

ABSTRACT
Adoptive transfer of T cells expressing chimeric antigen receptors (CARs) is an effective immunotherapy for B-cell malignancies but has failed in some solid tumors clinically. Intracerebral tumors may pose challenges that are even more significant. In order to devise a treatment strategy for patients with glioblastoma (GBM), we evaluated CARs as a monotherapy in a murine model of GBM. CARs exhibited poor expansion and survival in circulation and failed to treat syngeneic and orthotopic gliomas. We hypothesized that CAR engraftment would benefit from host lymphodepletion prior to immunotherapy and that this might be achievable by using temozolomide (TMZ), which is standard treatment for these patients and has lymphopenia as its major side effect. We modelled standard of care temozolomide (TMZSD) and dose-intensified TMZ (TMZDI) in our murine model. Both regimens are clinically approved and provide similar efficacy. Only TMZDI pretreatment prompted dramatic CAR proliferation and enhanced persistence in circulation compared to treatment with CARs alone or TMZSD + CARs. Bioluminescent imaging revealed that TMZDI + CARs induced complete regression of 21-day established brain tumors, which correlated with CAR abundance in circulation. Accordingly, TMZDI + CARs significantly prolonged survival and led to long-term survivors. These findings are highly consequential, as it suggests that GBM patients may require TMZDI as first line chemotherapy prior to systemic CAR infusion to promote CAR engraftment and antitumor efficacy. On this basis, we have initiated a phase I trial in patients with newly diagnosed GBM incorporating TMZDI as a preconditioning regimen prior to CAR immunotherapy (NCT02664363).
KEYWORDS: glioma, glioblastoma, brain tumor, immunotherapy, lymphopenia, temozolomide, adoptive transfer, chimeric antigen receptor
Introduction
Chimeric antigen receptors (CARs) are recombinant transmembrane proteins comprised of an antibody-derived single-chain variable fragment coupled to the intracellular signaling components of naturally occurring T cell receptors (TCRs).1 CARs can be retrovirally expressed on the cell surface of polyclonal T cells to redirect them against target antigens independent of native TCR specificity or major histocompatibility complex (MHC) expression by target tumor cells.2 The adoptive cell transfer of CARs has elicited complete remission rates of up to 90% in patients with refractory hematological cancers where CARs have targeted CD19,3-5 raising interest in adapting this technology for use against solid tumors including glioblastoma (GBM).6-12
GBM is the most common and deadly primary malignant brain tumor. Despite an aggressive multimodal standard of care, prognosis and quality of life remain exceptionally poor and patients experience a 5-year survival rate of just 5.1%.13 Recent preclinical studies have shown that CARs targeting the tumor-specific driver mutation EGFRvIII are curative in mice bearing established syngeneic GBM tumors after total body irradiation (TBI) pre-treatment.9 Preparative host lymphodepletion is believed to enhance adoptive T-cell immunotherapy through the upregulation and improved bioavailability of homeostatic gamma chain cytokines (e.g. IL-7 and IL-15), which become available for CAR consumption in the absence of competition from endogenous lymphocytes.14,15 This, in turn, promotes CAR survival and homeostatic proliferation during the recovery phase from lymphopenia. TBI, however, risks significant collateral toxicity without necessarily providing a direct antitumor benefit. Nonetheless, host preconditioning remains a critical component to this strategy as intravenously delivered CARs fail to treat established tumors in the absence of lymphodepletion.9
Patients with GBM are currently treated with temozolomide (TMZ) chemotherapy under the clinical standard of care,16,17 which is accompanied by lymphodepletion as its major side effect.18 In the present study, we evaluated TMZ regimens that mimic the standard (TMZSD) and dose-intensified (TMZDI) regimens administered in the clinic based on the lymphocytic nadir and kinetics of endogenous immune cell rebound observed in patients with GBM. We show that greater lymphodepletion induced by TMZDI is necessary to stimulate the proliferation and persistence of CARs in circulation, as CAR abundance in blood correlated with tumor regression.
Results
Systemic CARs fail as a monotherapy and require preparative host lymphodepletion for efficacy against established syngeneic and orthotopic glioblastoma
We established a protocol for efficient retroviral transduction of the EGFRvIII-CAR transgene and confirmed CAR expression on the cell-surface of CD3+ T cells as previously described (Fig. 1A).19 Next, we sought to establish a relevant treatment strategy against a tumor model that adequately recapitulates the highly invasive features of GBM within the context of a widely available syngeneic mouse model. KR158B is a murine cell line derived from a spontaneous glioma in Trp53/Nf1 double-mutant mice.20 This cell line was previously engineered to express firefly luciferase (KLuc), permitting a non-invasive approach to monitor tumor progression and responsiveness to therapy using bioluminescent imaging. We genetically engineered the KLuc cell line to express a murine homologue of the tumor-specific driver mutation, EGFRvIII, which is expressed exclusively by GBM and other cancers but not normal tissue.21-23 We evaluated the expression of EGFRvIII in KLuc-EGFRvIII tumor cells by flow cytometry (Fig. 1B) and by immunohistochemistry after tumor establishment in vivo (Fig. 1C). We also confirmed that this GBM model exhibits the classical and hallmark histopathological features of infiltrative GBM by H/E analysis (Fig. 1D).
Figure 1.

Systemic CARs fail as a monotherapy against orthotopic and syngeneic murine GBM. Flow cytometry analyses display (A) CAR expression on the cell surface of CD3+ T cells two days after retroviral transduction and (B) EGFRvIII expression by KLuc-EGFRvIII tumor cells compared to control. (C) 5 × 104 KLuc or KLuc-EGFRvIII tumor cells were implanted in C57 BL/6 mice and harvested to evaluate EGFRvIII expression by immunohistochemistry and (D) tumor characteristics by standard hematoxylin and eosin stain. (E) CARs were incubated with target tumor cells radiolabeled with Cr51 and display antigen-specific cytotoxicity in vitro (n = 3) (% specific lysis against KLuc-vIII vs. KLuc, t-test P = 0.0002). (F) CARs were infused intravenously into mice bearing 7-day established KLuc-EGFRvIII tumors as monotherapy (n = 5–7). Survival was monitored overtime as approved under Duke IACUC guidelines. Survival comparisons between CAR treated mice and control tumor alone did not reach statistical significance by Log-Rank statistical analysis, (Log-Rank, 1e6 CARs vs. tumor alone, P = 0.1955; 1e7 CARs vs. tumor alone, P = 0.5269; 1e8 CARs vs. tumor alone, P = 0.8155). Data are representative of at least 2 independent experiments.
Next, we evaluated the capacity of CARs to elicit antigen-specific cytotoxicity in vitro in a standard Cr51 release assay (Fig. 1E). CARs induced robust cell death and specific lysis against tumor cells expressing EGFRvIII at an effector:target ratio of 3:1 (t-test, P = 0.0002). Based on these data, we performed a dose-escalation study to evaluate the antitumor efficacy of CARs as a monotherapy against established brain tumors. We adoptively transferred 1 × 106, 1 × 107 or 1 × 108 CARs intravenously into mice bearing 7-day established KLuc-EGFRvIII tumors (Fig. 1F). CARs failed to induce a survival advantage in treated mice compared to controls at even the highest dose, which approaches the limit of what is logistically feasible (Log-Rank, 1e6 CARs vs. tumor alone, P = 0.1955; 1e7 CARs vs. tumor alone, P = 0.5269; 1e8 CARs vs. tumor alone, P = 0.8155). This finding, however, was unsurprising as we have previously reported the failure of systemic CARs as a monotherapy in the SMA560 GBM cell line that is syngeneic to the VMD/k murine model.9
We and others have previously shown that adoptive T-cell transfer strategies require preparative host lymphodepletion to achieve antitumor responses in both mice and humans.9,24-26 In order to corroborate these findings here, we pursued proof-of-principle studies using two separate experimental models. We evaluated the antitumor efficacy of CARs in RAG1−/− hosts (Fig. 2A), which are incapable of producing mature T and B cells and are therefore in a persistent state of lymphopenia, as well as in C57 BL/6 mice who were subjected to lymphodepleting 5 Gy total body irradiation (TBI) immediately prior to CAR infusion. In both models, CARs were infused intravenously into mice bearing these 7-day established radio-insensitive tumors. Strikingly, we found that CARs were capable of inducing long-term cures in a high proportion of RAG1−/− hosts (Log-Rank, P = 0.0361) and in C57 BL/6 mice who were pre-treated with lymphodepleting radiation (Log-Rank, P = 0.0004) (Fig. 2B). These findings collectively show that host lymphocyte reduction achieved through genetic or exogenous means enhances CAR efficacy against established brain tumors.
Figure 2.
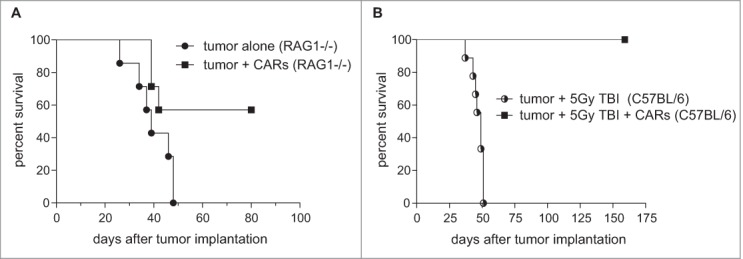
Host lymphopenia potentiates intravenous CAR immunotherapy against brain tumors. (A) CARs were infused intravenously into RAG1−/− mice bearing 7-day established KLuc-EGFRvIII tumors as monotherapy (n = 5–7) (Log Rank, P = 0.0361). (B) C57 BL/6 mice bearing 7-day established tumors were subjected to 5 Gy TBI and were either left untreated or received 1 × 107 CARs intravenously (n = 5–7) (Log Rank, P = 0.0004). Data are representative of at least 2 independent experiments.
Lymphodepletive temozolomide can be leveraged to enhance CAR persistence in peripheral blood circulation
Given the added risk of toxicity with TBI and its infrequent use in the clinic, we explored alternative preconditioning strategies with the goal of implementing CAR therapy for patients with newly diagnosed GBM. These patients routinely receive temozolomide (TMZ) chemotherapy concurrent with radiation and as maintenance therapy thereafter, which can induce varying grades of lymphopenia depending on the dose and schedule prescribed.27 TMZ is most commonly administered in either a standard (TMZSD; 200 mg/m2/day x 5 days per 28-day cycle) or dose-intensified (TMZDI; 100 mg/m2/day x 21 days per 28-day cycle) regimen that are equally efficacious but have different effects on the depletion and subsequent rebound of immune cells.28
We recently completed a phase I clinical trial in which newly diagnosed GBM patients were treated with TMZSD or TMZDI prior to and concurrent with immunotherapy (NCT00639639).29 We monitored patients with the goal of understanding which TMZ regimen, if any, would sufficiently precondition hosts prior to adoptive cell transfer with CARs. We studied the impact of TMZ on absolute counts of total lymphocytes and CD3+ T cells in patient blood over time in order to make a determination on the relative lymphotoxicity of either TMZ regimen. Patients who received TMZSD exhibited a significant decline from baseline in total lymphocyte counts that approached grade 3 lymphopenia (<500 cells/µL) after a single TMZ cycle, but this effect was found to be short lived (Fig. 3A). In contrast, TMZDI depressed total lymphocyte counts from baseline more severely when compared to TMZSD and induced a grade 3 lymphopenia through TMZ cycles 3 and 4. Similarly, we found that TMZDI had a more profound and sustained effect on CD3+ T cell depletion when compared to TMZSD (Fig. 3B). Importantly, the severity of total lymphocyte and CD3+ T cell depletion was exacerbated with successive cycles of TMZDI, whereas cell counts rebounded after completion of TMZSD cycle 2.
Figure 3.

Temozolomide chemotherapy sufficiently preconditions hosts to enhance CAR expansion and persistence in peripheral blood. (A) Peripheral blood sampling in two patient cohorts with newly diagnosed GBM receiving either TMZSD (200 mg/m2/day x 5 days) or TMZDI (100 mg/m2/day x 21 days) prior to and concurrent with immunotherapy (NCT00639639). Patient blood was monitored for absolute counts of lymphocytes and (B) CD3+ T cells over time, showing that for both absolute lymphocyte (Mixed model, P = 0.0029) and CD3+ counts (Mixed model, P = 0.0012), TMZDI induced a more profound and sustained depletion with continuous cycles compared to TMZSD. (C) Non-tumor bearing C57 BL/6 mice (n = 5) were either left untreated, subjected to 5 Gy TBI, or received an intraperitoneal injection of TMZSD (60 mg/kg/day x 5 days) or TMZDI (400 mg/kg/day x 1 day) where indicated. Lymphocyte counts were monitored by CBC 48 hours after treatment (Exact Wilcoxon of TBI vs. TMZDI, P = 0.1270; Exact Wilcoxon of TMZSD vs. TMZDI, P = 0.0317) and (D) over time (P < 0.0001). (E) Similarly, the effect of treatment on CD3+ T cell counts was determined by flow cytometry 24 hours after treatment (Exact Wilcoxon of TBI vs. TMZDI, P = 0.0079); Exact Wilcoxon of TMZSD vs. TMZDI, P = 0.0079) and (F) over time (P = 0.0161). (G) 1 × 107 CARs were administered intravenously 24 h after treatment, and CARs were monitored serially in peripheral blood circulation over time (n = 5). Data are representative of at least 2 independent experiments. Dotted lines represent grade 2 (<800 cells/µL) and grade 3 (<500 cells/µL) lymphopenia.
Next, we performed experiments in our mouse model to determine whether the lymphotoxic effects of TMZ could be leveraged to favorably attenuate CAR persistence during recovery from lymphodepletion. We established TMZ doses to mimic relative differences in the lymphocytic/CD3+ T cell nadir and rebound kinetics characteristic of TMZSD and TMZDI in the clinic. Non-tumor bearing mice were administered TMZ, and lymphocyte/CD3+ T cell counts were measured in peripheral blood circulation over time. By 48 hours after treatment, mice exhibited an average lymphocyte count of 1,036 cells/µL following TMZSD and 302 cells/µL following TMZDI, the latter of which was comparable to the nadir induced by TBI (394 cells/µL) (Exact Wilcoxon of TMZSD vs. TMZDI, P = 0.0317; Exact Wilcoxon of TBI vs. TMZDI, P = 0.1270) (Fig. 3C). Lymphocyte rebound kinetics were also assessed in blood circulation over time at 2, 14, and 28 days after TMZ or TBI. Lymphocyte counts in TMZSD-treated mice partially rebounded within a 14-day timeframe, whereas depletion was stained in TMZDI−treated mice in a manner virtually identical to mice who received TBI (Fig. 3D). These relative relationships also held true for CD3+ T cell nadirs (TMZSD 591 cells/µL; TMZDI, 146 cells/µL; and TBI, 24 cells/µL) (Exact Wilcoxon of TMZSD vs. TMZDI, P = 0.0079; Exact Wilcoxon of TBI vs. TMZDI, P = 0.0079) (Fig. 3E). CD3+ counts remained relatively unchanged in all cohorts for up to 14 days (Fig. 3F). Notably, CD3+ counts rebounded by day 28 in TBI-treated mice whereas counts remained depressed in TMZDI-treated mice.
Next, we explored how these two different lymphodepletive profiles would impact CAR numbers in blood compared to CARs administered alone or after TBI, which served as negative and positive controls, respectively. Non-tumor bearing mice were pre-treated with vehicle, TMZSD, TMZDI, or TBI followed by adoptive intravenous transfer of 1 × 107 CARs. As expected, CARs administered into mice pre-treated with vehicle declined within 7 days. Pre-treatment with TMZSD did not prompt an improvement in CAR numbers over time. In contrast, however, CAR numbers in mice pre-treated with TMZDI did not decline, persisted in blood circulation for up to 28 days, and exhibited similar counts over time in mice pre-treated with TBI (Fig. 3G).
Dose-intensified temozolomide plus CAR therapy significantly prolongs survival against a highly stringent 21-day established model of glioblastoma
In order to determine whether TMZ preconditioning would augment CAR efficacy against advanced disease, we performed experiments in mice bearing 21-day established KLuc-EGFRvIII brain tumors, which are chemotherapy-insensitive. A total of 5 × 104 tumor cells were implanted on day 0, and mice received a treatment schedule according to the schema shown (Fig. 4A).
Figure 4.
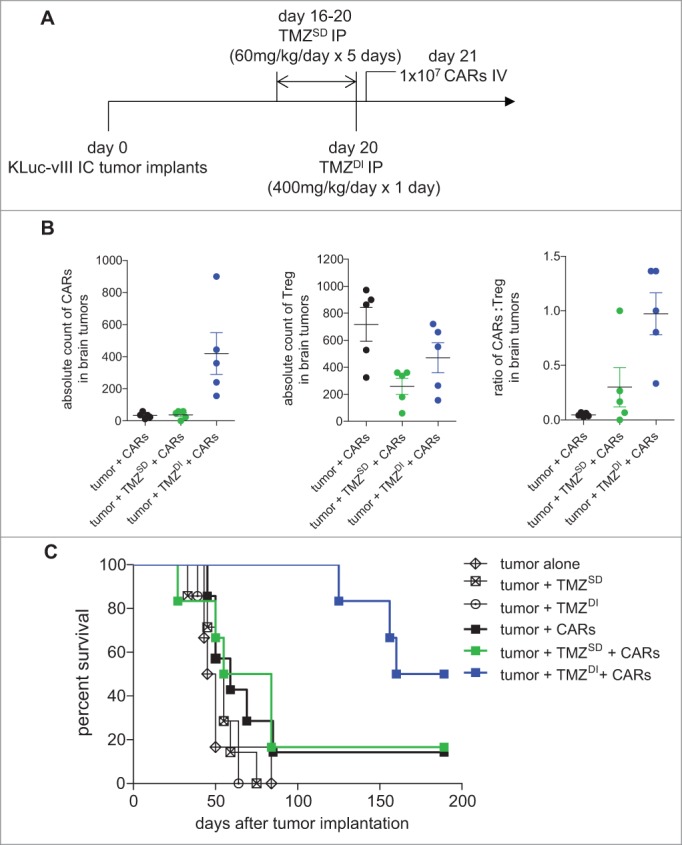
Dose-intensified temozolomide plus CAR immunotherapy significantly enhances survival against 21-day established glioblastoma. (A) C57 BL/6 mice were implanted with 5 × 104 KLuc-EGFRvIII tumor cells intracranially and allowed to engraft for 21 days prior to the adoptive intravenous transfer of 1 × 107 EGFRvIII-specific CARs. Where indicated, mice received TMZ intraperitoneally according to the treatment schema shown. (B) Mice were sacrificed 7 days after treatment in order to enumerate CARs and Treg in brain tumor specimens (n = 5) (Exact Wilcoxon; CARs: TMZSD vs. TMZDI, P = 0.0119; Tregs: TMZSD vs. TMZDI, P = 0.2948; CARs: Treg ratio: TMZSD vs. TMZDI, P = 0.0465) (C) Mice were screened for a minimum tumor burden of 1 × 106 p/s/cm2/sr on day 21 prior to the start of immunotherapy to ensure advanced disease. Mice were followed for survival (n = 6–7) (Log Rank, P = 0.0208). Data are representative of at least 2 independent experiments.
First, we studied whether TMZ enhanced CAR numbers in the brain and whether it altered regulatory T cell (Treg) numbers, which are otherwise elevated in GBM and have been previously shown to functionally inhibit CARs through both direct and indirect mechanisms. To do this, we sacrificed mice 7 days after intravenous CAR infusion (i.e. 28 days after tumor implantation and 8 days after TMZ administration) and enumerated CARs and Tregs in brain tumor specimens. CARs were significantly elevated in brain tumors in mice pretreated with TMZDI (Exact Wilcoxon of Control vs. TMZDI, P = 0.0119; TMZSD vs. TMZDI, P = 0.0119;), whereas CAR numbers were unaffected by TMZSD in comparison to control mice (Exact Wilcoxon of Control vs. TMZSD, P = 0.7496) (Fig. 4B). In contrast, TMZ pretreatment had no statistically significant impact on brain infiltrating Tregs when compared to controls (Exact Wilcoxon of Control vs. TMZSD, P = 0.0556; Control vs. TMZDI, P = 0.2222; TMZSD vs. TMZDI, P = 0.2948). Next, we sought to determine the relative ratios of CAR: Treg in tumor specimens based on previous evidence showing that higher ratios of effector T cells: Treg associate with favorable outcomes in cancer.31-33 Our analyses revealed that mice treated with TMZDI + CARs exhibited significantly higher ratios in the brain when compared to mice receiving treatment with CARs alone or TMZSD + CARs (Exact Wilcoxon of Control vs. TMZSD, P = 0.2222; Control vs. TMZDI, P = 0.0079; TMZSD vs. TMZDI, P = 0.0465).
We next tested the hypothesis that TMZDI would sufficiently potentiate CAR immunotherapy to provide a survival advantage within the context of these advanced tumors. KLuc-EGFRvIII tumor cells are genetically modified to express firefly luciferase, which permits sensitive in vivo bioluminescent tracking of disease burden upon the in vivo catalysis of the substrate luciferin.30 Mice were imaged to confirm tumor burden prior to the initiation of TMZ and again on day 21 to screen for a minimum threshold of disease. All mice in the study recorded tumor burden signals of ≥1 × 106 p/s/cm2/sr on day 21 prior to the start of immunotherapy in order to ensure the evaluation of CARs against advanced disease. Immunotherapy with CARs alone or after preconditioning with TMZSD failed to elicit a survival advantage when compared to untreated mice or mice treated with TMZ alone, although both groups contained a single long-term survivor. In marked contrast, treatment with TMZDI + CARs led to a median survival of 174.5 days compared to 47.5-69.5 days in the other cohorts shown (Log-Rank, P = 0.0208) (Fig. 4C). Mice were followed for a total of 189 days after initial tumor implantation, and we observed a 50% (3/6) long-term survival rate in the TMZDI + CARs treatment cohort.
Tumor regression correlates with CAR abundance in blood circulation
Next, we attempted to understand the kinetics of tumor regression by visualizing and non-invasively monitoring tumor responsiveness to therapy over time. B6(Cg)-Tyrc-2J/J (B6 albino) mice received 5 × 104 intracranial implants of KLuc-EGFRvIII tumor cells, and disease burden was evaluated prior to TMZ initiation and on day 21 prior to CAR immunotherapy to screen for threshold tumor burden, as described above. Mice were then imaged serially every 7 days (Fig. 5A). Importantly, the vast majority of tumors in control mice and mice treated with CARs alone or TMZSD + CARs grew over time unabated. However, we observed complete tumor regression in 5/8 mice (62.5%) who received TMZDI + CARs, with dramatic signal reductions occurring within 7–14 days after treatment.
Figure 5.

CAR abundance in peripheral blood is enhanced by TMZ and associated with reduced tumor burden. (A) B6(Cg)-Tyrc-2J/J (B6 albino) mice (n = 7–8) received 5 × 104 intracranial implants of KLuc-EGFRvIII tumor cells and were non-invasively monitored via bioluminescent imaging to track tumor burden and responsiveness to therapy over time. Mice were imaged and randomized on day 21 prior to CAR immunotherapy for a baseline measurement of tumor burden. 1 × 107 CARs were infused intravenously on day 21, and mice were serially imaged every 7 days afterward. (B, C) Mice were bled retro-orbitally to enumerate CARs in peripheral blood circulation by absolute count (day 28; Exact Wilcoxon TMZSD vs TMZDI, P = 0.0006, Exact Wilcoxon CARs alone vs TMZDI, P = 0.0002). (D) Signal measurements of tumor burden in individual mice on the log10 scale were compared to the corresponding values of CAR counts on the log10 scale obtained from blood circulation day 28 (Pearson coefficient = −0.8261, P < 0.0001). Data are representative of at least 2 independent experiments.
We enumerated CAR abundance in blood circulation over time and found that TMZDI prompted a dramatic expansion of CARs that persisted for several weeks (Peak analysis; Exact Wilcoxon of CARs alone vs. TMZDI at peak, P = 0.0002; Exact Wilcoxon of TMZSD vs. TMZDI at peak, P = 0.0011) (Fig. 5B). CAR counts on day 28 were significantly higher in TMZDI + CAR treated mice compared to all other groups (TMZDI + CARs, mean of 102 cells/µL blood; tumor + CARs, mean of 12.9 cells/µL blood; tumor + TMZSD + CARs, mean of 23.1 cells/µL blood) (Exact Wilcoxon of CARs alone vs. TMZDI on day 28, P = 0.0002; Exact Wilcoxon of TMZSD vs. TMZDI on day 28, P = 0.0006) (Fig. 5C). We compared absolute CAR counts in individual mice on the log10 scale with their corresponding bioluminescent signals on the log10 scale at day 28, and importantly, found that higher CAR numbers corresponded with lower tumor burden (Pearson coefficient = −0.8261, P < 0.0001) (Fig. 5D).
Discussion
TMZ became a routine component of clinical management for GBM in 2005 after it was shown to extend survival in patients with newly diagnosed disease.16,17 Lymphotoxicity is commonly observed as a byproduct of TMZ use, and the recovery phase from lymphopenia has been previously exploited to potentiate vaccine responses against GBM in mice and humans.27,33,34 This phenomenon, however, has never been studied in the context of CAR therapy, which offers an advantage over active immunotherapy as it incorporates the ex vivo activation and expansion of tumor-specific T cells without necessarily relying on this activity to occur in vivo. The promise of CAR therapy is underscored by the outstanding clinical success of anti-CD19 CARs targeting B-cell malignancies in recent years.3-5 As such, there has been a push to translate this strategy into the clinic for GBM on the heels of highly encouraging preclinical data,7-10 and several trials are currently ongoing (NCT02442297, NCT02209376,10 NCT02844062, NCT01109095, NCT01454596,6 NCT022 0836211). The purpose of this study was to identify a strategy for the optimal and rational integration of CARs into current clinical treatment for GBM.
Our work shows that preparative host lymphodepletion may be required for effective CAR immunotherapy targeting GBM, and importantly, that TMZ can be strategically leveraged for this purpose. Lymphodepletion improves the in vivo engraftment and functionality of transferred T cells through reduced competition for gamma chain cytokines14,15 and the depletion of inhibitory immune cells (e.g. Treg) that otherwise counteract productive anti-cancer immune responses.24,26 Although several agents have been previously studied for the purpose of host conditioning, none of them are routinely administered to GBM patients as standard of care. Our goal was to determine whether existing clinical treatment for GBM could establish a favorable in vivo environment to maximize CAR efficacy in order to avoid introducing an additional preparatory regimen for this purpose. We show that TMZ-induced lymphopenia induces both a dramatic proliferation of CARs in vivo and enhances CAR persistence in blood circulation, which was associated with reduced tumor burden.
It is important to note that although these data are promising, it remains unclear if this strategy is equally beneficial against tumors in settings where the target antigen, EGFRvIII, is heterogeneously expressed. Previous studies combining lymphodepletive total body irradiation (TBI) with CAR therapy have demonstrated evidence of epitope spreading and long-term immunological protection against antigen-negative tumors, suggesting that an acute depletion of host lymphocytes does not necessarily preempt the generation of de novo immune responses. We are currently evaluating the short- and long-term immunological effects of a TMZDI + CAR treatment strategy against heterogeneous GBM using our model systems.
In the present study, CARs were evaluated using a fully syngeneic, orthotopic and TMZ-resistant mouse model of GBM after allowing brain tumors to engraft for a total of 21 days. We chose this model in order to mimic the extent of disease encountered at clinical presentation as closely as possible, as mice bearing these 21-day established tumors are on the cusp of becoming neurologically symptomatic. CARs infused as a monotherapy or in combination with TMZSD failed to elicit a therapeutic impact on tumor burden or survival, whereas TMZDI + CARs induced dramatic tumor regression within 7 days of treatment, significantly enhanced survival, and led to long-term survivors. Despite these promising data, however, we observed instances in which tumors either did not respond to treatment or recurred after an initial response. We believe these outliers can be explained by either an insufficient penetration of CARs into tumor microenvironments, an abnormally short lifespan, or by reduced functionality owing to immunosuppression. These studies are ongoing.
On the basis of the data presented here, we have rationally designed and initiated a phase I clinical trial incorporating TMZDI as pre-treatment prior to the intravenous infusion of EGFRvIII-specific CARs in patients with newly diagnosed EGFRvIII-positive GBM (NCT02664363). To our knowledge, this is the first report demonstrating the critical need to substitute TMZSD with TMZDI in order to sufficiently precondition hosts prior to CAR immunotherapy. The work reported here is encouraging as it reveals an opportunity to incorporate CARs into current clinical treatment, mitigates the need for an additional cyto-reductive preconditioning measure, and provides a logical path forward to fully leveraging the therapeutic effects of CARs in brain tumor patients.
Materials and methods
CAR generation
The EGFRvIII-specific CAR is a third generation vector defined by inclusion of the CD28, 4-1BB, and CD3ζ intracellular signaling moieties on the CAR transgene. The single-chain variable fragment is derived from human monoclonal antibody (mAb) 139 and has been previously described.9 To produce CARs, retroviral supernatant was produced by co-transfection of HEK 293 T cells using Lipofectamine 2000 Transfection Reagent (Invitrogen), the CAR retrovirus, and pCL-Eco helper plasmid (Imgenex). On the same day as transfection, fresh spleens were isolated from donor C57 BL/6NCr mice (Charles River Laboratories) and manually disrupted on a 10 cm dish using a plunger. Red blood cells (RBCs) were lysed, and splenocytes were cultured in mouse T-cell media supplemented with 50 U/mL IL-2 and 2.5 µg/mL Concanavalin A. After 48 hours, splenic T cells were transduced with retroviral supernatant (CAR alone or 1:1 with CAR and firefly luciferase) on non-tissue-culture 24-well plates previously coated with 0.5 mL of RetroNectin (Clontech) at a concentration of 25 µg/mL in PBS. Cells were plated at a density of 1 × 106/mL in viral supernatant supplemented with 50 U/mL IL-2. Cells were split every 24 hours for two days.
Generation of the KLuc-EGFRvIII murine glioma cell line
KR158B-Luciferase (KLuc) is a C57 BL/6 murine malignant glioma cell line that was kindly provided by Tyler Jacks from the Massachusetts Institute of Technology.20 This cell line was retrovirally engineered to stably express murine EGFRvIII using a 2nd generation lentivirus with no antibiotic resistance. Briefly, parental KLuc glioma cells were transduced with lentivirus generated with the following plasmids: pMD2G, psPax2 and pak19-FUGW containing murine EGFRvIII (Addgene 12259, 12260 and 14883, respectively). After lentiviral transduction, cells were single-cell sorted into 96-well plates based on EGFRvIII surface expression detected with the ch-LA84 anti-EGFRvIII antibody and a secondary fluorescent anti-human IgG antibody. Single cell clones were allowed to grow and then re-screened for EGFRvIII expression. The highest EGFRvIII-expressing clones underwent a second round of single-cell cloning using limiting dilution. The final stable EGFRvIII-expressing cell clone (KLuc-EGFRvIII) was then selected based on EGFRvIII surface expression levels, in vivo tumorigenicity experiments, and cell line authentication which verified the cell lines species and tissue origin and lack of cross-contamination and also compared molecular satellite markers with those in the parental KLuc cell line.
Chromium release assay
To evaluate CAR reactivity and antigen-specific cytotoxicity, CARs were co-incubated with 1 × 105 KLuc-EGFRvIII or KLuc tumor cells previously radiolabeled with chromium51 (Cr51). Briefly, tumor cells were harvested, washed twice in culture medium, counted, and resuspended at a concentration of 1 × 107/mL. Tumor cells were labeled with 100 µCi of radioactive Cr51 for 90 minutes, washed twice in R10 mouse T-cell media, and plated on a 96-well round bottom plate. CARs were plated in a serial dilution to generate various effector:target ratios and were incubated for a total of 4 hours.
TMZ administration and radiation
TMZ was dissolved in a solution of 85% saline and 15% dimethyl sulfoxide (DMSO). Briefly, saline and TMZ + DMSO were warmed separately in a water bath until TMZ was completely dissolved in DMSO. Saline was added to the dissolved solution and vortexed thoroughly. C57 BL/6 mice (Charles River Laboratories) were weighed and injected intraperitoneally at the calculated doses, as indicated. TMZDI was defined as a single dose of 400 mg/kg/day x 1 day; TMZSD was defined as 60 mg/kg/day x 5 days. TMZ administration concluded 24 hours prior to CAR therapy. For experiments requiring radiation, mice were subjected to 5 Gy TBI 24 hours before CAR therapy.
GBM patient selection and immune monitoring
Absolute lymphocyte and CD3+ cell counts were sampled at various time points under an IRB-approved protocol for patients with glioblastoma receiving dendritic cell vaccination therapy with either cycles of TMZSD or TMZDI. The clinical protocol and informed consent were approved by the U.S. Food and Drug Administration and Institutional Review Board (IRB) at Duke University for this study (FDA-IND-BB-12839, Duke IRB Pro00003877, NCT00639639). Prior to the study drug regimen, patients under this protocol with histologically confirmed, newly diagnosed GBM were treated with standard of care therapy including gross total resection followed by a six week course of standard external beam radiation therapy (XRT) at 60 Gy with concurrent TMZ at a targeted daily dose of 75 mg/m2/d. Prior to XRT/TMZ, all patients underwent initial leukapheresis with peripheral blood sampling for baseline lymphocyte/CD3+ T cell counts. At four weeks following standard XRT/TMZ, patients in separate cohorts either underwent the first cycle of TMZSD at (200 mg/m2/day for five days) or TMZDI (100 mg/m2/day for 21 days) as conditioning regimens for antigen-specific dendritic cell (DC) vaccination given on Day 21 ± 2 of a 28-day cycle or Day 23 ± 1 of a 28-day cycle, respectively. Peripheral blood samples were obtained on the days of DC vaccination prior to vaccination to evaluate lymphopenic effects from either TMZ regimen. From TMZ cycle 2, patients received monthly DC vaccines in conjunction with subsequent TMZ cycles every 5 ± 1 weeks for a total of 6 to 12 cycles if patients had not progressed, but all patients received a minimum of four monthly TMZ cycles. Peripheral blood samples were collected in ACD tubes, and cell counts were determined by CBC with differential for lymphocyte percent and counts (1 × 106/mL) and flow cytometry of sampled blood for CD3+ cell percent and counts (cells/µL) from the Duke University Clinical Laboratory. For absolute lymphocyte counts, the standard grading scheme was utilized to classify grade 2 lymphopenia (< 800 cells/mL) and grade 3 lymphopenia (< 500 cells/mL) according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.0. Sampling at each time point (baseline and TMZ cycles 1–4) was comprised of available patient samples reaching no less than 90% of each cohort.
CAR enumeration in murine peripheral blood
CARs were monitored in blood circulation by retro-orbitally bleeding mice at indicated time points. Whole blood was collected into heparinized tubes, and 50 µl was stained using standard flow cytometry staining procedures.9 Briefly, whole blood was stained using anti-CD4 FITC, anti-CD3 PE, anti-CD8 PerCPCy5.5, and a custom EGFRvIII-specific CAR-AlexaFluor 647 multimer developed in our laboratory. Whole blood was stained in the antibody cocktail for 20 minutes in 150 µl FACS buffer (2% BSA in PBS) hidden from light. RBCs were lysed using 1 mL 1x BD FACS Lysing Solution (BD Bioscience, Cat# 349202). All samples were analyzed on a FACSCalibur flow cytometer (BD Biosciences). Absolute cell counts were enumerated using Flow-Count Fluorospheres from Beckman Coulter (Cat# 7547053) according to manufacturer instructions.
CAR enumeration in murine brain tumors
Tumors were minced, incubated in 100 units/mL collagenase IV (Thermo Fisher Scientific, Waltham, MA) and RPMI supplemented with 10% FBS for 30 minutes at 37°C in a Stomacher machine set at normal speed, washed through 40-micron nylon cell strainers (Falcon; BD Biosciences) in PBS with 2% FBS. After washing, cells were lysed using 1X BD Pharm Lyse Buffer (BD Biosciences). Cells were immediately stained and subsequently analyzed by flow cytometry (Live/Dead-PacOrange, Invitrogen; CD4-PE, H129.19, BD Biosciences; CD3-AF488, 145-2C11, BD Biosciences; CD8-PerCP-Cy5.5, 53–6.7, BD Biosciences; FOXP3-APC, FJK-16 s, eBioscience; EGFRvIII-specific CAR-AlexaFluor 647 multimer). Intracellular staining was performed for FOXP3 using FOXP3 Transcription Factor Staining Buffer Kit (ThermoFisher Scientific; Invitrogen).
Bioluminescent imaging
Bioluminescence was detected using a Xenogen IVIS Lumina XR Imaging System (Perkin Elmer), as previously described.7,35 Single mice were imaged 5–6 min after an intraperitoneal injection of D-luciferin (150 mg/kg). Images were acquired for 15 seconds. Data were analyzed using Living Image software (Perkin Elmer) by measuring signal intensity through region of interest analysis.
Statistical analyses
Statistical differences in cell counts among groups were evaluated using the exact Wilcoxon rank sum test. The Kaplan-Meier estimator was used to generate survival curves, and differences in survival distributions were assessed using a Log-Rank test. Differences in patterns of change in cell counts over time were assessed using a mixed model following a 1st degree autoregressive model for the covariance that included a time interaction with treatment. The correlation between the log-transformed CAR cell count and log-transformed bioluminescence was assessed using the Pearson coefficient. P-values have not been adjusted for multiple testing.
Author contributions
Conception and experimental design: C.M. Suryadevara, R. Desai, J.E. Herndon II, B.D. Choi, J.H. Sampson, L. Sanchez-Perez. Acquisition of Data: C.M. Suryadevara, R. Desai, M. L. Abel, K. Riccione, K.A. Batich, S.H. Shen, P.C. Gedeon, P. Chongsathidkiet, A.A. Elsamadicy, D.J. Snyder, B.D. Choi. Analysis and data interpretation: C.M. Suryadevara, R. Desai, K.A. Batich, P. Chongsathidkiet, A.A. Elsamadicy, J.E. Herndon II, P. Healy, B.D. Choi, G.E. Archer, P.E. Fecci, J.H. Sampson, L. Sanchez-Perez. Administrative, technical, and material support: D.J. Snyder, J.E. Herndon II, G.E. Archer, P.E. Fecci, J.H. Sampson, L. Sanchez-Perez. Study supervision: D.J. Snyder, G.E. Archer, P.E. Fecci, J.H. Sampson, L. Sanchez-Perez. Manuscript preparation: all authors.
Funding Statement
This work was supported by the HHS | National Institutes of Health (NIH) (5R01-NS085412-04) HHS | National Institutes of Health (NIH) (5R01-CA177476-04) HHS | National Institutes of Health (NIH) (5R01-NS086943-03) HHS | National Institutes of Health (NIH) (T32-CA009111) HHS | National Institutes of Health (NIH) (5U01-NS090284-02) HHS | National Institutes of Health (NIH) (5R25-NS065731-08) HHS | National Institutes of Health (NIH) (5P50-CA190991-02) HHS | National Institutes of Health (NIH) (4P01-CA154291-05).
Disclosure of potential conflicts of interest
The authors have no potential conflicts of interest to disclose.
Acknowledgments
The authors thank Steven A. Rosenberg and Richard A. Morgan of the Surgery Branch at the National Cancer Institute for providing us with the EGFRvIII CAR retroviral construct.
References
- 1.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–4. doi: 10.1073/pnas.90.2.720. PMID:8421711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566–81. doi: 10.1038/nrc.2016.97. PMID:27550819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al.. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England journal of medicine. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. PMID:25317870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al.. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28. doi: 10.1016/S0140-6736(14)61403-3. PMID:25319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al.. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science translational medicine. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. PMID:24553386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H, et al.. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23:1043–53. doi: 10.1089/hum.2012.041. PMID:22780919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G Sayour EJ, McLendon R, 2nd Herndon JE, Healy P, et al.. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One. 2014;9:e94281. doi: 10.1371/journal.pone.0094281. PMID:24722266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi BD, Suryadevara CM, Gedeon PC, Herndon JE, nd 2, Sanchez-Perez L, Bigner DD, Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J Clin Neurosci. 2014;21:189–90. doi: 10.1016/j.jocn.2013.03.012. PMID:24054399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, Schmittling RJ, Nair SK, Reap EA, Norberg PK, et al.. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20:972–84. doi: 10.1158/1078-0432.CCR-13-0709. PMID:24352643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, et al.. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Science translational medicine. 2015;7:275ra22. doi: 10.1126/scitranslmed.aaa4963. PMID:25696001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al.. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. The New England journal of medicine. 2016;375:2561–9. doi: 10.1056/NEJMoa1610497. PMID:28029927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sengupta S, Mao G, Gokaslan ZS, Sampath P.. Chimeric antigen receptors for treatment of glioblastoma: a practical review of challenges and ways to overcome them. Cancer Gene Ther. 2017;24:121–129. doi: 10.1038/cgt.2016.46. PMID:27767090. [DOI] [PubMed] [Google Scholar]
- 13.Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS, Statistical Report CBTRUS: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015;17(Suppl 4):iv1–iv62. doi: 10.1093/neuonc/nov189. PMID:26511214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, et al.. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–12. doi: 10.1084/jem.20050732. PMID:16203864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez-Perez L, Suryadevara CM, Choi BD, Reap EA, Sampson JH. Leveraging chemotherapy-induced lymphopenia to potentiate cancer immunotherapy. Oncoimmunology. 2014;3:e944054. doi: 10.4161/21624011.2014.944054. PMID:25610727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al.. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. PMID:19269895. [DOI] [PubMed] [Google Scholar]
- 17.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al.. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. PMID:15758009. [DOI] [PubMed] [Google Scholar]
- 18.Su YB, Sohn S, Krown SE, Livingston PO, Wolchok JD, Quinn C, Williams L, Foster T, Sepkowitz KA, Chapman PB, Selective CD4+ lymphopenia in melanoma patients treated with temozolomide: a toxicity with therapeutic implications. J Clin Oncol. 2004;22:610–6. doi: 10.1200/JCO.2004.07.060. PMID:14726505. [DOI] [PubMed] [Google Scholar]
- 19.Riccione K, Suryadevara CM, Snyder D, Cui X, Sampson JH, Sanchez-Perez L. Generation of CAR T cells for adoptive therapy in the context of glioblastoma standard of care. J Vis Exp. 2015;16(96). doi: 10.3791/52397. PMID:25741761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–13. doi: 10.1038/79075. PMID:10973261. [DOI] [PubMed] [Google Scholar]
- 21.Wikstrand CJ, Hale LP, Batra SK, Hill ML, Humphrey PA, Kurpad SN, McLendon RE, Moscatello D, Pegram CN, Reist CJ. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer research. 1995;55:3140–8. PMID:7606735. [PubMed] [Google Scholar]
- 22.Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, Biegel JA, Hayes RL, Wong AJ. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995;55:5536–9. PMID:7585629. [PubMed] [Google Scholar]
- 23.Sampson JH, Crotty LE, Lee S, Archer GE, Ashley DM, Wikstrand CJ, Hale LP, Small C, Dranoff G, Friedman AH, et al.. Unarmed, tumor-specific monoclonal antibody effectively treats brain tumors. Proc Natl Acad Sci U S A. 2000;97:7503–8. doi: 10.1073/pnas.130166597. PMID:10852962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al.. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. PMID:12242449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, et al.. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–9. doi: 10.1200/JCO.2008.16.5449. PMID:18809613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gattinoni L, Powell DJ Jr., Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–93. doi: 10.1038/nri1842. PMID:16622476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, McLendon RE, et al.. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13:324–33. doi: 10.1093/neuonc/noq157. PMID:21149254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi M, Jaeckle KA, et al.. RTOG 0525: A randomized phase III trial comparing standard adjuvant temozolomide (TMZ) with a dose-dense (dd) schedule in newly diagnosed glioblastoma (GBM). Journal of Clinical Oncology. 2011;29(5). doi: 10.1200/jco.2011.29.15_suppl.2006.. [DOI] [Google Scholar]
- 29.Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, Congdon KL, Reap EA, Archer GE, Desjardins A, et al.. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366–9. doi: 10.1038/nature14320. PMID:25762141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greer LF, 3rd Szalay AA. Imaging of light emission from the expression of luciferases in living cells and organisms: a review. Luminescence. 2002;17:43–74. doi: 10.1002/bio.676. PMID:11816060. [DOI] [PubMed] [Google Scholar]
- 31.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, et al.. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:18538–43. doi: 10.1073/pnas.0509182102. PMID:16344461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116:1935–45. doi: 10.1172/JCI27745. PMID:16778987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez-Perez LA, Choi BD, Archer GE, Cui X, Flores C, Johnson LA, Schmittling RJ, Snyder D, 2nd Herndon JE, Bigner DD, et al.. Myeloablative temozolomide enhances CD8(+) T-cell responses to vaccine and is required for efficacy against brain tumors in mice. PLoS One. 2013;8:e59082. doi: 10.1371/journal.pone.0059082. PMID:23527092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heimberger AB, Sun W, Hussain SF, Dey M, Crutcher L, Aldape K, Gilbert M, Hassenbusch SJ, Sawaya R, Schmittling B, et al.. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro Oncol. 2008;10:98–103. doi: 10.1215/15228517-2007-046. PMID:18079360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santos EB, Yeh R, Lee J, Nikhamin Y, Punzalan B, Punzalan B, La Perle K, Larson SM, Sadelain M, Brentjens RJ. Sensitive in vivo imaging of T cells using a membrane-bound Gaussia princeps luciferase. Nat Med. 2009;15:338–44. doi: 10.1038/nm.1930. PMID:19219023. [DOI] [PMC free article] [PubMed] [Google Scholar]