

Abstract
Aims
Alpha‐2 agonists are direct peripheral vasoconstrictors, which achieve these effects by activating vascular smooth muscle alpha‐2 adrenoceptors. The impact of this response during dexmedetomidine infusion remains poorly quantified. Our goal was to investigate the pharmacokinetic (PK) and pharmacodynamic (PD, vasoconstriction) effects of a computer‐controlled dexmedetomidine infusion in healthy volunteers.
Methods
After local ethics committee approval, we studied 10 healthy volunteers. To study the peripheral vasoconstrictive effect of dexmedetomidine without concurrent sympatholytic effects, sympathetic fibres were blocked with a brachial plexus block. Volunteers received a dexmedetomidine target‐controlled infusion for 15 min, to a target concentration of 0.3 ng ml–1. Arterial blood samples were collected during and for 60 min after dexmedetomidine infusion for PK analysis. Peripheral vasoconstriction (PD) was assessed using finger photoelectric plethysmography. PK/PD analysis was carried out using nonlinear mixed‐effect models.
Results
We found that the computer‐controlled infusion pump delivered mean concentrations greater than 0.3 ng ml–1 over the 15‐min infusion duration. The peripheral vasoconstrictive effect correlated with dexmedetomidine plasma concentrations during and after the infusion. A three‐compartment model provided a better fit to the data than a two‐compartment model.
Conclusions
We found that dexmedetomidine‐induced vasoconstriction is concentration dependent over time. Dexmedetomidine PK were best estimated by a three‐compartment model with allometric scaling. Our results may contribute to future modelling of dexmedetomidine‐induced haemodynamic effects.
Keywords: dexmedetomidine, pharmacodynamics, pharmacokinetics, vasoconstriction
What is Already Known about this Subject
Dexmedetomidine is a highly selective, nonsubtype‐specific alpha‐2 adrenergic agonist that is approved for clinical use for its sedative properties.
Alpha‐2 adrenergic agonists mediate haemodynamic effects by several different mechanisms. They mediate sympatholytic effects through activation of centrally and peripherally located alpha‐2 adrenoceptors. They also activate vascular smooth muscle alpha‐2 adrenoceptors, which are responsible for direct peripheral vasoconstrictive effects.
What this Study Adds
Dexmedetomidine‐induced peripheral vasoconstrictive effects were found to correlate with dexmedetomidine plasma concentrations over time.
This vasoconstrictive effect was more rapid (half‐life for equilibration 2.16 min) than most other dexmedetomidine‐mediated effects, such as its sedative and sympatholytic effects.
These results may contribute to the future modelling of more complex dexmedetomidine‐induced haemodynamic effects.
Introduction
Alpha‐2 agonists, through activation of centrally located http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=4, mediate sympatholytic and mild vagomimetic effects that are responsible for decreases in heart rate and blood pressure. They are also direct peripheral vasoconstrictors. Activation of vascular smooth muscle alpha‐2 adrenoceptors mediate a direct peripheral vasoconstrictive effect 1, 2, 3. This can be observed clinically as an increase in blood pressure and a reflex decrease in heart rate when alpha‐2 agonists are administered rapidly, resulting in high alpha‐2 agonist plasma concentrations.
In our previous studies, using computer‐controlled drug infusions that target specific drug plasma concentrations, we observed dose‐dependent peripheral vasoconstriction induced by two different alpha‐2 agonists – clonidine and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=521 4, 5, 6. We also observed that with both alpha‐2 agonists, the vasoconstriction was consistently more profound during the first 3–5 min of drug infusion and then reached a pseudo steady state during the next 10–15 min 4, 5, 6. This direct vasoconstrictor response has been characterized in children after bolus dose administration 7. The impact of this response in adults during infusion remains poorly quantified. Our goal was to investigate the pharmacokinetic (PK) and pharmacodynamic (PD; vasoconstriction) effects of a 15‐min computer‐controlled infusion of a highly selective alpha‐2 agonist, dexmedetomidine, in healthy volunteers.
Methods
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee, and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. With the approval of the institutional Review Board of the University of California, San Francisco, we enrolled 10 healthy volunteers. Informed consent was obtained from all individual participants included in the study. We excluded individuals who had a history of cardiac, pulmonary, hepatic or renal disease, or of alcohol or drug abuse; those taking prescription medications; those older than 45 years and those weighing more than 130% of normal. Our drug/molecular target nomenclature conforms to the British Journal of Pharmacology's Concise Guide to PHARMACOLOGY 2017/18 8.
Subjects rested in the supine position in a temperature‐controlled (22–23°C) anaesthesia study room. A catheter was inserted into a vein in the foot, to enable administration of intravenous fluids and dexmedetomidine on the morning of study. Lactated Ringer's solution (5 ml kg–1) was administered before drug administration, and 1.5 ml kg–1 h–1 was administered thereafter until the end of the study. After production of local anaesthesia with lignocaine, a cannula was placed into the radial artery of the right arm, to enable repeated blood sampling for PK analysis and measurement of arterial blood pressure. A pulse oximeter probe to measure finger blood volume (by photoplethysmography) was attached to the left hand, as described below. To minimize locally mediated vasomotor activity in response to changes in body temperature, subjects were covered with blankets during the study.
In order to study the peripheral vasoconstrictive effect of dexmedetomidine without its concurrent sympatholytic effects, approximately 15 min after application of all monitors the sympathetic fibres of the left arm were blocked by administration of 30 ml of 1% mepivacaine for production of an axillary perivascular brachial plexus block. Successful block was confirmed approximately 25 min later by testing the motor and sensory function of the left hand. Thirty minutes after axillary block, baseline measurements of haemodynamics and finger blood volume were obtained and a baseline arterial blood sample was collected. We then determined finger blood volume (vasoconstriction) and dexmedetomidine plasma concentration responses during and after a 15‐min dexmedetomidine infusion.
Dexmedetomidine infusion
A computer‐controlled infusion pump (Harvard Apparatus 22; Harvard Apparatus, South Natick, MA, USA) was used to infuse dexmedetomidine (4 μg ml–1) (Precedex, 100 μg ml–1 dexmedetomidine hydrochloride, Abbott Laboratories Inc.; North Chicago, IL, USA) for 15 min to target plasma concentrations of 0.3 ng ml–1. The pump was controlled by STANPUMP software (obtained from Professor Steven Shafer MD, Department of Anesthesia, Stanford University, Palo Alto, CA, USA), which adjusted and recorded the rate of infusion every 10 s. We used the same dexmedetomidine PK parameters as we had used in our previous studies (i.e. a central volume of distribution of 0.427 l kg–1, and elimination and intercompartment rate constants of k10 = 0.0212 min−1, k12 = 0.0744 min−1 and k21 = 0.0264 min−1; where k10 is the micro rate constant for those processes acting through biotransformation or elimination that irreversibly remove drug from the central compartment, and the intercompartmental micro rate constants (k12, k21, etc.) describe the exchange of drug between the central and peripheral compartments).
Photoelectric plethysmography
Blood volume in the finger was assessed using photoelectric plethysmography, which measures infrared light transmitted through a fingertip. The absolute level of transmitted light was determined by pulse oximeter (Nellcor N200; Nellcor Inc., Hayward, CA, USA), for which we placed a sensor (Nellcor D25; Nellcor Inc., Pleasanton, CA, USA) on the ring finger of the left hand.
The pulse oximeter consists of two parts – a sensor and a monitor. The sensor, which is applied to the tip of a finger, contains a low‐voltage, low‐intensity, light‐emitting diode that is supplied with constant drive current and emits infrared light (at a wavelength of approximately 920 nm). A detector photodiode in the sensor receives transmitted light and generates an electrical current proportional to the amount of light received. Data on electrical current thus generated were transmitted to a monitor, passed through an analogue‐to‐digital converter (ADC), sampled every 10 s and recorded by a computer. During vasoconstriction, blood volume in the finger decreases, and more light is transmitted through the finger. Thus, the ADC values increase in response to vasoconstriction. During vasodilation, blood volume in the finger increases, increasing tissue volume and decreasing the amount of light transmitted by the finger, resulting in a decrease in ADC values. During changes in blood vessel diameter (vasoconstriction or vasodilation), the change in light transmittance through the finger is rapid (seconds). This ADC measurement served as the qualitative measure of blood volume and, hence, vasoconstriction in the fingertip. ADC unit values were recorded for 3 min before and throughout the 15‐min dexmedetomidine infusion and for 60 min after the end of the infusion.
Dexmedetomidine plasma concentrations
Arterial blood samples (5 ml) were collected using the intra‐arterial cannula at baseline; 1, 2, 3, 4, 5, 7.5 10 and 15 min after the beginning of the dexmedetomidine infusion; and 15, 30 and 60 min after the end of the infusion. Blood samples were immediately placed on ice and plasma was subsequently separated in a refrigerated centrifuge. Plasma samples were stored at −70°C until analysis.
Concentrations of dexmedetomidine (reference standard: dexmedetomidine hydrochloride, Fermion Oy, Oulu, Finland) in human ethylenediamine tetra‐acetic acid plasma were determined using high‐performance liquid chromatography–tandem mass spectrometry after solid phase extraction with Sep‐Pak®tC18 96‐well extraction plates (Waters Co., Milford, MA, USA) with 2H6‐medetomidine (ORM‐14385, Orion Pharma, Espoo, Finland) as an internal standard. After separation with a Gemini® column (2 × 150 mm, 5 μm, Phenomenex, Torrance, CA, USA) and 0.1% formic acid in water and a mixture of methanol and acetonitrile as solvents, quantitative detection was performed in multireaction monitoring mode with a triple quadruple mass spectrometer (API 4000, MDS Sciex, Concord, Ontario, Canada). For dexmedetomidine and the internal standard, the precursor ions (m/z) were 201.2 and 208.2, respectively. The fragment ions (m/z) monitored and used for quantitation were 95.1 for dexmedetomidine and 97.1 for the internal standard. The chromatograms were processed using Applied Biosystems/MDS Sciex software (Analyst 1.4.1). Calibration standards with eight nonzero concentrations and quality controls samples with three different concentration levels (low, medium and high) were included in all assays. The linear concentration range was from 0.02 ng ml–1 to 10.0 ng ml–1. The interassay accuracies of the quality control samples (0.03, 0.9 and 8.0 ng ml–1) were 89%, 89% and 101.5%, respectively. This assay has a lower limit of quantification of 17 pg ml–1 and coefficient of variation of 5.7% in the relevant concentration range.
Haemodynamic variables and haemoglobin oxygen saturation
Systolic and diastolic blood pressure were measured via the radial artery cannula, and heart rate was measured noninvasively (Propaq 106; Protocol Systems, Beaverton, OR, USA). Haemoglobin oxygen saturation (SpO2) was measured continuously noninvasively by pulse oximeter (Nellcor N200; Nellcor Inc., Hayward, CA, USA). Haemodynamic and SpO2 data were recorded at baseline; 5, 10 and 15 min after the beginning of the dexmedetomidine infusion; and 15, 30 and 60 min after the end of the infusion.
PKPD analysis
The PK and PD data were initially analysed simultaneously using nonlinear mixed‐effects models (NONMEM 7.3, ICON Development Solutions, Ellicott City, MD 21076, USA). This method can distort PK model parameter estimates when PD or effect compartment model misspecification is present 8, 9. Consequently, a second modelling process was undertaken, whereby the PK parameters were estimated separately and then fixed in the PKPD model (known as the population PK parameters and data method) 9, 10. A three‐compartment (central and two peripheral) disposition linear model was used for the PK data. The model was parameterized in terms of elimination clearance, two intercompartment clearances, a central volume and two peripheral volumes. The parameter values were standardized for a body weight of 70 kg using the allometric model 11, 12.
where P i is the parameter in the ith individual, W i is the weight in the ith individual and P STD is the parameter in an individual with a weight (W STD) of 70 kg. The PWR exponent was 0.75 for clearance, 0.25 for half‐life (t½) values and 1 for distribution volumes 12.
An additional effect compartment was incorporated into the model in order to describe the delayed peripheral vasoconstrictor response quantified by photoplethysmography data. An equilibration rate constant (keo) characterizing the temporal relationship between the effect compartment and plasma was parameterized as an equilibration t½ (t½ keo):
The relationship between vasoconstriction and concentration was described using a sigmoid E MAX model:
where E 0 is the baseline effect, Ce is the concentration in the vasoconstrictor effect compartment, E MAX is the maximum response for vasoconstriction, C 50 is the concentration in the effect compartment producing 50% of E MAX and N is the Hill coefficient defining the steepness of the concentration–response curve.
The population parameter variability in model parameters was modelled by a proportional variance model. An additive (ERR ADD) and a proportional term (ERR PROP) characterized the residual unknown variability. The variances of the residual unidentified variability (ηRUV,i) were estimated. The population mean parameters, between subject variance and residual variance, were estimated using the first‐order conditional interaction estimate method, using ADVAN13 TOL9 (differential equation solver) of NONMEM VII. Convergence criterion was three significant digits. A Compaq Digital FORTRAN Version 6.6A compiler with Intel Celeron 333 MHz CPU (Intel Corp., Santa Clara, CA, USA) under MS WINDOWS XP was used to compile NONMEM.
The population parameter variability is modelled in terms of random‐effect (η) variables. Each of these variables was assumed to have mean of 0 and a variance denoted by ω2, which is estimated. The covariance between two elements of η (e.g. C 50 and E MAX) is a measure of statistical association between these two variables. Their covariance is related to their correlation (R) – i.e.:
The covariance of E MAX and C 50 variability was incorporated into the model.
Quality of fit
Models were nested and an improvement in the objective function was referred to the chi‐squared distribution to assess significance – e.g. an objective function change (ΔOBJ) of 3.84 is significant at α = 0.05. The quality of fit of the PK model to the data was sought by NONMEM's objective function and by visual examination of plots of observed vs. predicted concentrations. Nonparametric bootstrap methods provided a means to evaluate parameter uncertainty 13. A total of 1000 simulations was used to estimate confidence intervals. A visual predicted check 14, a modelling tool that estimates the prediction intervals and graphically superimposes these intervals on observed data after a standardized dose, was used to evaluate how well the model predicted the distribution of observed ADC measurements.
In any model, the quality of the individual parameter estimate will depend heavily on the observed data available. For example, sparse data can result in reduced variance (ω2) of parameter estimates and distortions of the distribution shape. If no data are available on a particular individual, the individual's estimate will be equal to the population value; the variance is shrinking towards zero as the quantity of information at the individual level diminishes, a phenomenon defined as η‐shrinkage (shε). This was calculated by:
where SD approximates the standard deviation. When there is no shrinkage, the model is correct and individual data are sufficiently abundant for individual parameter estimation. Data contain virtually no information about these parameters when shrinkage is 100% and the individual parameter values approach the typical parameter value.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 15, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 8.
Results
Table 1 shows the demographics of the 10 subjects, five of whom were male and five female. Table 2 shows dexmedetomidine plasma concentrations, ADC unit and haemodynamic values during and after dexmedetomidine infusion. Figure 1 illustrates the association between plasma dexmedetomidine concentrations and the percentage change in ADC unit values. Figure 2 shows the changes in ADC unit values during and for 60 min after dexmedetomidine infusion. PK data were available for 10 adult volunteers, with 120 concentrations. The mean age of the volunteers was 29 (SD 4, range 21–36) years, the mean weight was 72 (SD 13, range 52–89) kg, the mean height was 174 (SD 9, range 160–183) cm and the mean body mass index was 23 (SD 2, range 20–27) kg m–2. The cumulative dose of dexmedetomidine administered was 0.28 μg kg–1. There were no adverse effects during the study.
Table 1.
Demographics of the 10 study subjects
| Variable | |
|---|---|
| Age (years) | 29 ± 4 (21–36) |
| Height (cm) | 174 ± 9 (160–183) |
| Weight (kg) | 72 ± 13 (52–89) |
| BMI | 23 ± 2 (20–27) |
| Male/female | 5/5 |
Data are presented as mean ± standard deviation (range). BMI, body mass index
Table 2.
Dexmedetomidine plasma concentration, analogue‐to‐digital converter (ADC) unit and haemodynamic values during and after dexmedetomidine infusion
| Dexmedetomidine infusion | Post infusion | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 min | 1 min | 2 min | 3 min | 4 min | 5 min | 7.5 min | 10 min | 15 min | 15 min | 30 min | 60 min |
| Dexmedetomidine plasma concentration (ng ml –1 ) | |||||||||||
| 0 | 0.39 (0.26) | 1.11 (0.37) | 0.63 (0.17) | 0.52 (0.12) | 0.48 (0.13) | 0.44 (0.11) | 0.41 (0.09) | 0.43 (0.13) | 0.16 (0.04) | 0.12 (0.02) | 0.09 (0.02) |
| ADC units (% change from baseline) | |||||||||||
| 0 | 0 (3) | 22 (13) | 31 (15) | 29 (15) | 27 (15) | 25 (14) | 23 (14) | 23 (14) | 12 (9) | 9 (8) | 8 (6) |
| SBP (mmHg) | |||||||||||
| 125 (10) | 118 (10) | 114 (12) | 109 (12)* | 103 (14)* | 105 (13)* | 108 (13)* | |||||
| DBP (mmHg) | |||||||||||
| 65 (9) | 59 (11) | 61 (9) | 57 (10) | 55 (10) | 55 (10) | 58 (10) | |||||
| HR (bpm) | |||||||||||
| 67 (11) | 64 (8) | 67 (13) | 64 (11) | 65 (10) | 64 (11) | 63 (12) | |||||
Data are presented as mean (standard deviation). DBP, diastolic blood pressure; HR. heart rate; SBP, systolic blood pressure
Haemodynamic values that are significantly different from baseline (0 min) values (analysis of variance with Dunnett's post hoc test)
Figure 1.
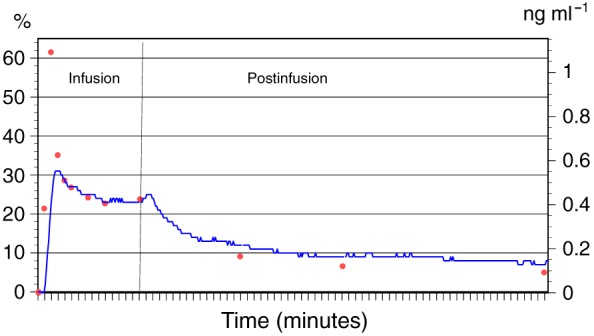
Mean dexmedetomidine plasma concentrations (red dots, right‐hand axis) and mean percentage change in analogue‐to‐digital converter units (blue line, left‐hand axis) during dexmedetomidine infusion and for 60 min after the infusion
Figure 2.
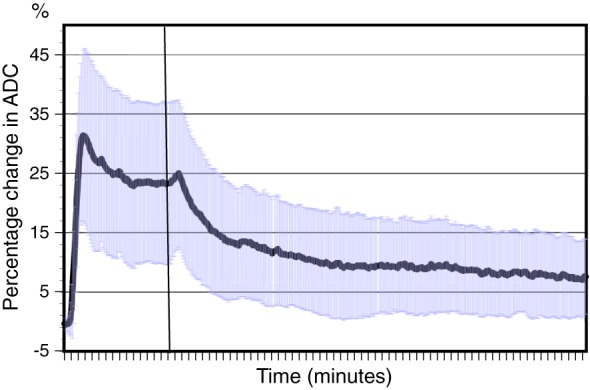
Mean and standard deviation of percentage change in analogue‐to‐digital converter units during dexmedetomidine infusion and for 60 min after the infusion
Nested models revealed that a three‐compartment model (Table 3A) provided a better fit to the data than a two‐compartment model (two extra parameters, ΔOBJ 12.919, P < 0.005). The correlation of between‐subject variability with PK parameters is shown in Table 3B. There were 4560 observations available for the PD analysis, and the final consequent parameter estimates are shown in Table 4A. The correlation of between‐subject variability with PD parameters is shown in Table 4B. A visual predictive check comparing the distribution of simulated PK data with observed data are shown in Figure 3; those for simulated PD data are shown in Figure 4. Shrinkage was acceptable for all model parameters. The effect site concentrations and their nonlinear relationship with vasoconstriction are shown in Figure 5.
Table 3A.
Pharmacokinetic parameter estimates for the three‐compartment disposition model
| Parameter | Estimate | PPV | SE% | Shrinkage% | Bootstrap estimate | 95% CI |
|---|---|---|---|---|---|---|
| CL (l min –1 70 kg –1 ) | 0.751 | 0.158 | 4.6 | 5.7 | 0.712 | 0.107, 0.872 |
| Q2 (l min –1 70 kg –1 ) | 1.01 | 0.590 | 11.2 | 9.4 | 0.96 | 0.531, 1.816 |
| Q3 (l min –1 70 kg –1 ) | 1.03 | 3.7 | 1.06 | 0.674, 1.39 | ||
| V1 (l 70 kg –1 ) | 12 | 0.339 | 6.3 | 4.6 | 12.8 | 9.570, 16.9 |
| V2 (l 70 kg –1 ) | 11.7 | 0.868 | 13.1 | 5.9 | 13.1 | 6.369, 35.61 |
| V3 (l 70 kg –1 ) | 47.2 | 11.2 | 48.4 | 35.295, 217.025 | ||
| ERR ADD (μg l −1 ) | 0.011 | ηRUV 0.66 |
5.4 |
0.010 | 0.001, 0.132 | |
| ERR PROP (%) | 10.4 | 11.8 | 7.02, 19.36 |
ηRUV, variance of the residual unidentified variability; CI, confidence interval; CL, clearance; ERRADD, additive residual error; ERRPROP, proportional residual error; PPV, apparent coefficient of variation of between‐subject variability, estimated using an exponential random‐effect model [sqrt(NONMEM Omega)]; Q2 and Q3, intercompartment clearances; SE, standard error of the structural parameter estimate; V1, central volume; V2 and V3, peripheral volumes
Table 3B.
Correlation of between‐subject variability with pharmacokinetic parameters
| CL | V1 | Q2 | V2 | |
|---|---|---|---|---|
| CL | 1 | |||
| V1 | 0.420 | 1 | ||
| Q2 | 0.685 | −0.367 | 1 | |
| V2 | 0.971 | 0.210 | 0.833 | 1 |
CL, clearance; Q2, intercompartment clearance; V1, central volume; V2, peripheral volume
Table 4A.
Parameter estimates for an effect compartment concentration–response relationship using an EMAX model (BSV is the between subject parameter variability, CI is the confidence interval presented as 5th, 95th)
| Parameter | Estimate | PPV | Shrinkage% | Bootstrap estimate | 95% CI |
|---|---|---|---|---|---|
| E 0 (ADC units) | 5860 | 0.462 | 11.3 | 5666 | 4549, 7527 |
| E MAX (ADC units) | 3090 | 1.349 | 13 | 3160 | 1540, 7777 |
| C 50 (μg l −1 ) | 0.233 | 1.628 | 28.6 | 0.211 | 0.098, 0.390 |
| Hill coefficient | 2.83 | 3.28 | 1.70, 4.35 | ||
| t 1/2 keo (min) | 2.16 | 0.841 | 18.2 | 2.01 | 1.37, 2.90 |
| ERRADD (ADC units) | 113 | ηRUV 0.442 |
29.2 |
103 | 72, 138 |
| ERRPROP (%) | 61.8 | 61.8 | 36.4, 326 |
ηRUV, variance of the residual unidentified variability; C50, concentration in the effect compartment producing 50% of EMAX; CI, confidence interval; EMAX, maximum response for vasoconstriction; E0, baseline effect; ERRADD, additive residual error; ERRPROP, proportional residual error; t½ keo, half‐life for equilibration; PPV, apparent coefficient of variation of between‐subject variability, estimated using an exponential random‐effect model [sqrt(NONMEM Omega)]
Table 4B.
The correlation of between subject‐variability with PD parameters
| E 0 | E MAX | C 50 | t 1/2 keo | |
|---|---|---|---|---|
| E 0 | 1 | |||
| E MAX | 0.116 | 1 | ||
| C 50 | 0.374 | 0.897 | 1 | |
| t½ keo | −0.458 | −0.716 | −0.913 | 1 |
C50, concentration in the effect compartment producing 50% of EMAX; EMAX, maximum response for vasoconstriction; E0, baseline effect; t½ keo, half‐life for equilibration
Figure 3.
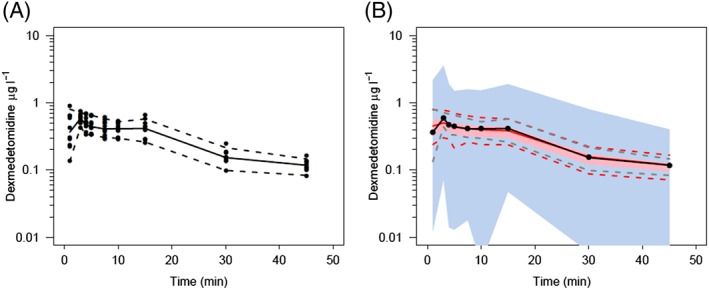
Visual predictive check for the pharmacokinetic model. All plots show median and 90% confidence intervals (solid and dashed lines, respectively). (A) Prediction‐corrected observed concentrations. (B) Prediction‐corrected percentiles (10%, 50% and 90%) for observations (black dashed lines) and predictions (pink dashed lines), with 95% confidence intervals for prediction percentiles (median, pink shading; 5th and 95th percentiles, blue shading). There is a mismatch between the observed and predicted concentrations in the first 2 min
Figure 4.
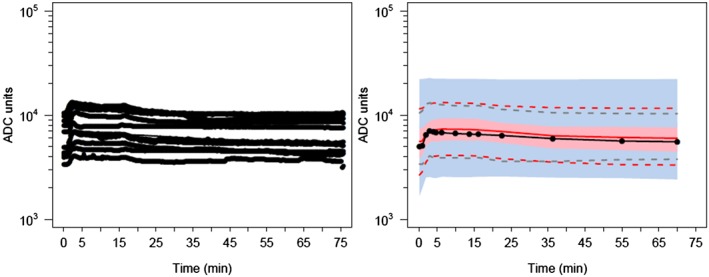
Visual predictive check for the pharmacodynamic model. All plots show median and 90% confidence intervals (solid and dashed lines, respectively). (A) All prediction‐corrected observed concentrations. (B) Prediction‐corrected percentiles (10%, 50%, and 90%) for observations (black dashed lines) and predictions (pink dashed lines) with 95% confidence intervals for prediction percentiles (median, pink shading; 5th and 95th percentiles, blue shading). ADC, analogue‐to‐digital converter
Figure 5.
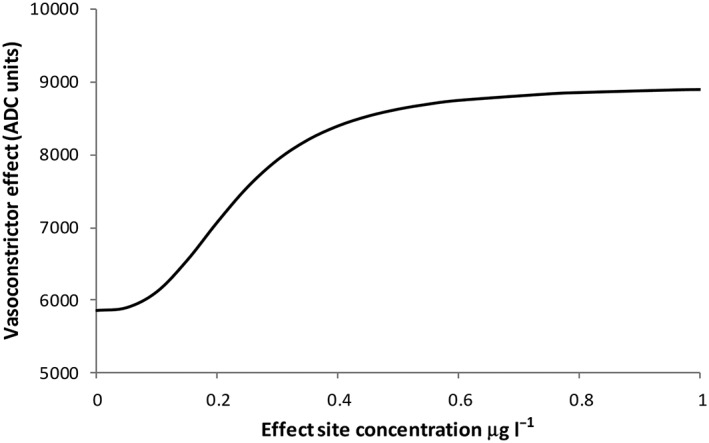
A final model of absolute analogue‐to‐digital converter (ADC) units vs. dexmedetomidine plasma concentration, illustrating a nonlinear relationship between effect site concentrations and vasoconstriction response
Discussion
We investigated the dynamic vasoconstrictive effects of computer‐controlled dexmedetomidine infusions. We found that the computer‐controlled infusion pump delivered mean concentrations greater than 0.3 ng ml–1 over the 15‐min infusion duration; a peak concentration of 1.1 ng ml–1 at 2 min decreased to 0.4 ng ml–1 at 15 min The alpha‐2 agonist‐induced peripheral vasoconstrictive effect correlated with dexmedetomidine plasma concentrations during and after the infusion. This verified that dexmedetomidine‐induced vasoconstriction is dose dependent over time. The estimated three‐compartment model PK parameters were consistent with several of those published previously 16, 17.
Several of our previous studies that investigated alpha‐2 agonist‐induced peripheral vasoconstriction used computer‐controlled alpha‐2 agonist infusions targeting a wide range of clonidine and dexmedetomidine plasma concentrations 4, 5, 6, 18. In these studies, we consistently observed an initial short‐term peak in alpha‐2 agonist‐induced vasoconstriction, followed by pseudo steady state of vasoconstriction 4, 5, 6, 18. These vasoconstrictive effects were associated with concomitant increases in intra‐arterial blood pressure and decreases in heart rate 4, 5, 6, 18. In our studies that used multistep infusions targeting increasing clonidine or dexmedetomidine plasma concentrations, we observed similar dynamic vasoconstrictive effects at the beginning of each infusion step 4, 5, 6, 18. As these dynamic vasoconstrictive effects were identical with two different alpha‐2 agonists (clonidine and dexmedetomidine) with independently derived PK models, we hypothesized that the initial 3–5‐min peak in vasoconstriction might have been due to PD effects. Theoretically, it seemed feasible that the alpha‐2 agonist‐induced vasoconstriction might have been rapidly attenuated either by alpha‐2 agonist‐mediated release of nitric oxide from endothelial cells or by the release of other compensatory vasodilators. We had previously shown that dexmedetomidine‐induced nitric oxide release attenuates dexmedetomidine‐induced vasoconstriction by 19% in healthy volunteers, further supporting the theory of compensatory vasodilation 19. As there was no available high‐frequency data on the performance of the clonidine and dexmedetomidine PK models during the beginning of the computer‐controlled infusion, we also considered the possibility that our observations of dynamic vasoconstrictive effects at the beginning of the infusion might have been due to plasma concentration changes. The results of the current study suggested that the observed dynamic peripheral vasoconstriction is attributable to rapid‐onset concentration‐dependent direct effects on the vasculature.
Our study was unique in performing PD modelling of dexmedetomidine using a continuously measured observation (vasoconstriction). Dexmedetomidine‐induced vasoconstriction is mediated by vascular smooth muscle alpha‐2B receptors 3. This vasoconstrictive effect is rapid (t½ for equilibration 2.16 min). Most other alpha‐2 agonist‐mediated effects, such as sedation and sympatholytic effects, may take 10–15 min to approach their maximum 20, 21. Blood pressure is complicated to model as it is a sum of several PD effects, such as peripheral vasoconstriction and centrally and peripherally mediated sympatholytic effects and central vagomimetic effects, all of which may have varying time courses. Our study adds to the understanding of dexmedetomidine‐induced vasoconstriction, and may further help in modelling other, more complex haemodynamic effects such as arterial blood pressure, which is a product of complex interactions of dexmedetomidine‐induced central sympatholytic, vagomimetic and imidazoline receptor‐mediated effects and peripherally mediated vasoconstrictive effects. The use of PKPD modelling in volunteers with sympathectomized limbs helps our understanding of the vasoconstrictor response. The use of target‐controlled infusions without assay of plasma dexmedetomidine concentrations can result in incorrect assumptions of plasma drug concentrations and introduce inaccuracies to PK and PD analysis. Dexmedetomidine PK parameter estimates from one population, when used in a different cohort, may not be applicable. Predicted concentrations used in any analysis may not be true. Front‐end kinetics at the beginning of an infusion can also result in misinterpretation. Arterial sampling for early drug assay (0–2 min) may be high (Figure 2); PK analysis relies on a ‘well stirred’ model, and that is not achievable in the first few minutes within blood. This false assumption of ‘instantaneous mixing’ in PK analysis could contribute to the initial peak in peripheral vasoconstriction observed in the present study; this artefact is particularly true of drugs with a rapid distributional t½. Others have modelled the pulse‐like changes in concentration reflected in early arterial assay samples using models of greater complexity (e.g. semiparametric approaches, physiological‐based models, more input compartments or sinusoidal models) 22, 23. Slower infusions, the use of sampling times that avoid this initial ‘unmixed’ phase and censoring of early assays have also been employed to reduce this artefact 24. Despite this impediment, modelling shows consistently raised concentrations above 0.3 ng ml–1 that decrease over the subsequent 15 min. The cohort studied was small and healthy. Covariates such as gender, age or pre‐existing disease processes (e.g. hypertension) could not be investigated.
In patients, dexmedetomidine causes a biphasic response after intravenous administration; there is an initial vasoconstriction, followed by delayed vasodilation. The initial vasoconstriction is mediated by activation of vascular smooth muscle alpha‐2 adrenoceptors and is associated with an increase in blood pressure and baroreflex‐mediated bradycardia. As a result of this vasoconstrictor effect, the dexmedetomidine loading dose should be administered over 10 min (to avoid high plasma drug concentrations and to allow time for central vasodilator effects). Once dexmedetomidine reaches the central nervous system, it mediates a slowly evolving (10–15 min) sympatholytic effect that is associated with a reduction in blood pressure and heart rate. We have quantified the effects of increasing plasma dexmedetomidine concentrations on initial vasoconstriction in vivo. At increasing dexmedetomidine concentrations, the central sympatholytic effect will be maximized, whereas the vasoconstrictive effects will continue to increase, resulting in an increase in systemic vascular resistance and potentially reduced organ blood flow 25.
In conclusion, we found that dexmedetomidine‐induced vasoconstriction is dose dependent over time and that our earlier findings of stronger than expected peripheral vasoconstriction at the beginning of dexmedetomidine infusion can be explained by dexmedetomidine PK. We also found that dexmedetomidine PK were best described by a three‐compartment model. Our results may contribute to the future modelling of dexmedetomidine‐induced haemodynamic effects.
Competing Interests
The authors declare no competing interests.
The study was funded by departmental funds. The authors wish to thank Dr Jacqueline A. Hannam for her help in data analysis and manuscript editing.
Contributors
P.T. designed the study and performed the research. B.J.A. analysed the data. PT. and B.J.A. wrote the manuscript.
Talke, P. , and Anderson, B. J. (2018) Pharmacokinetics and pharmacodynamics of dexmedetomidine‐induced vasoconstriction in healthy volunteers. Br J Clin Pharmacol, 84: 1364–1372. doi: 10.1111/bcp.13571.
References
- 1. Timmermans PB, van Zwieten PA. Vasoconstriction mediated by postsynaptic alpha 2‐adrenoceptor stimulation. Naunyn Schmiedebergs Arch Pharmacol 1980; 313: 17–20. [DOI] [PubMed] [Google Scholar]
- 2. Bylund DB, Regan JW, Faber JE, Hieble JP, Triggle CR, Ruffolo RR. Vascular alpha‐adrenoceptors: from the gene to the human. Can J Physiol Pharmacol 1995; 73: 533–543. [DOI] [PubMed] [Google Scholar]
- 3. Link RE, Desai K, Hein L, Stevens ME, Chruscinski A, Bernstein D, et al Cardiovascular regulation in mice lacking alpha2‐adrenergic receptor subtypes b and c. Science 1996; 273: 803–805. [DOI] [PubMed] [Google Scholar]
- 4. Talke P, Lobo E, Brown R. Systemically administered α2‐agonist–induced peripheral vasoconstriction in humans. Anesthesiology 2003; 99: 65–70. [DOI] [PubMed] [Google Scholar]
- 5. Talke PO, Lobo EP, Brown R, Richardson CA. Clonidine‐induced vasoconstriction in awake volunteers. Anesth Analg 2001; 93: 271–276. [DOI] [PubMed] [Google Scholar]
- 6. Talke P, Stapelfeldt C, Lobo E, Brown R, Scheinin M, Snapir A. Effect of alpha2B‐adrenoceptor polymorphism on peripheral vasoconstriction in healthy volunteers. Anesthesiology 2005; 102: 536–542. [DOI] [PubMed] [Google Scholar]
- 7. Potts L, Anderson BJ, Holford NH, Vu TC, Warman GR. Dexmedetomidine hemodynamics in children after cardiac surgery. Paediatr Anaesth 2010; 20: 425–433. [DOI] [PubMed] [Google Scholar]
- 8. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best‐case performance. J Pharmacokinet Pharmacodyn 2003; 30: 387–404. [DOI] [PubMed] [Google Scholar]
- 10. Zhang L, Beal SL, Sheinerz LB. Simultaneous vs. sequential analysis for population PK/PD data II: robustness of methods. J Pharmacokinet Pharmacodyn 2003; 30: 405–416. [DOI] [PubMed] [Google Scholar]
- 11. Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet 1996; 30: 329–332. [DOI] [PubMed] [Google Scholar]
- 12. Anderson BJ, Holford NH. Mechanism‐based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 2008; 48: 303–332. [DOI] [PubMed] [Google Scholar]
- 13. Efron B, Tibshirani R. Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat Sci 1986; 1: 54–77. [Google Scholar]
- 14. Post TM, Freijer JI, Ploeger BA, Danhof M. Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn 2008; 35: 185–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acid Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weerink MAS, Struys MMRF, Hannivoort LN, Barends CRM, Absalom AR, Colin P. Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin Pharmacokinet 2017; 56: 893–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hannivoort LN, Eleveld DJ, Proost JH, Reyntjens KM, Absalom AR, Vereecke HE, et al Development of an optimized pharmacokinetic model of dexmedetomidine using target‐controlled infusion in healthy volunteers. Anesthesiology 2015; 123: 357–367. [DOI] [PubMed] [Google Scholar]
- 18. Talke PO, Caldwell JE, Richardson CA, Heier T. The effects of clonidine on human digital vasculature. Anesth Analg 2009; 91: 793–797. [DOI] [PubMed] [Google Scholar]
- 19. Snapir A, Talke P, Posti J, Huiku M, Kentala E, Scheinin M. Effects of nitric oxide synthase inhibition on dexmedetomidine‐induced vasoconstriction in healthy human volunteers. Br J Anaesth 2009; 102: 38–46. [DOI] [PubMed] [Google Scholar]
- 20. Kallio A, Scheinin M, Koulu M, Ponkilainen R, Ruskoaho H, Viinamäki O, et al Effects of dexmedetomidine, a selective alpha 2‐adrenoceptor agonist, on hemodynamic control mechanisms. Clin Pharmacol Ther 1989; 46: 33–42. [DOI] [PubMed] [Google Scholar]
- 21. Scheinin M, Kallio A, Koulu M, Viikari J, Scheinin H. Sedative and cardiovascular effects of medetomidine, a novel selective alpha 2‐adrenoceptor agonist, in healthy volunteers. Br J Clin Pharmacol 1987; 24: 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barbour AM, Fossler MJ. Infusions are the optimal dosing method in intravenous ADME studies rather than bolus dosing. J Clin Pharmacol 2018; 58: 25–28. [DOI] [PubMed] [Google Scholar]
- 23. Bjornsson MA, Norberg AA, Kalman S, Karlsson MO. Simonsson US. A two‐compartment effect site model describes the bispectral index after different rates of propofol infusion. J Pharmacokinet Pharmacodyn 2010; 37: 243–255. [DOI] [PubMed] [Google Scholar]
- 24. Jacobs JR, Nath PA. Compartment model to describe peripheral arterial‐venous drug concentration gradients with drug elimination from the venous sampling compartment. J Pharm Sci 1995; 84: 370–375. [DOI] [PubMed] [Google Scholar]
- 25. Ebert TJ, Hall JE, Barney JA, Uhrich TD, Colinco MD. The effects of increasing plasma concentrations of dexmedetomidine in humans. Anesthesiology 2000; 93: 382–394. [DOI] [PubMed] [Google Scholar]