

Abstract
Hematopoietic stem cells (HSCs) are the self‐renewing multipotent progenitors to all blood cell types. Identification and isolation of HSCs for study has depended on the expression of combinations of surface markers on HSCs that reliably distinguish them from other cell types. However, the increasing number of markers required to isolate HSCs has made it tedious, expensive, and difficult for newcomers, suggesting the need for a simpler panel of HSC markers. We previously showed that phenotypic HSCs could be separated based on expression of CD11a and that only the CD11a negative fraction contained true HSCs. Here, we show that CD11a and another HSC marker, endothelial protein C receptor (EPCR), can be used to effectively identify and purify HSCs. We introduce a new two‐color HSC sorting method that can highly enrich for HSCs with efficiencies comparable to the gold standard combination of CD150 and CD48. Our results demonstrate that adding CD11a and EPCR to the HSC biologist's toolkit improves the purity of and simplifies isolation of HSCs. stem cells translational medicine 2018;7:468–476
Keywords: Hematopoietic stem cells, Bone marrow transplantation, Cell surface markers, long‐term repopulation
Significance Statement.
The study of hematopoietic stem cells (HSCs) and their purification for transplantation requires a panel of surface markers that can be used to distinguish HSCs from other cell types. The number of markers necessary to identify HSCs continues to grow, making it increasingly difficult to identify HSCs by flow cytometry. In this study, the authors identified a combination of two surface markers, CD11a and endothelial protein C receptor, to enrich for HSCs in the mouse bone marrow without the need for additional markers. This simplified panel could aid HSC research by reducing the number of markers necessary to identify and isolate HSCs.
Introduction
Hematopoietic stem cells (HSCs) are the self‐renewing, multipotent, and engraftable cells of the blood system 1. Successful HSC transplantation (HSCT) can potentially treat any disorder inherent to the hematopoietic system by ablation of the defective blood system followed by reconstitution by healthy donor HSCs 2. However, HSCT is reserved only for high‐risk patients due to the dangers of HSCT‐related complications, including graft rejection, graft failure, and Graft‐versus‐Host Disease 2. Transplantation of sufficient numbers of pure HSCs can bypass many of these HSCT‐related complications 3, 4, 5. Therefore, much effort has been invested in strategies to improve the purity of donor HSCs.
HSCs are identified by their expression of a combination of molecules on their cell surface called surface markers. In mice, the ever‐growing list of surface markers whose positive or negative expression marks HSCs includes CD34, Kit, Sca‐1, Lineage (a cocktail of markers of mature lineages), CD27, CD48, CD150, FLK2, CD9, EPCR, and many others 6, 7, 8, 9. The marker combination Kit+ Lineage– Sca‐1+, which defines the “KLS” population (also called “LSK” or “KSL”), contains all HSCs and multipotent progenitors in the bone marrow (BM). To isolate long‐term HSCs within the KLS population, additional marker combinations are needed such as (i) Flk2+ CD34–, (ii) CD48– CD150+, or (iii) CD150+ CD34– 6, 10, 11. However, the increasing number of markers needed to purify HSCs (currently around 6–8), the nuances of each of the fluorochromes and antibodies required for optimal staining and gating, and the long and expensive assays needed for gating validation have made it difficult for newcomers to properly identify and sort HSCs. Furthermore, many of these markers can change expression during stressful conditions such as upon inflammation or after irradiation, making many of them unreliable for identifying HSCs in these contexts 12, 13, 14. Therefore, there remains a need for simpler and more inclusive strategies for marking and identifying HSCs in healthy and challenged BM.
We previously introduced CD11a as a new marker to isolate HSCs. CD11a (integrin alpha L, or Itgal) heterodimerizes with the β2‐integrin CD18 to form LFA1. LFA1 interacts with ICAM‐1 and has roles in transendothelial migration, activation, and differentiation of lymphocytes 15, 16, 17, 18. We found that while CD11a is expressed on nearly all hematopoietic lineages, it is downregulated on HSCs 19. We showed that stringently‐gated adult HSCs can be separated into CD11a+ and CD11a– fractions, with only the CD11a– fraction showing long‐term engraftment upon transplantation. This was not due to antibody binding to the CD11a+ cells (potentially blocking LFA1‐mediated migration), as the CD11a antibody itself had no effect on either BM homing or long‐term engraftment of HSCs. These findings suggested that CD11a should be added to the marker panel when isolating HSCs at the highest level of purity. Here, we introduce an alternative strategy for identification and sorting of HSCs with the use of CD11a and EPCR (endothelial protein C receptor, Procr, CD201) as another efficient HSC marker, and compare this strategy to those using classical markers. We show that CD11a and EPCR can be used with classical HSC markers to purify HSCs, but furthermore, can be used alone as a simple two‐color method to highly enrich for HSCs.
Materials and Methods
Mice
C57Bl/6 (stock no. 00664) and mT/mG (stock no. 007576 20) strains from Jackson Laboratory (Bar Harbor, ME) were used as donors/recipients/helpers. CFP mice (Rosa‐ECFP aka TM5) mice were generously donated by Dr. Irving Weissman 21. All strains were maintained at the Gross Hall and Med Sci A vivarium facilities at UCI and fed with standard chow and water. All animal procedures were approved by the International Animal Care and Use Committee (IACUC) and University Laboratory Animal Resources (ULAR) of University of California, Irvine.
Antibodies
For list of antibodies, refer to Table S1 (“Antibodies Table”) in Supporting Information.
Cell Sorting
For flow cytometry, BM was harvested from tibias and femurs by flushing with ice‐cold fluorescence activated cell sorting (FACS) buffer (phosphate buffered saline (PBS) + 2% fetal bovine serum) followed by red blood cell lysis by ACK lysis buffer and filtration through a 70 μ mesh. BM was harvested from donor mice by crushing leg bones in ice‐cold FACS buffer followed by red blood cell lysis by ACK lysis buffer and filtration through a 70 μ mesh to remove debris. Where indicated, BM was Kit enriched using anti‐Kit (anti‐CD117) microbeads on an AutoMACS (Miltenyi Biotec, Somerville, MA). Cells were stained with antibodies listed in Supporting Information Table S1 (“Antibodies Table”) in FACS buffer. Cells were sorted on a BD FACS‐Aria II (Becton Dickinson, Franklin Lakes, NJ) into ice‐cold FACS buffer for transplantation.
Transplantation, and Blood and BM Analysis
Defined numbers of HSCs (as indicated in each experiment) were transplanted by retro‐orbital injection into lethally‐irradiated isoflurane‐anesthetized recipients alongside helper BM from congenically distinguishable C57BL/6 mice. Lethal doses of x‐ray irradiation were 800 Rads for single dose, or 950 Rads split dose (XRAD 320, Precision X‐ray, North Branford, CT). Transplanted recipients were fed an antibiotic chow of Trimethoprim Sulfa (Uniprim, Envigo, East Millstone, NJ) for 4 weeks post transplantation to prevent potential bacterial infections. For peripheral blood analysis, blood was obtained from the tail vein of transplanted mice at various time points, and red blood cells were depleted using ACK lysis buffer. For BM analysis, BM was harvested from tibias and femurs by flushing with ice‐cold FACS buffer followed by ACK lysis and filtration. Cells were stained with lineage antibodies and analyzed on the BD FACS‐Aria II. For a comprehensive list of markers used for identification of each population, refer to Table S2 (“Marker definitions of populations analyzed”) in Supporting Information. FlowJo software (Tree Star) was used for data analysis.
LPS‐, Poly(I:C)‐, and Irradiation‐Induced BM Injury
For LPS and poly(I:C) treatments, 10‐week‐old C57BL/6 mice were injected intraperitoneally (i.p.) with 2 mg/kg of LPS (lipopolysaccharides from Escherichia coli 0111:B4; Sigma‐Aldrich, St. Louis, MO, catalog no. L4391) or 5 mg/g of HMW pol(I:C) (InvivoGen, San Diego, CA; catalog no. 31852‐29‐6). Injected mice were sacrificed after 24 hours and bone marrow was analyzed by flow cytometry. For irradiation‐induced BM stress, 10‐week‐old C57BL/6 mice were sublethally irradiated with 6 Gy. BM analysis was performed 48 hours post irradiation.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism 5 software (La Jolla, CA).
Results
CD11a and EPCR in Combination with Classical HSC Markers Reveal a Distinct Population with Enriched HSC Activity
CD11a and EPCR have each been shown independently to increase HSC purity when used with conventional HSC markers 19, 22, 23. To assess the efficiency of purifying HSCs using CD11a and EPCR together, we first examined their expression in the KLS population, which contains all hematopoietic stem and multipotent progenitor cells and is often referred to as “HSPCs” (Fig. 1). KLS is traditionally defined as Kit+ Lin– Sca‐1+, but we substituted CD27 for the Lineage (Lin) cocktail, an expensive combination of markers (e.g., CD3, CD4, CD8, B220, Mac‐1, Gr1, Ter119, NK1.1, etc.) for mature hematopoietic lineages. CD27 is expressed on HSCs and MPPs, and together with the red blood cell marker Ter119, can be used in place of Lin 14, 24, 25. Because this population (CD27+ Ter119– Kit+ Sca‐1+) is identical to the original KLS population (Lin‐ Kit+ Sca‐1+), we keep the nickname “KLS” for simplicity. Within the KLS population, we identified two distinct fractions: a CD11a– EPCR+ population and a CD11a+ population (Fig. 1A). While the CD11a+ fraction could be further subdivided into EPCR+ and EPCR– fractions, we pooled all CD11a+ cells together because our previous work showed that there were few, if any, HSCs in the CD11a+ fraction 19. We sorted these two populations (from CFP+ donor mice) and transplanted them into lethally irradiated B6 adult recipients to determine which population contained long‐term engraftable HSCs. We transplanted roughly 1,500 CD11a– EPCR+ KLS cells and 10,000 CD11a+ KLS cells to maintain their physiological ratios. Five hundred thousand BM cells from Tomato+ mice were co‐transplanted as “helper” BM to protect the recipients from hematopoietic failure following irradiation. Recipients were bled and analyzed for donor chimerism in different blood lineages at 4‐week intervals (Supporting Information Fig. S1). Donor chimerism of total blood cells (CD45+) was significantly higher from the CD11a– EPCR+ KLS population than the CD11a+ KLS population, and this difference increased over time (Fig. 1B). Because granulocytes are short‐lived, granulocyte chimerism in the peripheral blood is a more accurate indicator of HSC chimerism in the BM compared with total CD45+ blood cells, which includes long‐lived lymphocytes that may have come from lymphoid progenitors or multipotent progenitors. The difference in granulocyte chimerism between CD11a– EPCR+ KLS cells and CD11a+ KLS was even more pronounced than total blood chimerism (Fig. 1C). Furthermore, when examining the BM of the recipients, the CD11a–EPCR+ KLS population had higher donor HSPCs (Fig. 1D). As only HSCs are capable of serial transplantation, we next transplanted whole BM from the primary recipients into secondary hosts. Only the BM of recipients of CD11a– EPCR+ KLS cells gave rise to robust donor chimerism in the secondary hosts, indicating that nearly all HSCs are contained in this population (Fig. 1E). Thus, CD11a and EPCR can be used to isolate HSCs within the KLS fraction of BM.
Figure 1.
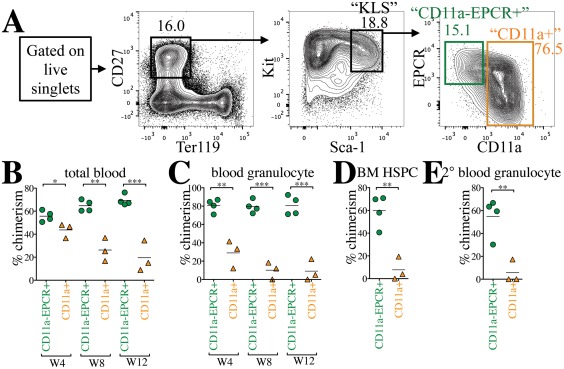
CD11a and EPCR inclusion enriches for HSCs within KLS population. (A): Representative sorting scheme of CD11a– EPCR+ KLS (green) and CD11a+ KLS (orange) populations from Kit‐enriched CFP+ BM. Each sorted population (1,500 CD11a– EPCR+ cells or 10,000 CD11a+ cells per recipient) was transplanted into lethally irradiated B6 recipients along with 500,000 Tomato+ WBM helper cells. (B, C): Time‐course analysis of donor chimerism in blood. Total (B) and granulocyte (C) blood chimerism from CD11a– EPCR+ KLS (“CD11a‐EPCR+”) and CD11a+ KLS (“CD11a+”) sources in primary recipients at weeks (W) 4, 8, and 12 post‐transplant. Total blood was defined as CD45+ and granulocytes as CD45+ Gr1+ Mac‐1+. (D): Donor chimerism of HSPCs in the BM of primary recipients 13 weeks post‐transplant. HSPCs are defined as Ter119– CD27+ Sca‐1+ Kit+. (E): Blood granulocyte chimerism in secondary recipients 6 weeks post‐secondary transplant. Secondary transplants were done using 1 × 106 WBM harvested from primary recipients that received “CD11a‐EPCR+” or “CD11a+” donor cells. *, p ≤ .05; **, p ≤ .01; ***, p ≤ .001 (Student's unpaired t test). Abbreviations: BM, bone marrow, EPCR, endothelial protein C receptor; HSPC, hematopoietic stem and progenitor cells.
CD11a– EPCR+ KLS Directly Outcompetes CD11a+ KLS in a Competitive Transplantation Assay
To directly compare HSC activity between the CD11a– EPCR+ and CD11a+ subsets of KLS cells, we performed a competitive transplantation, in which both populations are co‐transplanted into the same recipients. In this strategy, recipient mice receive both populations at their physiological ratios, providing a direct comparison of the engraftment efficiency of each. Also, because all KLS cells fall within one fraction or the other, all potential sources of HSCs in the BM are sorted and transplanted. To distinguish the two populations, we sorted one population from CFP‐expressing BM, and the other population from Tomato‐expressing BM and co‐transplanted them along with 100,000 unlabeled B6 helper BM cells (Fig. 2A). In the peripheral blood of the primary recipients, drastically higher percentages of donor total CD45+ cells and granulocytes were derived from the CD11a– EPCR+ KLS compared with the CD11a+ KLS source (Fig. 2B, 2C). Higher donor chimerism of HSCs in the BM compartment of primary recipients as well as blood granulocyte chimerism in secondary recipients also originated only from the CD11a– EPCR+ KLS donor source, indicating that this population contained all the HSCs (Fig. 2D, 2E). The mT/mG strain used for Tomato+ donor cells was originally on a mixed background, and therefore could lead to engraftment differences. However, in a competitive setting we observed no differences in engraftment based on the strain of the animal, only the population transplanted.
Figure 2.
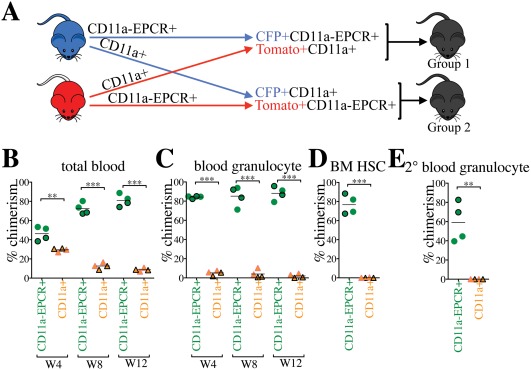
CD11a‐EPCR+ KLS cells outcompete CD11a+ counterparts in competitive transplants. (A): Representation of competitive transplant system. Group 1 recipients (outlined symbols in B–E) received CFP+ CD11a– EPCR+ KLS and Tomato+ CD11a+ KLS, and Group 2 (borderless symbols in B–E) received Tomato+ CD11a– EPCR+ KLS and CFP+ CD11a+ KLS. 3,000 CD11a– EPCR+ KLS and 10,000 CD11a+ KLS sorted cells (physiological ratios) along with 100,000 helper WBM (from Wt B6 mice) were co‐transplanted into each lethally irradiated B6 recipient. (B, C): Time‐course analysis of donor chimerism in blood. Total (B) and granulocyte (C) blood chimerism from CD11a– EPCR+ KLS (“CD11a– EPCR+”) and CD11a+ KLS (“CD11a+”) sources in primary recipients at weeks (W) 4, 8, and 12 post‐transplant. (D): Donor chimerism of HSCs in the BM of primary recipients 13 weeks post‐transplant. HSCs are defined as Ter119– CD27+ Sca‐1+ Kit+ CD11a– EPCR+. (E): Blood granulocyte chimerism in secondary recipients 6 weeks post‐secondary transplant. Secondary transplants were done using 1 × 106 WBM harvested from primary recipients 13 weeks after the primary transplantation. **, p ≤ .01; ***, p ≤ .001 (Student's unpaired t test). Abbreviations: BM, bone marrow, HSCs, hematopoietic stem cells.
We also examined the distribution of lineages derived from the two populations (Supporting Information Fig. S2). CD11a+ KLS‐derived cells showed a significantly higher production of B cells than other lineages when compared with CD11a– EPCR+ KLS and nontransplanted controls. While this may suggest that CD11a+ KLS cells have a lymphoid bias, it is more likely a byproduct of the fact that lymphocytes live longer than myeloid cells. This population likely contains short‐lived multipotent progenitors, which give rise to a brief outburst of myeloid and lymphoid populations. While the myeloid populations are quickly expended, the lymphocytes remain, producing the appearance of a lymphoid bias. The CD11a– EPCR+ KLS cells produced a much more balanced lineage distribution, further supporting the notion that all HSCs reside within this population.
CD11a and EPCR Alone Can Enrich HSCs Without the Need for Other HSC Markers
We next tested whether CD11a and EPCR alone were sufficient to sort HSCs in the absence of all other HSC markers. We used only these two markers for a competitive transplantation assay, and did not include any other HSC markers. We sorted CD11a– EPCR+ (“11a/EPCR”) cells into one tube, and all other live cells (referred to as “Not 11a/EPCR”) into another tube, then co‐transplanted one population from CFP+ BM and the other population from Tomato+ BM into the same recipient mice. By sorting all cells outside of the CD11a– EPCR+ gate, we could ensure that any potential HSCs that fall outside of the CD11a– EPCR+ population would be transplanted in the “Not 11a/EPCR” fraction. Because of the rarity of the CD11a– EPCR+ fraction (∼0.17% of whole BM; WBM) compared with the “Not 11a/EPCR” fraction (∼99.1% of WBM), we sorted and transplanted them in such numbers as to maintain their physiological ratios. For each transplant, we sorted 500,000 total BM cells into CD11a– EPCR+ fraction and “Not 11a/EPCR” fraction. We then mixed the CD11a– EPCR+ fraction from one reporter (e.g., Tomato+) and the “Not 11a/EPCR” fraction from the other reporter (e.g., CFP) and co‐transplanted them into the same recipient (Fig. 3A). Thus, the transplanted cells are the equivalent of 500,000 WBM cells, with the CD11a– EPCR+ cells distinguishable from the rest of the BM cells by CFP or Tomato expression. In the recipient mice, we found that only the CD11a– EPCR+ donor source showed donor chimerism in primary and secondary recipients (Fig. 3B, 3C). We also examined the BM and found that donor HSPCs were only derived from the CD11a– EPCR+ source (Fig. 3D). These results indicate that all HSCs are present in the rare CD11a– EPCR+ fraction of BM, and that CD11a and EPCR together are sufficient to sort an enriched HSC population.
Figure 3.
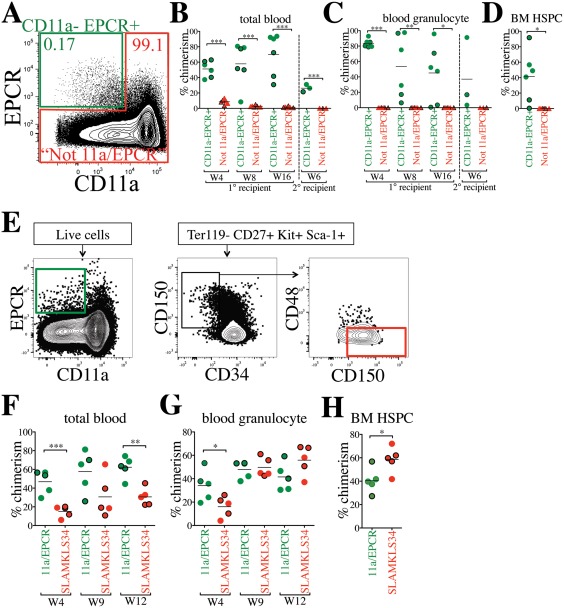
CD11a and EPCR alone are sufficient to sort a rare population enriched for HSCs. (A): Sorting strategy using only CD11a and EPCR as HSC markers. CFP+ CD11a– EPCR+ and Tomato+ “Not 11a/EPCR” (not CD11a– EPCR+) cells (and vice versa) were sorted and co‐transplanted in a competitive setting at physiological ratios. 850 CD11a– EPCR+ and 500,000 Not 11a/EPCR were transplanted into each recipient. Percentages of cells within each gate are shown. (B, C): Time‐course analysis of total blood (B) and blood granulocyte (C) chimerism from CD11a– EPCR+ and Not 11a/EPCR sources in primary recipients 4, 8, and 16 weeks (W) post‐transplant, and in secondary recipients (separated by vertical dashed line) at W6 following secondary transplant. CFP+ donor‐derived cells are represented by outlined symbols and Tomato+ donor‐derived cells with borderless symbols. Not all primary recipients were selected for secondary transplantation. (D): Donor chimerism of HSPCs in the BM of primary recipients transplanted with “CD11a– EPCR+” and “Not 11a/EPCR” sorted cells 17 weeks post‐transplant. HSPCs are defined as Ter119– CD27+ Sca‐1+ Kit+. CFP+ donor‐derived cells are represented by outlined symbols and Tomato+ donor‐derived cells with borderless symbols. (E): Sorting strategy to compare CD11a– EPCR+ (11a/EPCR) to Ter119– CD27+ Kit+ Sca‐1+ CD34– CD150+ CD48– (SLAMKLS34). 680 CFP‐expressing 11a/EPCR and 60 Tomato‐expressing SLAMKLS34 (and vice versa) were sorted and co‐transplanted in a competitive setting. 250,000 nonlabeled WBM was used as helper for each recipient. (F–H): Time‐course analysis of total blood (F) and blood granulocyte (G) chimerism from 11a/EPCR and SLAMKLS34 sources in primary recipients 4, 9, and 12 weeks (W) post‐transplant and HSPC chimerism (H) at W13. CFP+ donor‐derived cells are represented by outlined symbols and Tomato+ donor‐derived cells with borderless symbols. *, p ≤ .05; **, p ≤ .01; ***, p ≤ .001 (Student's unpaired t test). “Not 11a/EPCR” = not CD11a‐EPCR+. Abbreviations: BM, bone marrow, EPCR, endothelial protein C receptor; HSPC, hematopoietic stem and progenitor cells.
Because we found all HSCs were contained within the CD11a– EPCR+ (11a/EPCR) population in a two‐color sorting method (Fig. 3A–3D), we next investigated the efficiency of this method in comparison to an HSC population sorted with an extremely stringent method. Cells were sorted from either (a) 11a/EPCR two‐color method (CD11a– EPCR+) or (b) the “SLAMKLS34” population (defined as Ter119– CD27+ Kit+ Sca‐1+ CD34– CD150+ CD48–) (Fig. 3E). We sorted these populations at physiological ratios from a total of 500,000 WBM, and transplanted the sorted populations in a competitive setting and along with helper BM cells. Although we detected a significantly higher total blood chimerism from the 11a/EPCR source, levels of blood granulocyte chimerism were comparable after 3 months between the two methods (Fig. 3F, 3G). Higher 11a/EPCR‐derived total blood chimerism highlights the higher fraction of non‐HSC progenitors (e.g., MPP, CLP) when only these two markers are used. In the BM, we found slightly higher HSPC chimerism from the SLAMKLS34 population, though an average of ∼40% of the HSPCs were derived from 11a/EPCR (Fig. 3H). These data further demonstrate that all HSCs can be sorted using the 11a/EPCR method, although HSC purity in this population is lower than using a more stringent multi‐color approach.
“11a/EPCR” Two‐Color Sorting Method Produces Similar Purity of HSCs as the “SLAM” Method
While all HSCs are contained within the CD11a– EPCR+ fraction, it does not mean the population contains only HSCs. When examining BM cells gated on CD11a– EPCR+, approximately 81% are Kit+ and Sca‐1+, but only 12% are CD150+ CD48– (Supporting Information Fig. S3A). It was previously shown that HSCs are CD150+ CD48–, and thus there are likely non‐HSCs within the CD11a– EPCR+ fraction. The SLAM markers CD150 and CD48 have also been shown to be sufficient for two‐color sorting of HSCs 6. However, when examining the SLAM fraction (CD150+ CD48–) of BM, only 11% of these cells were Kit+ Sca‐1+, and only 7.2% were CD11a– EPCR+, suggesting this two‐color method may also be contaminated with non‐HSCs. To confirm the efficiency of the SLAM method to purify HSCs in our hands, we transplanted CD150+ CD48– (SLAM) and “Not” CD150+ CD48– (referred to as “Not SLAM”) in a competitive setting, and found that only the SLAM cells were able to engraft long‐term (Supporting Information Fig. S3B–S3D).
To directly compare our “11a/EPCR” two‐color sorting method with the SLAM method, CD11a– EPCR+ (11a/EPCR) and CD150+ CD48– (SLAM) cells were sorted, mixed, and co‐transplanted into recipients in a competitive setting (Fig. 4A, Group 1 recipients). Equal numbers of each population (380 cells) were transplanted, allowing us to directly compare which population contained the most HSCs. We did not detect any statistically significant difference in granulocyte chimerism between 11a/EPCR and SLAM populations in primary and secondary recipients (Fig. 4Bi). Analysis of HSPCs in the BM also confirmed comparable engraftability between the two populations (Fig. 4Bii). Total blood chimerism and lineage distribution were also not significantly different between the two sorting methods in primary and secondary recipients (Supporting Information Fig. S4A, S4B).
Figure 4.
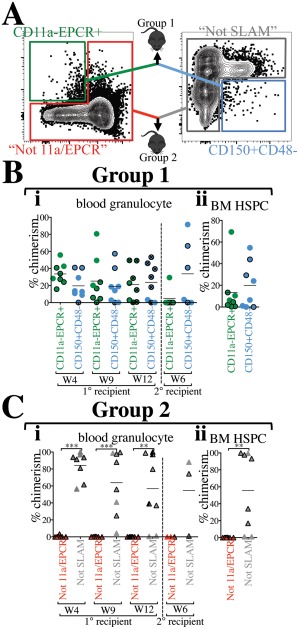
“11a/EPCR” two‐color sorting is as efficient as using the “SLAM” method. (A): Representation of direct comparison of two‐color sorting methods. BM from CFP or Tomato mice was sorted using the combination of CD11a and EPCR only or the combination of CD150 and CD48 only. Approximately 380 cells from each of CD11a– EPCR+ and CD150+ CD48– gates were sorted, mixed, and co‐transplanted with added 250,000 helper/competitor WBM (Group 1 recipients). Percentages of cells within each gate was kept consistent between the two methods. 200,000 cells from outside of the CD11a– EPCR+ gate (“Not 11a/EPCR”) and outside of the CD150+ CD48– gate (“Not SLAM”) were also mixed and co‐transplanted (Group 2 recipients). In (B, C), CFP+ donor‐derived cells are represented by outlined symbols and Tomato+ donor‐derived cells with borderless symbols. (Bi): Time‐course analysis of blood granulocyte chimerism from CD11a– EPCR+ and CD150+ CD48– sources in primary recipients 4, 9, and 12 weeks (W) post‐transplant, and in secondary recipients (separated by vertical dashed line) at week 6 following secondary transplant. Primary recipients used for secondary transplants are marked with an “x” inside circles at the 12‐week timepoint. (Bii): Donor chimerism of HSPCs in the BM of primary recipients transplanted with CD11a– EPCR+ and CD150+ CD48– sorted cells 13 weeks post‐transplant. (Ci): Time‐course analysis of blood granulocyte chimerism from “Not 11a/EPCR” and “Not SLAM” sources in primary recipients 4, 9, and 12 weeks (W) post‐transplant, and in secondary recipients (separated by vertical dashed line) at week 6 following secondary transplant. The primary recipients used for secondary transplant is marked (half shaded black) at the 12‐week timepoint. (Cii): Donor chimerism of HSPCs in the BM of primary recipients transplanted with “Not 11a/EPCR” and “Not SLAM” sorted cells 13 weeks post‐transplant. Number of experiments = 2. **, p ≤ .01; ***, p ≤ .001 (Student's unpaired t test). “Not 11a/EPCR” = not CD11a‐EPCR+; “Not SLAM” = not CD150+CD48–. Abbreviations: BM, bone marrow, EPCR, endothelial protein C receptor; HSPC, hematopoietic stem and progenitor cells.
To determine whether any HSCs resided outside of the CD11a– EPCR+ or the CD150+ CD48– gates, we sorted each of the “Not” populations (“Not 11a/EPCR” and “Not SLAM”) and co‐transplanted them into recipient mice (Fig. 4A, Group 2 recipients). We detected significantly higher “Not SLAM”‐derived granulocytes and HSPCs compared with “Not 11a/EPCR” in primary recipients, as well as secondary engraftment, suggesting the presence of HSCs outside of the SLAM gate (Fig. 4Ci, 4C ii). We also found higher total, macrophage and lymphocyte chimerism from the Not SLAM population. (Supporting Information Fig. S4C). Our data indicate that although both two‐color strategies are effective at sorting HSCs, HSCs are detectable outside of the SLAM gate, but not the 11a/EPCR gate. Taken together, these experiments suggest that the SLAM two‐color method is less contaminated with non‐HSCs than the 11a/EPCR two‐color method, but some HSCs fall outside of the SLAM gates, whereas with the 11a/EPCR method, all HSCs are sorted within the CD11a– EPCR+ gate, but also many downstream progenitors are included.
CD11a/EPCR Gating Identifies Phenotypic HSCs Following Irradiation and Poly(I:C) Treatment
Many common HSC markers change their expression when challenged, such as during an inflammatory response. Thus, the phenotypic definition of HSCs can change depending on the context. We sought to determine the expression levels of CD11a and EPCR and their ability to mark HSCs after a variety of types of challenges: irradiation, poly(I:C) treatment, and LPS treatment (Fig. 5). First, we sublethally irradiated (6 Gy) B6 mice and examined their BM 48 hours post‐irradiation. Consistent with previous observations, we detected a dramatic decrease in Kit expression 12, 14, reducing the frequency of KLS cells (Fig. 5A). The percentage of CD11a+ cells appeared to increase slightly after irradiation, though EPCR expression appeared unchanged (Fig. 5B, Supporting Information Fig. S5A). Other HSC markers also appeared unchanged, with the exception of CD48 which decreased after irradiation (Supporting Information Fig. S5A). Although CD11a expression appeared to increase overall in the irradiated BM, phenotypic HSCs were still found in the CD11a– EPCR+ fraction, suggesting these markers could still identify HSCs after irradiation (b).
Figure 5.
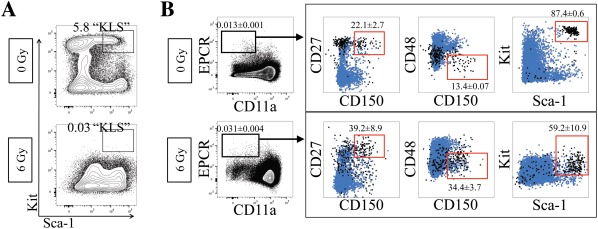
Efficacy of CD11a/EPCR combination post‐irradiation injury. (A): Expression of Sca‐1 and Kit on Ter119– CD27+ BM cells in nonirradiated controls (top) compared with 48 hours after 6 Gy irradiation (bottom). Percentages of KLS cells are shown. (B): Expression of HSC markers on Ter119– CD11a– EPCR+ without (0 Gy; n = 3) and with (6 Gy; n = 5) irradiation‐induced BM injury. Numbers shown are percentages of cells within CD11a– EPCR+ gate ± SD. Boxed plots show Ter119– CD11a– EPCR+ gated cells (black) and total Ter119– cells (blue). Gates (red) show phenotypic HSCs and what percentage of Ter119– CD11a– EPCR+ cells fall within those gates. Abbreviation: EPCR, endothelial protein C receptor.
We also examined CD11a and EPCR expression after two forms of inflammation, induced by injection of either poly(I:C), a TLR3 agonist 26, or endotoxin (lipopolysaccharide, LPS), a TLR4 agonist 27. Both are known to upregulate Sca‐1 and therefore make HSC identification more difficult using standard markers 28, 29, 30. After poly(I:C) injection, Sca‐1 expression dramatically increased in BM cells (Supporting Information Fig. S6). However, most other HSC markers remained unchanged including CD11a and EPCR, suggesting these markers may still identify HSCs following poly(I:C) treatment. Conversely, while CD11a expression appeared unchanged in LPS‐treated BM, EPCR expression changed significantly, and thus we were unable to use this marker combination to identify phenotypic HSCs in this context (Supporting Information Fig. S7). CD11a could still be combined with other HSC markers that appeared unchanged in LPS‐treated mice, including CD27 and Kit. Future transplantation experiments would be required to confirm whether the phenotypic HSCs identified in either irradiation or inflammation are in fact functional HSCs.
Discussion
Here, we demonstrate a novel strategy for a simplified, reproducible, and efficient way for HSC sorting with the use of CD11a and EPCR. Our transplantation strategy used direct competition between the two KLS fractions as the primary method to evaluate which fraction contained the most HSCs. Methods like limit dilution assays and single cell transplantation assays can provide quantitative estimates of the number of HSCs in a population. While we did not perform those types of assay, in our system we transplanted all possible sources of HSCs at their physiologic proportions into each recipient. Thus, if more HSCs existed outside of the CD11a– EPCR+ fraction, then the “Not CD11a– EPCR+” fraction would have shown higher donor chimerism. The fact that little granulocyte chimerism was found outside of the CD11a– EPCR+ fraction indicates that this population contained all HSCs.
We found that the 11a/EPCR method was comparable to the SLAM method of two‐color HSC sorting. This is despite the fact that there appeared to be very little overlap between the two populations, with only 12% of CD11a– EPCR+ cells falling within the CD150+ CD48– gate and only 7.2% of CD150+ CD48– cells falling within the CD11a– EPCR+ gate. While some HSCs were found outside of the SLAM gates (Fig. 4C), this did not happen in every experiment (Supporting Information Fig. S3) and is likely an infrequent event. Because both methods have been shown to contain nearly all HSCs, this means that likely nearly all HSCs exist within the overlapping population, which would be CD11a– EPCR+ CD150+ CD48–. This also indicates that both populations contain many non‐HSCs, as expected from a two‐color approach. For the CD11a– EPCR+ fraction, the contaminating cells are highly enriched for MPPs, as nearly all the cells (81%) were Kit+ Sca‐1+. For the SLAM fraction, most of the contaminating cells were CD11a+ and possibly lymphoid or myeloid cells. Therefore, both strategies have their strengths and weaknesses for use as two‐color method, and the user should select whether they would rather have MPPs in their sort (11a/EPCR method) or other contaminating cells which are likely not progenitors (SLAM method).
Use of CD11a and EPCR to identify HSCs after irradiation or induction of inflammation gave mixed results. The combination appeared to work after irradiation, although total levels of CD11a were upregulated. CD11a upregulation may be involved in HSC differentiation to progenitors, which could be necessary to replenish hematopoietic populations depleted by irradiation, and thus the true undifferentiated HSCs remain CD11a–. As part of LFA1, CD11a upregulation may also be involved in the migration of HSCs out of their niche and into circulation. We previously found precursors to HSCs, “pre‐HSCs,” to be contained within the CD11a– fraction of progenitors during early embryonic development 31. Yet later in embryonic development and during expansion of mature HSCs in the fetal liver, a group of CD11a+ progenitors also show long‐term engraftment capacity 19. Interestingly, these CD11a+ fetal liver HSCs downregulate CD11a after seeding the BM and remain negative for CD11a until differentiation into downstream multi‐potent progenitors. These findings may suggest a role for downregulating CD11a in HSCs during homeostasis as a means to prevent the migration of these cells out of their BM niche and into the circulation. On the other hand, EPCR has been suggested to play an active role in retention of HSCs in their niche. Whereas “pre‐HSCs” are CD11a–, they express high levels of EPCR 32. EPCR+ HSCs in the fetal liver interact with the perisinusoidal niche, and the interaction between EPCR+ HSCs and niche cells seems to persist into the BM where EPCR shedding from HSCs has been correlated with mobilization of these cells into the circulation 23, 33.
Conclusion
Efficient sorting of mouse HSCs allows in‐depth molecular and functional characterization and contributes greatly to our understanding of the biology of these cells. CD11a and EPCR can now be added to the pantheon of available markers for stringent HSC purification, but also as an alternative method for two‐color enrichment of HSCs. A method of HSC sorting using metal conjugated antibodies and a magnet (e.g., MACS) could potentially be used to enrich HSCs using antibodies for either EPCR (positive enrichment) or CD11a (depletion). While both methods would potentially work, the enriched fractions would be significantly contaminated with non‐HSCs. The CD11a/EPCR two‐color method is not as simple as one that is MACS‐based, but would likely be purer and would not miss any HSCs. Last, while we did not address CD11a expression on human HSCs, EPCR has recently been used for purification of in vitro expanded human HSCs 34. Whether or not CD11a can similarly be used for human HSC identification is of great translational interest, and merits further examination.
Author Contributions
A.K.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; V.M.S.: collection and assembly of data, administrative support, data analysis; E.V. and C.C.: conception and design, collection and assembly of data; K.G.: collection and assembly of data; J.W.F.: conception and design, data analysis and interpretation; D.A.F. and T.S.: conception and design, data analysis and interpretation, manuscript writing; M.A.I.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
J.F. declared employment position with Novartis and Kirin; ownership opportunity with Pfizer, BMS, Novartis, AstraZeneca. D.F. declared research funding from Revolution Medicines. The other authors indicated no potential conflicts of interest.
Supporting information
Supporting Information
Acknowledgments
We thank Martina Sassone‐Corsi, Melissa Lodoen, Craig Walsh, and Matthew Blurton‐Jones for technical assistance and helpful discussion, and Yasamine Ghorbanian, Ankita Shukla, and Tannaz Faal of the Inlay lab for helpful comments and discussion. This study was supported by National Institutes of Health grant R56HL133656 (to M.A.I.), American Cancer Society ACS/IRG Seed Grant IRG 98‐27‐10 (to M.A.I.), and California Institute for Regenerative Medicine grant CL1–00520‐1.2 (to V.S. and the UC Irvine Stem Cell FACS Core). A.K. was supported in part from National Institutes of Health grant R01AG055524, and E.V. was supported by a gift from the Oxnard Foundation.
References
- 1. Seita J, Weissman IL. Hematopoietic stem cell: Self‐renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med 2010;2:640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de la Morena MT, Nelson RP Jr. Recent advances in transplantation for primary immune deficiency diseases: A comprehensive review. Clin Rev Allergy Immunol 2014;46:131–144. [DOI] [PubMed] [Google Scholar]
- 3. Boitano AE, Wang J, Romeo R et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010;329:1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brunstein CG, Gutman JA, Weisdorf DJ et al. Allogeneic hematopoietic cell transplantation for hematologic malignancy: Relative risks and benefits of double umbilical cord blood. Blood 2010;116:4693–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hagedorn EJ, Durand EM, Fast EM et al. Getting more for your marrow: Boosting hematopoietic stem cell numbers with PGE2. Exp Cell Res 2014;329:220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kiel MJ, Yilmaz ÖH, Iwashita T et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005;121:1109–1121. [DOI] [PubMed] [Google Scholar]
- 7. Balazs AB. Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood 2006;107:2317–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karlsson G, Rörby E, Pina C et al. The tetraspanin CD9 affords high‐purity capture of all murine hematopoietic stem cells. Cell Rep 2013;4:642–648. [DOI] [PubMed] [Google Scholar]
- 9. Wiesmann A, Phillips RL, Mojica M et al. Expression of CD27 on murine hematopoietic stem and progenitor cells. Immunity 2000;12:193–199. [DOI] [PubMed] [Google Scholar]
- 10. Chen JY, Miyanishi M, Wang SK et al. Hoxb5 marks long‐term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature 2016;530:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Challen GA, Boles N, Lin KK‐Y et al. Mouse hematopoietic stem cell identification and analysis. Cytometry A 2009;75:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simonnet AJ, Nehmé J, Vaigot P et al. Phenotypic and functional changes induced in hematopoietic stem/progenitor cells after gamma‐ray radiation exposure. Stem Cells 2009;27:1400–1409. [DOI] [PubMed] [Google Scholar]
- 13. Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol 2011;32:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vazquez SE, Inlay MA, Serwold T. CD201 and CD27 identify hematopoietic stem and progenitor cells across multiple murine strains independently of Kit and Sca‐1. Exp Hematol 2015;43:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol 2005;5:546–559. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y, Wang H. Integrin signalling and function in immune cells. Immunology 2012;135:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lum AFH, Green CE, Lee GR et al. Dynamic regulation of LFA‐1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM‐1) in shear flow. J Biol Chem 2002;277:20660–20670. [DOI] [PubMed] [Google Scholar]
- 18. Shamri R, Grabovsky V, Gauguet J‐M et al. Lymphocyte arrest requires instantaneous induction of an extended LFA‐1 conformation mediated by endothelium‐bound chemokines. Nat Immunol 2005;6:497–506. [DOI] [PubMed] [Google Scholar]
- 19. Fathman JW, Fernhoff NB, Seita J et al. Upregulation of CD11A on hematopoietic stem cells denotes the loss of long‐term reconstitution potential. Stem Cell Reports 2014;3:707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muzumdar MD, Tasic B, Miyamichi K et al. A global double‐fluorescent Cre reporter mouse. Genesis 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- 21. Ueno H, Weissman IL. Clonal analysis of mouse development reveals a polyclonal origin for yolk sac blood islands. Dev Cell 2006;11:519–533. [DOI] [PubMed] [Google Scholar]
- 22. Benz C, Copley MR, Kent DG et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 2012;10:273–283. [DOI] [PubMed] [Google Scholar]
- 23. Iwasaki H, Arai F, Kubota Y et al. Endothelial protein C receptor‐expressing hematopoietic stem cells reside in the perisinusoidal niche in fetal liver. Blood 2010;116:544–553. [DOI] [PubMed] [Google Scholar]
- 24. Serwold T, Ehrlich LI, Weissman IL. Reductive isolation from bone marrow and blood implicates common lymphoid progenitors as the major source of thymopoiesis. Blood 2009;113:807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Inlay MA, Bhattacharya D, Sahoo D et al. Ly6d marks the earliest stage of B‐cell specification and identifies the branchpoint between B‐cell and T‐cell development. Genes Dev 2009;23:2376–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsumoto M, Seya T. TLR3: Interferon induction by double‐stranded RNA including poly(I:C). Adv Drug Deliv Rev 2008;60:805–812. [DOI] [PubMed] [Google Scholar]
- 27. Schuettpelz LG, Link DC. Regulation of hematopoietic stem cell activity by inflammation. Front Immunol 2013;4:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang P, Nelson S, Bagby GJ et al. The lineage‐c‐Kit+Sca‐1+ cell response to Escherichia coli bacteremia in Balb/c mice. Stem Cells 2008;26:1778–1786., [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Esplin BL, Shimazu T, Welner RS et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol 2011;186:5367–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pietras EM, Lakshminarasimhan R, Techner J‐M et al. Re‐entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med 2014;211:245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Inlay MA, Serwold T, Mosley A et al. Identification of multipotent progenitors that emerge prior to hematopoietic stem cells in embryonic development. Stem Cell Reports 2014;2:457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou F, Li X, Wang W et al. Tracing haematopoietic stem cell formation at single‐cell resolution. Nature 2016;533:487–492. [DOI] [PubMed] [Google Scholar]
- 33. Gur‐Cohen S, Itkin T, Chakrabarty S et al. PAR1 signaling regulates the retention and recruitment of EPCR‐expressing bone marrow hematopoietic stem cells. Nat Med 2015;21:1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fares I, Chagraoui J, Lehnertz B et al. EPCR expression marks UM171‐expanded CD34+ cord blood stem cells. Blood 2017;129:3344–3351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information