

ABSTRACT
A systematic search for anticancer agents that may induce the release of high mobility group box 1 (HMGB1) protein from cells into the extracellular space has led to the identification of several drugs capable of elevating plasma HMGB1 levels in vivo, in mice. Such agents include bona-fide immunogenic cell death inducers such as oxaliplatin, as well as a series of epigenetic modifiers, namely azacitidine, decitabine, and suberoylanilide hydroxamic acid (SAHA).
KEYWORDS: biotin, cancer, chemotherapy, immunosurveillance, RUSH
It is nowadays commonly acknowledged that chemotherapeutic agents, which allow for tumor control beyond treatment discontinuation achieve such long-term effects through the induction of anticancer immune responses.1 One mechanism through which such immunostimulatory effects are achieved consists in the induction of immunogenic cell death (ICD), meaning that the cytotoxic drug kills cancer cells in a way that they become recognizable to the immune system.2 The immunogenicity of cancer cells results from a combination of antigenicity (i.e. the presence of antigenic proteins/peptides in malignant cells that are absent in normal circumstances) and adjuvanticity (i.e. the presence of danger-associated molecular patterns acting on pattern recognition receptors of the innate immune system). ICD is mostly coupled to an increase in adjuvanticity.2,3 One of the most important adjuvant signals that is emitted by cancer cells succumbing to ICD inducers is the release of high mobility group box1 (HMGB1). HMGB1 is the most abundant non-histone chromatin binding protein and hence is usually present in the nucleus. However, in response to ICD inducers, HMGB1 can leave the nucleus to relocalize in the cytoplasm, presumably as a result of changes in its charge (e.g. due to deacetylation of the protein) and loss of the integrity of the nuclear envelope.2,4 Upon plasma membrane permeabilization, which marks the final step of the apoptotic or necrotic process, HMGB1 then is released from the cells into the extracellular fluid where it can interact with a number of additional soluble molecules (such as RNA, DNA, bacterial lipopolysaccharide etc.) as well as with receptors (such as Toll-like receptor 4, TLR4, and advanced glycosylation end-product specific receptor, AGER).4 By virtue of the activation of TLR4, HMGB1 then stimulates the maturation of dendritic cells increasing their capacity to cross-present tumor-associated antigens to cytotoxic T cells.2 Importantly, loss of HMGB1 expression by cancer cells or loss-of-function mutations of TLR4 both have a negative impact on breast cancer prognosis,5 underscoring the functional importance of the interaction between HMGB1 and TLR4.
Based on these considerations, we decided to design a screen that would allow to accurately determine the nuclear release of HMGB1 induced by anticancer agents.6 For this, we used the “retention using selective hooks” (RUSH) system, in which a streptavidin-binding peptide (SBP) fused with HMGB1 and green fluorescent protein (GFP) was sequestered by streptavidin-NLS3 fusion protein in the nucleus. In cells expressing both the HMGB1-SBP-GFP fusion protein and the streptavidin-NLS3 hook, the GFP-dependent fluorescent signal is strictly confined to the nucleus, in punctiform structures. Upon addition of biotin, which competitively disrupts the interaction between HMGB1-SBP-GFP and streptavidin-NLS3, HMGB1-SBP-GFP remains in the nucleus, yet changes from a punctiform to a diffuse distribution. However, it is only after addition of ICD inducers such as anthracyclines, digoxigen, doxorubicin and mitoxantrone that HMGB1-SBP-GFP redistributed from the nucleus to the cytoplasm. (Fig. 1)6 This system then was used to identify HGMB1 releasing agents among approximately 2000 drugs and drug candidates. In the top list of agents causing nuclear HMGB1 release, we found several epigenetic modifiers (azacitidine, decitabine and suberoylanilide hydroxamic acid, SAHA), microtubule inhibitors (docetaxel, paclitaxel and nocodazole) as well as several anthelmintic agents (albendazole, fenbendazole, flubendazole, mebendazole, oxibendazole) that all are known to inhibit microtubule formation.6 Importantly, we could subsequently validate that intraperitoneal injection of azacitidine, decitabine and SAHA as well as that of anthelmintics induced the appearance of circulating HMGB1 in the plasma from mice. Thus, pharmacologically meaningful concentrations of these drugs stimulate the cellular release of HMGB1.
Figure 1.
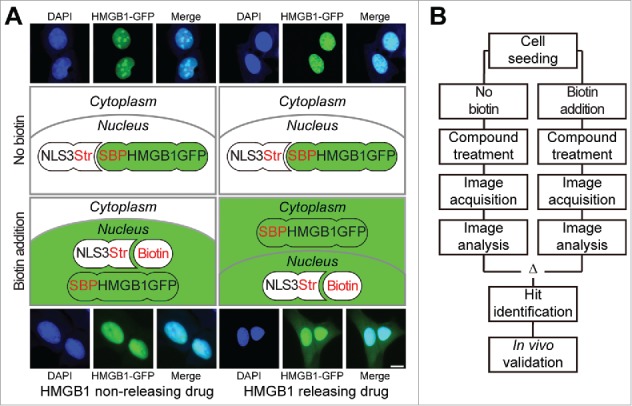
Workflow of the screening procedure leading to the identification of epigenetic modifiers as HMGB1 releasing agents. Cells stably expressing the two components of the HMGB1-Retention Using Selective Hooks (RUSH) assay were used for chemical drug screening. (A) Through the interaction between streptavidin (Str) in the Str-NLS3 hook with the streptavidin binding protein (SBP) in the HMGB1-SBP-GFP reporter the latter is retained in the nucleus. Exclusively in the presence of biotin, that competes with the Str-SBP mediated retention, the reporter diffuses in the nucleus and translocates to the cytoplasm in response to the treatment with HMGB1 releasing agents, such as oxaliplatin, and epigenetic modifiers such as azacitidine, decitabine and suberoylanilide hydroxamic acid (SAHA). Representative images show control and SAHA-treated cells in the presence and absence of biotin. The sizebar equals 10 μm. (B) A schematic workflow is depicted entailing the initial high content compound screen, the data acquisition and analysis, the hit identification and in vivo validation steps.
We were particularly intrigued by the capacity of epigenetic drugs to stimulate HMGB1 release. The epigenetic modifiers identified in this screen may induce the release of HMGB1 through on-target effects. Indeed, SAHA caused histone hyperacetylation, which may favor HMGB release. Moreover, the knockdown of the two DNA methyl transferases DNMT3a and DNMT3b, which are targets of azacitidine and decitabine induced the nucleo-cytoplasmic translocation of GFP-HMGB1.
Epigenetic modifiers including histone deacetylase inhibitors may have some immuno-oncologically relevant effect. Thus, SAHA induces calreticulin exposure, one of the hallmarks of ICD in brain tumor cells7 and stimulates anticancer immune responses in rodent models8,9 Moreover, hypomethylating agents such as azacitidine and decitabine have broad immunomodulatory effects.10 It will be important to understand to which extent the immunostimulatory effects of such drugs may be attributed to the release of HMGB1. Moreover, it will be interesting to explore the capacity of anthelmintics to boost anticancer immunity in experimental and clinical settings.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
PL is supported by the Chinese Scholarship Council (CSC). LZ received a fellowship from la Ligue contre le cancer and was supported by the Taxe d'apprentisage grant 2017 from the Gustave Roussy Cancer Center. GK is supported by the Ligue contre le Cancer Comité de Charente-Maritime (équipe labelisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; the European Commission (ArtForce); the European Research Council (ERC); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
References
- 1.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer cell. 2015;28:690–714. doi: 10.1016/j.ccell.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nature reviews Immunology. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 3.Bloy N, Garcia P, Laumont CM, Pitt JM, Sistigu A, Stoll G, Yamazaki T, Bonneil E, Buqué A, Humeau J, et al. Immunogenic stress and death of cancer cells: Contribution of antigenicity vs adjuvanticity to immunosurveillance. Immunol Rev. 2017;280:165–74. doi: 10.1111/imr.12582. [DOI] [PubMed] [Google Scholar]
- 4.Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol Res. 2016;111:534–44. doi: 10.1016/j.phrs.2016.06.031. [DOI] [PubMed] [Google Scholar]
- 5.Kroemer G, Senovilla L, Galluzzi L, Andre F, Zitvogel L. Natural and therapy-induced immunosurveillance in breast cancer. Nat Med. 2015;21:1128–38. doi: 10.1038/nm.3944. [DOI] [PubMed] [Google Scholar]
- 6.Liu P, Zhao L, Loos F, Iribarren K, Lachkar S, Zhou H, Gomes-da-Silva LC, Chen G, Bezu L, Boncompain G, et al. Identification of pharmacological agents that induce HMGB1 release. Sci Rep. 2017;7:14915. doi: 10.1038/s41598-017-14848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sonnemann J, Gressmann S, Becker S, Wittig S, Schmudde M, Beck JF. The histone deacetylase inhibitor vorinostat induces calreticulin exposure in childhood brain tumour cells in vitro. Cancer Chemother Pharmacol. 2010;66:611–6. doi: 10.1007/s00280-010-1302-4. [DOI] [PubMed] [Google Scholar]
- 8.Christiansen AJ, West A, Banks KM, Haynes NM, Teng MW, Smyth MJ, Johnstone RW. Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proc Natl Acad Sci U S A. 2011;108:4141–6. doi: 10.1073/pnas.1011037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, Johnstone RW. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer research 2013;73:7265–76. doi: 10.1158/0008-5472.CAN-13-0890. [DOI] [PubMed] [Google Scholar]
- 10.Lindblad KE, Goswami M, Hourigan CS, Oetjen KA. Immunological effects of hypomethylating agents. Expert Rev Hematol. 2017;10:745–52. doi: 10.1080/17474086.2017.1346470. [DOI] [PMC free article] [PubMed] [Google Scholar]