

Abstract
Background and Purpose
N‐type voltage‐gated calcium (Cav2.2) channels are critical determinants of increased neuronal excitability and neurotransmission accompanying persistent neuropathic pain. Although Cav2.2 channel antagonists are recommended as first‐line treatment for neuropathic pain, calcium‐current blocking gabapentinoids inadequately alleviate chronic pain symptoms and often exhibit numerous side effects. Collapsin response mediator protein 2 (CRMP2) targets Cav2.2 channels to the sensory neuron membrane and allosterically modulates their function. A 15‐amino‐acid peptide (CBD3), derived from CRMP2, disrupts the functional protein–protein interaction between CRMP2 and Cav2.2 channels to inhibit calcium influx, transmitter release and acute, inflammatory and neuropathic pain. Here, we have mapped the minimal domain of CBD3 necessary for its antinociceptive potential.
Experimental Approach
Truncated as well as homology‐guided mutant versions of CBD3 were generated and assessed using depolarization‐evoked calcium influx in rat dorsal root ganglion neurons, binding between CRMP2 and Cav2.2 channels, whole‐cell voltage clamp electrophysiology and behavioural effects in two models of experimental pain: post‐surgical pain and HIV‐induced sensory neuropathy induced by the viral glycoprotein 120.
Key Results
The first six amino acids within CBD3 accounted for all in vitro activity and antinociception. Spinal administration of a prototypical peptide (TAT‐CBD3‐L5M) reversed pain behaviours. Homology‐guided mutational analyses of these six amino acids identified at least two residues, Ala1 and Arg4, as being critical for antinociception in two pain models.
Conclusions and Implications
These results identify an antinociceptive scaffold core in CBD3 that can be used for development of low MW mimetics of CBD3.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Abbreviations
- CBD3
calcium channel‐binding domain 3
- CRMP2
collapsin response mediator protein 2
- gp120
glycoprotein of 120 kDa
- HIV
human immunodeficiency virus
- HIV‐SN
HIV‐induced sensory neuropathy
- PLA
proximity ligation assay
- TAT
HIV‐1 transactivator of transcription domain
Tables of Links
| TARGETS |
|---|
| Voltage‐gated ion channels |
| http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=533 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Improving the current chronic pain management strategies currently available could benefit over 20% of the worldwide population (Vink and Alewood, 2012). At present, only 30–40% of patients experience greater than 40–50% of pain relief with existing analgesics (Backonja and Woolf, 2010). Despite their limited utility and numerous side effects (Gray et al., 2014), opioids are the most frequently used drugs for neuropathic pain syndromes (Hassett et al., 2014; Dowell et al., 2016). Opioid receptor agonists inhibit pain neurotransmission by activating postsynaptic K+ currents and by inhibiting presynaptic N‐type voltage‐gated calcium (Cav2.2) channels (Malmberg and Yaksh, 1995; Bourinet et al., 1996). Cav2.2 channels are critical determinants of increased neuronal excitability and neurotransmission accompanying persistent neuropathic pain (Cizkova et al., 2002). Expressed in the presynaptic termini of primary afferent nociceptors in the spinal cord (Westenbroek et al., 1992), the Cav2.2 channels represent possible control points for aberrant synaptic activity. The therapeutic potential of targeting Cav2.2 channels has been demonstrated with blockade of these channels with conotoxins, for allodynia (Scott et al., 2002) and the altered pain behaviours of Cav2.2 channel knockout mice (Hatakeyama et al., 2001; Kim et al., 2001). Although Cav2.2 channel antagonists are recommended as first‐line treatment for neuropathic pain (Attal et al., 2010; Dowell et al., 2016), calcium current‐blocking gabapentinoids inadequately alleviate chronic pain symptoms and often are compromised by numerous and serious side effects (Dworkin et al., 2007, Zamponi et al., 2015). Another Cav2.2 channel‐targeted drug, ω‐conotoxin MVIIA (ziconotide/Prialt®), is used clinically to treat neuropathic pain (Wallace, 2006), but its use is hampered by difficult dosing regimens, a narrow therapeutic window and numerous side effects (Thompson et al., 2006; Schmidtko et al., 2010). The discovery of an opioid‐resistant Cav2.2 splice isoform in sensory neurons (Jiang et al., 2013) further emphasizes the necessity of developing alternatives to opioids.
In this context, rather than inhibiting the Cav2.2 channels themselves, targeting the regulators of its trafficking has emerged as a promising approach (Feldman and Khanna, 2013). The membrane localization of Cav2.2 channels is dependent on their interaction with the collapsin response mediator protein 2 (CRMP2) (Brittain et al., 2009; Brittain et al., 2011b; Moutal et al., 2016b). CRMP2, a determinant of axonal growth (Fukata et al., 2002), interacts with the first intracellular loop and the C‐terminal domain of Cav2.2 channels (Brittain et al., 2009; Brittain et al., 2011b). Further mapping identified a short peptide within CRMP2 that uncoupled the CRMP2–Cav2.2 channel interaction (Brittain et al., 2011b). This calcium channel‐binding domain 3 (CBD3) peptide inhibited Cav2.2 channel activity by inducing its relocalization away from the sensory neuron membrane (Francois‐Moutal et al., 2015; Xie et al., 2016). When conjugated to various cell‐penetrating motifs [e.g. HIV‐1 transactivator of transcription domain (TAT), Brittain et al., 2011b; Piekarz et al., 2012; nona‐arginine, Ju et al., 2012; or myristate, Francois‐Moutal et al., 2015], CBD3 inhibited the CRMP2– Cav2.2 channel interaction, decreased Cav2.2 channel surface trafficking and, consequently, inhibited the release of the peptide neurotransmitter calcitonin gene‐related peptide (Brittain et al., 2011b). These CRMP2 peptides demonstrated antinociceptive properties in models of post‐operative pain (Francois‐Moutal et al., 2015), neuropathic pain induced by antiretroviral drug treatment (Ripsch et al., 2012) or focal nerve demyelination (Wilson et al., 2011), as well as neuroprotective properties in models of excitotoxicity and stroke (Brittain et al., 2011a; Brittain et al., 2012a). Notably, the peptides did not affect memory, motor functions or anxiety/depression and did not produce any addictive behaviours (Brittain et al., 2011b; Francois‐Moutal et al., 2015; Xie et al., 2016). Thus, while the CBD3 peptides themselves add to the armamentarium of possible newer therapeutics, here we set out to map the functional requirements for the anti‐nociceptive specificity of CRMP2‐derived peptides. Once an anti‐nociceptive scaffold core in CBD3 has been identified, a pharmacophore can be constructed and, in turn, low MW mimetics of CBD3 can be designed computationally.
Methods
Animals
All animal care and experimental procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health and the ethical guidelines of the International Association for the Study of Pain, and were approved by the Institutional Animal Care and Use Committee of the University of Arizona. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Pathogen‐free, adult female and male Sprague–Dawley rats (initial weight 225–250 g; Harlan Laboratories, Houston, TX, USA) were housed in temperature (23 ± 3°C) and light (12 h light:12 h dark cycle; lights on at 07:00 h) controlled rooms with standard rodent chow and water available ad libitum. There are very few studies using female animals in preclinical settings, despite many chronic pain conditions involving more female patients than male counterparts. Historically, we have used mostly female rats for behavioural studies and male rats for in vitro studies. Here, we have switched the genders in a type of ‘crossover’ approach and used male rats for behaviour and females for in vitro work. The data presented in this manuscript demonstrate congruence between genders in various assays. Animals were randomly assigned to treatment or control groups and experimenters were blinded to the grouping information, until the analysis was completed.
Purification of recombinant CRMP2‐6XHis
CRMP2‐6XHis fusion protein was purified as described previously (Francois‐Moutal et al., 2015). Briefly, following a 19‐h induction at 16°C with 0.5 mM Isopropyl β‐D‐1‐thiogalactopyranoside, BL21 Escherichia coli cells (Thermo Fisher Scientific, Waltham, MA) expressing recombinant CRMP2‐6XHis were resuspended in 50 mM NaH2PO4, pH 7.5, 500 mM NaCl, 10% glycerol (vol./vol/), 0.5 mM Tris(2‐carboxyethyl)phosphine hydrochloride (TCEP), supplemented with complete EDTA‐free protease inhibitors (Roche, Basel, Switzerland). Disruption of the bacteria was performed by two rounds of high‐pressure homogenization at 10 000 PSI with a LM10 microfluidizer (Microfluidics, Westwood, MA, USA), and the lysate was centrifuged at 4°C for 45 min at 4500× g. The supernatant was loaded on a HisTrap HP column (GE Healthcare, Uppsala, Sweden) equilibrated with 50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 0.5 mM TCEP. After a washing step with 50 mM HEPES, pH 7.5, 20 mM imidazole, 300 mM NaCl, 10% glycerol, 0.5 mM TCEP, CRMP2‐6XHis was eluted with an imidazole gradient. The fractions of interest were desalted using a PD10 desalting column (GE Healthcare) in a 50 mM NaH2PO4, pH 7, 10% glycerol, 0.5 mM TCEP and then loaded on a HiPrep Q XL column (GE Healthcare). Elution was carried out with a NaCl gradient between 50 mM NaH2PO4, pH 7.5 10% glycerol, 0.5 mM TCEP and 50 mM NaH2PO4, 1 M NaCl, 10% glycerol, 0.5 mM TCEP. Finally, the fractions were loaded on a HiLoad Superdex size exclusion column (GE Healthcare) and eluted with 50 mM NaH2PO4, pH 7.5, 10% glycerol, 0.5 mM TCEP. The eluted protein was concentrated with Amicon Ultra 15 centrifugal filters (Regenerated cellulose 10 000 NMWL; Merck Millipore, Darmstadt, Germany), aliquoted and flash‐frozen on dry ice and stored at −80°C until use. Protein concentration was determined by a Pierce assay using BSA as a standard. The purity of the protein was verified with SDS‐PAGE and structure/activity by checking its ability to bind tubulin (data not shown).
Purification of Cav2.2 fragments
Plasmids expressing GST‐fused intracellular loop 1 (Loop 1 or L1; amino acids 356–483 of the rat sequence) or distal C‐terminus (C‐term or Ct; amino acids 6400–7044 of the rat sequence) fragments of Cav2.2 (Brittain et al., 2009) were transformed into BL21 E. coli and grown in Luria‐Bertani media until reaching an optical density of 0.6–0.8, and then protein synthesis was induced with 0.5 mM IPTG during ~19 h at 16°C. Bacterial cells were resuspended in 20 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Triton X‐100 and 20% glycerol, supplemented with complete EDTA‐free protease inhibitors (Roche, Basel, Switzerland). Disruption of the bacteria was performed by two rounds of high‐pressure homogenization at 10 000 PSI with an LM10 microfluidizer (Microfluidics), and the lysate was centrifuged 1 h at 130 000× g at 4°C. Supernatant was aliquoted, flash‐frozen and kept at −80°C until use.
elisa‐based CRMP2–Cav2.2 fragment binding assay
Plates (96‐well, Nunc Maxisorp; Thermo Fisher Scientific, Waltham, MA, USA) were coated with anti‐GST antibody (200 ng per well) and incubated at room temperature overnight. The following day, the plates were washed and blocked with 3% BSA in PBS to minimize non‐specific adsorptive binding to the plates. Lysates from bacteria expressing L1 or Ct were added to the plates. As a negative control, some wells received PBS containing 3% BSA. The plates were incubated at room temperature with shaking for 3 h. Next, the plates were washed with PBS containing 0.1% Tween‐20 to eliminate unbound protein. Then, 1 μM CRMP2 was added to the plates in the presence of the indicated peptides (10 μM) or DMSO (0.1%) as a control and incubated for an hour at room temperature. After three washes with PBS containing 0.1% Tween‐20, bound CRMP2 was detected by anti‐His probe HRP (Cat#15165; Thermo Fisher Scientific). Tetramethylbenzidine (R&D Systems, St. Louis, MO) was used as the colorimetric substrate. The optical density of each well was determined immediately, using a microplate reader (Multiskan Ascent; Thermo Fisher Scientific) set to 450 nm with a correction wavelength of 570 nm. Data were analysed by linear regression analysis using GraphPad Prism 7 (GraphPad, San Diego, CA, USA).
Differential scanning fluorimetry
For the differential scanning fluorimetry (DSF) experiments analysing thermal stability, 10 μM of indicated peptide was added to a buffer consisting of 10 mM Tris, pH 7.4, 50 mM NaCl, to which 5 μL of 250‐times diluted SYPRO Orange stock (5000×) (Cat#S6650; Thermo Fisher Scientific) was added. Changes in the fluorescence intensity of the fluorophore (excitation and emission were 450/490 and 510/530 nm, respectively) were determined in a 96‐well thin‐wall PCR plate using a CFX Connect Real‐Time System (Bio‐Rad Laboratories, Hercules, CA, USA). The temperature of the samples was changed from 25 to 90°C at a heating rate of 0.5°C·min−1 and the fluorescence recorded every 0.5°C. The melting points of the peptides were determined using a Boltzmann model using GraphPad Prism 7.
Culturing of rat primary DRG neurons
Rat dorsal root ganglia (DRG) neurons were isolated from Sprague–Dawley rats (150–174 g ) using previously developed procedures (Moutal et al., 2016a). In brief, removing dorsal skin and muscle and cutting the vertebral bone processes parallel to the dissection stage exposed DRGs. DRGs were then collected, trimmed at their roots and digested in 3 mL bicarbonate‐free, serum‐free, sterile DMEM (Cat#11965; Thermo Fisher Scientific) solution containing neutral protease (3.125 mg·mL−1, Cat#LS02104, Worthington, Lakewood, NJ, USA) and collagenase Type I (5 mg·mL−1, Cat#LS004194, Worthington) and incubated for 60 min at 37°C under gentle agitation. Dissociated DRG neurons (~1.5 × 106) were then gently centrifuged to collect cells and washed with DRG media DMEM containing 1% penicillin/streptomycin sulfate from 10 000 μg·mL−1 stock, 30 ng·mL−1 nerve growth factor and 10% fetal bovine serum (Hyclone) before plating onto poly‐d‐lysine‐ and laminin‐coated glass 12 or 15 mm coverslips. Small diameter neurons were selected to target Aδ‐ and c‐fibre nociceptive neurons. For rat DRG cultures, small cells were considered to be approximately <30 μm. All cultures were used within 48 h.
Calcium imaging in acutely dissociated DRG neurons
DRG neurons were loaded for 30 min at 37°C with 3 μM Fura‐2 Low Affinity (K D = 25 μM, λex 340, 380 nm/λemi 512 nm) to follow changes in intracellular calcium ([Ca2+]c) in a standard bath solution containing 139 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM Na HEPES, pH 7.4, 5 mM glucose, exactly as previously described (Brittain et al., 2011b). Fluorescence imaging was performed with an inverted microscope, Nikon Eclipse TE2000‐U (New York, NY, USA), using objective Nikon Super Fluor 20× 0.75 NA and a Photometrics cooled CCD camera CoolSNAPHQ (Roper Scientific, Tucson, AZ, USA) controlled by MetaFluor 6.3 software (Molecular Devices, Downingtown, PA, USA). The excitation light was delivered by a Lambda‐LS system (Sutter Instruments, Novato, CA, USA). The excitation filters (340 ± 5 and 380 ± 7) were controlled by a Lambda 10‐2 optical filter change (Sutter Instruments). Fluorescence was recorded through a 505 nm dichroic mirror at 535 ± 25 nm. For minimization of photobleaching and phototoxicity, the images were taken every ~5 s during the time course of the experiment using the minimal exposure time that provided acceptable image quality. The changes in [Ca2+]c were monitored by following a ratio of F340/F380, calculated after subtracting the background from both channels.
Proximity ligation assay
The proximity ligation assay (PLA) was performed to visualize protein–protein interactions by microscopy. This assay is based on paired complementary oligonucleotide‐labelled secondary antibodies that can hybridize and amplify a red fluorescent signal only when bound to two corresponding primary antibodies whose targets are in close proximity. DRG neurons were incubated overnight with 10 μM of the indicated peptides or DMSO as a control before fixation using 4% paraformaldehyde for 20 min at room temperature. Blocking and permeabilization were done by incubating the cells with PBS, 0.1% Triton X‐100 with 3% BSA for 30 min at room temperature. Primary antibodies were incubated for 1 h at room temperature in PBS, 0.1% triton X‐100, 3% BSA before three washes in PBS, 0.1% Triton for 5 min at room temperature. The proximity ligation reaction and amplification of signal were performed according to the manufacturer's protocol using the Duolink Detection Kit with PLA PLUS and MINUS probes for mouse and rabbit antibodies (Duolink; Sigma). DAPI stain was used to detect cell nuclei. Immunofluorescent micrographs were acquired on a Nikon Eclipse Ti/U microscope with a photometrics cooled CCD camera CoolSNAP ES2 (Roper Scientific, Planegg, Germany) controlled by NIS Elements software (version 4.30; Nikon Instruments), using a 60X plan Apo 1.40 numerical aperture objective. Image J was used to count the number of PLA dots per cell, which was normalized to the area analysed.
Whole‐cell voltage clamp electrophysiology
Recordings were obtained from acutely dissociated DRG neurons as described above. For isolation of calcium currents, Na+ and K+ currents were blocked with 500 nM tetrodotoxin (TTX; Alomone Laboratories, Jerusalem, Israel) and 30 mM tetraethylammonium chloride (TEA‐Cl; Sigma). Extracellular recording solution (at ~315 mOsm) consisted of the following (in mM): 110 N‐methyl‐d‐glucamine (NMDG), 10 BaCl2, 30 TEA‐Cl, 10 HEPES, 10 glucose, pH at 7.4, 0.001 TTX, 0.01 nifedipine. The intracellular recording solution (at ~315 mOsm) consisted of the following (in mM): 150 CsCl2, 10 HEPES, 5 Mg‐ATP, 5 BAPTA, pH at 7.4. Fire‐polished recording pipettes, 2 to 5 MΩ resistance, were used for all recordings. Whole‐cell recordings were obtained with a HEKA EPC‐10 USB (HEKA Instruments Inc., Bellmore, NY, USA); data were acquired with a Patchmaster (HEKA) and analysed with a Fitmaster (HEKA). Capacitive artefacts were fully compensated, and series resistance was compensated by ~70%. Recordings made from cells with greater than a 5 mV shift in series resistance compensation error were excluded from analysis. All experiments were performed at room temperature (~23°C).
Indwelling intrathecal (i.t.) catheter
Rats were anaesthetized (ketamine/xylazine anaesthesia, 80/12 mg·kg−1 i.p.; Sigma) and placed in a stereotaxic head holder. The cisterna magna was exposed and incised, and an 8 cm catheter (PE‐10, Stoelting Co., Wood Dale, IL, USA) was implanted as previously reported, terminating in the lumbar region of the spinal cord (Yaksh and Rudy, 1976). Catheters were sutured (3‐0 silk suture) into the deep muscle and externalized at the back of the neck; skin was closed with autoclips, and hind paw incision was performed after a 5–7 day recovery period.
Measurement of thermal withdrawal latency
The method of Hargreaves et al. (1988) was used. Rats were acclimated within Plexiglas enclosures on a clear glass plate maintained at 30°C. A radiant heat source (high‐intensity projector lamp) was focused onto the plantar surface of the hind paw. When the paw was withdrawn, a motion detector halted the stimulus and a timer. A maximal cut‐off of 33.5 s was used to prevent tissue damage.
Testing of allodynia
The assessment of tactile allodynia (i.e. a decreased threshold to paw withdrawal after probing with normally innocuous mechanical stimuli) consisted of testing the withdrawal threshold of the paw in response to probing with a series of calibrated fine (von Frey) filaments. Each filament was applied perpendicularly to the plantar surface of the paw of rats held in suspended wire mesh cages. Withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength (the ‘up‐and‐down’ method), and data were analysed with the nonparametric method of Dixon, as described by Chaplan et al. (1994), and expressed as the mean withdrawal threshold.
Paw incision model of post‐operative pain
An animal model of surgical pain was generated by plantar incision as previously described (Brennan et al., 1996). Male Sprague–Dawley rats were anaesthetized with isoflurane vaporized through a nose cone. The plantar aspect of the left hind paw was scrubbed with betadine and 70% alcohol three times. A 1‐cm‐long incision, starting 0.5 cm from the heel and extending toward the toes, was made with a number 11 blade, through the skin and fascia of the plantar aspect of the left hind paw including the underlying muscle. The plantaris muscle was then elevated and longitudinally incised, leaving the muscle origin and insertion intact. After haemostasis with gentle pressure, the skin was closed with two mattress sutures of 5‐0 nylon on a curved needle. Rats received an injection of gentamicin (1 mL·kg−1 of 8 mg·mL−1 solution, s.c.) and were allowed to recover from the anaesthesia before returning to their home cage. Sham animals were anaesthetized, and the left hind paw was scrubbed with betadine three times, then 70% ethanol, but no incision was made. Animals were allowed to recover for 24 h, and then paw withdrawal thresholds were measured at 24 h after surgery.
HIV‐induced sensory neuropathy
Mechanical allodynia is produced by i.t. administration of the human immunodeficiency virus‐1 (HIV‐1) envelope glycoprotein, gp120 (Milligan et al., 2001). Seven days after implantation of an i.t. catheter, baseline behavioural measurements were obtained and then rats were randomly assigned to two groups. On days 10, 12 and 14, rats were injected i.t. with 300 ng of gp120 (Cat#4961, HIV‐1 BaL gp120 recombinant protein, NIH‐AIDS Reagent program) in a final volume of 20 μL in 0.9% saline and 0.1% BSA.
Pharmacophore modelling
CBD3 and other mutant peptides were constructed and refined using Maestro molecular modelling programme available from Schrodinger LLC (New York, NY, USA). Pharmacophore elements were manually defined. These peptides were superimposed using common structural features and using structure–activity relationship data on these peptide molecules. These manual overlays were used to create pharmacophore models. Arginine residues were used to create positive charged site and hydrophobic region, while alanine residues were used to create excluded volume site on the potential receptor. All the figures were created using Maestro viewer.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). In these studies, we performed a minimum of five independent experiments, where individual data points were based on at least six replicates each. All data were first tested for a Gaussian distribution using a Shapiro–Wilk test (Graphpad Prism 7 Software). The statistical significance of differences between means was determined either by parametric ANOVA followed by post hoc comparisons (Tukey) using GraphPad Prism 7 Software or by nonparametric Kruskall–Wallis test followed by post hoc comparisons (Dunnett's test; nparcomp plugin, R software; Konietschke et al., 2012). All behavioural data were analysed by a two‐way ANOVA (post hoc: Student–Neuman–Keuls) in FlashCalc (Dr Michael H. Ossipov, University of Arizona, Tucson, AZ, USA). AUCs were compared by one‐way ANOVA. Differences were considered to be significant if P ≤ 0.05. All data were plotted in GraphPad Prism 7. No outlier data were removed.
Materials
All peptides (sequences in Table 1) were synthesized and HPLC‐purified (>95% purity) by Genscript Inc. (Piscataway, NJ, USA). Scramble and random sequence‐based peptides conjugated to various cargoes as controls have been previously studied as controls in molecular, biochemical and behavioural assays and demonstrated to have no effects (Brittain et al., 2011b; Ju et al., 2012; Piekarz et al., 2012; Francois‐Moutal et al., 2015). All chemicals, unless noted, were purchased from Sigma (St. Louis, MO, USA). Fura‐2 Low Affinity was obtained from Teflabs (Austin, TX, USA). Antibodies were purchased from various vendors: anti‐CRMP2 C4G monoclonal antibody (Cat#11096; Immuno‐Biological Laboratories, Minneapolis, MN, USA), anti‐ Cav2.2 polyclonal antibody (Cat#TA308673; Origene Technologies, Inc., Rockville, MD, USA) and anti‐glutathione S transferase (GST) (Cat#SAB4301139, Sigma).
Table 1.
Sequences of peptides used in this study
| Peptide | Sequence (N ➔ C‐termini) | Molecular weight (g·mol−1) |
|---|---|---|
| TATa‐CBD3 | ARSRLAELRGVPRGL | 3192.75 |
| TAT‐CBD3Δ1‐6 | ELRGVPRGL | 2538.01 |
| TAT‐CBD3Δ7‐15 | ARSRLA | 2214.62 |
| TAT‐CBD3‐A1I | IRSRLA | 2256.68 |
| TAT‐CBD3‐S3N | ARNRLA | 2241.63 |
| TAT‐CBD3‐S3R | ARRRLA | 2283.71 |
| TAT‐CBD3‐R4K | ARSKLA | 2186.59 |
| TAT‐CBD3‐L5V | ARSRVA | 2200.57 |
| TAT‐CBD3‐L5M | ARSRMA | 2232.64 |
| TAT‐CBD3‐A6F | ARSRLF | 2290.7 |
| TAT‐CBD3‐A1I,R4K,L5V,A6F | IRSKVF | 2290.74 |
| TAT‐CBD3‐S3R,L5M | ARRRMA | 2273.73 |
Residues in boldface font represent mutations compared with the truncated N‐terminal parental CBD3 sequence.
TAT = YGRKKRRQRRR.
Results
Identification of the minimal functional domain of CBD3
We previously reported that CBD3, the 15‐amino‐acid CRMP2‐derived peptide, uncoupled the CRMP2–Cav2.2 channel interaction to curb Ca2+ influx, thereby blunting transmitter release to ultimately reverse nociceptive behaviours in preclinical models of experimental pain (Feldman and Khanna, 2013). CBD3 is a linear peptide, predicted to fold into an α helix (Figure 1A). As only the first six amino acids are present in the available crystal structure of CRMP2 (Stenmark et al., 2007), we split CBD3 into two fragments: a peptide encompassing the first six (CBD3Δ7‐15) or last nine (CBD3Δ1‐6) amino acids (Figure 1A). We also added the cell‐penetrating TAT sequence to both peptides to ensure their intracellular delivery. We then tested the peptides in calcium imaging experiments in rat DRG neurons. Depolarizing cells with a high KCl trigger elicited a transient rise in intracellular calcium ([Ca2+]c), which was blunted, in a concentration‐dependent manner, by the peptide harbouring the N‐terminus of CBD3 peptide (i.e. TAT‐CBD3Δ7–15; Figure 1B). Consistent with previous results in DRGs from female rats (Ju et al., 2012; Francois‐Moutal et al., 2015), TAT‐CBD3 also inhibited depolarization‐evoked increase in [Ca2+]c in DRGs from male rats. In contrast, TAT‐CBD3Δ1‐6, the peptide encompassing the C‐terminus of CBD3, failed to elicit any inhibition as did full‐length CBD3 without the cell‐penetrating TAT motif (Figure 1B). Because the molecular basis for this inhibition is to the CBD3‐mediated uncoupling of the CRMP2–Cav2.2 channel interaction via the first intracellular loop (L1) and C‐terminus of the channel (Brittain et al., 2011b), we examined if the truncated peptides disrupted the interaction of CRMP2 interaction with these intracellular regions. Throught the use of elisa, CRMP2 binding to the L1 or C‐terminus fragments of Cav2.2 was inhibited by TAT‐CBD3Δ7‐15 to a similar level as TAT‐CBD3; however, TAT‐CBD3Δ1‐6 peptide failed to inhibit these interactions (Figure 1C). Finally, we tested if the truncated peptides could uncouple the CRMP2–Cav2.2 channel interaction directly in sensory neurons using PLA. This assay couples the detection specificity of an antibody with the amplification power of PCR to permit detection and quantification of protein complexes in situ. When proteins are within 30 nm of each other, a PLA signal (fluorescent amplification product) is generated that can be interpreted as the detection of a protein complex. Direct protein interactions between CRMP2 and Cav2.2 channels in DRG neurons were evident with PLA (Figure 1D). Treatment of DRG neurons with TAT‐CBD3 decreased the PLA signal by ~60 ± 3.2%, while those treated with TAT‐CBD3Δ7‐15 had an ~52 ± 4.2% inhibition of PLA signal compared with vehicle‐treated neurons (Figure 1E). Collectively, these results demonstrate that peptide TAT‐CBD3Δ7‐15, which harbours the first six amino acids of CBD3, is sufficient for disruption of the CRMP2‐ Cav2.2 channel interaction and, consequently, for a decrease in Ca2+ influx through inhibition of Cav2.2 channel activity.
Figure 1.

Identification of a minimal functional domain within the CBD3 peptide. (A) Predicted α‐helical model of the secondary structure of the parental Ca2+ CBD3 peptide. Sequence of the parental and two truncated peptides are shown. (B) Bar graphs show the K+‐evoked peak fluorescence response (adjusted for background) of DRG neurons incubated overnight with increasing concentrations (1, 3, 10 or 30 μM) of the indicated peptides or 0.1% DMSO as the vehicle control. Values represent the mean ± SEM, normalized to the DMSO level within each experiment. n = 39 to 58 cells per condition from five independent experiments. *P < 0.05, significantly different from control; one‐way ANOVA with Tukey's post hoc analysis. (C) Bar graph showing normalized CRMP2 binding to the first intracellular loop (Loop 1; left) or the C‐terminus (Ct; right) of Cav2.2. The indicated peptides (10 μM) or DMSO (0.1%) was added to CRMP2 prior to their addition on a 96‐well assay plate where Loop 1 or Ct fragments had been immobilized. TAT‐CBD3 and the N‐terminal containing region of CBD3 (i.e. TAT‐CBD3Δ7‐15) inhibited CRMP2 binding to Cav2.2 fragments (n = 12). *P < 0.05, significantly different from DMSO; one‐way ANOVA with Tukey's post hoc analysis). (D) Representative pseudocolour inverted micrographs showing PLA signal (fluorescent amplification product; black dots) for CRMP2‐ Cav2.2 channel interaction in sensory neurons treated with 0.1% DMSO or 10 μM of TAT‐CBD3Δ7‐15. DAPI (yellow) signal shows the nucleus of the cell. Scale bar is 10 μm. (E) Summary of the number of PLA puncta, normalized to the area of the cell analysed (n = 13–18 cells each from at least five independent experiments). Data shown are means ± SEM. *P < 0.05; significantly different from DMSO;, one‐way ANOVA with Tukey's post hoc analysis.
Rationale for homology‐guided mutagenesis of N‐terminus of CBD3
Having mapped the functional region of the CBD3 peptide to the six N‐terminal amino acids (i.e. ARSRLA), we next asked which of these were key to this function of CBD3. As the CRMP protein family consists of five members, with CRMPs 1–4 sharing more than 70% sequence homology (Charrier et al., 2003), we aligned the N‐terminus of CBD3 across these four CRMPs. CRMP5 was not used in this alignment because it is not expressed in adult DRG (Ricard et al., 2001). Only the arginine residue, at the second position, was conserved among all CRMP isoforms, while one (in CRMP3), three (in CRMP4) or four (in CRMP1) amino acids were different in comparison with the CRMP2 sequence (Figure 2A). We designed six‐mer peptides, modelled as α helices, bearing combinational mutations corresponding to the variable residues found in the alignment (Figure 2B). The mutations did not result in discernible structural changes as assessed by DSF: no differences were observed in the melting temperatures – an index of structural stability, between any of the novel peptides and the parent‐truncated (i.e. CBD3Δ7‐15) peptide (Figure 2C).
Figure 2.
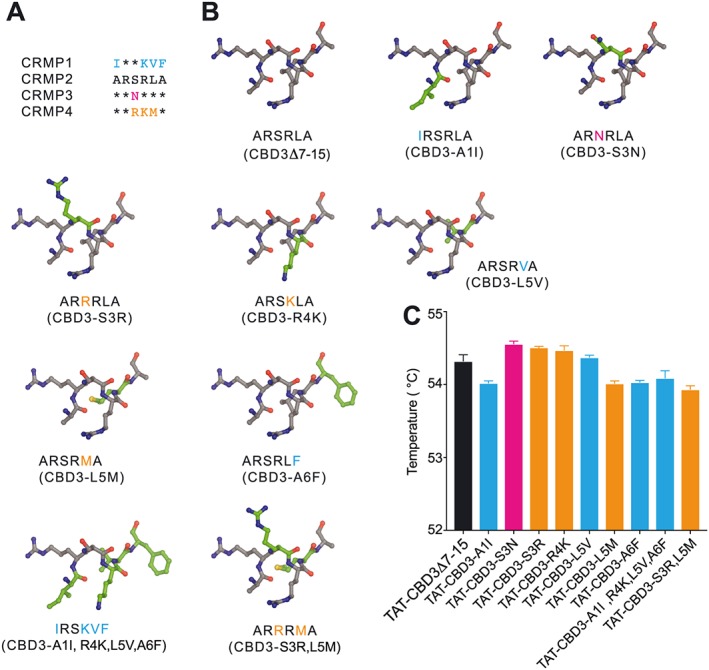
Homology‐guided design of N‐terminal region of CBD3 peptides. (A) Amino acid alignment of the six N‐terminal residues of the CBD3 peptide across CRMPs 1–4. Asterisks denote amino acids that are conserved/identical between the CRMPs. Residues that differ from the CRMP2 sequence are shown in coloured text. (B) Mutant CBD3 peptides are shown in capped‐stick representation. The coordinates for the six‐amino‐acid fragment were extracted from chain A of human CRMP2 (Protein Data Base ID:2GSE; Stenmark et al., 2007). PyMol (PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC) was used to model each peptide using the mutation wizard and choosing the most common rotamer without clashes with adjacent amino acids. The mutated amino acids are shown in green on the peptide structure. The sequence of each peptide is shown below the model with the coloured residues corresponding to the CRMP isoform that the mutation is originating from (see panel A). (C) Bar graphs showing the melting temperature, measured by DSF, of the indicated peptides. The bars are colour‐coded according to the CRMP protein from which the mutation is derived. No significant difference was observed between any of the peptides (n = 6).
Homology‐guided mutant peptides of the N‐terminus of CBD3 – biochemical and functional mapping
We first investigated which amino acids within the six‐mer N‐terminus of CBD3 were important for biochemically uncoupling the CRMP2–Cav2.2 channel interaction. As earlier, we used elisa to quantify possible inhibition of the interaction of CRMP2 with the L1 and C‐terminal intracellular regions of Cav2.2 channels. Incubating peptides CBD3‐S3N, CBD3‐S3R and CBD3‐L5M (10 μM) significantly decreased the binding of CRMP2 to L1 and C‐terminus of Cav2.2 channels when compared with DMSO (Figure 3A, B). Peptides CBD3‐R4K and CBD3‐L5V decreased the binding of CRMP2 to the C‐terminus, but not L1, of Cav2.2 channels (Figure 3B). The other peptides did not alter the interaction of CRMP2 with either region of the Cav2.2 channel (Figure 3A, B).
Figure 3.

elisa‐based analyses of the ability of CBD3‐derived peptides to inhibit the CRMP2– Cav2.2 channel interaction. Bar graph showing normalized CRMP2 binding to Loop 1 (A) or C‐terminus (B) of Cav2.2. The indicated peptides (10 μM) or DMSO (0.1%) was added to CRMP2 prior to their addition on a 96‐well assay plate where Loop 1 or Ct fragments had been immobilized. The dotted line shows the level of CRMP2 binding in the presence of 10 μM of TAT‐CBD3. TAT‐CBD3‐S3N, TAT‐CBD3‐S3R, TAT‐CBD3‐R4K, TAT‐CBD3‐L5V and TAT‐CBD3‐L5M inhibited CRMP2 binding to Cav2.2 fragments. The bars are colour‐coded according to the CRMP protein from which the mutation is derived. Data shown are means ± SEM (n = 12) *P < 0.05; significantly different from DMSO; Kruskal–Wallis test with pairwise comparison with Dunnett's post hoc analysis).
A functional consequence of disrupting the biochemical CRMP2–Cav2.2 channel interaction is the inhibition of depolarization‐evoked increase in [Ca2+]c; therefore, we tested if the mutant peptides affected calcium influx. Sensory neurons were treated with increasing concentrations of the mutant peptides, and K+‐evoked increase in [Ca2+]c was interrogated. Peptides CBD3‐S3N, CBD3‐S3R, CBD3‐L5M and CBD3‐A6F inhibited calcium influx in a concentration‐dependent manner (Figure 4), demonstrating that these mutations preserve the activity present within the N‐terminus of CBD3. Mutating peptides at residue 1 (CBD3‐A1L) or 4 (CBD3‐R4K) caused inhibition of calcium influx in a non‐concentration‐dependent manner, while simultaneously mutating more than one residue (CBD3‐A1L,R4K,L5V,A6F and CBD3‐S3R,L5M) completely eliminated their ability to inhibit the calcium influx (Figure 4). Thus, the N‐terminal CBD3 peptides retained activity if residues at positions 1, 2 and 4 were not mutated. The reason for an increase in calcium influx seen with higher concentrations of TAT‐CBD3‐S3N and TAT‐CBD3‐A6F is at present unclear but may involve actions on other off‐targets, such as the sodium–calcium exchanger (Brustovetsky et al., 2014); this is another reason why this peptide was not used in behavioural experiments. Consequently, we selected to further characterize four peptides (CBD3‐S3N, CBD3‐S3R, CBD3‐L5M and CBD3‐A6F), which demonstrated both an inhibition of the CRMP2–Cav2.2 channel interaction and a reduction in depolarization‐evoked Ca2+ influx.
Figure 4.

CBD3‐derived peptides inhibit depolarization‐evoked Ca2+ influx in DRG neurons. Bar graphs showing the K+‐evoked peak fluorescence response (adjusted for background) of DRGs incubated overnight with increasing concentrations (1, 3, 10 or 30 μM) of the indicated peptides or DMSO (0.1%) as the vehicle control. The dotted line shows the peak fluorescence response in the presence of 30 μM of TAT‐CBD3 (from Figure 1B). Values represent the average ± SEM, normalized to the DMSO level within each experiment. n = 37 to 68 cells per condition from five independent experiments. *P < 0.05, significantly different from control; one‐way ANOVA with Tukey's post hoc analysis).
Mutant peptides of the N‐terminus of CBD3 that disrupt the CRMP2– Cav2.2 channel complex also reduce Ca2+ currents
We next examined the effects of the four mutant CBD3 peptides (CBD3‐S3N, CBD3‐S3R, CBD3‐L5M and CBD3‐A6F) on Ca2+ currents in sensory neurons. Treatment with peptides (10 μM) resulted in >55% inhibition of total Ca2+ current in sensory neurons incubated with CBD3‐S3N, CBD3‐S3R and CBD3‐L5M; peptide CBD3‐A6F was ineffective (Figure 5A, B), and no changes were observed in the activation or inactivation of these currents (Table 2). Consistent with the electrophysiology data, peptides CBD3‐S3N, CBD3‐S3R and CBD3‐L5M also decreased the PLA signal, providing confirmation of the ability of these peptides to inhibit the CRMP2– Cav2.2 channel interaction in situ (Figure 5C). Not surprisingly, peptide CBD3‐A6F failed to inhibit the CRMP2– Cav2.2 channel interaction as assessed by no significant change in PLA signal compared with control cells (Figure 5C). Together, these results show that introducing mutations at position 3 or 5 of the N‐terminus of CBD3 are permissive for maintaining complex uncoupling and functional inhibition of calcium influx.
Figure 5.

CBD3‐derived peptides inhibit Ca2+ currents and the CRMP2– Cav2.2 channel interaction in sensory neurons. (A) Representative family of current traces are illustrated from sensory neurons treated with DMSO (0.1%; vehicle control) or treated with 10 μM of the indicated peptides. (B) Peak current density, at +10 mV, for the indicated conditions. n = 8–32 cells from five independent experiments. Data shown are means ± SEM. *P < 0.05, significantly different from DMSO; one‐way ANOVA with Tukey's post hoc analysis. (C) Summary of the number of PLA dots, normalized to the area of the cell analysed (n = 11–23 cells each from five independent experiments). The dotted line shows the mean PLA signal in the presence of 10 μM of TAT‐CBD3 or TAT‐CBD3Δ7‐15 (from Figure 1E). Data shown are means ± SEM. *P < 0.05, significantly different from DMSO; one‐way ANOVA with Tukey's post hoc analysis.
Table 2.
Comparison of calcium channel currents following treatment with CBD3 peptides in DRG cells
| Peak currents (nA) | Conductance (pF) | Voltage dependence of activation | Voltage dependence of inactivation | |||
|---|---|---|---|---|---|---|
| V1/2 (mV) | Slope (mV/e‐fold) | V1/2 (mV) | Slope (mV/e‐fold) | |||
| Vehicle (0.1% DMSO) | −9.4 ± 2.1 (19) | 67.2 ± 6.2 (19) | 8.5 ± 3.1 (21) | 10.5 ± 1.2 (21) | −23.3 ± 1.8 (21) | 9.8 ± 1.3 (20) |
| TAT‐CBD3‐S3N | −4.3 ± 0.1 (8) * | 59.8 ± 4.3 (8) | 9.4 ± 2.9 (7) | 8.5 ± 1.8 (7) | −32.9 ± 2.1 (6) | 10.5 ± 1.5 (6) |
| TAT‐CBD3‐S3R | −2.9 ± 0.0 (8) * | 69.9 ± 11.2 (8) | 9.1 ± 0.7 (7) | 11.9 ± 0.9 (7) | −23.2 ± 5.8 (5) | 13.3 ± 4.1 (5) |
| TAT‐CBD3‐L5M | −3.6 ± 0.6 (17) * | 66.1 ± 5.6 (17) | 10.2 ± 1.4 (15) | 7.2 ± 3.5 (15) | −20.0 ± 1.6 (15) | 10.3 ± 1.0 (15) |
| TAT‐CBD3‐A6F | −4.6 ± 0.6 (15) | 67.0 ± 7.5 (15) | 8.5 ± 3.2 (27) | 10.5 ± 1.2 (27) | −13.0 ± 4.1 (20) | 9.8 ± 2.1 (20) |
n values shown in parentheses. Comparative calcium current statistics from DRG cells treated overnight with 10 μM of the indicated peptides. Comparative current densities and Boltzmann properties of half‐maximal effect (V1/2) and slope (k) for activation and fast inactivation are presented. Data shown are means ± SEM.
P < 0.05; Kruskal–Wallis test with pairwise comparisons.
Peptide CBD3‐L5M suppresses post‐surgical pain
Targeting Cav2.2 channels with gabapentin (Field et al., 1997) or with the conotoxin peptide Prialt (Atanassoff et al., 2000) has been demonstrated to be analgesic in rats and humans, respectively, in models of acute post‐operative pain. We previously reported that inhibiting the CRMP2–Cav2.2 channel interaction with a membrane‐tethered CBD3 peptide was antinociceptive in rats with paw incision (Francois‐Moutal et al., 2015). Therefore, we tested the antinociceptive potential of CBD3‐L5M on thermal hyperalgesia and tactile allodynia induced by an incision of the plantaris muscle of the rat hind paw (Brennan et al., 1996); this peptide was selected on the basis of being among the most potent in blocking calcium influx (Figure 4). An incision of the rat plantaris muscle led to a reduction of paw withdrawal latency (thermal hyperalgesia; Figure 6A, B) and of paw withdrawal threshold (mechanical allodynia; Figure 6C, D). Both nociceptive responses peaked within 24 h after surgery and were maintained during the 5 h experimental period in vehicle‐treated animals. Spinal administration of TAT‐CBD3‐L5M (20 μg/5 μL) blocked the development of thermal hypersensitivity for 5 h (Figure 6A, B) and mechanical hypersensitivity for at least 4.5 h (Figure 6C, D). We determined the AUC to assess effects over the full experimental duration. AUC analysis confirmed the reversal of mechanical allodynia and thermal hyperalgesia compared with sham‐injured and vehicle‐treated injured animals (Figure 6B, C). Thus, the peptide CBD3‐L5M can produce pain relief in an animal model of post‐operative pain.
Figure 6.

TAT‐CBD3‐L5M peptide reduces incision‐ and gp120‐induced nociceptive behaviours. Rats received a plantar incision on the left hind paw. Paw withdrawal latencies (PWLs) were significantly decreased 24 h after incision. (A) TAT‐CBD3‐L5M (20 μg/5 μL) or vehicle (saline) was injected into the i.t. space and PWLs measured. Paw withdrawal latencies were significantly reversed at the indicated times after injection of TAT‐CBD3‐L5M. Data shown are means ± SEM (n = 5–6). *P < 0.05; significantly different from pre‐incision value; two‐way ANOVA with a Student–Neuman–Keuls post hoc test) where time was treated as ‘within subjects’ factor, whereas treatment was treated as ‘between subjects’ factor. Likewise, paw withdrawal thresholds (PWTs) were significantly decreased 24 h after incision. (C) Injection of TAT‐CBD3‐L5M significantly reversed PWTs at the indicated times. Data shown are means ± SEM (n = 5–6). *P < 0.05; significantly different from pre‐incision value; two‐way ANOVA with a Student–Neuman–Keuls post hoc test). AUC, using the trapezoid method, for PWL (B; summary for data shown in A) and PWT (D; summary for data shown in C) are shown. Data shown are means ± SEM. *P < 0.05, significantly different as indicated; one‐way ANOVA with Tukey's post hoc analysis. (E) PWTs were significantly reduced 15 days after three injections of gp120 in the i.t. space. (F) Injection of TAT‐CBD3‐L5M (20 μg in 5 μL) significantly reversed PWTs at the indicated times. Data shown are means ± SEM (n = 5). *P < 0.05; significantly different from pre‐incision value; two‐way ANOVA with a Student–Neuman–Keuls post hoc test). (G) Bar graph showing the AUC, using the trapezoid method for the data shown in (F). *P < 0.05, significantly different from vehicle; one‐way ANOVA with Tukey's post hoc analysis.
Effect of CBD3‐L5M on HIV‐induced sensory neuropathy
HIV‐induced sensory neuropathy (HIV‐SN) is a frequently occurring neurological complication of HIV infection (Manji, 2000). Gabapentin targets the Cav2.2 channel and has been reported to reduce pain in patients with HIV‐SN (Newshan, 1998). Therefore, we tested the antinociceptive potential of TAT‐CBD3‐L5M on mechanical allodynia induced by i.t. injections of the HIV‐1 envelope glycoprotein (gp120), which results in robust mechanical allodynia (Milligan et al., 2001; Yuan et al., 2014). Spinal (i.t.) injections of gp120 led to the gradual lowering of paw withdrawal thresholds, compared with baseline (i.e. pre‐gp120) (Figure 6E). This mechanical allodynia was reversed by a single injection of TAT‐CBD3‐L5M (20 μg/5 μL) for at least 4 h (Figure 6F, G). These results suggest that targeting the CRMP2–Cav2.2 channel interaction can reduce pain behaviours in a rat model of HIV‐SN.
Discussion
Chronic pain is a devastating global epidemic in an urgent need of novel therapeutics. Although the recommended firstline of treatment involves nonsteroidal anti‐inflammatory drugs, the failure of such drugs to alleviate neuropathic pain leaves physicians with limited options that include anticonvulsants, antidepressants and opioids (Hassett et al., 2014). Only a third of patients experience greater than 40–50% of pain relief with currently available analgesics (Backonja and Woolf, 2010). Opioids, despite their limited utility and numerous side effects (Gray et al., 2014), are among the most commonly used drugs for neuropathic pain syndromes (Hassett et al., 2014; Dowell et al., 2016). Opioids act on the μ‐opioid receptor, which is a G‐protein coupled receptor, to inhibit presynaptic Cav2.2 channels (Malmberg and Yaksh, 1995; Bourinet et al., 1996). These channels are critical determinants of increased neuronal excitability and neurotransmission accompanying persistent neuropathic pain (Cizkova et al., 2002; Cao, 2006; Park and Luo, 2010). Targeting Cav2.2 channels, both directly (Prialt®) and indirectly (gabapentin), has shown clinical promise, but both drugs exhibit side effects that include dizziness, nausea, nystagmus, memory impairment and hallucinations (Thompson et al., 2006; Skov et al., 2007; Rauck et al., 2009; Schmidtko et al., 2010; Moore et al., 2011). Another gabapentinoid (pregabalin) also indirectly acts on Cav2.2 channels by impairing anterograde trafficking of the α(2)δ‐1 channel subunit, resulting in its decrease in presynaptic terminals, which translates into a reduction in neurotransmitter release and spinal sensitization, factors important in the maintenance of neuropathic pain (Bauer et al., 2010). The results presented here demonstrate that the indirect inhibition of Cav2.2 channels via uncoupling of the interaction between Cav2.2 channels and its regulator CRMP2 is a potential novel approach to address this medical shortcoming.
Recent findings have highlighted the importance of modulating Cav2.2 channel activity via its trafficking (Heblich et al., 2008; Hendrich et al., 2008; Brittain et al., 2011b; Feldman and Khanna, 2013). For instance, CRMP2 expression correlated directly with surface expression of Cav2.2 channels and current density (Brittain et al., 2009; Chi et al., 2009). Interestingly, antagonism of the CRMP2–Cav2.2 channel interaction with CRMP2‐derived peptides (designated as CBD3) was shown to limit the movement of Cav2.2 channels to the neuronal surface (as demonstrated by loss of plasma membrane localization through confocal microscopy and biotinylation), resulting in reduced Cav2.2 current density, and a diminution of transmitter release, which accounted for relief of pain behaviours in experimental models of acute, inflammatory, post‐surgical and drug‐ and injury‐induced neuropathic pain (Wilson et al., 2011, 2012; Brittain et al., 2011b; Feldman and Khanna, 2013; Francois‐Moutal et al., 2015). A similar mechanistic convergence was observed with a peptide derived from Cav2.2, which also uncoupled the CRMP2–Cav2.2 channel interaction to limit surface trafficking, current density and transmitter release, resulting in pain relief (Wilson et al., 2012). Adenoviral injection of CBD3 into the lumbar DRG of rats protected them from nerve injury‐induced neuropathic pain (Fischer et al., 2014). Limited mutational analyses of the 15‐amino‐acid parental CBD3 peptide revealed the presence of key residues that could be ‘tailored’ to alter the antinociceptive properties of the peptides. For example, a mutant CBD3 peptide with a lysine instead of alanine (A6K) demonstrated suppression of nociceptor excitability and inhibition of several high and low voltage‐activated Ca2+ channels and was efficacious in reversing neuropathic pain induced by antiretroviral drug treatment (Piekarz et al., 2012). Another mutant CBD3 peptide with a phenylalanine instead of glycine (G14F) inhibited capsaicin‐induced meningeal vascular responses related to headache pain in addition to in reversing neuropathic pain induced by antiretroviral drug treatment (Ripsch et al., 2012). Finally, although mice lacking the Crmp2 gene do not have an analgesic phenotype (Nakamura et al., 2016), mice with a knock‐in mutation of the Cdk5 phosphorylation site in CRMP2 (S522A) develop an antinociceptive phenotype after spinal cord injury (Nagai et al., 2016). The functional coupling between Cav2.2 channels and CRMP2 is reduced by loss of CRMP2 phosphorylation by Cdk5 (Brittain et al., 2012b; Moutal et al., 2016a, 2016b), supporting the idea that antagonism or deletion of CRMP2 is anti‐nociceptive. Overall, these observations offer support to the notion of peptide‐mediated suppression of the CRMP2–Cav2.2 channel interaction as an important and novel antinociceptive strategy.
Here, we have presented evidence that the N‐terminal six amino acids of CBD3 are sufficient for conferring biochemical and functional activity. These six residues are present in the crystal structure of CRMP2 (Stenmark et al., 2007). This suggests that the ‘active fragment’ of the CBD3 peptide is the only structured part of the peptide and that the remaining nine amino acids of CBD3 are not likely to be folded. The N‐terminal amino acids of CBD3 are conserved across various species (Supporting Information Fig. S1), except for the sequence of CRMP3, which has an asparagine (N) instead of serine (S) at position 3 of the six‐mer peptide. This CRMP3 peptide (ARNRLA) also uncoupled the CRMP2–Cav2.2 channel interaction and inhibited calcium influx and currents. These results are in agreement with previously reported regulation of Cav2.2 channels by CRMP3 in hippocampal neurons (Quach et al., 2011). When mapping residues within the truncated peptides important for biochemical and functional activities, we considered a homology‐guided approach. Only two amino acids were similar in the homologous six‐mer region of the related CRMP1 protein; mutating any amino acid from this six‐mer CBD3 sequence of CRMP2 to the corresponding position in CRMP1 compromised the ability of CBD3 to inhibit the CRMP2– Cav2.2 channel interaction or to inhibit calcium influx. The peptide CBD3‐R4K retained some capability to inhibit the CRMP2– Cav2.2 channel interaction but failed to inhibit the channel functionally, suggesting that the arginine residue in position 4, which has an additional amine group compared with the isoleucine in CRMP1, is important for the functional activity of the peptide. Another inference from this result is that CRMP1 probably does not interact with Cav2.2 channels and therefore is highly unlikely to regulate the channel. The six‐mer CBD3 sequence of CRMP2 is different at three amino acids in CRMP4. While single mutants (S3R or L5M) were still functional, mutating both residues simultaneously, which adds a significant amount of charge, was not permissive for the activity of the peptide. These results suggest that the amino acid differences between CRMP4 and CRMP2 are also likely to prevent it from binding to and regulating the activity of Cav2.2 channels. Further mutagenesis studies may be required to optimize the specificity and selectivity of the six‐mer CBD3 peptides.
An additional important finding here is the efficacy of the peptide CBD3‐L5M in a rat model of HIV‐induced sensory neuropathy (HIV‐SN). It has been long known that gp120 enhances calcium influx through voltage‐dependent and glutamate‐gated ion channels to induce its neurotoxicity (Lipton, 1994; Lipton and Nicotera, 1998). Blocking calcium entry restores calcium homeostasis and alleviates neuronal damage (Dreyer et al., 1990). The Cacna1b gene encoding Cav2.2 is highly up‐regulated in a rodent model of HIV‐SN treated with gp120 and the antiretroviral agent zalcitabine (2′,‐3′,‐dideoxycytidine, ddC), compared with sham‐treated controls (Maratou et al., 2009). CRMP2 levels are also increased in brains from gp120 transgenic mice (Crews et al., 2011) and in hippocampi of patients with HIV (Zhou et al., 2010; Crews et al., 2011). Gabapentin targets the Cav2.2 channel complex and this drug reversed mechanical hypersensitivity in ddC‐, gp120‐ or gp120 + ddC‐treated rats (Wallace et al., 2007, Nasirinezhad et al., 2015). These results triangulate to establish CRMP2 antagonism of Cav2.2 channels as a novel therapeutic signalling hub for HIV‐SN.
The short CBD3 peptides reported here may represent a unique approach of suppressing Cav2.2 channel function in neuropathic pain. Homology‐guided mutational analysis of these peptides identified key residues that will be instructive in future studies aimed at designing pharmacophores to mimic these peptides. For instance, mutating the first residue (alanine to isoleucine) destroyed the resulting peptide's activity, suggesting that this amino acid is likely to be at the interface of the protein–protein interaction with either L1 or C‐terminus domain of the channel as isoleucine may have disrupted the interaction because of a steric clash. The arginine located in the second position was conserved among all CRMPs and is therefore not likely to be important for interactions with the calcium channel. The third amino acid position from the CBD3 peptide was able to withstand being changed to a larger side chain and the addition of a charge (mutations to arginine or asparagine). Thus, this amino acid is also likely not directly involved in the interface with Cav2.2 channels. Similarly, when the leucine residue in the fifth position was mutated to a methionine, the peptide was still able to disrupt the CRMP2‐ Cav2.2 channel interaction, inhibit the Ca2+ currents and reverse pain behaviour in a model of post‐operative pain and in a model of HIV‐induced neuropathy.
The N‐terminal six residues of the CBD3 peptide are sufficient for inhibition of the CRMP2–Cav2.2 channel interaction, and furthermore, residues alanine (at position 1) and arginine (at position 4) are key mediators of this interaction. In light of this, pharmacophore models for CBD3Δ7‐15 peptide were created using structure activity data on mutant peptides. As shown in Figure 7, both models consist of three basic pharmacophore elements – (i) a positively charged site corresponding to a guanidine group in arginine at position 4 of the six‐mers; (ii) a hydrophobic region corresponding to the arginine residue at position 4 of the six‐mers; and (iii) a hydrogen bond acceptor based on the alanine at the first position within CBD3Δ7‐15. The second model (Figure 7, right) also consists of an excluded hydrophobic volume region (on the receptor for CBD3) corresponding to a binding site for alanine at first position of CBD3Δ7‐15. These pharmacophore models focus on the interface of the alanine (at position 1) and arginine (at position 4) residues of the six‐mer CBD3 peptide. Further model refinement and computational screening of ligands and molecules are expected to identify small molecules that will biochemically and functionally target the CRMP2– Cav2.2 channel interaction and reverse neuropathic pain.
Figure 7.

Pharmacophore models for the N‐terminal six‐mer peptide of CBD3. Two models, based on the biochemical and functional data on mutant peptides, are illustrated. P17 (blue) is a positively charged site, H15 (green) is a hydrophobic region and A2 (orange) is a hydrogen bond acceptor. The yellow sphere in the right panel represents excluded volume corresponding to alanine at the first position of the six‐mer N‐terminal CBD3 peptide.
Author contributions
R.K. and M.K. designed the research study; A.M., W.J., L.F.‐M. and S. P‐M. performed the in vitro experiments; W.L. and S.C. performed the electrophysiology experiments; Y.W., S.L. and J.H. performed the in vivo experiments; V.G. and E.T.D. performed the in silico modelling experiments; A.M. and R.K. prepared the manuscript; all authors approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 The CRMP2‐derived CBD3 peptide sequence is evolutionarily conserved. The six‐amino‐acid residue sequence ‘ARSRLA’ is perfectly conserved within a variety of species of mammals, reptiles, birds, frogs, and fish. CRMP2 Mus musculus (NM_009955.3) sequence was analysed using the National Center for Biotechnology Information (NCBI) protein blast suite and a phylogenetic tree was created using ‘Interactive Tree of Life’ (iTOL2.0) software (Letunic & Bork, 2007; Letunic & Bork, 2011). Select images and common names are colour matched to nearby scientific names. Major branches are labelled by common name of lowest shared taxonomic classification. Branch separation represents divergent evolutionary relationship and closely related organisms are grouped by colour.
Figure S1 The CRMP2‐derived CBD3 peptide sequence is evolutionarily conserved. The six‐amino‐acid residue sequence ‘ARSRLA’ is perfectly conserved within a variety of species of mammals, reptiles, birds, frogs, and fish. CRMP2 Mus musculus (NM_009955.3) sequence was analysed using the National Center for Biotechnology Information (NCBI) protein blast suite and a phylogenetic tree was created using ‘Interactive Tree of Life’ (iTOL2.0) software (Letunic & Bork, 2007; Letunic & Bork, 2011). Select images and common names are colour matched to nearby scientific names. Major branches are labelled by common name of lowest shared taxonomic classification. Branch separation represents divergent evolutionary relationship and closely related organisms are grouped by colour.
Acknowledgements
We thank Dr Tally Largent‐Milnes and Kerry Beth Gilbraith for guidance with gp120 experiments. M.K. was supported by a Career Development Award from the Arizona Health Science Center. This work is supported by a National Scientist Development grant SDG5280023 from the American Heart Association and a Neurofibromatosis New Investigator Award NF1000099 from the Department of Defence Congressionally Directed Military Medical Research and Development Program (R.K.) and a Young Investigator Award from the Children's Tumour Foundation (A.M.).
Moutal, A. , Li, W. , Wang, Y. , Ju, W. , Luo, S. , Cai, S. , François‐Moutal, L. , Perez‐Miller, S. , Hu, J. , Dustrude, E. T. , Vanderah, T. W. , Gokhale, V. , Khanna, M. , and Khanna, R. (2018) Homology‐guided mutational analysis reveals the functional requirements for antinociceptive specificity of collapsin response mediator protein 2‐derived peptides. British Journal of Pharmacology, 175: 2244–2260. doi: 10.1111/bph.13737.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanassoff PG, Hartmannsgruber MW, Thrasher J, Wermeling D, Longton W, Gaeta R et al. (2000). Ziconotide, a new N‐type calcium channel blocker, administered intrathecally for acute postoperative pain. Reg Anesth Pain Med 25: 274–278. [DOI] [PubMed] [Google Scholar]
- Attal N, Cruccu G, Baron R, Haanpaa M, Hansson P, Jensen TS et al. (2010). EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol 17: 1113–e1188. [DOI] [PubMed] [Google Scholar]
- Backonja M, Woolf CJ (2010). Future directions in neuropathic pain therapy: closing the translational loop. Oncologist 15 (Suppl 2): 24–29. [DOI] [PubMed] [Google Scholar]
- Bauer CS, Rahman W, Tran‐van‐Minh A, Lujan R, Dickenson AH, Dolphin AC (2010). The anti‐allodynic alpha(2)delta ligand pregabalin inhibits the trafficking of the calcium channel alpha(2)delta‐1 subunit to presynaptic terminals in vivo. Biochem Soc Trans 38: 525–528. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Stea A, Snutch TP (1996). Determinants of the G protein‐dependent opioid modulation of neuronal calcium channels. Proc Natl Acad Sci U S A 93: 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TJ, Vandermeulen EP, Gebhart GF (1996). Characterization of a rat model of incisional pain. Pain 64: 493–501. [DOI] [PubMed] [Google Scholar]
- Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM et al. (2011a). Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J Biol Chem 286: 37778–37792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL et al. (2011b). Suppression of inflammatory and neuropathic pain by uncoupling CRMP‐2 from the presynaptic Ca(2)(+) channel complex. Nat Med 17: 822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JM, Pan R, You H, Brustovetsky T, Brustovetsky N, Zamponi GW et al. (2012a). Disruption of NMDAR‐CRMP‐2 signaling protects against focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Channels (Austin) 6: 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R (2009). An atypical role for collapsin response mediator protein 2 (CRMP‐2) in neurotransmitter release via interaction with presynaptic voltage‐gated calcium channels. J Biol Chem 284: 31375–31390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JM, Wang Y, Eruvwetere O, Khanna R (2012b). Cdk5‐mediated phosphorylation of CRMP‐2 enhances its interaction with CaV2.2. FEBS Lett 586: 3813–3818. [DOI] [PubMed] [Google Scholar]
- Brustovetsky T, Pellman JJ, Yang XF, Khanna R, Brustovetsky N (2014). Collapsin response mediator protein 2 (CRMP2) interacts with N‐methyl‐D‐aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity. J Biol Chem 289: 7470–7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao YQ (2006). Voltage‐gated calcium channels and pain. Pain 126: 5–9. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55–63. [DOI] [PubMed] [Google Scholar]
- Charrier E, Reibel S, Rogemond V, Aguera M, Thomasset N, Honnorat J (2003). Collapsin response mediator proteins (CRMPs): involvement in nervous system development and adult neurodegenerative disorders. Mol Neurobiol 28: 51–64. [DOI] [PubMed] [Google Scholar]
- Chi XX, Schmutzler BS, Brittain JM, Hingtgen CM, Nicol GD, Khanna R (2009). Regulation of N‐type voltage‐gated calcium (CaV2.2) channels and transmitter release by collapsin response mediator protein‐2 (CRMP‐2) in sensory neurons. J Cell Sci 23: 4351–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizkova D, Marsala J, Lukacova N, Marsala M, Jergova S, Orendacova J et al. (2002). Localization of N‐type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp Brain Res 147: 456–463. [DOI] [PubMed] [Google Scholar]
- Crews L, Ruf R, Patrick C, Dumaop W, Trejo‐Morales M, Achim CL et al. (2011). Phosphorylation of collapsin response mediator protein‐2 disrupts neuronal maturation in a model of adult neurogenesis: implications for neurodegenerative disorders. Mol Neurodegener 6: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell D, Haegerich TM, Chou R (2016). CDC guideline for prescribing opioids for chronic pain–United States, 2016. JAMA 315: 1624–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer EB, Kaiser PK, Offermann JT, Lipton SA (1990). HIV‐1 coat protein neurotoxicity prevented by calcium channel antagonists. Science 248: 364–367. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, O'Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS et al. (2007). Pharmacologic management of neuropathic pain: evidence‐based recommendations. Pain 132: 237–251. [DOI] [PubMed] [Google Scholar]
- Feldman P, Khanna R (2013). Challenging the catechism of therapeutics for chronic neuropathic pain: targeting CaV2.2 interactions with CRMP2 peptides. Neurosci Lett 557 (Pt A): 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MJ, Holloman EF, McCleary S, Hughes J, Singh L (1997). Evaluation of gabapentin and S‐(+)‐3‐isobutylgaba in a rat model of postoperative pain. J Pharmacol Exp Ther 282: 1242–1246. [PubMed] [Google Scholar]
- Fischer G, Pan B, Vilceanu D, Hogan QH, Yu H (2014). Sustained relief of neuropathic pain by AAV‐targeted expression of CBD3 peptide in rat dorsal root ganglion. Gene Ther 21: 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois‐Moutal L, Wang Y, Moutal A, Cottier KE, Melemedjian OK, Yang X et al. (2015). A membrane‐delimited N‐myristoylated CRMP2 peptide aptamer inhibits Cav2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain 156: 1247–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T et al. (2002). CRMP‐2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 4: 583–591. [DOI] [PubMed] [Google Scholar]
- Gray D, Coon H, McGlade E, Callor WB, Byrd J, Viskochil J et al. (2014). Comparative analysis of suicide, accidental, and undetermined cause of death classification. Suicide Life Threat Behav 44: 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988). A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77–88. [DOI] [PubMed] [Google Scholar]
- Hassett AL, Aquino JK, Ilgen MA (2014). The risk of suicide mortality in chronic pain patients. Curr Pain Headache Rep 18: 436. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Wakamori M, Ino M, Miyamoto N, Takahashi E, Yoshinaga T et al. (2001). Differential nociceptive responses in mice lacking the alpha(1B) subunit of N‐type Ca(2+) channels. Neuroreport 12: 2423–2427. [DOI] [PubMed] [Google Scholar]
- Heblich F, Tran Van Minh A, Hendrich J, Watschinger K, Dolphin AC (2008). Time course and specificity of the pharmacological disruption of the trafficking of voltage‐gated calcium channels by gabapentin. Channels (Austin) 2: 4–9. [DOI] [PubMed] [Google Scholar]
- Hendrich J, Van Minh AT, Heblich F, Nieto‐Rostro M, Watschinger K, Striessnig J et al. (2008). Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci U S A 105: 3628–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YQ, Andrade A, Lipscombe D (2013). Spinal morphine but not ziconotide or gabapentin analgesia is affected by alternative splicing of voltage‐gated calcium channel CaV2.2 pre‐mRNA. Mol Pain 9: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, Li Q, Allette YM, Ripsch MS, White FA, Khanna R (2012). Suppression of pain‐related behavior in two distinct rodent models of peripheral neuropathy by a homopolyarginine‐conjugated CRMP2 peptide. J Neurochem 124: 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Jun K, Lee T, Kim SS, McEnery MW, Chin H et al. (2001). Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage‐dependent calcium channel. Mol Cell Neurosci 18: 235–245. [DOI] [PubMed] [Google Scholar]
- Konietschke F, Libiger O, Hothorn LA (2012). Nonparametric evaluation of quantitative traits in population‐based association studies when the genetic model is unknown. PLoS One 7: e31242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA (1994). AIDS‐related dementia and calcium homeostasis. Ann N YAcad Sci 747: 205–224. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Nicotera P (1998). Calcium, free radicals and excitotoxins in neuronal apoptosis. Cell Calcium 23: 165–171. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Yaksh TL (1995). Effect of continuous intrathecal infusion of omega‐conopeptides, N‐type calcium‐channel blockers, on behavior and antinociception in the formalin and hot‐plate tests in rats. Pain 60: 83–90. [DOI] [PubMed] [Google Scholar]
- Manji H (2000). Neuropathy in HIV infection. Curr Opin Neurol 13: 589–592. [DOI] [PubMed] [Google Scholar]
- Maratou K, Wallace VC, Hasnie FS, Okuse K, Hosseini R, Jina N et al. (2009). Comparison of dorsal root ganglion gene expression in rat models of traumatic and HIV‐associated neuropathic pain. Eur J Pain 13: 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, O'Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema RP et al. (2001). Intrathecal HIV‐1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. J Neurosci 21: 2808–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RA, Wiffen PJ, Derry S, McQuay HJ (2011). Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev : CD007938. doi:10.1002/14651858.CD007938.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutal A, Chew LA, Yang X, Wang Y, Yeon SK, Telemi E et al. (2016a). (S)‐lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain 157: 1448–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutal A, Francois‐Moutal L, Perez‐Miller S, Cottier K, Chew LA, Yeon SK et al. (2016b). (S)‐lacosamide binding to collapsin response mediator protein 2 (CRMP2) regulates CaV2.2 activity by subverting its phosphorylation by Cdk5. Mol Neurobiol 53: 1959–1976. [DOI] [PubMed] [Google Scholar]
- Nagai J, Owada K, Kitamura Y, Goshima Y, Ohshima T (2016). Inhibition of CRMP2 phosphorylation repairs CNS by regulating neurotrophic and inhibitory responses. Exp Neurol 277: 283–295. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Yamashita N, Kimura A, Kimura Y, Hirano H, Makihara H et al. (2016). Comprehensive behavioral study and proteomic analyses of CRMP2‐deficient mice. Genes Cells 21: 1059–1079. [DOI] [PubMed] [Google Scholar]
- Nasirinezhad F, Jergova S, Pearson JP, Sagen J (2015). Attenuation of persistent pain‐related behavior by fatty acid amide hydrolase (FAAH) inhibitors in a rat model of HIV sensory neuropathy. Neuropharmacology 95: 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newshan G (1998). HIV neuropathy treated with gabapentin. AIDS 12: 219–221. [PubMed] [Google Scholar]
- Park J, Luo ZD (2010). Calcium channel functions in pain processing. Channels (Austin) 4: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekarz AD, Due MR, Khanna M, Wang B, Ripsch MS, Wang R et al. (2012). CRMP‐2 peptide mediated decrease of high and low voltage‐activated calcium channels, attenuation of nociceptor excitability, and anti‐nociception in a model of AIDS therapy‐induced painful peripheral neuropathy. Mol Pain 8: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach TT, Wang Y, Khanna R, Chounlamountri N, Auvergnon N, Honnorat J et al. (2011). Effect of CRMP3 expression on dystrophic dendrites of hippocampal neurons. Mol Psychiatry 16: 689–691. [DOI] [PubMed] [Google Scholar]
- Rauck RL, Wallace MS, Burton AW, Kapural L, North JM (2009). Intrathecal ziconotide for neuropathic pain: a review. Pain Pract 9: 327–337. [DOI] [PubMed] [Google Scholar]
- Ricard D, Rogemond V, Charrier E, Aguera M, Bagnard D, Belin MF et al. (2001). Isolation and expression pattern of human Unc‐33‐like phosphoprotein 6/collapsin response mediator protein 5 (Ulip6/CRMP5): coexistence with Ulip2/CRMP2 in Sema3a‐sensitive oligodendrocytes. J Neurosci 21: 7203–7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripsch MS, Ballard CJ, Khanna M, Hurley JH, White FA, Khanna R (2012). A peptide uncoupling CRMP‐2 from the presynaptic Ca2+ channel complex demonstrate efficacy in animal models of migraine and AIDS therapy‐induced neuropathy. Transl Neurosci 3: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Lotsch J, Freynhagen R, Geisslinger G (2010). Ziconotide for treatment of severe chronic pain. Lancet 375: 1569–1577. [DOI] [PubMed] [Google Scholar]
- Scott DA, Wright CE, Angus JA (2002). Actions of intrathecal omega‐conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur J Pharmacol 451: 279–286. [DOI] [PubMed] [Google Scholar]
- Skov MJ, Beck JC, de Kater AW, Shopp GM (2007). Nonclinical safety of ziconotide: an intrathecal analgesic of a new pharmaceutical class. Int J Toxicol 26: 411–421. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark P, Ogg D, Flodin S, Flores A, Kotenyova T, Nyman T et al. (2007). The structure of human collapsin in response mediator protein 2, a regulator of axonal growth. J Neurochem 101: 906–917. [DOI] [PubMed] [Google Scholar]
- Thompson JC, Dunbar E, Laye RR (2006). Treatment challenges and complications with ziconotide monotherapy in established pump patients. Pain Physician 9: 147–152. [PubMed] [Google Scholar]
- Vink S, Alewood PF (2012). Targeting voltage‐gated calcium channels: developments in peptide and small‐molecule inhibitors for the treatment of neuropathic pain. Br J Pharmacol 167: 970–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MS (2006). Ziconotide: a new nonopioid intrathecal analgesic for the treatment of chronic pain. Expert Rev Neurother 6: 1423–1428. [DOI] [PubMed] [Google Scholar]
- Wallace VC, Blackbeard J, Segerdahl AR, Hasnie F, Pheby T, McMahon SB et al. (2007). Characterization of rodent models of HIV‐gp120 and anti‐retroviral‐associated neuropathic pain. Brain 130: 2688–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA (1992). Biochemical properties and subcellular distribution of an N‐type calcium channel alpha 1 subunit. Neuron 9: 1099–1115. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Brittain JM, Piekarz AD, Ballard CJ, Ripsch MS, Cummins TR et al. (2011). Further insights into the antinociceptive potential of a peptide disrupting the N‐type calcium channel‐CRMP‐2 signaling complex. Channels (Austin) 5: 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SM, Schmutzler BS, Brittain JM, Dustrude ET, Ripsch MS, Pellman JJ et al. (2012). Inhibition of transmitter release and attenuation of anti‐retroviral‐associated and tibial nerve injury‐related painful peripheral neuropathy by novel synthetic Ca2+ channel peptides. J Biol Chem 287: 35065–35077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JY, Chew LA, Yang X, Wang Y, Qu C, Wang Y et al. (2016). Sustained relief of ongoing experimental neuropathic pain by a CRMP2 peptide aptamer with low abuse potential. Pain 157: 2124–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA (1976). Chronic catheterization of the spinal subarachnoid space. Physiol Behav 17: 1031–1036. [DOI] [PubMed] [Google Scholar]
- Yuan SB, Shi Y, Chen J, Zhou X, Li G, Gelman BB et al. (2014). Gp120 in the pathogenesis of human immunodeficiency virus‐associated pain. Ann Neurol 75: 837–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A, Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67: 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Diefenbach E, Crossett B, Tran SL, Ng T, Rizos H et al. (2010). First evidence of overlaps between HIV‐Associated Dementia (HAD) and non‐viral neurodegenerative diseases: proteomic analysis of the frontal cortex from HIV+ patients with and without dementia. Mol Neurodegener 5: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The CRMP2‐derived CBD3 peptide sequence is evolutionarily conserved. The six‐amino‐acid residue sequence ‘ARSRLA’ is perfectly conserved within a variety of species of mammals, reptiles, birds, frogs, and fish. CRMP2 Mus musculus (NM_009955.3) sequence was analysed using the National Center for Biotechnology Information (NCBI) protein blast suite and a phylogenetic tree was created using ‘Interactive Tree of Life’ (iTOL2.0) software (Letunic & Bork, 2007; Letunic & Bork, 2011). Select images and common names are colour matched to nearby scientific names. Major branches are labelled by common name of lowest shared taxonomic classification. Branch separation represents divergent evolutionary relationship and closely related organisms are grouped by colour.
Figure S1 The CRMP2‐derived CBD3 peptide sequence is evolutionarily conserved. The six‐amino‐acid residue sequence ‘ARSRLA’ is perfectly conserved within a variety of species of mammals, reptiles, birds, frogs, and fish. CRMP2 Mus musculus (NM_009955.3) sequence was analysed using the National Center for Biotechnology Information (NCBI) protein blast suite and a phylogenetic tree was created using ‘Interactive Tree of Life’ (iTOL2.0) software (Letunic & Bork, 2007; Letunic & Bork, 2011). Select images and common names are colour matched to nearby scientific names. Major branches are labelled by common name of lowest shared taxonomic classification. Branch separation represents divergent evolutionary relationship and closely related organisms are grouped by colour.