

Summary
This is the second report of the United Kingdom Primary Immunodeficiency (UKPID) registry. The registry will be a decade old in 2018 and, as of August 2017, had recruited 4758 patients encompassing 97% of immunology centres within the United Kingdom. This represents a doubling of recruitment into the registry since we reported on 2229 patients included in our first report of 2013. Minimum PID prevalence in the United Kingdom is currently 5·90/100 000 and an average incidence of PID between 1980 and 2000 of 7·6 cases per 100 000 UK live births. Data are presented on the frequency of diseases recorded, disease prevalence, diagnostic delay and treatment modality, including haematopoietic stem cell transplantation (HSCT) and gene therapy. The registry provides valuable information to clinicians, researchers, service commissioners and industry alike on PID within the United Kingdom, which may not otherwise be available without the existence of a well‐established registry.
Keywords: autoimmunity, autoinflammatory disease, human, immunodeficiency diseases, transplantation
Introduction
Primary immunodeficiencies (PID) are rare diseases, with a reported prevalence of between 1 : 16 000 and 1 : 50 000 1. The small numbers of patients cared for by individual centres provides challenges to effective diagnosis, clinical care and research. National and international registries have sought to overcome these barriers by encouraging collaboration and providing valuable data sets to clinicians, researchers, pharmaceutical companies and health policymakers. The United Kingdom Primary Immunodeficiency (UKPID) registry has provided a unique repository of longitudinal UK data. It was established in 2008 and the first report was published in 2013, covering the first 4 years of activity (2008–12) 2. The registry has now expanded to 4758 patients from the 2229 patients in our first report, highlighting the success and efforts of the registry team and local collaborators. While much data overlap with the European Society for Immunodeficiencies (ESID) registry, establishing a standalone UKPID registry allows the addition of variables that are of importance to UK PID clinicians and researchers that may not otherwise be available from the ESID registry.
Improved recognition of PID and advances in molecular diagnostics have led to a significant increase in the numbers of individual PIDs being recognized, with nearly 300 genes identified 3. It is increasingly recognized that these PIDs not only present with increased susceptibility to infections, but also immune dysregulation, autoimmunity and an increased susceptibility to malignancy. In addition, an ever‐expanding range of treatment options are now available, resulting in improved patient outcomes. Reduced morbidity and mortality following haematopoietic stem cell transplantation (HSCT) means that clinicians are more willing to offer this therapy to a wider range of patients, including adults with PID, and to a greater range of PIDs in a bid for complete cure. Furthermore, new strategies such as gene therapy and newborn screening for severe combined immunodeficiency (SCID), molecular therapy (e.g. Janus kinase inhibitors) and monoclonal antibody therapy are now viable options to include within the UK health‐care system. Data from national registries provide vital information for clinicians and health policy planners in evaluating the merits of the potential introduction of such strategies.
Methods
The development, ongoing management and technical database structure of the registry was described in our first report 2, 4, 5, 6. Multicentre Research Ethics (MREC) approval was obtained in 2004 for the ESID online database (MREC number: 04/MRE07/68). Approvals have been amended to reflect the establishment of a UK‐based database.
A retrospective analysis of the registry data was performed. Minimum prevalence and incidence, as well as live birth data, were calculated using UK population data sourced from the Office for National Statistics estimates 7, 8, 9, 10. Annual incidence rates have been calculated per 100 000 UK population. Data relating to geographical, gender and sex distribution in addition to age of onset and diagnostic delay were analysed using parametric and non‐parametric analysis as appropriate. Where data were available for only a subset of the patients the denominator is stated within the text. The UKPID registry also collects data on patients with secondary antibody deficiency. These patients have been excluded from data pertaining to prevalence and incidence of PID as well as International Union of Immunological Sciences (IUIS) category breakdown. Their data have been included to demonstrate their diagnostic delay and immunoglobulin data due to the significant contribution this patient group make to the UK clinical immunology workload and as a comparator cohort for immunoglobulin‐treated patients with infection.
Data quality continues to be heavily reliant upon qualified users inputting data. Contributing centres are well established within the primary immunodeficiency field. Users must be approved by their head of department and are trained in the documentation of medical data. There is additional ongoing data monitoring by a registry co‐ordinator and a nominated person in each centre. The database itself has further features to assure data quality, e.g. mandatory fields and logic rules. New entries are reviewed by the registry co‐ordinator to ensure that no replication has occurred. In addition, the registry is interrogated on a regular basis to detect and correct any further duplicated entries.
Results
There are currently 38 recognized centres in the United Kingdom providing specialist immunology services, 37 of which (97%) are enrolling patients actively into the UKPID database, compared to 71% in 2012 (Fig. 1). As of August 2017, 4758 patients have been entered into the registry. Recruitment has increased significantly since our 2013 report, which included data on 2229 patients (Fig. 2); 4258 patients were alive and being followed‐up (89·5%). Excluding those patients with secondary antibody deficiency (n = 369), this equates to a minimum 2017 UK PID prevalence of 5·90/100 000. Three hundred (6·3%) patients have died since being entered into the database from the ESID database inception in 2004 and this UKPID registry in 2008. Antibody disorders make up the largest group of patients within the registry, with a minimum UK prevalence of 3·92/100 000 (n = 2589, 60%). Prevalence data for the nine IUIS classification categories 3 are shown in Table 1. There were 2399 females and 2359 males registered. Eight hundred and seven (17·0%) patients were aged 16 years or younger at the time of the latest data entry and collection.
Figure 1.
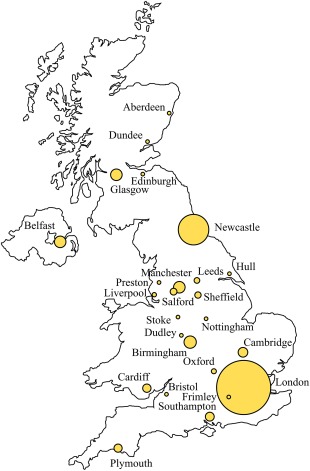
Geographical distribution of patients enrolled in the United Kingdom Primary Immune Deficiency (UKPID) Registry by city or town of documenting centre. The diameter of the circle is directly proportional to the number of patients enrolled in each centre.
Figure 2.
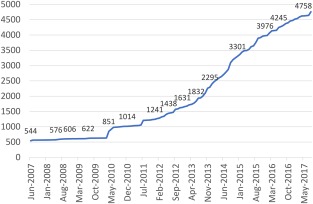
Recruitment of total patient numbers into the United Kingdom Primary Immunodeficiency Registry.
Table 1.
Frequency table for International Union of Immunological Sciences (IUIS) classification and minimum disease prevalence
| IUIS classification | n (alive patients) | UK prevalence/100 000 |
|---|---|---|
| Autoinflammatory disorders | 25 | 0·04 |
| Combined immunodeficiencies | 329 | 0·50 |
| Complement deficiencies | 559 | 0·85 |
| Defects in innate immunity | 39 | 0·06 |
| Diseases of immune dysregulation | 94 | 0·14 |
| Other well‐defined PIDs | 325 | 0·49 |
| Phagocytic disorders | 177 | 0·27 |
| Predominantly antibody disorders | 2589 | 3·92 |
| Unclassified immunodeficiencies | 160 | 0·24 |
Estimated minimum prevalence data for primary immunodeficiency (PID) in the United Kingdom is based on a national population of 66 029 990 (source: Office for National Statistics).
Consanguinity was reported in 118 of 4097 cases (2·9%), equal to the proportion in our previous report (2·9%). Nine hundred and sixty‐eight of 3971 available cases were identified as familial cases (24·4%), as per our previous report of 24·0%. One thousand and thirty‐five (21·8%) patients had a proven genetic defect underlying their PID. Of patients with agammaglobulinaemia, 5·7% (177) had a defect in BTK and one patient had a defect in the Immunoglobulin Heavy Constant Mu (IGHM) gene; 75·5% (n = 142) patients with severe combined immunodeficiency (SCID) had a proven genetic defect, with common gamma chain being the most common, accounting for 32·4% (n = 46) of cases. A full breakdown of the genetic defects found in the SCID registry patients is shown in Table 2. Of patients with chronic granulomatous disease (CGD), 66·6% (n = 96) had a proven genetic defect, with mutations in CYBB gene encoding the gp91‐phox protein accounting for the majority of cases (68·8%, n = 66). Eighteen (2·7%) of the 678 for whom data were available had their genetic defect diagnosed using whole exome sequencing.
Table 2.
Genetic defects in SCID registry patients
| Genetic defect | Number of cases | Proportion (%) |
|---|---|---|
| Common gamma chain (X‐linked) | 46 | 32·39 |
| ADA | 38 | 26·76 |
| IL‐7Rα | 14 | 9·86 |
| JAK3 | 11 | 7·75 |
| RAG1 | 15 | 10·56 |
| Artemis | 10 | 7·04 |
| RAG2 | 3 | 2·11 |
| IL‐21R | 3 | 2·11 |
| CD3e | 1 | 0·70 |
| LIG4 | 1 | 0·70 |
ADA = adenosine deaminase; IL = interleukin; JAK3 = Janus kinase 3; RAG = recombination activating; LIG4 = DNA ligase 4; SCID = severe combined immunodeficiency.
Antibody disorders continue to make up the largest group of all registered patients, accounting for 2821 (59·7%) of a total of 4727 registry patients for whom diagnosis was recorded. The most frequently reported PID is common variable immunodeficiency (CVID), accounting for 1404 patients (29·7%). The second most frequent diagnosis was hereditary angioedema (HAE) (n = 514, 10·9%). Secondary hypogammaglobulinaemia (n = 409, 8·7%), unclassified antibody deficiencies (n = 310, 6·6%), agammaglobulinaemia (n = 209, 4·4%), unclassified immunodeficiencies (n = 191, 4·0%), SCID (n = 188, 4·0%) and specific antibody deficiency (n = 165, 3·5%) were the next most frequent reported diagnoses. The minimum UK prevalence for CVID is 1·93/100 000 population, HAE 0·73/100 000, secondary hypogammaglobulinaemia 0·56/100 000, unclassified antibody deficiency 0·43/100 000, agammaglobulinaemia 0·30/100 000 and SCID 0·26/100 000. A full list of prevalence rates for all diseases recorded within the registry can be found in Supporting information, Table S1.
The median annual prevalence of PID from 2010 to 2015 was 0·38 new cases per 100 000 UK population (one per 270 270), peaking at 0·44 new cases per 100 000 UK population in 2012 (one per 227 518). The incidence per 100 000 UK live births is shown in Fig. 3. There is a clear rise in incidence per 100 000 live UK births from the mid‐1980s. This is likely to be due to an increased recognition of PID, resulting in more patients being entered into the registry enabling a truer reflection of incidence. In addition, with modern management, many patients are expected to live into adulthood, thereby increasing the number of cases of inherited PID in addition to any de‐novo genetic mutations. The apparent drop in incidence from 2000, seen in Fig. 3, is a result of cases born in this time‐period not yet diagnosed with PID (e.g. CVID). From 1980 to 2000 the minimum median incidence of PID was 7·60 cases per 100 000 UK live births or one per 13 157 births.
Figure 3.
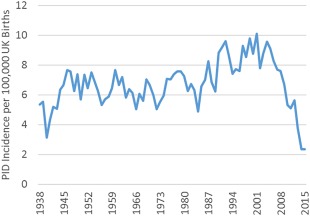
UK incidence of registered primary immunodeficiency (PID) per 100 000 live births.
Diagnostic delay can affect outcome negatively in PID. Prompt diagnosis improves outcomes following HSCT for SCID 11, 12, 13 and is recognized as an important prognostic indicator in antibody deficiencies 14, 15, 16. The current median diagnostic delay for SCID was 60 days [interquartile range (IQR) = 0–121]. The current median diagnostic delay in CVID was 4 years (IQR = 1–10). Spearman's correlation demonstrates a statistically significant but weak correlation for a decreasing diagnostic delay over time for CVID (r s = –0·0719, P = 0·0213). For agammaglobulinaemia the median delay is 1 year (IQR = 0–2). For the 3912 patients for whom data are available, the main presenting symptom is infection‐related, accounting for 76·8% of patients, followed by immune dysregulation with 8·1%. Presenting symptom and diagnostic delay by diagnosis and IUIS category are shown in Table 3.
Table 3.
Diagnostic delay and main presenting symptom for the most common primary immunodeficiencies (PIDs) and International Union of Immunological Societies (IUIS) category in years (median, 25th and 75th quartiles)
| Min | 25th quartile | Median | 75th quartile | Max | Immune dysregulation | Infections | Malignancy | Syndromal | Other | |
|---|---|---|---|---|---|---|---|---|---|---|
| PID | ||||||||||
| CVID | 0 | 1 | 4 | 10 | 69 | 5·1% | 93·7% | 0·1% | 0·1% | 1·0% |
| Hereditary angioedema | 0 | 0 | 2 | 10 | 55 | 0·0% | 0·3% | 0·0% | 2·0% | 97·7% |
| Secondary hypogammaglobulinaemia | 0 | 0 | 1 | 3 | 64 | 1·9% | 96·7% | 0·8% | 0·0% | 0·6% |
| Agammaglobulinaemia | 0 | 0 | 1 | 2 | 44 | 0·0% | 98·2% | 0·0% | 0·6% | 1·2% |
| Unclassified antibody deficiency | 0 | 1 | 2 | 5 | 61 | 3·0% | 93·7% | 0·3% | 0·3% | 2·6% |
| Age (years) | ||||||||||
| < 18 | 0 | 0 | 0 | 1 | 14 | 19·2% | 65·7% | 0·0% | 8·7% | 6·4% |
| Between 18 and 65 | 0 | 1 | 3 | 8 | 48 | 1·2% | 89·6% | 0·4% | 0·4% | 8·4% |
| > 65 | 0 | 1 | 3 | 10 | 69 | 7·0% | 76·2% | 0·3% | 1·6% | 14·9% |
| IUIS category | ||||||||||
| Autoinflammatory disorders | 0 | 2·5 | 6 | 10·5 | 33 | 81·8% | 0·0% | 0·0% | 9·1% | 9·1% |
| Combined immunodeficiencies | 0 | 0 | 0 | 1 | 47 | 17·6% | 77·9% | 0·0% | 1·5% | 3·1% |
| Complement deficiencies | 0 | 0 | 1 | 8 | 55 | 0·0% | 10·6% | 0·0% | 1·9% | 87·4% |
| Defects in innate immunity | 0 | 1 | 2 | 6 | 61 | 4·8% | 88·1% | 0·0% | 4·8% | 2·4% |
| Diseases of immune dysregulation | 0 | 0 | 1 | 4 | 43 | 67·0% | 26·2% | 0·0% | 4·9% | 1·9% |
| Other well‐defined PIDs | 0 | 0 | 1 | 3·5 | 66 | 19·9% | 50·7% | 0·0% | 27·2% | 2·2% |
| Phagocytic disorders | 0 | 0 | 1 | 3 | 37 | 14·5% | 83·6% | 0·0% | 1·2% | 0·6% |
| Predominantly antibody disorders | 0 | 1 | 3 | 8 | 69 | 3·5% | 94·3% | 0·4% | 0·1% | 1·6% |
| Unclassified immunodeficiencies | 0 | 0 | 2 | 9 | 66 | 14·8% | 80·0% | 0·7% | 2·2% | 2·2% |
| Presenting symptom | ||||||||||
| Immune dysregulation | 0 | 0 | 1 | 6 | 43 | |||||
| Infections | 0 | 1 | 2 | 6 | 67 | |||||
| Malignancy | 0 | 3 | 4 | 4·25 | 5 | |||||
| Syndromal | 0 | 0 | 1 | 8 | 55 | |||||
| Other | 0 | 0 | 0 | 2·25 | 11 | |||||
PID = primary immunodeficiency; CVID = common variable immunodeficiency; IUIS = International Union of Immunological Societies.
A total of 2836 patients are recorded to have received immunoglobulin replacement therapy (59·6% of the total 4758 registry patients); 1391 (49%) received this by intravenous route and 1440 (51%) by subcutaneous route; and 1669 (58·9%) received their infusion at home. The median dose of immunoglobulin was 514 mg/kg/month (IQR = 424–645), with a median interval of 3 weeks.
A total of 679 patients were recorded as having received an HSCT since 1973, with the majority (87·2%) transplanted after 2000 (Fig. 4). Three hundred and ten (45·7%) received their HSCT from donor blood marrow, 200 (29·5%) from peripheral blood stem cells, 59 (8·7%) from cord blood stem cells and in 110 (16·2%) the donor was not recorded. Two hundred and ninety‐four (43·3%) were matched unrelated donor (MUD), 167 (24·6%) matched sibling donor (MSD), 77 (11·3%) haploidentical, 73 (10·8%) mismatched unrelated donor (MMUD), two (0·3%) autologous and the source was unrecorded in 66 (9·7%). Autologous HSCT is not a standard of care in PID; there are no further data on these two cases recorded in this registry. The overall survival rate for HSCT in this registry is 83·8% with a mortality of 7·7% (8·5% are either discharged or lost to follow‐up). Since 2000, 26 patients have undergone gene therapy. The survival for gene therapy patients in the registry is currently 100%.
Figure 4.

Number of primary immunodeficiency (PID) patients undergoing haematopoietic stem cell transplantation (HSCT) or gene therapy.
Discussion
The UKPID registry celebrates its 10th birthday in 2018. During this decade almost all immunology centres in the United Kingdom have contributed actively to the database, and the number of recruited patients continues to grow each year. London and Newcastle (supraregional centres for transplantation of paediatric PID) continue to provide a large contribution to the database (accounting for 25·0% and 12·6% of the total registry, respectively). The wide geographical spread of actively recruiting centres should ensure that the registry reflects accurately the pattern of health‐care service access and delivery throughout the United Kingdom.
The UKPID registry allows easy‐to‐access and reliable data sets for clinicians and researchers. This enables assessment of patient outcomes to be performed in a timely and effective manner, such as that seen in the recent work from Stubbs et al. 17, suggesting that patients with agammaglobulinaemia in the United Kingdom suffer from deteriorating pulmonary health despite current therapies. Compiling such a body of work without the aid of the UKPID registry would result in considerable additional workload and time to the research process.
Since our first report, we estimated the number of patients with PID in the United Kingdom to be between 4000 and 5000. Our latest count of 4258 verified live patients is extremely encouraging. The minimum prevalence of PID in the United Kingdom with these latest data stands at 5·90/100 000 population. This is similar to the reported incidence in France of 6·06 per 100 000 and larger than Switzerland (4·16 per 100 000) and Germany (2·11 per 100 000) 1. These disparities are likely to be due to differences in reporting, as individual countries continue to develop their own reporting strategies. With the coverage of the UKPID registry (97% of immunology centres), we feel this minimum incidence is an accurate reflection of the burden of PID within the general population. It is possible that this is still an underestimate, with some patients not recruited to the registry and some patients being treated at hospitals not designated as immunology centres, but these numbers are likely to be small. However, a recent epidemiological field survey from Mahlaoui et al. 18 suggests that the true minimum prevalence of PID in France is actually 11 per 100 000 population, and may therefore mean that these numbers still underestimate significantly the true burden of PID within the population.
The expansion in registry patients also enables us to calculate a reliable estimate of PID incidence per UK annual live births. The data showed a median PID incidence from 1980 to 2000 of one in 13 157 births. This number is still likely to be an underestimate of the true value, with a significant proportion of patients in this period dying either before their PID is recognized or before the establishment of the UKPID registry. With the registry now firmly established, we hope to increase the accuracy of these data for future reports. The proportion of under 16‐year‐olds in the database is currently 17·0%, similar to the under 16‐year‐old proportion of the general UK population at 18·8% 7.
Antibody deficiencies continue to account for the largest group of PID cases within the registry (60%), has remained stable since our first report and is in keeping with other registries 1.
Clinicians strive continually to diagnose patients earlier to improve patient outcomes. Nearly a quarter of patients presented with symptoms other than recurrent infections. Non‐infectious presentations such as autoimmune cytopaenias, inflammatory bowel disease and malignancy are being recognized increasingly as possible presentations of PID 19, 20, 21, 22. The median diagnostic delay for patients who presented with malignancy is 4 years, the highest amongst the presenting symptoms recorded by our registry. Increased awareness of these facts as demonstrated by these data and those of others should hopefully result in reductions in diagnostic delay for future patients.
Increased awareness of the genetic basis of PID and thus the importance of screening newborn siblings of affected patients will help to reduce delays. Newborn screening for SCID by measuring T cell receptor excision circles (TRECs) on the newborn blood spot is due to start in the United Kingdom in 2018 under a pilot programme, which may offer significant improvements in event‐free survival for SCID patients in the United Kingdom. Diagnostic delay in the diagnosis of agammaglobulinaemia remains consistent at 1 year. Newborn screening for congenital B cell deficiencies is possible using a similar technique to SCID screening, by measuring kappa‐deleting recombination excision circles (KRECs) on the newborn blood spot. Some countries do, indeed, combine a TREC/KREC screening programme, but the effectiveness of a KREC screening programme is currently unknown.
Immunoglobulin therapy remains the mainstay of treatment for the vast majority of antibody deficiency syndromes. The proportion of those patients receiving intravenous immunoglobulin therapy (IVIG) has fallen from 60% in our previous report to an equal split in the cohort between intravenous and subcutaneous therapies (SCIG). For the 2836 patients recorded as receiving immunoglobulin therapy, more than half (59%) receive their therapy at home. These data highlight the patient preference for therapy at home, and should continue to be offered actively to all patients wherever possible.
Better understanding of, and access to, genetic testing can enable faster and more accurate diagnosis of PID leading to improved outcomes 23. Nearly a quarter of the registry patients have a proven gene defect underlying their PID, although the number of patients who had genetic testing but no defect found is unknown in this registry's data. In the previous report (2014) only 20 patients had a recorded genetic diagnosis; significant work to improve capture of genetic diagnoses has been undertaken. Diseases such as agammaglobulinaemia continue to show a high proportion of cases where a genetic defect is found (85%). However, common diseases such as CVID continue to show a low proportion of cases for which a genetic defect is found (1·78%). Next‐generation sequencing looks set to supersede conventional Sanger sequencing in the coming years, leading to a potentially higher proportion of patients for whom a genetic defect is known and to the discovery of new PIDs 24.
The UKPID registry is now established firmly within the United Kingdom and data are available for the majority of PID patients. This data set enables a relatively accurate estimate of disease burden of primary immunodeficiency within the United Kingdom. During the next 5–10 years we hope to continue this successful recruitment, as well as adding the next level of registry data encompassing more detailed diagnostic and follow‐up data; e.g. infection incidence, medication, vaccinations, lung function, laboratory values and quality of life. It is also planned to include further therapeutic data, most notably the use of biologicals and targeted therapy, for which this registry could provide a useful data source for surveying the use of these agents. These extra levels of detail will further enable accurate assessment of outcomes in PID to be performed quickly and with relative ease than would otherwise be possible without such a registry. As research in PID advances there is likely to be an increasing range of interventions available to patients. The ability to evaluate current outcomes in a timely manner will be vital to ensuring that patients are able to access the best possible care. We look forward to working with researchers and clinicians in providing reliable, detailed data on PID within the United Kingdom to aid research, rational resource allocation and improvements in clinical care.
Disclosure
B. S., C. B., D. G., A. G., M. S., M., A. H., P. A., H. A., S. S., S. P., H. B., S. N., P. Y., C. W., P K., R. H., J. B., G. H. and A. R. have no conflicts of interest to declare. H. L. has contributed as a research support, educational support, speaker's bureau, adviser to Biotest, CSL Behring, Shire (Baxalta) and as an adviser to Octapharma. J. D. M. E. has received fees for speaking (Shire Israel), and consulting (Shire UK, Octapharma UK, Grifols UK and CSL Behring UK). A. W. has worked as a paid consultant for Biotest UK, working on an advisory board for the use of CMV‐specific immunoglobulin. S. J. has received support for speaker, congress, advisory boards, clinical trials, DSMB, and projects from CSL Behring, LFB, Shire, Biotest, Grifols, BPL, Octapharma, UCB Pharma, Binding Site, Sanofi, GSK and SOBI. H. B. acted as paid consultant to Baxter and Biotest and has received reimbursement for symposium attendance by CSL Behring, Baxter, Biotest and Octopharma. D. K. has between 2013 and 2018, been funded to attend scientific meetings by CSL Behring and Shire and Biotest, and been paid for delivery of a lecture by Biotest and acted as a paid consultant to Shire. P. V. has received financial support from Binding Site and Shire to attend symposiums. M. B. has attended the global forum for HAE and ESID 2016 sponsored by Shire, and the Biotest Immunology Forum sponsored by Biotest. C. S. has received education grants from CLS Behring, Alk and Baxter. M. G. has received payments from BioCryst pharmaceuticals for advisory board work and educational grants from Novartis pharmaceuticals, Bristol Myers and has ongoing research trials with Merck and Viiv. C. C. has received reimbursement from CSL Behring to attend the large PID Congress (ESID Conference) in 2016 and 2017. T. G. is chair of UKPIN, has received financial support to attend conferences from CSL and Shire contributes to a paid advisory board and chairing work for CSL and Shire. M. B. has received funding for trips or meetings and/or consultancy from Shire, Octopharma, CSL Behring and Biotest.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Frequency table for individual diagnosis for live patients and disease prevalence. Estimated data for primary immunodeficiency (PID) in the United Kingdom based on national population of 66 029 900 (source: Office for National Statistics).
Acknowledgements
The authors thank the following individuals: D. Mullen (NHS Grampian); L. Lorenzo, J. Dempster, S. Grigoriadou (Barts Health NHS Trust); L. Devlin (Belfast Health and Social Care Trust); C. Jones, M. Kusano (Sandwell and West Birmingham Hospitals); J. Daglish, S. Onyango‐Odera, S. Hackett (Heart of England NHS Trust); E. Knight (University Hospitals Birmingham NHS Foundation Trust); F. Manyika (University Hospitals Bristol NHS Foundation Trust); L. Jennings, L. Smith (North Bristol NHS Trust); A. Manson, M. Fordham, A. Chandra, M. Krishna (Cambridge University Hospitals NHS Foundation Trust); K. Henderson, H. Gronlund (Papworth Hospital NHS Foundation Trust); E. Carne, C. Joyce, C. Kingdon, T. El‐Shanawany (Cardiff and Vale University Health Board); G. Menzies (NHS Tayside); G. Paul, D. Baxter (NHS Lothian); M. Milarionmayieka (Epsom and St Helier University Hospitals); C. Quinn (Frimley Health NHS Foundation Trust); M. Brownlie, H. Millar, S. Murng (NHS Greater Glasgow and Clyde), R. Savjani (Great Ormond Street Hospital for Children NHS Foundation Trust); J. Moor, B. Fish (Hull and East Yorkshire Hospitals NHS Trust); K. Ford, J. Toolan, P. Wood, G. Arumugakani (The Leeds Teaching Hospitals NHS Trust); J. Berry (The Royal Liverpool and Broadgreen University Hospitals); C. Beeson (Alder Hey Children's NHS Foundation Trust); B. Boardman, S. Hughes (Manchester University NHS Foundation Trust); T. Green, O. Grix, S. Elcombe, C. Stroud, P. Tierney, A. Cant (The Newcastle upon Tyne Hospitals NHS Foundation Trust); R. Weldon, E. Drewe, P. Madhuri Vaitla (Nottingham University Hospitals NHS Trust); A. Welby, R. Jain (Oxford University Hospitals NHS Foundation Trust); C. Symons, T. Trump, A. Whyte (Plymouth Hospitals NHS Trust); K. Haworth, A. Anantharachagan (Lancashire Teaching Hospitals NHS Foundation Trust); S. Workman, A. Symes (Royal Free London NHS Foundation Trust); L. Common, I. Jones, M. Fernandez, A. Herwadkar (Salford Royal NHS Foundation Trust); A. Ford, F. Shackley (Sheffield Children's NHS Foundation Trust); F. Ashworth, A. Shrimpton (Sheffield Teaching Hospitals NHS Foundation Trust); S. Fenton‐Edwards, W. Rae, E. Eren (University Hospital Southampton NHS Foundation Trust); C. Bowmar‐Scothern (St George's University Hospitals NHS Foundation Trust), the United Kingdom Primary Immunodeficiency Network.
References
- 1. Grimbacher B. The European Society for Immunodeficiencies (ESID) registry 2014. Clin Exp Immunol 2014; 178:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Edgar JDM, Buckland M, Guzman D et al The United Kingdom Primary Immune Deficiency (UKPID) Registry: report of the first 4 years' activity 2008–2012. Clin Exp Immunol 2014; 175:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Picard C, Al‐Herz W, Bousfiha A et al Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for primary immunodeficiency 2015. J Clin Immunol 2015; 35:696–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eades‐Perner A‐MM, Gathmann B, Knerr V et al The European internet‐based patient and research database for primary immunodeficiencies: results 2004‐06. Clin Exp Immunol 2007; 147:306–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gathmann B, Grimbacher B, Beauté J et al The European internet‐based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol 2009; 157:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guzman D, Veit D, Knerr V et al The ESID online database network. Bioinformatics 2007; 23:654–5. [DOI] [PubMed] [Google Scholar]
- 7.Office for National Statistics (ONS) . Overview of the UK population – Office for National Statistics. Office for National Statistics; 2017. Available at: https://www.ons.gov.uk/peoplepopulationandcommunity/populationandmigration/populationestimates/articles/overviewoftheukpopulation/july2017 (accessed 24 October 2017).
- 8. Office for National Statistics . England and Wales Live Births 1938–2015. Available at: https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/livebirths (accessed 16 June 2017).
- 9. Scotland National Register of Scottish Live Births 1918–2015. Available at: https://www.nrscotland.gov.uk/statistics-and-data/statistics/statistics-by-theme/vital-events/births (accessed 16 June 2017).
- 10. Northern Ireland Statistics and Research Agency . Northern Ireland Live Births 1887–2015. Available at: https://www.nisra.gov.uk/statistics/births-deaths-and-marriages/births (accessed 16 June 2017).
- 11. Chan A, Scalchunes C, Boyle M, Puck JM. Early vs. delayed diagnosis of severe combined immunodeficiency: a family perspective survey. Clin Immunol 2011; 138:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brown L, Xu‐Bayford J, Allwood Z et al Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood 2011; 117:3243–6. [DOI] [PubMed] [Google Scholar]
- 13. Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood 2002; 99:872–8. [DOI] [PubMed] [Google Scholar]
- 14. Wood P, Turner‐Stokes L, Higgins B. Primary antibody deficiencies: recognition, clinical diagnosis and referral of patients. Clin Med 2009; 9:595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wood P, Stanworth S, Burton J et al Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systematic review. Clin Exp Immunol 2007; 149:410–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spickett GP, Askew T, Chapel HM. Management of primary antibody deficiency by consultant immunologists in the United Kingdom: a paradigm for other rare diseases. Qual Saf Health Care 1995; 4:263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stubbs A, Bangs C, Shillitoe B et al Bronchiectasus and deteriorating lung function in agammaglobulinaemia despite immunoglobulin replacement therapy. Clin Exp Immunol 2018; 191:212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mahlaoui N, Jais J‐P, Brosselin P et al Prevalence of primary immunodeficiencies in France is underestimated. J Allergy Clin Immunol 2017; 140: 1731–3. [DOI] [PubMed] [Google Scholar]
- 19. Fischer A, Provot J, Jais J‐P, Alcais A, Mahlaoui N. Autoimmune and inflammatory manifestations occur frequently in primary immunodeficiencies. J Allergy Clin Immunol 2017; 140:1388–93.e8. [DOI] [PubMed] [Google Scholar]
- 20. Grimbacher B, Warnatz K, Yong PFK, Korganow A‐S, Peter H‐H. The crossroads of autoimmunity and immunodeficiency: lessons from polygenic traits and monogenic defects. J Allergy Clin Immunol 2016; 137:3–17. [DOI] [PubMed] [Google Scholar]
- 21. Seidel MG. Autoimmune and other cytopenias in primary immunodeficiencies: pathomechanisms, novel differential diagnoses, and treatment. Blood 2014; 124:2337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hauck F, Voss R, Urban C, Seidel MG. Intrinsic and extrinsic causes of malignancies in patients with primary immunodeficiency disorders. J Allergy Clin Immunol 2018; 141:59–68.e4. [DOI] [PubMed] [Google Scholar]
- 23. Raje N, Soden S, Swanson D, Ciaccio CE, Kingsmore SF, Dinwiddie DL. Utility of next generation sequencing in clinical primary immunodeficiencies. Curr Allergy Asthma Rep 2014; 14:468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Picard C, Fischer A. Contribution of high‐throughput DNA sequencing to the study of primary immunodeficiencies. Eur J Immunol 2014; 44:2854–61.] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Frequency table for individual diagnosis for live patients and disease prevalence. Estimated data for primary immunodeficiency (PID) in the United Kingdom based on national population of 66 029 900 (source: Office for National Statistics).