

ABSTRACT
The Chronic Myeloproliferative Neoplasms (MPN) are cancers characterized by hyperinflammation and immune deregulation. Concurrently, the expression of the immune check point programmed death ligand 1 (PD-L1) is induced by inflammation. In this study we report on the occurrence of spontaneous T cell responses against a PD-L1 derived epitope in patients with MPN. We show that 71% of patients display a significant immune response against PD-L1, and patients with advanced MPN have significantly fewer and weaker PD-L1 specific immune responses compared to patients with non-advanced MPN. The PD-L1 specific T cell responses are CD4+ T cell responses, and by gene expression analysis we show that expression of PD-L1 is enhanced in patients with MPN. This could imply that the tumor specific immune response in MPN could be enhanced by vaccination with PD-L1 derived epitopes by boosting the anti-regulatory immune response hereby allowing tumor specific T cell to exert anti-tumor immunity.
KEYWORDS: Myeloproliferative neoplasms, immune therapy, PD-L1, vaccination, T cells, immune check point
Introduction
There is compelling evidence describing a profound immune dysregulation in patients with Philadelphia chromosome negative chronic myeloproliferative neoplasms (MPN). The cytokine milieu, immune phenotype and gene expression profile is severely deregulated,1-8 and it is speculated, that MPN may evolve and progress due to a defect tumor immune surveillance.9 Tumor cells employ several immune suppressive mechanisms to subvert the anti-tumor immune response. One such well described mechanism is the binding of programmed death-1 receptor on activated T-cells to programmed death-1-ligand (PD-L1) on either tumor cells or cells in the tumor microenvironment, rendering tumor reactive T-cells anergic, hereby facilitating tumor immune escape.10 The targeting of the PD1/PD-L1 axis with blocking antibodies has shown remarkable results in both solid and hematological malignancies.11,12 However, the immune system harbors naturally occurring PD-L1 specific T-cells, and these T-cells are able to target and kill PD-L1 positive cells.13,14 Additionally, stimulation of T cells with PD-L1 derived epitopes has been shown to boost both anti-leukemic and anti-viral immune responses as well as immune responses against autologous dendritic cells (DC) transfected with the tumor associated antigens hTERT and survivin.15 As such, we are currently investigating safety and tolerability of a peptide vaccination with a PD-L1 derived epitope in patients with multiple myeloma (NCT03042793), as we expect the vaccine to enhance the tumor specific immune response in patients. In the setting of MPN, the identification of PD-L1 specific T cell responses is highly intriguing, as the hyperinflammatory state in MPN potentially enhances expression of PD-L1, which likely suppresses the neoantigen specific T cell responses in MPN just recently described by our group.16-18 We believe, that spontaneously occurring PD-L1 specific T cell responses in MPN might be enhanced by vaccination with the PD-L1 derived epitope used in the above mentioned myeloma vaccination, hereby generating an improved anti-neoplastic and anti-regulatory immune response. Hence, we investigated if patients with MPN harbor spontaneously occurring T cell responses against the PD-L1 derived epitope, and correlated the identified responses with clinical data.
Results
Patient characteristics
In total we enrolled 51 patients of whom 19 (37%) were males. The median age of diagnosis of the included patients was 55.9 years (range, 17–84), and the median age of the patients at time of study sampling was 66.9 years (range, 25 – 84 years). Clinical data of these patients is available in Table 1. In short, 14 patients (28%) were diagnosed with essential thrombocythemia (ET), 16 (31%) with polycythemia vera (PV), 7 (14%) with prefibrotic myelofibrosis (PreMF) and 14 (28%) with primary myelofibrosis (PMF). Twenty-four patients (47%) were JAK2V617F+, 23 (45%) patients were calreticulin (CALR) mutants, 3 patients (6%) were triple negative and one patient (2%) had a myeloproliferative leukemia virus (MPL) mutation. At the time of study blood sample, 16 patients (31%) were treated with interferon-alpha (IFN-α), 15 (29%) were treated with hydroxyurea (HU), 9 patients (18%) did not receive any kind of treatment, 5 (10%) were treated with anagrelide (ANA), 4 patients (8%) were treated with ANA and HU, 2 patients (4%) were receiving phlebotomies, and 1 patient was treated with rituximab for autoimmune thyroiditis. Of the 14 patients with myelofibrosis, 6 (43%) had DIPSS low risk, 7 (50%) had DIPSS intermediate-1 risk, and 1 patient (7%) had DIPSS intermediate-2 risk. None of the patients had DIPSS high risk PMF. Nine patients (18%) experienced progression of their MPN either before or after study sample, and 11 patients (22%) were diagnosed with a secondary malignancy either before or after study sample. Thirty-nine patients (77%) had a complete hematological response (CHR) to any therapy given at any time point both before and after blood sampling, whereas the remaining 12 patients (24%) either had a partial response, no response, or progressive disease. Patients with disease progression from ET or PV to PMF before or after blood sampling and patients with progression from MPN to acute myeloid leukemia (AML), after blood sampling were termed as having progressive disease.
Table 1.
Clinical data of patients included in the study.
| Variable | ||
|---|---|---|
| Total patients | 51 | |
| Male gender, n (%) | 19 (37%) | |
| Diagnosis at blood sample | ||
| ET | 14 (28%) | |
| PV | 16 (31%) | |
| PreMF | 7 (14%) | |
| PMF | 14 (28%) | |
| Driver mutation | ||
| JAK2V617F | 24 (47%) | |
| CALR | 23 (45%) | |
| Triple negative | 3 (6%) | |
| MPL | 1 (2%) | |
| Treatment at time of blood sampling | ||
| Interferon-alpha | 16 (31%) | |
| Hydroxyurea | 15 (29%) | |
| None | 9 (18%) | |
| Anagrelide | 5 (10%) | |
| Anagrelide and hydroxyurea | 4 (8%) | |
| Phlebotomy | 2 (4%) | |
| Rituximab | 1 (2) | |
| DIPSS score | ||
| Low-risk | 6 (43%) | |
| Intermediate-1 risk | 7 (50%) | |
| Intermediate-2 risk | 1 (7%) | |
| High-risk | 0 (0%) | |
| Transformation/progression, yes | 9 (18%) | |
| Secondary malignancy, yes | 11 (22%) | |
| Complete hematological response | 36 (71%) | |
| Age in years at diagnosis, median (min - max) | 55.9 (17–84) | |
| Age in years at draw of study sample, median (min - max) | 66.9 (25 – 84) | |
| DFR-defined response | 36 (71%) | |
| DFRx2 defined response | 19 (37%) |
Patients with MPN display frequent immune responses against the PD-L1Long1 epitope, and frequency and amplitude of the responses correlate with disease severity
Of the 51 patients, 36 (71%) displayed a statistical significant IFN-γ response according to the DFR-rule by Moodie et al.19 After applying the more conservative DFR(2x) rule, we identified 19 responders (37%). Both patients with ET, PV, PreMF and PMF displayed a PD-L1Long1 specific immune response (Fig. 1A-1D). Next, we compared the clinical data of DFR(2x) defined responders with non-responders (Table 2). As the immune dysregulation in MPN becomes more pronounced with disease progression, we hypothesized that the frequency of immune responses would be most frequent in patients with ET, second most frequent in patients with PV, and that patients with PreMF and PMF would harbor the lowest frequency of responses. This was confirmed as ET-patients harbored the most frequent responses, followed by PV, then Pre-MF and finally PMF (57% vs. 44% vs. 29% vs. 14%) (p = 0.107). In 41 patients, the ELISPOTs were performed with a concentration of 3 × 105 cells per well allowing us to compare the mean amount of PD-L1Long specific cells in these 41 subjects. We demonstrated that PBMC from patients with ET and PV secrete more IFN-γ upon stimulation with PD-L1Long1 peptide compared to PBMC from patients with PreMF and PMF (Fig. 1E) with the difference between patients with ET and PMF and PV and PMF being statistically significant (p = 0.018 and p = 0.042). Even more, we compared the amount of DFR(2x) defined responses in non-PMF patients (17 responders; 46% of patients) with PMF patients (2 responders; 14% of patients) (p = 0.053).
Figure 1.
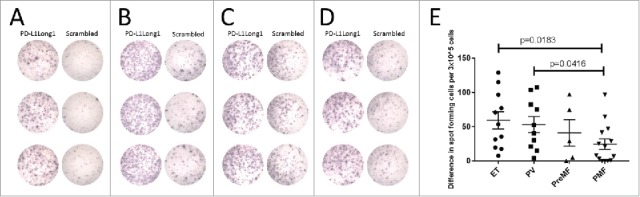
Patients with all stages of MPN display PD-L1 Long1 specific immune responses, but response amplitude declines with disease severity. (A) Example of an PD-L1 Long1 specific IFN-γ response in a patient with ET. (B) Example of a PD-L1 Long1 specific IFN-γ response in a patient with PV. (C) Example of a PD-L1 Long1 specific IFN-γ response in a patient with PreMF. (D) Example of a PD-L1 Long1 specific IFN-γ response in a patient with PMF. (E) IFN-γ ELISPOTs were performed with 3 × 10ˆ5 cells/well in 41 patients, and the response amplitude was calculated by subtracting the mean amount of spots in wells stimulated with negative control peptide from the mean amount of spots in wells stimulated with PD-L1 Long1 peptide. Bars represent mean and standard error of the mean. All experiments were performed in triplicates.
Table 2.
Correlation of clinical data to DFR(2x) defined ELISPOT responses. Categorical data were analysed using the chi-squared test or Fisher's exact test, where appropriate. Continous data were analyzed using the Mann-Whitney U-test.
| Variable | No response (DFRx2) | Response (DFRx2) | p-value | |
|---|---|---|---|---|
| Gender | 0.247 | |||
| Female | 18 (56%) | 14 (44%) | ||
| Male | 14 (74%) | 5 (26%) | ||
| Diagnosis | 0.107 | |||
| ET | 6 (43%) | 8 (57%) | ||
| PV | 9 (56%) | 7 (44%) | ||
| PreMF | 5 (71%) | 2 (29%) | ||
| PMF | 12 (86%) | 2 (14%) | ||
| Diagnosis (non-PMF vs. PMF) | 0.053 | |||
| Non-PMF | 20 (54%) | 17 (46%) | ||
| PMF | 12 (86%) | 2 (14%) | ||
| Mutation type | 1.00 | |||
| CALR | 16 (70%) | 7 (30%) | ||
| JAK2V617F | 16 (67%) | 8 (33%) | ||
| Transformation or progression | 0.128 | |||
| Yes | 8 (89%) | 1 (11%) | ||
| No | 24 (57%) | 18 (43%) | ||
| Secondary malignancy | 0.176 | |||
| Yes | 9 (82%) | 2 (18%) | ||
| No | 23 (58%) | 17 (43%) | ||
| IFN-α at blood sample | 0.549 | |||
| Yes | 11 (69%) | 5 (31%) | ||
| No | 21 (60%) | 14 (40%) | ||
| Hematological response | 0.171 | |||
| Complete hematological response | 20 (56%) | 16 (44%) | ||
| Did not obtain complete hematological response | 12 (80%) | 3 (20%) | ||
| Median age at diagnosis | 56.2 | 55.4 | 0.914 | |
| Median age at blood sampling | 67.3 | 66.9 | 0.914 |
Patients without complete hematological response, and patients with disease progression show less frequent PD-L1Long1 specific immune response
Sixteen patients (44%) who experienced a CHR also displayed a DFR(2x) defined immune response, whereas only 3 (20%) of patients with a non-CHR had a DFR(2x) defined response (p = 0.123). As disease progression was termed as having non-CHR, we analyzed if patients, that did not have disease progression, but still had non-CHR, displayed less frequent responses. Of the 7 patients with a non-CHR, that did not have disease progression, 3 (43%) had a DFR-defined response, whereas 28 patients (79%) with a CHR had a response (p = 0.081). Similarly, it is noteworthy that only 1 patient (11%) with disease progression displayed a DFR(2x) defined ELISPOT response, whereas 18 patients (43%) without progression displayed a response (p = 0.128). There was no difference between responders and non-responders according to gender, mutational status, DIPSS-score, treatment, occurrence of secondary malignancy, median age at diagnosis, median age at blood sampling or treatment at time of sampling of study sample.
PD-L1Long1 specific T cells are CD4+ T-cells
To clarify the phenotype of the IFN-γ producing cells, 13 patients with a solid IFN-γ ELISPOT response were analyzed using ICS. All analyzed patients displayed CD4+ T-cell responses against PD-L1Long1 peptide (Fig. 2A). We demonstrated a marked difference in production of IFN-γ and TNF-α between PD-L1Long1 stimulated cells and cells stimulated with scrambled negative control (Fig. 2B-2D). The median amount of IFN-γ+ CD4+ T-cells was 0.081% vs. 0.030% (p<0.0001), the median amount of TNF-α+/IFN-γ+ double positive CD4+ T-cells was 0.037% vs. 0.002% (p<0.0001) and the median amount of TNF-α+ positive CD4+ T-cells was 0.463% vs. 0.125% (p = 0.0001).
Figure 2.
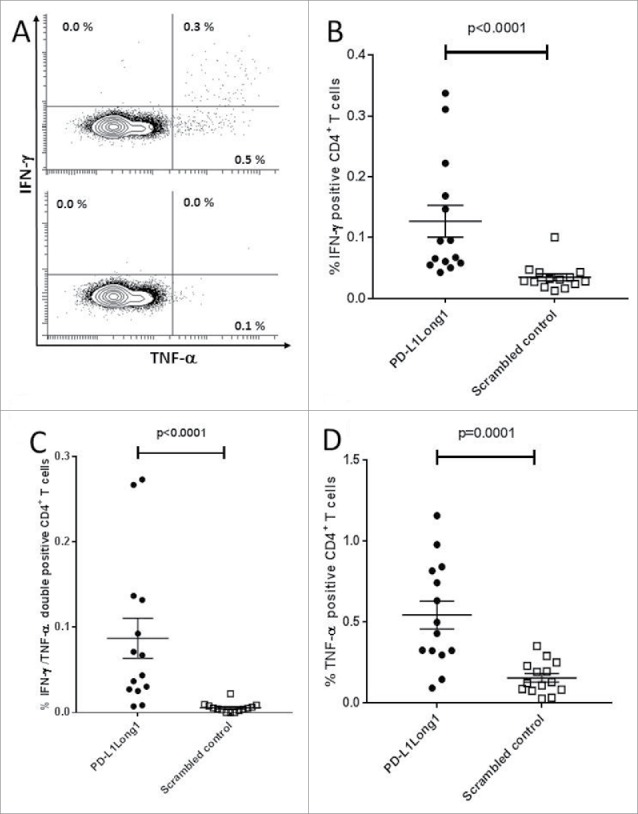
CD4+ T-cell responses against the PD-L1Long1 epitope. (A) Representative experiment of a PD-L1 Long1 specific immune response in CD4+ gated T cells with PD-L1 Long1 stimulated cells (top) and cells stimulated with negative scrambled control (bottom). (B) Plot of CD4+ T cells releasing IFN-γ upon stimulation with PD-L1 Long1. (C) Plot of IFN-γ+/TNF-α+ CD4+ T upon stimulation with PD-L1 Long1. (D) Plot of CD4+ T cells releasing TNF-α upon stimulation with PD-L1 Long1.
Patients with MPN have increased expression of PD-L1
As the MPNs are hyperinflammatory diseases, and PD-L1 expression is enhanced by inflammation, we speculated that the expression of PD-L1 is enhanced in MPN. Hence, we compared the gene expression in 69 MPN-patients to the gene expression in 21 healthy donors using materials and methods as previously described,20 and identified an enhanced expression of PD-L1 in peripheral blood in MPN-patients compared to healthy controls (fold change 1.2; p = 0.0003, false discovery rate (FDR) = 0.001). Data are available from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo; accession no. GSE26049).
Discussion
In the present study we have shown that patients with MPN harbor spontaneously occurring immune responses against a PD-L1 derived epitope. This implies that the immune system in MPN selectively targets regulatory cells and hereby enhance the anti-tumor immune response. It has been shown that the in-vitro immune response to a DC-based cancer vaccine was enhanced by in vitro co-stimulation with a PD-L1 derived peptide,15 and that stimulation of virus specific T-cell cultures with either PD-L1 derived epitopes or PD-L1 specific T-cells significantly enhanced the amount of virus-specific T-cells.21,22 Furthermore, PD-L1 specific T-cells have been described to kill leukemic cells including the JAK2V617F+ and PD-L1+ cancer cell line UKE-1.22,23 In addition, T-cell cultures specific for the JAK2V617F+ cancer cell line SET-2 display enhanced killing fraction of SET-2 specific T-cells after stimulation with PD-L1 peptide. This proves that PD-L1 stimulation of T-cells increases the anti-leukemic activity of the T-cells.21 Of note, both UKE-1 and SET-2 are JAK2V617F+ and are killed by JAK2V617F-specific cytotoxic T-cells.16 Thus, boosting PD-L1 specific T-cells could directly modulate immune regulation and potentially alter tolerance to tumor antigens. Concurrently, data from this study implies, that patients with advanced MPN display less frequent and weaker immune responses against the PD-L1Long1 epitope giving impetus to the notion, that patients with advanced cancer are immune suppressed and are not able to mount an effective tumor specific immune response. Of note, patients with CALR mutant PMF also show less frequent responses against CALR mutant epitopes compared to patients with CALR mutant ET.17 It should be noted however, that patients with MPN are in a hyperinflammatory state which could be the reason that some ELISPOT experiments show a high background, and in some cases this background could occlude an immune response. Although not statistically significant, the data generated by this study indicates, that patients that have a poor response to therapy are less prone to harbor PD-L1 specific immune responses. Concurrently, data from this study show, that patients with progression of MPN do not mount as frequent PD-L1 specific immune responses. Together this could imply, that an effective PD-L1 specific immune response is involved in obtaining a proper response to therapy and to prevent disease progression.
Recently, we described that both the JAK2V617F and CALR exon 9 mutations are recognized by specific T-cells, and thus are potential targets for cancer immune therapy.16-18 Given the strong and frequent immune responses against CALR mutant epitopes in patients with MPN, it is a paradox that especially CALR mutant patients are not able to spontaneously clear the CALR mutant cells. The increased levels of PD-L1 identified in this study could partially explain this, as tumor specific T cells may simply be rendered anergic, due to the enhanced levels of PD-L1 in patients. As such, vaccination combining the PD-L1 Long1 epitope used in the current study with CALR-mutant or JAK2-mutant epitopes would be easy to implement and is likely to be highly beneficial. PD-L1 specific T-cells can directly support anti-cancer immunity by killing target cells, as well as indirectly boost anti-cancer immunity by killing regulatory cells and by releasing pro-inflammatory cytokines. Concurrently, vaccination with JAK2-mutant/CALR-mutant peptides will induce and activate T cells specific for JAK2- and CALR-mutant cells. Cancer vaccines represent a promising means of eliminating minimal residual disease without inducing significant toxicity or secondary malignancies. However, to date, they have largely failed to significantly improve patient outcomes. This likely reflects malignant cells' ability to suppress the functions of the induced immune cells. As such, we think that the identification of PD-L1 specific immune responses in MPN hold great promises for the future treatment of MPN in the setting of cancer immune therapy. Consequently, we are initiating a phase-I clinical vaccination trial in MPN combining PD-L1 derived epitopes with the recently described JAK2-mutant epitopes.16
Methods
Patient material
The study was approved by the local ethics committee at Zealand Region (SJ-456), and all patients provided signed informed consent according to the Helsinki Declaration before entry in the study. Patient diagnosis followed the 2016 WHO classification of MPN-disease.24 Patient peripheral blood mononuclear cells (PBMCs) were isolated using Lymphoprep (Axis Shield, Oslo, Norway), and were frozen in fetal calf serum with 10% dimethyl sulfoxide (Sigma-Aldrich). Clinico-hematological response to therapy was evaluated as earlier described,25 however patients with progression of their MPN before or after study sampling were termed to have progressive disease.
Experimental procedures
Studies in immune responses against PD-L1 derived epitopes have previously been identified by the robust interferon-gamma (IFN-γ) Enzyme Linked ImmunoSPOT (ELISPOT)14 and we used the same experimental procedures in this study. We chose the 19 amino acid PD-L1 derived epitope PD-L1Long1 (PD-L19–27, FMTYWHLLNAFTVTVPKDL) that resides within the signal peptide of PD-L1. This epitope has been shown to elicit spontaneous immune responses in patients with solid malignancies14 and is now used in a clinical setting in the above mentioned myeloma vaccination trial. We used a scrambled control (GARVERVDFGNFVFNISVLW) as a negative control. To evaluate the phenotype of the IFN-γ producing cells, we employed intracellular cytokine staining. In short, after 7 days of in vitro culture, 106 PBMCs were stimulated with PD-L1Long1 peptide or negative control peptide. After one hour of incubation with peptide, the Golgi Transport inhibitor Brefeldin A (BD Biosciensen, San José, CA, USA) was added. After additional four hours of incubation, cells were washed twice in PBS with 2% fetal calf serum and then stained with the following surface markers 4 μl NIR, 10 μl CD4 PerCP, 2 μl CD8 Pacific Blue, and 10 μl CD3 FITC for 30 minutes on ice. Next, cells were washed once more and fixed/permeabilized with Fixation/Permeabilization Reagent and Permeabilization Buffer (both from eBiosciences). Next, cells were stained for 30 minutes on ice using the following intracellular antibodies: 2 μl anti-TNF-α and 2 μl anti-IFN-γ conjugated to either PE-Cy7 or APC. Next, cells were washed twice in Permeabilization buffer and then resuspended in PBS with 2% fetal calf serum. Cells were acquired with a FACS CANTO II (BD Bioscienses, San José, CA, USA) and analyzed using FACS Diva software version 8.0.1.
Statistical analysis
The distribution free resampling method (DFR) and the more conservative DFR(2x) rule were used for statistical analysis of the ELISPOT responses.19 The statistical software package R was used for the DFR analyses using the R code provided at (http://www.scharp.org/zoe/runDFR/). The Pearsson chi-squared test, Fisher's exact test and Mann-Whitney U test were used for statistical analyses of clinical data by using the statistical software package SPSS version 21.
Funding Statement
This work was supported by the Region Sjællands Sundhedsvidenskabelige Forskningsfond, 12-000095, Region Sjællands Sundhedsvidenskabelige Forskningsfond, 15-000342, Kræftens Bekæmpelse, R90-A6143-14-S2.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank laboratory technician Merete Jonassen for outstanding help teaching M.O.H to perform the immune cell assays, and the secretaries, nurses and laboratory technicians at Zealand University Hospital for organizing blood draws from patients. This study was supported in part by grant from Kræftens Bekæmpelse to H.C.H (grant number R90-A6143-14-S2) and by grants from Region Sjællands Sundhedsvidenskabelige Forskningsfond to M.O.H (grant numbers 12-000095 and 15-000342).
Support for this study
This study was supported in part by grant from Danish Cancer Society to H.C.H under grant R90-A6143-14-S2 and by grants from Region Sjællands Sundhedsvidenskabelige Forskningsfond to M.O.H grants 12-000095 and 15-000342 in addition to support from Herlev Hospital.
References
- 1.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J Clin Oncol. 2011;29:1356–63. doi: 10.1200/JCO.2010.32.9490. PMID:21300928. [DOI] [PubMed] [Google Scholar]
- 2.Pourcelot E, Trocme C, Mondet J, Bailly S, Toussaint B, Mossuz P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: clinical implications. Exp Hematol. 2014;42:360–8. doi: 10.1016/j.exphem.2014.01.006. PMID:24463275. [DOI] [PubMed] [Google Scholar]
- 3.Cervantes F, Hernández-Boluda JC, Villamor N, Serra A, Montserrat E. Assessment of peripheral blood lymphocyte subsets in idiopathic myelofibrosis. Eur J Haematol. 2000;65:104–8. doi: 10.1034/j.1600-0609.2000.90262.x. PMID:10966169. [DOI] [PubMed] [Google Scholar]
- 4.Barosi G. An Immune Dysregulation in MPN. Curr Hematol Malig Rep. 2014;9:331–9. doi: 10.1007/s11899-014-0227-0. PMID:25139710. [DOI] [PubMed] [Google Scholar]
- 5.Wang JC, Kundra A, Andrei M, Baptiste S, Chen C, Wong C. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk Res. 2016;43:39–43. doi: 10.1016/j.leukres.2016.02.004. PMID:26943702. [DOI] [PubMed] [Google Scholar]
- 6.Bjørn ME, Andersen CL, Jensen MK, Hasselbalch HC. Circulating YKL-40 in myelofibrosis a potential novel biomarker of disease activity and the inflammatory state. Eur J Haematol. 2014;93:224–8. doi: 10.1111/ejh.12332. PMID:24689875. [DOI] [PubMed] [Google Scholar]
- 7.Skov V, Larsen TS, Thomassen M, Riley CH, Jensen MK, Bjerrum OW, Kruse T, Hasselbalch HC. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: identification of deregulated genes of significance for inflammation and immune surveillance. Leuk Res. 2012;36:1387–92. doi: 10.1016/j.leukres.2012.07.009. PMID:22877729. [DOI] [PubMed] [Google Scholar]
- 8.Skov V, Riley CH, Thomassen M, Larsen TS, Jensen MK, Bjerrum OW, Kruse T, Hasselbalch HC. Whole blood transcriptional profiling reveals significant down-regulation of human leukocyte antigen class I and II genes in essential thrombocythemia, polycythemia vera and myelofibrosis. Leuk Lymphoma. 2013;54:2269–73. doi: 10.3109/10428194.2013.764417. PMID:23302045. [DOI] [PubMed] [Google Scholar]
- 9.Hasselbalch HC. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk Res. 2013;37:214–20. doi: 10.1016/j.leukres.2012.10.020. PMID:23174192. [DOI] [PubMed] [Google Scholar]
- 10.Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med. 2016;375:1767–78. doi: 10.1056/NEJMra1514296. PMID:27806234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al.. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin's Lymphoma. N Engl J Med. 2014;372:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al.. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627–39. doi: 10.1056/NEJMoa1507643. PMID:26412456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munir S, Andersen GH, Met Ö, Donia M, Frøsig TM, Larsen SK, Klausen TW, Svane IM, Andersen MH. HLA-restricted CTL that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res. 2013;73:1764–76. doi: 10.1158/0008-5472.CAN-12-3507. PMID:23328583. [DOI] [PubMed] [Google Scholar]
- 14.Munir S, Andersen GH, Svane IM, Andersen MH. The immune checkpoint regulator PD-L1 is a specific target for naturally occurring CD4(+) T cells. Oncoimmunology. 2013;2:e23991. doi: 10.4161/onci.23991. PMID:23734334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munir Ahmad S, Martinenaite E, Hansen M, Junker N, Borch TH, Met Ö, Donia M, Svane IM, Andersen MH. PD-L1 peptide co-stimulation increases immunogenicity of a dendritic cell-based cancer vaccine. Oncoimmunology. 2016;5:e1202391. doi: 10.1080/2162402X.2016.1202391. PMID:27622072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmström MO, Hjortsø MD, Ahmad SM, Met Ö, Martinenaite E, Riley C, Straten P, Svane IM, Hasselbalch HC, Andersen MH. The JAK2V617F mutation is a target for specific T cells in the JAK2V617F-positive myeloproliferative neoplasms. Leukemia. 2017;31:495–8. doi: 10.1038/leu.2016.290. PMID:27761006. [DOI] [PubMed] [Google Scholar]
- 17.Holmstrom MO, Riley CH, Svane IM, Hasselbalch HC, Andersen MH, Holmström MO, Riley CH, Svane IM, Hasselbalch HC, Andersen MH, et al.. The CALR exon 9 mutations are shared neoantigens in patients with CALR mutant chronic myeloproliferative neoplasms. Leukemia. 2016;30:2413–6. doi: 10.1038/leu.2016.233. PMID:27560107. [DOI] [PubMed] [Google Scholar]
- 18.Holmstrom MO, Martinenaite E, Ahmad SM, Met O, Friese C, Kjaer L, Riley CH, thor Straten P, Svane IM, Hasselbalch HC, et al.. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia. 2017. doi: 10.1038/leu.2017.214. [DOI] [PubMed] [Google Scholar]
- 19.Moodie Z, Price L, Gouttefangeas C, Mander A, Janetzki S, Löwer M, Welters MJP, Ottensmeier C, Van Der Burg SH, Britten CM. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother. 2010;59:1489–501. doi: 10.1007/s00262-010-0875-4. PMID:20549207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skov V, Larsen TS, Thomassen M, Riley CH, Jensen MK, Bjerrum OW, Kruse TA, Hasselbalch HC. Whole-blood transcriptional profiling of interferon-inducible genes identifies highly upregulated IFI27 in primary myelofibrosis. Eur J Haematol. 87:54–60. doi: 10.1111/j.1600-0609.2011.01618.x. PMID:21447007. [DOI] [PubMed] [Google Scholar]
- 21.Ahmad SM, Svane IM, Andersen MH. The stimulation of PD-L1-specific cytotoxic T lymphocytes can both directly and indirectly enhance antileukemic immunity. Blood Cancer J. 2014;4:e230. doi: 10.1038/bcj.2014.50. PMID:25036801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmad SM, Larsen SK, Svane IM, Andersen MH. Harnessing PD-L1-specific cytotoxic T cells for anti-leukemia immunotherapy to defeat mechanisms of immune escape mediated by the PD-1 pathway. Leukemia. 2014;28:236–8. doi: 10.1038/leu.2013.261. PMID:24091833. [DOI] [PubMed] [Google Scholar]
- 23.Munir S, Andersen GH, Woetmann A, Ødum N, Becker JC, Andersen MH. Cutaneous T cell lymphoma cells are targets for immune checkpoint ligand PD-L1-specific, cytotoxic T cells. Leukemia. 2013;27:2251–3. doi: 10.1038/leu.2013.118. PMID:23660624. [DOI] [PubMed] [Google Scholar]
- 24.Arber DA, Orazi A, Hasserjian R, Borowitz MJ, Beau MM Le, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–406. doi: 10.1182/blood-2016-03-643544. PMID:27069254. [DOI] [PubMed] [Google Scholar]
- 25.Kjær L, Cordua S, Holmström MO, Thomassen M, Kruse TA, Pallisgaard N, Larsen TS, de Stricker K, Skov V, Hasselbalch HC. Differential Dynamics of CALR Mutant Allele Burden in Myeloproliferative Neoplasms during Interferon Alfa Treatment. PLoS One. 2016;11:e0165336. doi: 10.1371/journal.pone.0165336. PMID:27764253. [DOI] [PMC free article] [PubMed] [Google Scholar]