐α effect in prostate cancer cells
We first determined TNF‐α and CCR7 mRNA levels in PC‐3, DU145, LNCaP, and LNCaP‐SF prostate cancer cell lines using RT‐PCR, which indicated that TNF‐α and CCR7 mRNA are expressed at detectable levels in all cell lines (Figure 1A). Additionally, Western blotting and immunocytochemistry revealed that TNF‐α and CCR7 are also expressed at the protein level in all cell lines (Figure 1B,C). Following exogenous stimulation with 10 ng/mL TNF‐α, TNF‐α protein levels in prostate cancer cells increased gradually for up to 6 hour in an autocrine fashion (Figure 1D). PC‐3 cells were also treated with TNF‐α at different concentrations for up to 3 days to assess whether TNF‐α induces prostate cancer cell proliferation. Although higher TNF‐α concentrations (100 and 300 ng/mL) significantly inhibited cell proliferation (Figure 1E, upper panel), lower TNF‐α concentrations (0, 5, 10, and 20 ng/mL) did not (Figure 1E, lower panel). Finally, prostate cancer cells were treated with 0, 5, 10, or 20 ng/mL TNF‐α for 6 hour to explore the role of TNF‐α in CCR7 expression through Western blotting. As shown in Figure 1F, CCR7 protein levels, which were increased in response to treatment with 5 or 10 ng/mL TNF‐α for 6 hour, did not differ from those in untreated cultures in response to treatment with 20 ng/mL TNF‐α.
Figure 1.
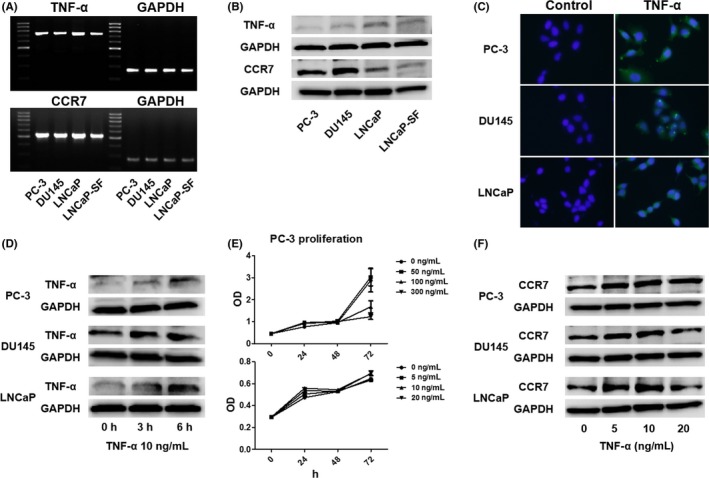
Low‐dose tumor necrosis factor‐α (TNF‐α) induces C‐C chemokine receptor 7 (CCR7) expression in prostate cancer cells. A,B, Total RNA and protein were extracted from prostate cancer cells, and their mRNA and protein levels were analyzed using RT‐PCR A and Western blotting B. C, Prostate cancer cells (1.0 × 105 cells/well) were seeded into 6‐well plates and cultured until they reached 60%‐70% confluence. The cells were incubated with a primary anti‐TNF‐α antibody, followed by incubation with a secondary antibody conjugated with FITC (green). Cells were counterstained with DAPI (blue). D, Changes in TNF‐α protein levels in prostate cancer cells after stimulation with exogenous TNF‐α (10 ng/mL) were determined by Western blotting. E, PC‐3 cell proliferation was determined with the WST‐1 assay using a range of TNF‐α concentrations. Although high TNF‐α concentrations (100 and 300 ng/mL, compared with 0 ng/mL, P < .01) significantly inhibited cell proliferation at 72 hour (upper panel), low concentrations did not lead to inhibition of cell proliferation (lower panel). Data are presented as mean ± SD. F, Prostate cancer cells were treated with TNF‐α at different concentrations for 6 hour, and Western blot analysis was used to detect CCR7
3.2. Tumor necrosis factor‐α augments CCL21‐mediated migration of prostate cancer cells
Transwell migration assay was undertaken to determine the functional role of CCL21/CCR7 signaling in prostate cancer cells. Although CCL21 led to an increase in the migration of PC‐3 and DU145 cells in a dose‐dependent manner, a similar increase in migration was not observed in LNCaP cells that expressed lower CCR7 compared with the PC‐3 and DU145 cells (Figure 2A). The pretreatment of prostate cancer cells with 10 ng/mL TNF‐α led to a significant increase in migration compared with the untreated cultures, even in LNCaP cells, which showed significantly increased migration with TNF‐α treatment (Figure 2B).
Figure 2.
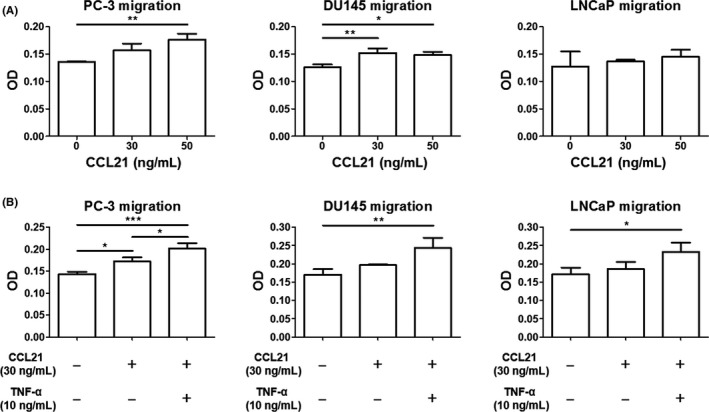
Tumor necrosis factor‐α (TNF‐α) augments C‐C chemokine ligand 21 (CCL21)‐induced prostate cancer cell migration. A, Prostate cancer cells were placed in Transwell inserts and treated with CCL21 (0, 30, or 50 ng/mL). After 24 hour (PC‐3 and DU145 cells) or 40 hour (LNCaP cells), cells that migrated through the membranes were stained with crystal violet. B, After pretreatment with 10 ng/mL TNF‐α for 6 hour, prostate cancer cells were placed in Transwell inserts and treated with CCL21 (30 ng/mL), followed by incubation for 24 hour (PC‐3 and DU145 cells) or 40 hour (LNCaP cells). The cells that migrated through the membranes were then stained. Mean optical density (OD) at 595 nm was determined using a microplate reader. Data are presented as means ± SD. All experiments were performed in triplicate. *P < .05; **P < .01; ***P < .01
3.3. Tumor necrosis factor‐α upregulates CCR7 through ERK signaling in prostate cancer cells
Next, we determined the mechanism of TNF‐α‐mediated CCR7 upregulation by Western blot analysis. The stimulation of PC‐3, DU145, and LNCaP cells with TNF‐α resulted in an increase in phosphorylation of ERK within 1 hour (Figure 3A). Conversely, pretreatment with the ERK inhibitor U0126 (10 μmol/L) significantly suppressed the phosphorylation of ERK with a concomitant decrease in the expression of TNF‐α‐mediated CCR7 induction (Figure 3B). These results indicate that TNF‐α upregulates CCR7 through the ERK signaling pathway.
Figure 3.
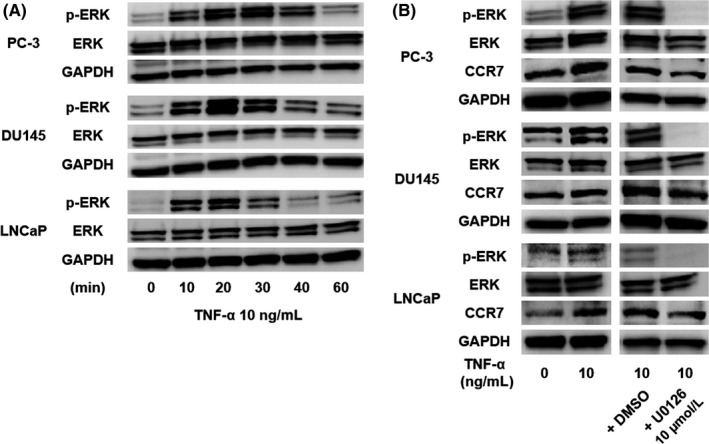
Tumor necrosis factor‐α (TNF‐α) upregulates C‐C chemokine receptor 7 (CCR7) through ERK signaling. A, Phosphorylation of ERK was determined by Western blotting of prostate cancer cells at 0, 10, 20, 30, 40, and 60 minutes after stimulation with TNF‐α. B, After pretreatment with the ERK inhibitor U0126 (10 μmol/L), phosphorylation of ERK and expression of CCR7 in prostate cancer cells were examined at 20 minutes after treatment with 10 ng/mL TNF‐α
3.4. C‐C chemokine ligand 21/CCR7 signaling promotes prostate cancer cell migration through activation of p38
The interaction between CCR7 and mitogen‐activated protein kinase in the lymphatic dissemination of breast cancer has been reported.18 Furthermore, we previously showed that chemokines increase cell migration and invasion ability through AKT activation.19, 20 Therefore, p38, ERK, and AKT were investigated as potential downstream effectors of CCL21/CCR7 signaling during prostate cancer cell migration. As shown in Figure 4(A), despite a gradual increase in p38 following stimulation with 30 ng/mL CCL21, we failed to detect phosphorylation of ERK or AKT (Figure 4B,C).
Figure 4.
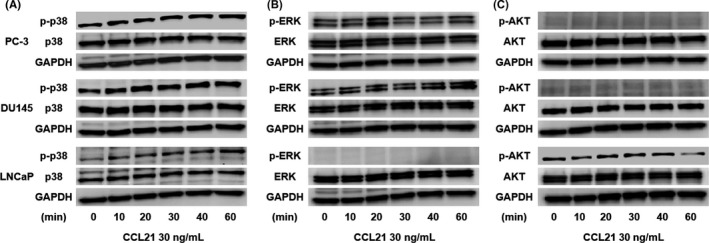
C‐C chemokine ligand 21 (CCL21)/C‐C chemokine receptor 7 (CCR7) signaling promotes prostate cancer cell migration through the activation of p38. Phosphorylation (p‐) of p38 A, ERK B, and AKT C in prostate cancer cells at the indicated times after CCL21 stimulation was assessed using Western blotting
3.5. Blockade of p38 inhibits CCL21/CCR7 signaling‐induced migration of prostate cancer cells
To determine whether p38 is responsible for the increase in the migration ability of prostate cancer cells, the p38 inhibitor SB203580 was used in the migration assay for prostate cancer cells. In the wound‐healing assay, SB203580 treatment led to a significant suppression of CCL21/CCR7‐mediated PC‐3 and DU145 cell migration (Figure 5). These results clearly indicate that p38 is a downstream target of CCL21/CCR7 signaling and a key factor in the acceleration of prostate cancer cell migration.
Figure 5.
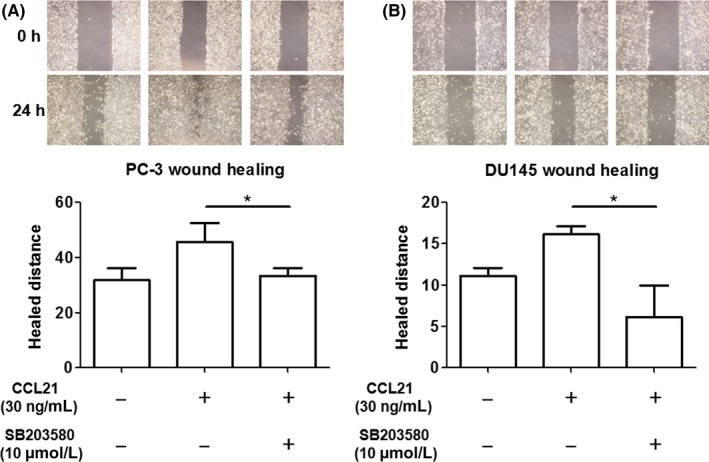
Blockade of p38 inhibits C‐C chemokine ligand 21 (CCL21)/C‐C chemokine receptor 7 (CCR7) signaling‐induced prostate cancer cell migration. The effect of UB203580 on CCL21/CCR7 signaling‐induced migration in PC‐3 A, and DU145 B, cells was assessed using the wound‐healing assay. Cells were pretreated with 10 μmol/L SB203580 for 1 hour, followed by incubation with 30 ng/mL CCL2 for 24 hour. Data are presented as means ± SD. All experiments were performed in triplicate. *P < .05
4. DISCUSSION
We revealed the presence of an autocrine TNF‐α loop in prostate cancer cells. Although high TNF‐α concentrations (≥100 ng/mL) inhibited PC‐3 proliferation, low TNF‐α concentrations (≤50 ng/mL) induced the upregulation of CCR7 and the phosphorylation of ERK. The upregulation of CCR7 was blocked by the ERK inhibitor, ascribing an important regulatory role to ERK in the TNF‐α‐mediated expression of CCR7. We also investigated the contribution of CCL21/CCR7 signaling in lymph node metastasis and revealed that CCL21 can enhance prostate cancer cell migration, particularly in prostate cancer cells with increased CCR7 levels in response to TNF‐α treatment. We found that p38 activation is involved in CCL21‐induced prostate cancer cell migration, as evidenced by the efficient inhibition of migration with the specific p38 inhibitor. Although CCL21 activated p38 in LNCaP cells, CCL21 alone could not induce LNCaP cell migration. This means that the activation of p38 by CCL21 stimulation alone is not sufficient to induce LNCaP cell migration. This is because the CCR7 expression level in LNCaP cells is naturally lower than that in other cell lines (Figure 1B). After the induction of CCR7 stimulated by TNF‐α, LNCaP cell migration could be induced by CCL21 (Figure 2B). C‐C chemokine ligand 21 certainly activates p38; however, TNF‐α is needed to sufficiently increase the CCR7 expression level to induce LNCaP cell migration. These findings support our hypothesis that TNF‐α induces the migration of prostate cancer cells in the microenvironment of lymph node metastasis through the TNF‐α/ERK/CCR7/p38 signaling pathway (Figure 6). In combination with androgen‐deprivation therapy, the CCL21/CCR7 axis might be a superior target compared with single androgen/androgen receptor signaling‐targeted therapy for the treatment of patients with prostate cancer and lymph node metastasis.
Figure 6.
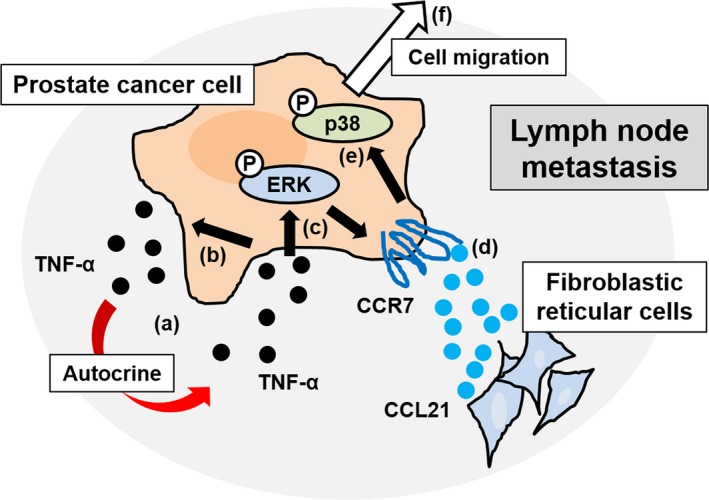
Schematic illustration of the mechanism of prostate cancer cell migration within the microenvironment of lymph node metastasis. (a), Tumor necrosis factor‐α (TNF‐α) is secreted from prostate cancer cells. (b), Low‐dose TNF‐α induces TNF‐α in an autocrine manner. (c), Low‐dose TNF‐α also induces the phosphorylation of ERK. (d), Phosphorylation of ERK increases C‐C chemokine receptor 7 (CCR7) expression. Given the abundance of C‐C chemokine ligand 21 (CCL21), the ligand for CCR7, in the microenvironment of lymph node metastasis, the CCL21/CCR7 signaling pathway is activated. (e), CCL21/CCR7 signaling subsequently induces the phosphorylation (P) of p38. (f), Phosphorylated p38 is a key molecule for prostate cancer cell migration within the microenvironment of lymph node metastasis. This mechanism could account for multiple lymph node metastases, from sentinel to more centrally located lymph nodes
Furthermore, the CCL21/CCR7 axis might be a novel biomarker for prostate cancer. The limitations of using prostate‐specific antigen alone as a biomarker for prostate cancer have been extensively reported, and the need for more specific and effective biomarkers is evident.21, 22 Previously, we reported that the serum CCL2 level in patients with prostate cancer is clearly predictive of the time interval for the tumor to become castration‐resistant as well as the overall and prostate cancer‐specific survival rates.23 C‐C chemokine ligand 2 is a secreted protein and can be measured easily in a patient's serum; CCL21 is also a secreted chemokine and is abundantly expressed on fibroblastic reticular cells in lymph node metastasis. However, additional clinical evidence is necessary to establish the superiority of the CCL21/CCR7 axis over the CCL2/CCR2 axis as a prostate cancer biomarker.
In conclusion, our data suggest that TNF‐α acts as a regulator of increased tumor cell migration through the upregulation of CCR7. Targeting TNF‐α and ultimately the CCL21/CCR7 axis is a novel therapeutic strategy for patients with prostate cancer.
CONFLICT OF INTEREST
The authors have no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by the Japan Society for the Promotion of Science (KAKENHI Grant No. 16K10998 to K. Izumi).
Maolake A, Izumi K, Natsagdorj A, et al. Tumor necrosis factor‐α induces prostate cancer cell migration in lymphatic metastasis through CCR7 upregulation. Cancer Sci. 2018;109:1524–1531. https://doi.org/10.1111/cas.13586
Funding information
Japan Society for the Promotion of Science.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen‐independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209‐9216. [DOI] [PubMed] [Google Scholar]
- 3. Cai T, Nesi G, Tinacci G, et al. Clinical importance of lymph node density in predicting outcome of prostate cancer patients. J Surg Res. 2011;167:267‐272. [DOI] [PubMed] [Google Scholar]
- 4. Fleischmann A, Schobinger S, Schumacher M, Thalmann GN, Studer UE. Survival in surgically treated, nodal positive prostate cancer patients is predicted by histopathological characteristics of the primary tumor and its lymph node metastases. Prostate. 2009;69:352‐362. [DOI] [PubMed] [Google Scholar]
- 5. Kadono Y, Nohara T, Ueno S, et al. Validation of TNM classification for metastatic prostatic cancer treated using primary androgen deprivation therapy. World J Urol. 2016;34:261‐267. [DOI] [PubMed] [Google Scholar]
- 6. Gandaglia G, Karakiewicz PI, Briganti A, et al. Impact of the site of metastases on survival in patients with metastatic prostate cancer. Eur Urol. 2015;68:325‐334. [DOI] [PubMed] [Google Scholar]
- 7. Goillot E, Combaret V, Ladenstein R, et al. Tumor necrosis factor as an autocrine growth factor for neuroblastoma. Cancer Res. 1992;52:3194‐3200. [PubMed] [Google Scholar]
- 8. Liu RY, Fan C, Mitchell S, Chen Q, Wu J, Zuckerman KS. The role of type I and type II tumor necrosis factor (TNF) receptors in the ability of TNF‐alpha to transduce a proliferative signal in the human megakaryoblastic leukemic cell line Mo7e. Cancer Res. 1998;58:2217‐2223. [PubMed] [Google Scholar]
- 9. Cassier PA, Treilleux I, Bachelot T, et al. Prognostic value of the expression of C‐Chemokine Receptor 6 and 7 and their ligands in non‐metastatic breast cancer. BMC Cancer. 2011;11:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cunningham HD, Shannon LA, Calloway PA, et al. Expression of the C‐C chemokine receptor 7 mediates metastasis of breast cancer to the lymph nodes in mice. Transl Oncol. 2010;3:354‐361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Du P, Liu Y, Ren H, et al. Expression of chemokine receptor CCR7 is a negative prognostic factor for patients with gastric cancer: a meta‐analysis. Gastric Cancer. 2017;20:235‐245. [DOI] [PubMed] [Google Scholar]
- 12. Hong H, Jiang L, Lin Y, et al. TNF‐alpha promotes lymphangiogenesis and lymphatic metastasis of gallbladder cancer through the ERK1/2/AP‐1/VEGF‐D pathway. BMC Cancer. 2016;16:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hong H, He C, Zhu S, et al. CCR7 mediates the TNF‐alpha‐induced lymphatic metastasis of gallbladder cancer through the “ERK1/2 ‐ AP‐1” and “JNK ‐ AP‐1” pathways. J Exp Clin Cancer Res. 2016;35:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Comerford I, Harata‐Lee Y, Bunting MD, Gregor C, Kara EE, McColl SR. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 2013;24:269‐283. [DOI] [PubMed] [Google Scholar]
- 15. Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50‐56. [DOI] [PubMed] [Google Scholar]
- 16. Kato M, Kitayama J, Kazama S, Nagawa H. Expression pattern of CXC chemokine receptor‐4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res. 2003;5:R144‐R150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Izumi K, Fang LY, Mizokami A, et al. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2‐induced STAT3 activation. EMBO Mol Med. 2013;5:1383‐1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weitzenfeld P, Kossover O, Korner C, et al. Chemokine axes in breast cancer: factors of the tumor microenvironment reshape the CCR7‐driven metastatic spread of luminal‐A breast tumors. J Leukoc Biol. 2016;99:1009‐1025. [DOI] [PubMed] [Google Scholar]
- 19. Maolake A, Izumi K, Shigehara K, et al. Tumor‐associated macrophages promote prostate cancer migration through activation of the CCL22‐CCR4 axis. Oncotarget. 2017;8:9739‐9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin TH, Izumi K, Lee SO, Lin WJ, Yeh S, Chang C. Anti‐androgen receptor ASC‐J9 versus anti‐androgens MDV3100 (Enzalutamide) or Casodex (Bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3‐CCL2 signaling. Cell Death Dis. 2013;4:e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Izumi K, Lin WJ, Miyamoto H, et al. Outcomes and predictive factors of prostate cancer patients with extremely high prostate‐specific antigen level. J Cancer Res Clin Oncol. 2014;140:1413‐1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Izumi K, Ikeda H, Maolake A, et al. The relationship between prostate‐specific antigen and TNM classification or Gleason score in prostate cancer patients with low prostate‐specific antigen levels. Prostate. 2015;75:1034‐1042. [DOI] [PubMed] [Google Scholar]
- 23. Izumi K, Mizokami A, Lin HP, et al. Serum chemokine (CC motif) ligand 2 level as a diagnostic, predictive, and prognostic biomarker for prostate cancer. Oncotarget. 2016;7:8389‐8398. [DOI] [PMC free article] [PubMed] [Google Scholar]