

ABSTRACT
Receptor activator of NF-κB ligand (RANKL) and its receptor RANK, are members of the tumor necrosis factor and receptor superfamilies, respectively. Antibodies targeting RANKL have recently been evaluated in combination with anti-CTLA4 in case reports of human melanoma and mouse models of cancer. However, the efficacy of anti-RANKL in combination with antibodies targeting other immune checkpoint receptors such as PD1 has not been reported. In this study, we demonstrated that blockade of RANKL improves anti-metastatic activity of antibodies targeting PD1/PD-L1 and improves subcutaneous growth suppression in mouse models of melanoma, prostate and colon cancer. Suppression of experimental lung metastasis following combination anti-RANKL with anti-PD1 requires NK cells and IFN-γ, whereas subcutaneous tumor growth suppression with this combination therapy is attenuated in the absence of T cells and IFN-γ. Furthermore, addition of anti-RANKL to anti-PD1 and anti-CTLA4 resulted in superior anti-tumor responses, irrespective of the ability of anti-CTLA4 isotype to engage activating FcR, and concurrent or delayed RANKL blockade was most effective. Early-during-treatment assessment reveals this triple combination therapy compared to dual anti-PD1 and anti-CTLA4 combination therapy further increased the proportion of tumor-infiltrating CD4+ and CD8+ T cells that can produce both IFN-γ and TNF. Finally, RANKL expression appears to identify tumor-specific CD8+ T cells expressing higher levels of PD1 which can be modulated by anti-PD1. These data set the scene for clinical evaluation of denosumab use in patients receiving contemporary immune checkpoint blockade.
KEYWORDS: CD8+ T cells, metastasis, RANK, RANKL, tumors
Introduction
Antibodies targeting the immune checkpoint receptor programmed death-1 (PD1) are indicated in the treatment of several malignancies.1,2,3,4,5 Importantly, studies in melanoma in both the high-risk adjuvant and advanced settings have demonstrated that anti-PD1 therapy is more efficacious and less toxic than an antibody targeting cytotoxic T-lymphocyte associated protein-4 (CTLA4).6,7,8 The combination of these two antibodies improves response rate and overall survival in melanoma, albeit at the expense of increased grade 3/4 immune related adverse events (irAEs).9 Nevertheless, there remains a proportion of patients who do not derive clinical benefit from anti-PD1 monotherapy or the combination of anti-PD1 and anti-CTLA4 due to primary resistance.10 Novel combinations in immunotherapy are therefore needed for these patients.
Receptor activator of NF-κB ligand (RANKL) and its receptor, RANK, are members of the tumor necrosis factor (TNF) and TNF receptor (TNFR) superfamilies, and their roles were initially defined in the context of T cell and dendritic cell (DC) interactions.11 However, their essential roles in bone homeostasis led to the widespread use of the human IgG2 anti-RANKL antibody, denosumab, as an anti-resorptive therapy in patients with conditions including bone metastasis.12,13,14 Intriguingly, two case reports reported that the concomitant treatment of patients with metastatic melanoma with denosumab and ipilimumab induced potent clinical efficacy above what was expected for ipilimumab alone, suggesting that RANKL on T cells may have immune checkpoint function.15,16 Recent preclinical data from our group evaluating this combination in mice supports an immune-based mechanism of the RANK-RANKL pathway in suppressing effector T cell function, independently of any effect on regulatory T cell populations, however these findings were only examined in the context of CTLA4 blockade.17 In addition, post-hoc analysis suggested improved overall survival in patients with bone metastatic NSCLC when treated with denosumab relative to zolendronate,18 with ongoing phase III trials assessing overall survival benefit with denosumab in advanced NSCLC (NCT02129699) and adjuvant high-risk breast cancer (NCT01077154).
Given the superior anti-tumor efficacy and safety profile of anti-PD1 therapies compared to anti-CTLA4, and the differential mechanism of action of PD1/PD-L1 blockade to CTLA4 blockade, the evaluation of anti-RANKL in combination with PD1 or PD-L1 blockade is of interest. Furthermore, the evaluation of whether clinical efficacy of anti-PD1 alone or in combination with anti-CTLA4 can be further improved with the addition of anti-RANKL is also of interest as this combination immunotherapy is the current standard of care for certain patients with melanoma. These questions are of particular current translational relevance, with a clinical trial in advanced melanoma set to assess the efficacy of combining denosumab with nivolumab (anti-PD1) and/or ipilimumab (anti-CTLA4) (NCT03161756). Preclinical insights into the mechanism of action of these duo and triple combination immunotherapies may inform future trial settings, as well as identify investigational biomarkers of efficacy. In this study, we demonstrate that the addition of anti-RANKL to anti-PD1/PDL1 or anti-PD1 and anti-CTLA4 improves anti-tumor efficacy in various mouse models of cancer which are largely resistant to single agent immunotherapy.
Results
Co-targeting RANKL and PD1/PD-L1 alone or with CTLA4 suppresses experimental lung metastases and subcutaneous tumor growth
Pre-treatment of wild type (WT) mice with anti-RANKL or anti-PD1 alone displayed modest protection from experimental B16F10 melanoma lung metastases, compared with the control immunoglobulin (cIg)-treated group, while combined anti-RANKL and anti-PD1 significantly improved metastatic control (Fig. 1A). A similar effect for this combination therapy was seen in WT mice injected intravenously with RM1 prostate carcinoma cells (Fig. 1B). Anti-RANKL also combined well with anti-PD-L1 to suppress both B16F10 and RM-1 experimental lung metastasis compared to monotherapy alone (Fig. 1 C–D).
Figure 1.
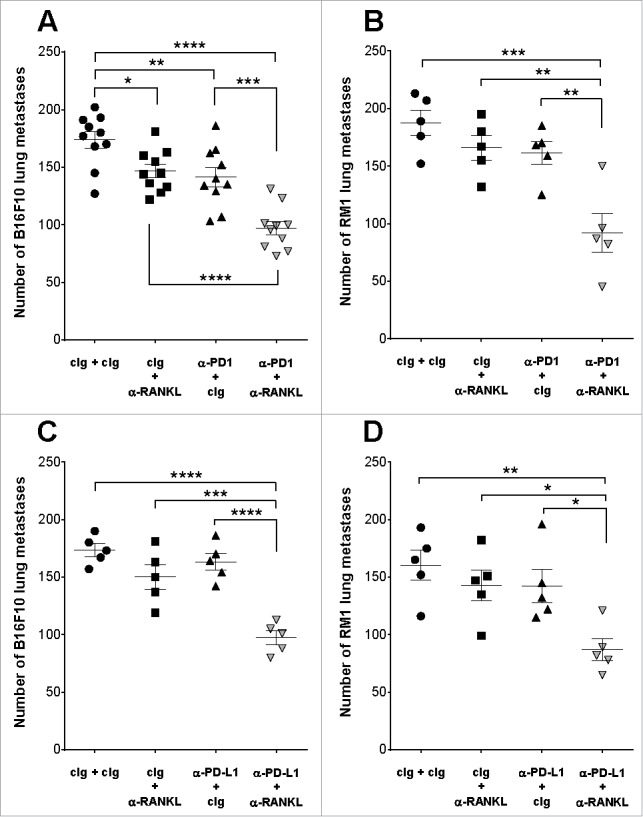
Co-targeting of RANKL and PD1or PD-L1 suppresses experimental lung metastasis. Groups of C57BL/6 wild type (WT) mice (n = 5/group) were injected i.v with 2 × 105 B16F10 melanoma cells (A, C) or RM-1 prostate carcinoma cells (B, D). Mice were treated on days -1, 0 and 2 (relative to tumor inoculation) with cIg, anti-RANKL, and/or anti-PD1 or anti-PD-L1 (200 µg i.p for each antibody) as indicated. Metastatic burden was quantified in the lungs after 14 days by counting colonies on the lung surface. Means ± SEM are shown. (A) is a pooled result from two independent experiments, all other experiments were performed once. Statistical differences between the indicated groups were determined by one way ANOVA with Tukey's post-test analysis (*p < 0.05, **p < 0.01, *** p < 0.001, **** p < 0.0001).
We next assessed the efficacy of dual blockade of PD-1 and RANKL in mice bearing s.c. CT26 colon or TRAMP-C1 prostate tumors (Fig. 2). In CT26 tumor-bearing mice, neither anti-RANKL nor anti-PD1 (250 μg) had significant effects as monotherapies, but significantly suppressed established tumor growth when combined (Fig. 2A). Given that combination immune checkpoint blockade (ICB) of PD1 and CTLA4 is an emerging standard of care in certain clinical contexts such as advanced melanoma,6 we next assessed if the addition of anti-RANKL could further improve the anti-tumor efficacy of anti-CTLA4 and anti-PD1 or anti-PD-L1 combination therapy (Fig. 2B–D). We first assessed anti-RANKL in combination using lower doses of anti-PD1 (100 μg) and the IgG2a isotype of anti-CTLA4 (clone 9D9)(50 μg) in the suppression of WT mice bearing established CT26 tumors (Fig. 2B). We and others previously demonstrated this IgG2a isotype of anti-CTLA4 (clone 9D9) depletes intratumor Tregs compared to other anti-CTLA4 isotypes19,20 and was the most efficacious in combination with anti-RANKL in the suppression of experimental lung metastases and subcutaneous tumor growth.17 Consistent with previous results, adding anti-RANKL to anti-PD1 significantly suppressed tumor growth, but triple combination therapy significantly suppressed growth of CT26 tumor-bearing mice compared to any dual combination therapy, and importantly, addition of anti-RANKL to anti-CTLA4+anti-PD1 improved the tumor rejection rate (Fig. 2B). Next, we assessed the efficacy of anti-PD-L1 in combination with anti-RANKL with or without anti-CTLA4 (mIgG2a) in the suppression of CT26 s.c. tumor growth (Fig. 2C). Compared with anti-PD-L1 alone, which (similarly to anti-RANKL and anti-PD1 monotherapies) has minimal efficacy, combination anti-PD-L1 and anti-RANKL significantly suppressed tumor growth (Fig. 2C). Additionally, triple combination of anti-PD-L1 and anti-RANKL with anti-CTLA4 (mIgG2a) was the most efficacious in suppression of CT26 s.c. growth; when this triple combination was specifically compared with dual ICB (anti-PD-L1 and anti-CTLA4), a small but significant difference was evident (Fig. 2C). Finally, the ability of triple combination therapy (anti-PD1+anti-CTLA4+anti-RANKL) to control tumor growth was also assessed in the autochthonous TRAMP transgenic mice,21 bearing Tramp-C1 subcutaneous tumor.22 In this setting where endogenous tumor-specific T cells may be tolerized, triple combination therapy was again most efficacious in controlling subcutaneous tumor growth with 15 out of 16 mice completely rejecting their tumors compared with select dual therapies and cIg (Fig. 2D).
Figure 2.
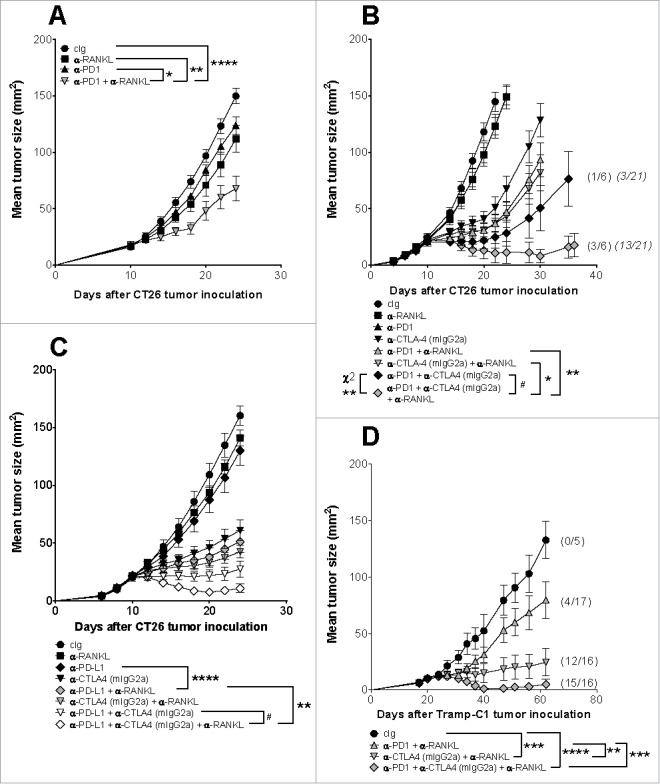
Co-targeting of RANKL with PD1/PD-L1 alone or in combination with CTLA4 suppresses subcutaneous tumor growth. Groups of BALB/c (A-C) wild type (WT) or TRAMP transgenic (D) mice (n = 5–17/group) were injected s.c. either with 1 × 105 CT26 (A-C), or with 1 × 106 Tramp-C1 prostate carcinoma (D) on day 0, and tumor growth was monitored. Mice were treated i.p. on (A-C) days 10, 14, 18 and 22 or (D) 20, 24, 28 and 32 (relative to tumor inoculation) with the following antibodies: cIg (to a total of 250–350 µg), anti-CTLA4 (9D9 mIgG2a, 50 µg), anti-PD1 (A, D: 250 µg; B: 100 µg), anti-PD-L1 (100 µg), anti-RANKL (200µg) or their combinations as indicated. Tumor sizes presented as mean ± SEM. (A, B) are representative of 2–3 independent experiments, all other experiments were performed once. Statistical differences between indicated groups were determined by one way ANOVA with Tukey's post-test analysis on the final day of measurement unless indicated otherwise (*p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). In (B), significant differences in tumor sizes between dual-antibody and triple-antibody combinations at day 30 were assessed; not shown on graph is the following comparison at day 22: anti-PD1 vs anti-PD1+anti-RANKL (****); #: at day 35, significant difference between the two remaining groups were determined by an unpaired t-test (* p < 0.05). In (C), # indicates significant difference for the indicated comparison determined by an unpaired t-test (*p< 0.05). In (B, D) parentheses: tumor rejection rates (no parentheses = no rejections). In (B), rejection rates for two identical experiments were pooled and presented in italicised parentheses; significant differences between rejection rates indicated groups were determined by Chi-squared (χ2) analysis (Fisher's exact test; **p < 0.01).
Various anti-CTLA4 isotypes combine activity with anti-RANKL and anti-PD1
We have previously shown that the mechanism of action of the anti-RANKL antibody is independent of activating Fc receptors (FcR),17 and this is also true for anti-PD1.23 Although in mice, full efficacy of anti-CTLA4 relies on FcR-mediated depletion of Tregs, the precise mechanism of action of the FDA-approved human IgG1 antibody, ipilimumab, remains controversial.20,24,25 Whether antibody-mediated depletion occurs, in addition to blockade of negative regulation of T-effector cell (Teff) function resulting from CTLA4 engagement by B7 ligands, is unclear. Multiple isotypes of the 9D9 clone of anti-CTLA4 have been generated, allowing assessment of whether depletion of Tregs is essential for optimal efficacy of combination anti-CTLA4 with anti-PD1 and anti-RANKL. The IgG1-D265A isotype of 9D9 contains a mutation which prevents its binding to all Fc-receptors and therefore cannot deplete Tregs by ADCC or ADCP.19 Therefore, we next assessed the anti-tumor efficacy of using the IgG1-D265A isotype of anti-CTLA4 (clone 9D9) in combination with anti-RANKL and anti-PD1, and compared these results to the IgG2a isotype of anti-CTLA4 (9D9) (Fig. 3). In WT mice bearing CT26 s.c. tumors, anti-RANKL added to anti-PD1 efficacy, but superior growth suppression was once again seen with the triple combination of anti-RANKL with anti-PD1 and anti-CTLA4, despite the use of the non-FcR-engaging IgG1-D265A isotype (Fig. 3A). However, no mice rejected CT26 s.c. tumor (0/10) in this experiment, which compares with >50% tumor rejection in similar experiments employing the anti-CTLA4 IgG2a isotype in triple combination (Fig. 2B), even despite later treatment in the experiment employing anti-CTLA4 (mIgG2a); this suggests that the Treg-depleting clone more successfully achieved tumor rejections in this tumor model. To confirm these results, the efficacy of triple combination anti-RANKL with anti-PD1 and anti-CTLA4 (9D9) of either mouse IgG2a or IgG1-D265A isotypes was assessed in subcutaneous B16F10 melanoma in WT mice. The triple combination utilizing either anti-CTLA4 isotype was significantly more efficacious than the respective dual combination of anti-CTLA4 and anti-PD1 (Fig. 3B), confirming that FcR engagement was not required for improved combinatorial efficacy. However, the depleting mouse IgG2a isotype of anti-CTLA4 once again seemed more efficacious in combination with anti-PD1 and anti-RANKL compared to the IgG1-D265A isotype, although relevant comparisons did not reach statistical significance (Fig. 3B).
Figure 3.

Various anti-CTLA4 isotypes have combined activity with anti-RANKL and anti-PD1, whereas sequencing of anti-RANKL has therapeutic impact. Groups of BALB/c (A, C) wild type (WT) or C57BL/6 WT (B) mice (n = 5–10/group) were injected s.c. with either 1 × 105 CT26 colon carcinoma (A, C) or B16F10 melanoma cells (B) and tumor growth was monitored. Mice were treated i.p. on (A) days 6, 9, 12 and 15 or (B) days 9, 12, 15 and 18 (relative to tumor inoculation) with the following antibodies: cIg (350 µg), anti-CTLA4 (9D9, mIgG2a or mIgG1-D265A isotypes as indicated, 50 µg), anti-PD1 (100 µg), anti-RANKL (200 µg), or the indicated combinations. In (C), mice were treated concurrently i.p. on days 8, 12, 16 and 20 (relative to tumor inoculation) with the following antibodies: cIg (200 µg), anti-CTLA4 (9D9 mIgG2a, 50 µg), anti-PD1 (100 µg), anti-RANKL (100 µg); or were treated sequentially i.p. on days 8, 12 followed by 16, 20 (order of treatment indicated by arrow, →) with the following antibodies: cIg (200 µg), anti-CTLA4 (9D9 mIgG2a, 100 µg), anti-PD1 (200 µg), anti-RANKL (200 µg). Tumor sizes presented as mean ± SEM. (A, C) were performed once while (B) is representative of two independent experiments. Statistical differences between groups were determined by one way ANOVA with Tukey's post-test analysis on (A) day 22, (B) day 26, (C) day 30 data for groups remaining in experiment (* p < 0.05, *** p < 0.001, **** p < 0.0001). In (B), # indicates significant differences at day 28 between anti-PD1+anti-CTLA4 (mIgG2a) vs anti-PD1+anti-CTLA4 (mIgG2a)+anti-RANKL as determined by an unpaired t-test. (*p < 0.05). In (C), # indicates the following additional significant comparisons from one way ANOVA at day 30: concurrent anti-PD1+anti-CTLA4 vs sequential anti-RANKL→ anti-PD1+anti-CTLA4 (**), sequential anti-PD1+anti-CTLA4 → cIg vs sequential anti-RANKL→ anti-PD1+anti-CTLA4 (**); Ф indicates the following additional significant comparison from one way ANOVA at day 24: concurrent anti-RANKL vs sequential anti-RANKL → anti-PD1+anti-CTLA4 (**).
Anti-RANKL is optimally administered concurrently with or following dual immune checkpoint blockade
We were next interested in assessing the optimal sequence of antibody therapy. Using the more effective anti-CTLA4 isotype (mIgG2a), we compared concurrent antibody therapy (antibody treatment on days 8, 12, 16, 20 after tumor inoculation) with sequential therapy (equivalent total dose of antibody on days 8, 12 or 16, 20) in s.c. growth suppression of CT26. Significantly superior growth suppression was achieved when anti-RANKL was administered concurrently with, or following, dual ICB therapy (Fig. 3C). Compared with concurrent anti-RANKL monotherapy, sequential anti-RANKL followed by dual ICB significantly suppressed tumor growth; however, this sequence was less efficacious than concurrent dual ICB alone, suggesting that early ICB treatment is optimal (Fig. 3C).
Efficacy of PD1, CTLA4 and RANKL blockade in tumor-bearing mice is dependent on either NK cells or T cells and IFNγ
We next assessed the mechanism of action of combining anti-RANKL with anti-PD1 alone or in combination with anti-CTLA4 in the suppression of experimental lung metastases or subcutaneous tumor growth (Fig. 4). In mice bearing B16F10 lung metastases, NK cells and IFNγ were critical for the efficacy of the anti-RANKL and anti-PD1 combination therapy as depletion of NK cells or neutralization of IFNγ in these treated mice resulted in their inability to suppress experimental lung metastases (Fig. 4A). In contrast, the efficacy of the anti-RANKL and anti-PD1 combination therapy did not depend on T cells as depletion of CD4+ or CD8+ T cells in treated mice did not attenuate the level of experimental lung metastases (Fig. 4A). The lack of effect of T cells is consistent with the early/innate therapeutic effect of many immunotherapies in protecting against experimental lung metastases.
Figure 4.
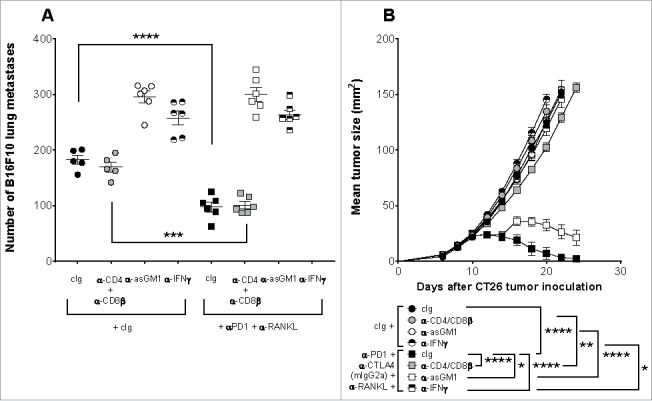
Mechanism of action of anti-tumor efficacy from combination anti-RANKL and anti-PD1. (A): Groups of C57BL/6 wild type (WT) mice (n = 5–6/group) were injected i.v. with 2 × 105 B16F10 melanoma cells. Metastatic burden was quantified in the lungs after 14 days by counting colonies on the lung surface. Mice were treated i.p on days -1, 0 and 2, relative to tumor inoculation with either cIg (400 µg) or anti-PD1 (200 µg) plus anti-RANKL (200 µg). Further, indicated groups were additionally treated i.p with anti-asGM1 (50 µg), anti-CD4 (100 µg) plus anti-CD8β (100 µg), anti-IFN-γ (250 µg), or cIg (100 µg) on days -2, -1 and 6, relative to tumor inoculation. Means ± SEM are shown. (B): Groups of BALB/c WT mice (n = 5/group) were injected s.c. with 1 × 105 CT26 colon cancer cells and tumor growth was monitored. Mice were treated i.p. on days 10, 14, 18 and 22 (relative to tumor inoculation) with cIg (350 µg) or anti-PD1 (100 µg), anti-CTLA4 (9D9, mIgG2a, 50 µg) and anti-RANKL (200 µg). Further, indicated groups were additionally treated i.p. on days 9, 10 and 17 (relative to tumor inoculation) with anti-asGM1 (50 µg), anti-CD4 (100 µg) plus anti-CD8β (100 µg), anti-IFN-γ (250 µg), or cIg (100 µg). Tumor sizes presented as mean ± SEM. In (A) and (B), statistical differences between treatment groups were determined by one way ANOVA with Tukey's post-test analysis (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). In (B), ANOVA was performed on day 20 tumor sizes. Experiments performed once.
Unlike the metastases models, T cells were required for the efficacy of anti-RANKL in combination with anti-PD1 and anti-CTLA4 (IgG2a) in WT mice bearing subcutaneous CT26 tumors, as there was a complete loss of growth suppression in treated mice depleted of CD4+ and CD8+ T cells (Fig. 4B). Similar to the metastases setting, the efficacy of the triple combination needed IFNγ as growth suppression was completely lost in treated mice neutralized of IFNγ (Fig. 4B). Interestingly, a minor although significant increase in tumor size was noted in anti-asGM1-treated mice treated with the triple combination therapy suggesting a minor role for NK or other asGM1+ cells (Fig. 4B). It is possible that such immune cells are not critical but contribute to the major effect of CD8+ T cells.
Triple blockade of PD1, CTLA4 and RANKL improves T cell effector function in tumor-bearing mice
Given that triple combination therapy targeting RANKL, CTLA4 and PD1 suppressed s.c. tumor growth better than any dual combination therapy, we next assessed if T cell effector function was increased with combination therapy (Fig. 5). Clinically, early-during-treatment (EDT) immune changes following immunotherapy have been reported to predict positive responses.26,27 We therefore assessed EDT immune cell changes in CT26 tumor-bearing mice following one dose of treatment. On day 10, tumor-bearing mice were allocated to groups with equivalent tumor size (20–30 mm2) and treated with a single dose of control, dual or triple combination therapy containing the IgG2a isotype of anti-CTLA4 as this clone resulted in the best suppression of tumor growth. Three days later, tumors were harvested for FACs analysis of tumor infiltrating leukocytes (TILs). Interestingly, even at this early time point, significant suppression of tumor growth, as reflected in wet mass of tumor, was noted with the most efficacious dual and triple combination therapies when compared to cIg, anti-RANKL, or anti-PD1 alone (Supp. Fig. S1A). At this early time point, we did not observe any significant changes in the proportion or numbers of tumor infiltrating CD8+ T cells between any of the cIg or therapy treated groups (Supp. Fig. S2A) (data not shown). A trend towards an increase in proliferative status of CD8+ TILs as measured by Ki67 staining was seen in monotherapy-treated mice and this became more pronounced in any dual or triple combination therapy containing anti-CTLA4, but this effect was not statistically significant (Supp. Fig. S2B). TILs from this experiment were next stimulated ex vivo, and interestingly, we noted tumor-infiltrating CD8+ and CD4+ T cells that were treated with the triple combination therapy compared to anti-CTLA4 (mIgG2a) + anti-PD1 dual combination therapy displayed a significant increase in Th1-type cytokine polyfunctionality as reflected in their co-expression of IFNγ and TNFα (Fig. 5A–B, Supp. Fig. S1B-C). This increase in effector function was only observed in the tumor and not the spleen of triple combination therapy-treated mice (data not shown).
Figure 5.
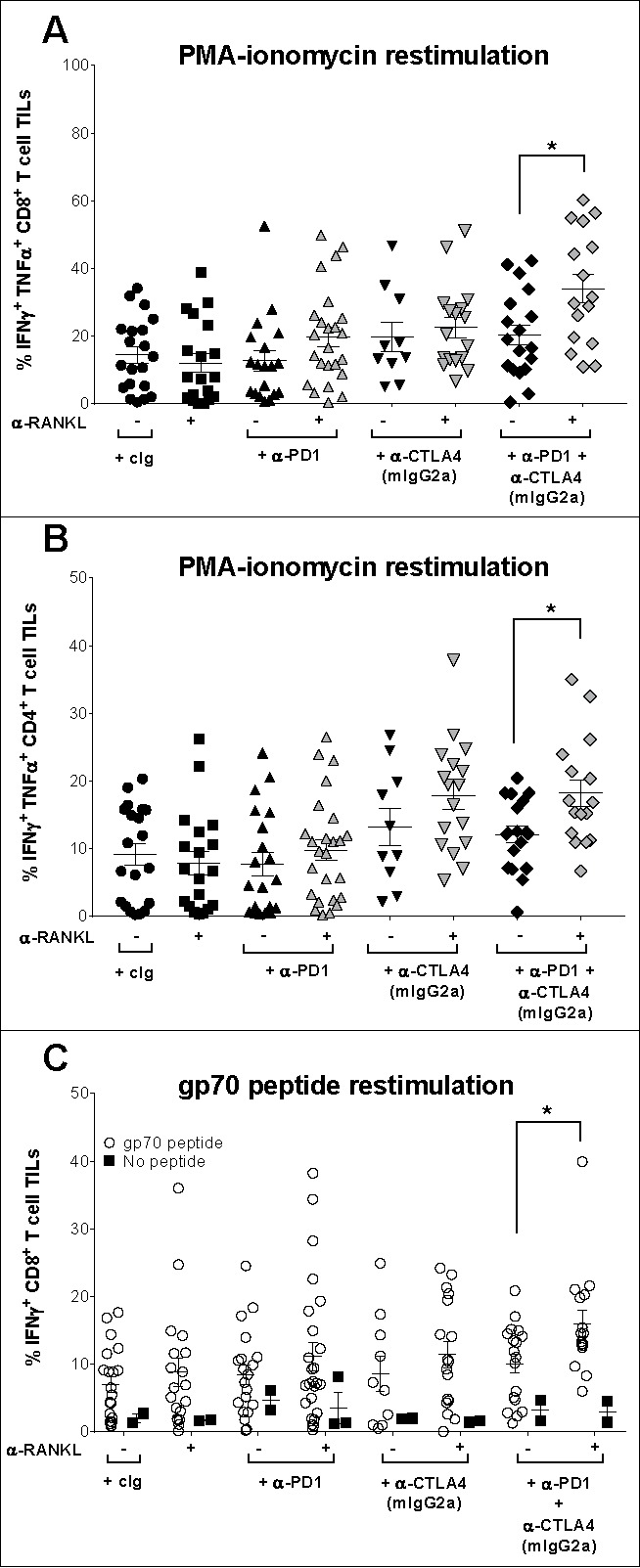
Blockade of PD1, CTLA4 and RANKL improves T cell effector function in tumor-bearing mice. Groups of BALB/c wild type (WT) mice (n = 5–10/group) were injected s.c. with 2 × 105 CT26 colon carcinoma cells. On day 10 after tumor inoculation, mice were randomized into groups bearing equivalent median tumor size and were treated i.p with a single dose of antibody as indicated: cIg (200 µg), anti-CTLA4 (9D9, mIgG2a isotype, 50 µg), anti-PD1 (200 µg), anti-RANKL (200 µg) or the indicated combinations. Three days after treatment, tumors were harvested and processed for flow cytometry gating on live CD45.2+ cells of leukocyte morphology. Mean ± SEM of the proportion of tumor-infiltrating leukocytes (TILs) that were (A) CD8+ T cells or (B) CD4+ T cells co-staining for IFN-γ and TNFα after ex vivo stimulation with PMA-ionomycin. (C) Mean ± SEM proportion of tumor-infiltrating CD8+ T cells expressing IFN-γ when co-cultured ex vivo in the presence or absence of gp70 peptide. Statistical differences between indicated groups were determined by a Mann-Whitney U test (* p < 0.05). Data pooled from three independent experiments.
Given that the gp70 envelope glycoprotein encoded by the endogenous murine leukemia virus (MuLV) is expressed by many cell lines including CT26, we therefore had the ability to assess the antigen specificity of gp70 CD8+ T cell responses in this same experiment. TILs from each of the control- or therapy-treated groups were cultured with or without the addition of the immunodominant gp70 peptide to restimulate gp70-specific CD8 T cell responses.28 Following co-culture with the gp70 peptide, there was a significant increase in the proportion of IFNγ producing CD8+ TILs cells from triple combination therapy treated mice compared to dual anti-CTLA4 and anti-PD1 combination-treated mice (Fig. 5C, Supp. Fig. S1D). In the absence of gp70 peptide, the proportion of CD8+ TILs expressing IFNγ was similar between all treatment groups. Although triple combination therapy increased the cytokine polyfunctionality of T cells, it did not increase the proportion or numbers of gp70 tetramer-specific CD8+ T cells (Supp. Fig. S2C). Altogether, these data suggest that adding anti-RANKL to the combination of anti-PD1 and anti-CTLA4 increases the effector function of both CD4+ and CD8+ T cells in the tumor.
Favorable early alterations in the TME after first treatment of anti-RANKL with ICB
To further assess mechanisms by which anti-RANKL was improving ICB therapy in our models, we set up a similar experiment as in Fig. 5 where tumors were harvested 3 days after first antibody treatment for FACs analysis (Fig. 6). We first assessed the proportion of CD8+ T cells that expressed RANKL (Fig. 6A). In cIg-treated mice, approximately 5% expressed RANKL but this increased to over 10% following anti-PD1 monotherapy. The mechanisms driving enhanced RANKL expression following anti-PD1 therapy remain unclear. Furthermore, RANKL expression was enriched amongst the subset of gp70-reactive CD8+ T cell TILs (almost 15% in cIg-treated mice), and was significantly increased in tumors which had received dual ICB as compared with cIg (Fig. 6B). Although a lower proportion of CD4+ T cell TILs express RANKL compared with CD8+ T cell TILs, ICB (particularly anti-PD1) similarly increased RANKL expression (Fig. 6C). Through upregulating the main intratumor source of RANKL expression, administration of ICB possibly primed the TME to respond to RANKL blockade (Fig. 6A–C).
Figure 6.

Favorable early alterations in the TME after first treatment of anti-RANKL with ICB. (A-E) Groups of BALB/c wild type (WT) mice (n = 5–10/group) were injected s.c. with 2 × 105 CT26 colon carcinoma cells. On day 10 after tumor inoculation, mice were randomized into groups bearing equivalent median tumor size and were treated i.p with a single dose of antibody as indicated: cIg (200 µg), anti-CTLA4 (9D9, mIgG2a isotype, 50 µg), anti-PD1 (200 µg) or the indicated combinations. Three days after treatment, tumors were harvested and processed for flow cytometry gating on (A-D) live CD45.2 cells of leukocyte morphology, or (E) on live single CD45.2+ cells excluded from the lymphocyte gate. (A) Proportion of CD8+ T cell TILs expressing RANKL, (B) proportion of gp70-specific CD8+ T cell TILs expressing RANKL, and (C) proportion of CD4+ T cell TILs expressing RANKL displayed for indicated treatment groups. (D) Geometric mean fluorescent intensity (gMFI) PD1 expression by gp70-specific CD8+ T cell TILs shown for indicated treatment groups. (E) Proportion of cells expressing PD-L1 shown for indicated treatment groups. Means ± SEM are shown. Statistical differences were determined by one way ANOVA with Tukey's post-test analysis, except in (A), where Kruskal-Wallis test with Dunn's post-test analysis was used, and in (E), where statistical differences were determined by Mann-Whitney test for indicated comparisons (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). Additionally, in (D), statistical difference in gMFI for indicated comparison of anti-PD1 alone or combined with anti-RANKL was determined by Mann-Whitney test (#p < 0.05). Two-three experiments were pooled in (A-D).
We have previously reported that tumor infiltrating PD1hi T cells were insensitive to anti-PD1 therapy and displayed an exhausted phenotype. However, some immunotherapies such as anti-CTLA4 or anti-CD40 can lower the levels of PD1 on PD1hi T cells to resensitize them to PD1 blockade.23 Consistent with this, although anti-PD1 monotherapy significantly attenuated PD1 expression compared with cIg, administration of anti-PD1+anti-RANKL further decreased PD1 expression in gp70-specific CD8+ T cell TILs (Fig. 6D), as well as unselected CD8+ T cell TILs (data not shown). Interestingly, PD1 expression by gp70-specific CD8+ T cell TILs was not further reduced with the further addition of anti-CTLA4 to anti-PD1 and anti-RANKL. Concurrently, expression of PD-L1 (a ligand for PD1) in the non-lymphoid CD45.2+ components of the tumor was assessed (Fig. 6E). In keeping with adaptive immune resistance secondary to ICB23,26 it was noted that the proportion of such cells expressing PD-L1 was slightly increased after a single dose of anti-PD1, but this was mitigated when anti-RANKL was given with anti-PD1 (Fig. 6E, Supp. Fig. S3). Similar results were seen with combination anti-PD1 and anti-CTLA4 with or without anti-RANKL, where triple combination therapy seemed to reduce PD-L1 expression levels below the baseline level in cIg-treated mice (Fig. 6E). These PD-L1+ cells were not further characterised, but may include myeloid cells.
Expression of another T-cell immune checkpoint, CTLA4, was also assessed. Amongst all CD8+ T cell TILs as well as the gp70-reactive subset, CTLA4 expression was low overall. Administration of ICB or combination did not generally alter CTLA4 expression, with the exception of combination anti-CTLA4+anti-RANKL, which resulted in significantly lower CTLA4 expression compared with anti-CTLA4 alone amongst gp70-specific CD8+ T cell TILs (data not shown). In unselected CD4+ T cell TILs, much lower expression of immune checkpoints PD1 and CTLA4 were seen overall compared with CD8+ counterparts. Only minor changes in expression pattern were seen with ICB with or without anti-RANKL, except for CTLA4 expression which was reduced where anti-CTLA4 (mIgG2a) was given consistent with that antibody's known mechanism of action (depletion of Tregs) (data not shown).
Thus, minor but significant changes seen early after the first dose of antibody therapy suggest several favorable alterations in the TME consistent with increased RANKL expression and reduction in PD1/PD-L1 expression.
RANKL identifies PD1hi expressing T cells whose PD1 expression can be modulated
The phenotype of RANKL+ T cell TILs have not been previously described. In cIg-treated mice, almost all CD8+RANKL+ T cell TILs (>90%) co-expressed PD1; in comparison, less than 40% CD8+RANKL− T-cell TILs were PD1 positive (Supp. Fig. S4A). Furthermore, the MFI of PD1 was at least 3-fold higher on CD8+RANKL+ compared to CD8+RANKL− T cells identifying the former as PD1hi cells (Supp. Fig. S4B). Three days following anti-PD1 or anti-CTLA4 mono or dual therapy, the PD1 MFI on CD8+RANKL+ T cells was reduced approximately 2-fold (Supp. Fig. S4B) indicating responsiveness to PD1 blockade. In the TILs of cIg-treated mice, we also observed that a greater proportion of CD8+RANKL+ T cells (∼ 95%) were proliferative as assessed by Ki67 staining compared to CD8+RANKL− T cells (∼70%) (Supp. Fig. S4C). We assessed expression of another immune checkpoint, CTLA4, and compared this expression between RANKL+ and RANKL− CD8+ T cell TILs; reassuringly, CTLA4 expression was not significantly higher in RANKL+CD8+ T cells compared with their RANKL− counterparts and was not significantly altered by ICB (data not shown). CTLA4 expression, regardless of therapy administered, was low in comparison to PD1 expression (Supp. Fig. S5).
Similar to trends seen in unselected CD8+ T cell TILs, gp70-specific CD8+RANKL+ T cells were almost all PD1 positive (>95%) compared to gp70 specific CD8+RANKL− T cells where only 80–85% expressed PD1 in cIg-treated mice (data not shown). Akin to our observations in total CD8+ T cells, the PD1 MFI was higher on gp70-specific CD8+ that were RANKL+ compared to those that were RANKL− in TILs from cIg-treated mice (Supp. Fig. S4D). Once again anti-PD1 or anti-CTLA4 mono or dual therapy was able to attenuate PD1 expression on gp70 specific CD8+RANKL+ T cells (Supp. Fig. S4D).
When assessing CD4+ T cell TILs, proportion expressing PD1 and expression level of PD1 in CD4+RANKL+ T cell TILs is higher than CD4+RANKL− T-cell TILs, but expression level was generally lower than in CD8+ TILs (Supp. Fig. S4E-F). Interestingly, PD1 expression was not significantly attenuated in RANKL+ CD4+ T cell TILs in response to anti-PD1 therapy, probably because PD1 levels (assessed by gMFI) were already quite low (Supp. Fig. S4E). A small significant reduction in proportion of CD4+ T cell TILs expressing PD1 was seen with anti-CTLA4 (mIgG2a), which could be due to Treg depletion (Supp. Fig. S4 F).
Discussion
We have shown that combining anti-RANKL and anti-PD1 or anti-PD-L1 improved anti-tumor efficacy against subcutaneous tumor growth and experimental lung metastases which were partially resistant to anti-PD1 or anti-PD-L1 monotherapy. Furthermore, the anti-tumor efficacy of anti-PD1 or anti-PD-L1 and anti-CTLA4 could be further improved by the addition of RANKL blockade. In mouse tumor models, RANKL expression on CD8+ T cells was readily identifiable and defined a subset of PD1hi TILs which respond to ICB through attenuation of PD1 expression. In particular anti-PD1-containing treatments increased RANKL expression on infiltrating T-cell TILs. Dual anti-RANKL and anti-PD1 suppression of metastasis required NK cells and IFNγ, whereas the optimal triple anti-RANKL/anti-PD1/anti-CTLA4 suppression of subcutaneous tumor growth was abrogated in the absence of T cells and IFNγ. This distinction is consistent with the critical role of NK cells in suppressing experimental lung metastases, and the role of T cells in controlling subcutaneous tumor transplants.17,29,30,31 Early-during-treatment assessment revealed a triple anti-RANKL/anti-PD1/anti-CTLA4 combination therapy compared to dual anti-PD1 and anti-CTLA4 combination therapy further increased the proportion of tumor-infiltrating CD4+ and CD8+ T cells that can produce both IFNγ and TNFα. Favorable TME changes ascribable to anti-RANKL combined with ICB included attenuation of PD1/PD-L1 expression. This new data generalizes the potential clinical importance of denosumab use in patients receiving contemporary immune checkpoint blockade.
Reactive changes in the TME to anti-cancer therapies such as anti-PD1 antibody have been previously described in early-during-treatment (EDT) biopsies, such as adaptive immune resistance, whereby factors such as increased production of IFNγ induces higher PD-L1 expression on tumor or myeloid cells.23,26 Higher PD-L1 expression has also been suggested as a possible resistance mechanism to anti-PD1 treatment in human melanoma.32 Additionally, it has been previously demonstrated in preclinical models that PD1 expression by T cell TILs above a threshold level can result in resistance to anti-PD1 antibodies, and strategies (such as combination with alternative immunotherapies) to reduce expression below this level results in therapeutic benefit.23,15 In this study, therapeutic sensitivity of dual anti-RANKL and anti-PD1 treatment was associated with favourable changes to the tumor microenvironment including both reduction of PD1 expression by T cells, and reduction in PD-L1 expression. These alterations were significant but relatively small, likely due to the nature of EDT assessment, where initial changes are assessed early after the first antibody therapy. The precise mechanism of these TME alterations remains unclear. We have previously shown that the efficacy of the anti-RANKL antibody IK22/5 is independent of activating FcR, and is unlikely to be depleting.17 A possible explanation is that anti-RANKL is acting directly on non-lymphoid components of the TME (e.g. myeloid cells) to reverse a suppressive phenotype, as reflected in reduced PD-L1 expression. This would be consistent with the improved antigen-specific T cell effector function noted here on EDT assessment. Interestingly, in a previous study where myeloid cells derived from peripheral blood of multiple myeloma patients were cultured with RANKL and M-CSF, these myeloid cells assumed a pre-osteoclastic morphology and immunosuppressive phenotype, as reflected through expression of factors such as IDO and IL-10, and in vitro suppression of cytotoxic T cell proliferation and killing; these effects were partially reversible with ICB (anti-PD-L1).33 The role and function of RANK-expressing myeloid cells in tumor TME, and functional changes after anti-RANKL therapy, should next be assessed.
Despite an enrichment for PD1 co-expression, the characteristics of RANKL+CD8+ T cell TILs in the present model is more consistent with an activated rather than exhausted phenotype, given that RANKL-expressing cells generally were more proliferative and had low expression of another immune checkpoint, CTLA4.34,35 Recently, adverse outcomes in a mouse model of breast carcinoma were reported when anti-PD1 was administered simultaneously with an agonist antibody targeting a different TNF receptor, OX40, with a mechanism suggesting compensatory increases in alternative immune checkpoints CTLA4 and Tim3.36 Reassuringly, similar increases were not noted with the combination of anti-PD1 and anti-RANKL in this study. Furthermore, we have shown that antibody sequencing may impact on therapeutic efficacy of anti-RANKL with ICB, in that optimal tumor growth suppression was seen when anti-RANKL was given concurrently or following ICB therapy. These data suggest that in clinical studies, denosumab given prior to ICB may not yield the same clinical benefit as could be seen with concurrent or subsequent therapy. Our mechanism of action studies suggested that RANKL expression was increased upon anti-PD-1 therapy, so one possible mechanism of concurrent or anti-PD1-anti-RANKL sequential therapy is to relieve RANKL-RANK mediated immune suppression that occurs post anti-PD1 treatment.
In hallmark oncology studies, no immune-related toxicities of denosumab were reported, although any toxicity in combination with ICB has not been assessed.14,37,38 Furthermore, with respect to sequencing of anti-RANKL, we have shown that ICB (anti-PD1 and anti-PD1+anti-CTLA4 combination) can itself increase RANKL expression by tumor-infiltrating T cells, which is the main intratumor source of RANKL in this model. In the event that this mechanism primes the TME to respond to RANKL blockade, this may provide a biological rationale for superior efficacy noted with anti-RANKL given concurrently with or following ICB. It is unclear why anti-CTLA4 treatment alone did not also modify RANKL expression levels in this study. One explanation relies on the observation that RANKL is generally upregulated early after activation of T cells, particularly in a tolerogenic context.39 It is possible that, given the Treg-depleting clone of anti-CTLA4 that was used, the dynamics of T cell activation and consequent RANKL expression is earlier than is seen with anti-PD1 monotherapy. In this case, an earlier time point than 72 hours post-antibody administration could be assessed to capture upregulation of RANKL expression.
Defining the characteristics of RANKL and RANK expression on the immune component in human cancers is critical to advance this research into the translational setting. An important variable that modulates RANKL expression by T cells is their activation status, whereby RANKL expression is rapidly increased after stimulation via T cell receptor (TCR) and CD3 ligation, and to a lesser extent by alternative stimulation methods such as IL2 exposure.40,41 Our results suggest that RANKL expression may also be modified through therapies such as ICB, presumably indirectly via activation-induced expression. We have previously shown that T cell expression of RANKL in subcutaneous mouse melanoma tumors is higher at an earlier time point in tumor growth,17 which may also be related to activation status. Factors such as activation status of T cells (as influenced by administration of therapies such as ICB) should be considered in future analysis of immune cell RANKL expression. Additionally, it is possible that sources of RANKL other than infiltrating T cells are important for tumor immunity in humans. Alternative possibilities include lymph node stromal subsets such as marginal reticular cells, which constitutively express RANKL,42 and RANKL expression by stroma in certain sites such as lung, breast and bone or tumor cells themselves.43,44,45 The relative contributions of different RANKL sources to tumor immunity remain unknown.
This study extends the preclinical evidence for anti-RANKL in combination immunotherapy to combinations including targeting the PD1/PD-L1 pathway, as well as dual immune checkpoint blockade. RANKL expression by tumor-infiltrating T cells in mouse defines a proliferative subset enriched for PD1 expression but which pharmacodynamically responds to checkpoint blockade. Whether RANKL therapy might be more effective in the setting of an immunogenic tumor (i.e. already infiltrated by T cells either prior to or in the setting of dual ICB) versus in the setting of a poorly immunogenic tumor (i.e. as a means to overcome T cell exclusion) is not yet clear. However, the former would seem to be better supported from our mechanism of action findings which were conducted in relatively immunogenic tumor models. Further assessment of RANKL and RANK expression in human tumors should now be undertaken. Importantly, these results also suggest that in translational studies of denosumab in combination with anti-PD1-based ICB strategies, relative dynamics of PD1 and RANKL expression on tumor-infiltrating and perhaps circulating CD8+ T cells would be of interest.
Materials and methods
Cell culture
Mouse melanoma cell line B16F10 (ATCC, 2007), colon cancer cell line CT26 (Robert Wiltrout, National Cancer Institute Frederick, 1991), and prostate cancer cell lines RM-1 (Pam Russell, University of Sydney, 1996)46 and Tramp-C1 (Norman Greenberg, Baylor College of Medicine, 2000)22 were injected, maintained and monitored as previously described.17 All cell lines were never passaged for more than 14 days and were routinely tested negative for Mycoplasma, but cell line authentication was not routinely performed.
Mice
C57BL/6 or BALB/c wild-type (WT) mice were bred-in-house at QIMR Berghofer Medical Research Institute or purchased from the Walter and Eliza Hall Institute for Medical Research. TRAMP transgenic mice were bred-in-house.47,48 Groups of 5 to 17 mice were used for subcutaneous growth and experimental lung metastases models. All experiments were approved by the QIMRB Animal Ethics Committee.
Antibodies
Purified anti-mouse anti-RANKL (IK22–5; rat IgG2a),49 anti-PD-L1 (10F.9G2) and control antibodies (1–1 or 2A3, rat IgG2a) purchased from BioXcell (West Lebanon, NH). Anti-PD1 clone RMP1–14 was purchased from Leinco (St Louis, MO). Anti-CTLA4 (clone 9D9 of IgG2a or IgG1-D265A isotype), and control antibody (1D12; mouse IgG2a), were supplied by Bristol-Myers Squibb. Antibodies to deplete NK cells (anti-asialoGM1, rabbit polyclonal, Wako), CD4 (GK1.5, BioXcell) and CD8 T cells (53.5.8, BioXcell), or to neutralise IFNγ (H22, Leinco) were administered at the dose and schedule as indicated in the Fig. legends.
Subcutaneous tumor models
For B16F10 (1 × 105), CT26 (1 × 105) and Tramp-C1 (1 × 106) tumor formation, cells were inoculated subcutaneous (s.c.) into the abdominal flank of female (B16F10) or male (CT26, Tramp-C1) mice. Therapeutic antibody treatment commenced as indicated on day 6–10 after tumor inoculation and was given every 3–4 days for 4 doses, except for Tramp-C1, where treatment started at day 20 after tumor inoculation and was given every 4 days for 4 doses. Additionally, where indicated, anti-asGM1, anti-CD4/CD8β and anti-IFNγ were administered on days 9, 10 and 17, relative to tumor inoculation. Digital callipers were used to measure the perpendicular diameters of the tumors. The tumor size was calculated as the product of the two measurements and is presented as mean ± SEM.
Experimental lung metastasis models
Single-cell suspensions of B16F10 (2 × 105) or RM-1 (2 × 105) were injected i.v. into the tail vein of the indicated strains of mice. Lungs were harvested on day 14, and surface tumor nodules were counted under a dissection microscope. Antibody treatments were as indicated, with anti-PD1/anti-PD-L1 and/or anti-RANKL mAbs administered on days -1, 0 and 2, relative to tumor inoculation, and anti-asGM1, anti-CD4/CD8β and anti-IFNγ administered on days -2, -1 and 6, relative to tumor inoculation.
Flow cytometry
Tumor-bearing mice were inoculated s.c. with CT26 (2 × 105 cells) and were treated with a single dose of cIg or therapeutic antibody (anti-RANKL, anti-CTLA4, anti-PD1 and combinations) on day 10 before being sacrificed on day 13. Single-cell suspensions were generated as previously described for FACs analysis.50
The following antibodies (all from Biolegend or eBioscience) were used: CD4-BV605 (RM4–5), CD8-BV711 (53–6.7), PD-L1-BV421 (10F.9G2) and Zombie Aqua live/dead dye; TCRβ-PerCP-Cy5.5 (H57–597), CD45.2-A780 (104), CD69-PECy7 (H1.2F3), PD1-FITC (J43, which does not cross-react with the clone used therapeutically23). To identify gp70-specific CD8 T cells, the following reagents were obtained from the NIH Tetramer Core Facility: H-2Ld MuLV gp70 Tetramer-SPSYVYHQF-PE and -APC. For intracellular cytokine staining (ICS), cells were suspended in complete RPMI media and either stimulated for 4 hours with Cell Stimulation Cocktail (plus protein transport inhibitors) (1/1000) (eBioscience) containing PMA-ionomycin and brefeldin A with monensin, or cultured for 2 hours with gp70 peptide (SPSYVYHQF)(GenScript, Hong Kong) in the presence of brefeldin A and monensin (1/1000)(BioLegend). Cells were then surface stained as described above before being fixed/permeabilized with Cytofix/Cytoperm (BD) and stained with IFNγ-AF488 or IFNγ-APC (Biolegend) or TNFα-PE (BD). For intracellular transcription factor staining, cells were fixed and permeabilized after surface staining using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience), according to the manufacturer's protocol and in some cases was stained with Ki67-EF450 (Sol185) (eBioscience). For cytoplasmic staining, CTLA4-APC (UC10.4B9) (eBioscience) was used after permeabilization. All data were collected on a Fortessa 4 (BD Biosciences) flow cytometer and analyzed with FlowJo v10 software (Tree Star, Inc.).
Statistical analysis
GraphPad Prism software was used for statistical analysis. For column analyses, Brown-Forsythe test was used to assess equal variances. If non-significant, one-way ANOVA with Tukey's post-test for multiple comparisons was used. In the event of unequal variances between groups, Kruskal-Wallis analysis with Sidak's or Dunnett's multiple comparisons were employed as appropriate. Mann-Whitney or unpaired t-test as appropriate was used when comparing two groups. Data were considered to be statistically significant where the P value was less than 0.05.
Supplementary Material
Funding Statement
M.J.S. was supported by a National Health and Medical Research Council (NH&MRC) Senior Principal Research Fellowship (1078671) and The Cancer Council of Queensland (1102242). E.A. and was supported by a University of Queensland (UQ) Australian Postgraduate Award (APA). H.H. was supported by a UQ International Postgraduate Research Scholarship, a UQ APA, and a QIMR Berghofer Top-Up award. J. S. O'D was supported by a UQ APA and a QIMR Berghofer Top-Up award.
Acknowledgments
The authors wish to thank Liam Town and Kate Elder for breeding, genotyping, maintenance and care of the mice used in this study.
Competing financial interests
M.J. Smyth has research agreements with Bristol Myers Squibb, Aduro Biotech, and Tizona Therapeutics. W.C. Dougall has received a speaker's honorarium from AMGEN. The remaining authors report no competing financial interest.
Author contributions
Conception and design: W. C. Dougall, M. W. L. Teng & M.J. SmythDevelopment of methodology: E. Ahern, W. C. Dougall, M. W. L. Teng, M. J. SmythAcquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): E. Ahern, H. Harjunpää, S. AllenAnalysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): E. Ahern, J. S. O'Donnell, W. C. Dougall, M. W. L. Teng, M. J. Smyth.Writing, review, and/or revision of the manuscript: E. Ahern, H. Harjunpää, J. S. O'Donnell, S. Allen, W. C. Dougall, M. W. L. Teng, M. J. Smyth.Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): E. Ahern, H. Harjunpää, S. Allen.Study supervision: W. C. Dougall, M. W. L. Teng, M.J. Smyth.
References
- 1.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–13. doi: 10.1056/NEJMoa1510665. PMID:26406148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, et al.. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–65. doi: 10.1016/S1470-2045(16)30066-3. PMID:27247226. [DOI] [PubMed] [Google Scholar]
- 3.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al.. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311–9. doi: 10.1056/NEJMoa1411087. PMID:25482239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al.. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30. doi: 10.1056/NEJMoa1412082. PMID:25399552. [DOI] [PubMed] [Google Scholar]
- 5.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al.. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med. 2016;375:1823–33. doi: 10.1056/NEJMoa1606774. PMID:27718847. [DOI] [PubMed] [Google Scholar]
- 6.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al.. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. PMID:26027431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al.. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–32. doi: 10.1056/NEJMoa1503093. PMID:25891173. [DOI] [PubMed] [Google Scholar]
- 8.Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, et al.. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–84. doi: 10.1016/S1470-2045(15)70076-8. PMID:25795410. [DOI] [PubMed] [Google Scholar]
- 9.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al.. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345–56. doi: 10.1056/NEJMoa1709684. PMID:28889792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Donnell J, Long G, Scolyer R, Teng M, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2016;52:71–81. doi: 10.1016/j.ctrv.2016.11.007. PMID:27951441. [DOI] [PubMed] [Google Scholar]
- 11.Lacey DL, Boyle WJ, Simonet WS, Kostenuik PJ, Dougall WC, Sullivan JK, San Martin J, Dansey R. Bench to bedside: elucidation of the OPG–RANK–RANKL pathway and the development of denosumab. Nat Rev Drug Discov. 2012;11:401–19. doi: 10.1038/nrd3705. PMID:22543469. [DOI] [PubMed] [Google Scholar]
- 12.Henry DH, Costa L, Goldwasser F, Hirsh V, Hungria V, Prausova J, Scagliotti GV, Sleeboom H, Spencer A, Vadhan-Raj S, et al.. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate Cancer) or multiple Myeloma. J Clin Oncol. 2011;29:1125–32. doi: 10.1200/JCO.2010.31.3304. PMID:21343556. [DOI] [PubMed] [Google Scholar]
- 13.Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, Miller K, Sieber P, Karsh L, Damião R, et al.. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet. 2012;379:39–46. doi: 10.1016/S0140-6736(11)61226-9. PMID:22093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stopeck AT, Lipton A, Body J-J, Steger GG, Tonkin K, Boer RHd, Lichinitser M, Fujiwara Y, Yardley DA, Viniegra M, et al.. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol. 2010;28:5132–9. doi: 10.1200/JCO.2010.29.7101. PMID:21060033. [DOI] [PubMed] [Google Scholar]
- 15.Smyth MJ, Ngiow SF, Ribas A, Teng MWL. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13:143–58. doi: 10.1038/nrclinonc.2015.209. PMID:26598942. [DOI] [PubMed] [Google Scholar]
- 16.Bostwick AD, Salama AK, Hanks BA. Rapid complete response of metastatic melanoma in a patient undergoing ipilimumab immunotherapy in the setting of active ulcerative colitis. J Immunother Cancer. 2015;3:19. doi: 10.1186/s40425-015-0064-2. PMID:25992290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahern E, Harjunpaa H, Barkauskas D, Allen S, Takeda K, Yagita H, Wyld D, Dougall WC, Teng MWL, Smyth MJ, et al.. Co-administration of RANKL and CTLA4 antibodies enhances lymphocyte-mediated anti-tumor immunity in mice. Clin Cancer Res. 2017;23:5789–801. doi: 10.1158/1078-0432.CCR-17-0606. PMID:28634284. [DOI] [PubMed] [Google Scholar]
- 18.Scagliotti GV, Hirsh V, Siena S, Henry DH, Woll PJ, Manegold C, Solal-Celigny P, Rodriguez G, Krzakowski M, Mehta ND, et al.. Overall survival improvement in patients with lung cancer and bone metastases treated with denosumab versus zoledronic acid: subgroup analysis from a randomized phase 3 study. J Thorac Oncol. 2012;7:1823–9. doi: 10.1097/JTO.0b013e31826aec2b. PMID:23154554. [DOI] [PubMed] [Google Scholar]
- 19.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al.. Anti-CTLA-4 antibodies of IgG2 a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. PMID:24777248. [DOI] [PubMed] [Google Scholar]
- 20.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al.. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–710. doi: 10.1084/jem.20130579. PMID:23897981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma P, Schreiber-Agus N. Mouse models of prostate cancer. Oncogene. 1999;18:5349–55. doi: 10.1038/sj.onc.1203037. PMID:10498888. [DOI] [PubMed] [Google Scholar]
- 22.Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–30. PMID:9269988. [PubMed] [Google Scholar]
- 23.Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, Smyth MJ. A threshold level of intratumor CD8+ T cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res. 2015;75:3800–11. doi: 10.1158/0008-5472.CAN-15-1082. PMID:26208901. [DOI] [PubMed] [Google Scholar]
- 24.Furness AJS, Vargas FA, Peggs KS, Quezada SA. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol. 2014;35:290–8. doi: 10.1016/j.it.2014.05.002. PMID:24953012. [DOI] [PubMed] [Google Scholar]
- 25.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti–CTLA-4 antibodies. J Exp Med. 2009;206:1717–25. doi: 10.1084/jem.20082492. PMID:19581407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vilain RE, Menzies AM, Wilmott JS, Kakavand H, Madore J, Guminski A, Liniker E, Kong BY, Cooper AJ, Howle JR, et al.. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in melanoma. Clin Cancer Res. 2017;23(17):5024–5033. doi: 10.1158/1078-0432.CCR-16-0698. PMID:28512174. [DOI] [PubMed] [Google Scholar]
- 27.Chen P-L, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al.. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6:827–837. doi: 10.1158/2159-8290.CD-15-1545. PMID:27301722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selby MJ, Engelhardt JJ, Johnston RJ, Lu LS, Han M, Thudium K, Yao D, Quigley M, Valle J, Wang C, et al.. Preclinical development of ipilimumab and nivolumab combination immunotherapy: mouse tumor models, in vitro functional studies, and cynomolgus Macaque toxicology. PLoS One. 2016;11:e0161779. doi: 10.1371/journal.pone.0161779. PMID:27610613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mittal D, Vijayan D, Putz EM, Aguilera AR, Markey KA, Straube J, Kazakoff S, Nutt SL, Takeda K, Hill GR, et al.. Interleukin-12 from CD103(+) Batf3-dependent dendritic cells required for NK-cell suppression of metastasis. Cancer Immunol Res. 2017;5:1098–108. doi: 10.1158/2326-6066.CIR-17-0341. PMID:29070650. [DOI] [PubMed] [Google Scholar]
- 30.Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, Aguilera AR, Miles JJ, Lutzky VP, de Andrade LF, et al.. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016;6:446–59. doi: 10.1158/2159-8290.CD-15-0944. PMID:26787820. [DOI] [PubMed] [Google Scholar]
- 31.Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, et al.. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26:923–37. doi: 10.1016/j.ccell.2014.10.018. PMID:25465800. [DOI] [PubMed] [Google Scholar]
- 32.Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw RPM, Thompson JF, Wilmott JS, Long GV, Scolyer RA, et al.. Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod Pathol. 2017;30:1666–76. doi: 10.1038/modpathol.2017.89. PMID:28776578. [DOI] [PubMed] [Google Scholar]
- 33.An G, Acharya C, Feng X, Wen K, Zhong M, Zhang L, Munshi NC, Qiu L, Tai YT, Anderson KC. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: therapeutic implication. Blood. 2016;128:1590–603. doi: 10.1182/blood-2016-03-707547. PMID:27418644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–9. doi: 10.1038/ni.2035. PMID:21739672. [DOI] [PubMed] [Google Scholar]
- 35.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. PMID:19043418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, Gough MJ, Urba WJ, Fox BA. Timing of PD-1 blockade is critical to effective combination immunotherapy with anti-OX40. Clin Cancer Res. 2017;23:6165–77. doi: 10.1158/1078-0432.CCR-16-2677. PMID:28855348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fizazi K, Lipton A, Mariette X, Body JJ, Rahim Y, Gralow JR, Gao G, Wu L, Sohn W, Jun S. Randomized phase II trial of denosumab in patients with bone metastases from prostate cancer, breast cancer, or other neoplasms after intravenous bisphosphonates. J Clin Oncol. 2009;27:1564–71. doi: 10.1200/JCO.2008.19.2146. PMID:19237632. [DOI] [PubMed] [Google Scholar]
- 38.Lipton A, Steger GG, Figueroa J, Alvarado C, Solal-Celigny P, Body JJ, de Boer R, Berardi R, Gascon P, Tonkin KS, et al.. Randomized active-controlled phase II study of denosumab efficacy and safety in patients with breast cancer-related bone metastases. J Clin Oncol. 2007;25:4431–7. doi: 10.1200/JCO.2007.11.8604. PMID:17785705. [DOI] [PubMed] [Google Scholar]
- 39.Hochweller K, Anderton SM. Kinetics of costimulatory molecule expression by T cells and dendritic cells during the induction of tolerance versus immunity in vivo. Eur J Immunol. 2005;35:1086–96. doi: 10.1002/eji.200425891. PMID:15756642. [DOI] [PubMed] [Google Scholar]
- 40.Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, Kalachikov S, Cayani E, Bartlett FS 3rd, Frankel WN, et al.. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190–4. doi: 10.1074/jbc.272.40.25190. PMID:9312132. [DOI] [PubMed] [Google Scholar]
- 41.Wang R, Zhang L, Zhang X, Moreno J, Celluzzi C, Tondravi M, Shi Y. Regulation of activation-induced receptor activator of NF-κB ligand (RANKL) expression in T cells. Eur J Immunol. 2002;32:1090–8. doi: 10.1002/1521-4141(200204)32:4%3c1090::AID-IMMU1090%3e3.0.CO;2-P. PMID:11920576. [DOI] [PubMed] [Google Scholar]
- 42.Chang JE, Turley SJ. Stromal infrastructure of the lymph node and coordination of immunity. Trends Immunol. 2015;36:30–9. doi: 10.1016/j.it.2014.11.003. PMID:25499856. [DOI] [PubMed] [Google Scholar]
- 43.Hu H, Wang J, Gupta A, Shidfar A, Branstetter D, Lee O, Ivancic D, Sullivan M, Chatterton RT Jr, Dougall WC, et al.. RANKL expression in normal and malignant breast tissue responds to progesterone and is up-regulated during the luteal phase. Breast Cancer Res Treat. 2014;146:515–23. doi: 10.1007/s10549-014-3049-9. PMID:25007964. [DOI] [PubMed] [Google Scholar]
- 44.Jones DH, Kong Y-Y, Penninger JM. Role of RANKL and RANK in bone loss and arthritis. Ann Rheum. 2002;61:ii32–ii9. doi: 10.1136/ard.61.suppl_2.ii32.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dougall WC, Holen I, Gonzalez Suarez E. Targeting RANKL in metastasis. BoneKEy Rep. 2014;3:519. doi: 10.1038/bonekey.2014.14. PMID:24795813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baley PA, Yoshida K, Qian W, Sehgal I, Thompson TC. Progression to androgen insensitivity in a novel in vitro mouse model for prostate cancer. J Steroid Biochem Mol Biol. 1995;52:403–13. doi: 10.1016/0960-0760(95)00001-G. PMID:7538321. [DOI] [PubMed] [Google Scholar]
- 47.Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, Cunha GR, Donjacour AA, Matusik RJ, Rosen JM. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A. 1995;92:3439–43. doi: 10.1073/pnas.92.8.3439. PMID:7724580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hurwitz AA, Foster BA, Allison JP, Greenberg NM, Kwon ED. The TRAMP mouse as a model for prostate cancer. Curr Protoc Immunol. 2001;Chapter 20:Unit 20.5. PMID:18432778. [DOI] [PubMed] [Google Scholar]
- 49.Kamijo S, Nakajima A, Ikeda K, Aoki K, Ohya K, Akiba H, Yagita H, Okumura K. Amelioration of bone loss in collagen-induced arthritis by neutralizing anti-RANKL monoclonal antibody. Biochem Biophys Res Commun. 2006;347:124–32. doi: 10.1016/j.bbrc.2006.06.098. PMID:16815304. [DOI] [PubMed] [Google Scholar]
- 50.Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, Huang Q, Liu J, Takeda K, Teng MWL, et al.. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell. 2016;30:391–403. doi: 10.1016/j.ccell.2016.06.025. PMID:27622332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.