

Abstract
Aims
This study aimed to analyse the effects of genetic polymorphisms in drug transporters and metabolizing enzymes, and clinical laboratory data on the pharmacokinetic parameters of apixaban.
Methods
Data were collected from 81 Japanese patients with atrial fibrillation. Pharmacogenomic data were stratified by ABCB1, ABCG2 and CYP3A5 polymorphisms. The pharmacokinetic profile of apixaban was described by a one‐compartment model with first‐order absorption. Population pharmacokinetic analysis was conducted using a nonlinear mixed effect modelling (NONMEM™) program.
Results
The nonlinear relationship between oral clearance (CL/F) of apixaban and creatinine clearance (Ccr) was observed. The population mean of CL/F for a typical patient (Ccr value of 70 ml min−1) with the CYP3A5*1/*1 and ABCG2 421C/C or C/A genotypes was estimated to be 3.06 l h−1. When Ccr values were set to the typical value, the population mean of CL/F was 1.52 times higher in patients with the CYP3A5*1/*1 genotype compared with patients with the CYP3A5*1/*3 or *3/*3 genotype, while the population mean of CL/F was 1.49 times higher in patients with the ABCG2 421C/C or C/A genotype compared with patients with the ABCG2 421A/A genotype. However, no covariates affected the population mean of the apparent volume of distribution (Vd/F) of apixaban. The population mean of Vd/F was estimated to be 24.7 l.
Conclusion
The present study suggests that the ABCG2 421A/A and CYP3A5*3 genotypes and renal function are intrinsic factors affecting apixaban pharmacokinetics. These findings may provide useful information for precision medicine using apixaban, to avoid the risk of adverse reactions.
Keywords: cytochrome P450, drug transporters, genetic polymorphism, pharmacogenomics, population analysis
What is Already Known about this Subject
The pharmacokinetic variability of apixaban is related to covariates such as renal function, sex, body weight, age and race.
There is little information concerning the relationship between the pharmacokinetic variability of apixaban and genetic polymorphisms of drug metabolizing enzymes and transporters.
What this Study Adds
The population pharmacokinetic and pharmacogenomic model of apixaban in Japanese patients with atrial fibrillation is developed to estimate apixaban concentrations appropriately.
The ABCG2 421A/A and CYP3A5*3 genotypes as well as renal function can explain the inter‐individual variability in apixaban pharmacokinetics in patients with atrial fibrillation.
Introduction
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6390 is a direct and selective inhibitor of activated coagulation factor X (FXa), which exists as a key enzyme positioned at the confluence of intrinsic and extrinsic coagulation pathways 1. Apixaban is orally administered for the prevention of stroke or systemic embolism in patients with nonvalvular atrial fibrillation (AF) 2, 3. The ARISTOTLE clinical trial demonstrated that in AF patients, efficacy and safety of apixaban was better than those of warfarin, which has been the most commonly used anticoagulant therapy 3, 4, 5. Therefore, the number of prescriptions for direct FXa inhibitors such as apixaban and rivaroxaban are tending to increase while the number of prescriptions for warfarin have decreased 6, 7, 8. However, the apixaban package insert indicates that there is no way to adjust its dosage according to monitoring anticoagulant activities such as prothrombin time (PT), international normalized ratio of PT (PT‐INR), and anti‐FXa activity because, unlike warfarin, the apixaban therapeutic window remains unclear 9, 10, 11. Recent exposure–response studies showed that the area under the plasma concentration–time curve (AUC) for apixaban was related to the incidence of bleeding and venous thromboembolism 12, 13. Additionally, underdosing anticoagulants in AF patients partly caused a high risk for stroke and thromboembolic events in the clinic 14. Thus, monitoring the apixaban plasma concentrations in AF patients may lead to safer and more effective therapeutic dose adjustments.
Urinary unchanged apixaban represented approximately 25% of oral dose in healthy subjects, and its total metabolites represented approximately 25% of oral dose in healthy subjects 11, 15. Apixaban is reported to be transported by the ATP‐binding cassette multidrug transporters P‐glycoprotein (ABCB1) and breast cancer resistance protein (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=792) in the small intestine, liver, and kidney, and is predominantly metabolized by intestinal and hepatic http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=263 11, 16, 17. Therefore, individual expression and/or function of these proteins can explain the pharmacokinetic variability of apixaban.
The clinical effectiveness of anticoagulants has been examined in relation to drug transporter and metabolizing enzyme single nucleotide polymorphisms (SNPs) 18, 19, 20, 21, 22, 23. Recent studies suggested that precision medicine using vitamin K epoxide reductase complex 1 (VKORC1) and CYP2C9 genotypes is clinically useful for pharmacotherapy of warfarin 18, 19, 20. For dabigatran, a direct thrombin inhibitor, an SNP of carboxyesterase 1, which rapidly hydrolyses the prodrug (dabigatran etexilate) to dabigatran, is related to low trough concentrations of dabigatran and the risk of bleeding 21. For both rivaroxaban and edoxaban, which are FXa inhibitors, no ABCB1 polymorphisms affect these pharmacokinetics 22, 23. However, our previous pharmacogenomic study indicated that trough concentrations of apixaban are associated with the ABCG2 421A/A and CYP3A5*3 genotypes 24. These results also suggest that ABCG2 and CYP3A5 genotypes can be good predictors of apixaban pharmacokinetics.
A few population pharmacokinetic studies of apixaban have suggested that the pharmacokinetic variability of apixaban was mainly explained by renal function, sex, body weight, age, race, venous thromboembolism, and patient and subject having a history of orthopaedic surgery 12, 13, 25. However, a population pharmacokinetic analysis in Japanese AF patients has not been reported. Additionally, there is little information concerning the relationship between the pharmacokinetic variability of apixaban and genetic polymorphisms of drug metabolizing enzymes and transporters.
In this study, we conducted a population pharmacokinetic analysis of apixaban in Japanese AF patients and examined the effect of ABCB1, ABCG2 and CYP3A5 polymorphisms and clinical laboratory data on the apixaban pharmacokinetic parameters.
Methods
Ethics
This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Review Board of Ritsumeikan University Biwako‐Kusatsu Campus (Approval number BKC‐IRB‐2014‐021) and the Ethics Boards of Shiga University of Medical Science (Approval number 26‐116). Written informed consent was obtained from all patients prior to enrolment.
Patients and study design
Japanese adult inpatients and outpatients with AF who received apixaban at the Shiga University of Medical Science Hospital from February 2015 to May 2016 were enrolled in this study. All patients took oral apixaban tablets (Eliquis®, Bristol‐Myers Squibb, Princeton, NJ, USA and Pfizer Inc., Groton, CT, USA) twice daily at a dose of 5−20 mg day−1. For inpatients, blood samples were drawn in 3.2% citrated tubes at three time points: trough sampling and serial sampling at 0.5−2 h and 9−12 h after last apixaban dose. For outpatients, blood samples were also drawn in 3.2% citrated tubes at a single point during each hospital visit (0.3–16 h after last apixaban dose). The demographic and clinical laboratory data such as sex, age, body weight, serum creatinine (Scr), creatinine clearance (Ccr), aspartate amino transferase (AST), alanine amino transferase (ALT), and concomitant CYP3A4 and/or P‐glycoprotein inhibitors or inducers were retrospectively collected from electronic medical records. Ccr was calculated using the Cockcroft–Gault equation 26. Physicians and pharmacists asked patients about drug compliance to confirm drug exposure at each hospitalization or hospital visit. According to their assessment, we judged drug compliance of the patients. Patients who had no record of these measurements or who exhibited poor drug compliance were excluded from the analyses.
Apixaban assay
Plasma samples from patients were separated by centrifugation at 2500 g for 15 min at 4°C and stored at −80°C until analysis. Plasma concentrations of apixaban were analysed using liquid chromatography with electrospray ionization tandem mass spectrometry (LC/MS/MS), according to our previous study 24. The calibration curve of apixaban was linear from 2.5 to 500 ng ml−1, and the lower limit of quantification was 2.5 ng ml−1. All plasma samples were handled in this study because their concentrations of apixaban were higher than the lower limit of quantification. When plasma concentration of apixaban was more than 500 ng ml−1, the sample was diluted with blank plasma and reanalysed. The coefficient of deviation of the intra‐ and inter‐day precision were from 1.1 to 8.0% and from 2.0 to 8.2%, respectively. The accuracy value was from 96.3 to 103.7%.
Genotyping
After extracting genomic DNA from blood samples using a DNA Extract All Reagents Kit (Applied Biosystems, Waltham, MA, USA), the ABCB1 1236C>T (rs1128503), 2677G>T/A (rs2032582), 3435C>T (rs1045642), ABCG2 421C>A (rs2231142), and CYP3A5 6986A>G (rs776746; *3) polymorphisms were analysed using a real‐time PCR system (StepOnePlus™; Applied Biosystems), as described previously 24.
Population pharmacokinetic analysis
A population pharmacokinetic analysis was conducted using a nonlinear mixed‐effects modelling (NONMEM) program version 7.3.0 (Icon Development Solutions, Ellicott City, MD), using the first‐order conditional estimation method with interaction. The plasma concentration–time profiles of apixaban after oral administration were analysed using a 1‐compartment model with first‐order absorption (ADVAN2 TRANS2). The pharmacokinetic parameters of apixaban were estimated as apparent oral clearance (CL/F) and apparent volume of distribution (Vd/F), while the absorption rate constant (ka) was fixed at the reported value of 0.42 h−1 13 because of a lack of data on the absorption phase. The relationship between the observed and predicted apixaban concentrations was described by following an exponential error model, taking into account intra‐individual variability (ε):
| (1) |
where Cij and designate the observed and predicted apixaban concentrations in the jth record of ith patient, respectively. The parameters CL/F, Vd/F and Ka are then described using the population mean parameters (θ), as in following equations:
| (2) |
| (3) |
| (4) |
where η 1 and η 2 designate the intra‐individual variabilities for CL/F and Vd/F, respectively. Hereafter, the combination of these equations was selected as a basic model for the subsequent covariant analysis.
To develop the population pharmacokinetic model of apixaban, the influence of Ccr was first assessed in the covariate model, because renal function was known to be related to the CL/F of apixaban 12, 13, 25, 27, 28. The CL/F of apixaban was described as the sum of apparent renal (CLR/F) and apparent nonrenal (CLNR/F) clearances as follows 13, 25:
| (5) |
To assess the influence of Ccr on CLR/F, a covariate analysis was conducted using the two following equations:
| (6) |
| (7) |
where CCRi and CCRmedian denote the Ccr of the ith patients and the median Ccr, and θ 4 is the population mean estimate. The covariate models incorporating the Ccr value were selected based on the lowest objective function value (OBJ) that was calculated using the NONMEM program.
After conducting the covariate analysis for Ccr, the effects of continuous covariates such as AST, ALT, body weight, and age of patients on the parameters CLR/F, CLNR/F, and Vd/F were evaluated using the following equations:
| (8) |
| (9) |
where Pi is a pharmacokinetic parameter of the ith patients, COVi and COVmedian denote the covariate of the ith patients and median of the covariate, and θ 5 and θ 6 are population mean estimates. The effects of categorical covariates such as concomitant CYP3A4 and/or P‐glycoprotein inhibitors, co‐morbidities and genetic polymorphisms in ABCB1, ABCG2 and CYP3A5 were also evaluated using the following equation:
| (10) |
where θ 7 is the population mean estimate, and the dichotomous parameter A is equal to 1 if the categorical covariate is present, and 0 if it was not present. For example, in the case of genetic polymorphisms, the parameter A is equal to 1 if a patient had a specific genotype, otherwise it was set to 0. Covariates were added to the basic model or the model involving Ccr using a forward stepwise inclusion method and considered statistically significant if the OBJ decreased more than 3.84 (P < 0.05 with 1 degree of freedom). Subsequently, covariates were removed from the full model using a backward stepwise deletion method and considered statistically significant if the OBJ increased more than 6.63 (P < 0.01 with 1 degree of freedom).
Model evaluation
The following goodness‐of‐fit plots were used to investigate the models: the relationship between observed (OBS) and population predicted value (PRED) or individual predicted value (IPRED), and the relationship between conditional weighted residuals (CWRES) and time after the last dose or PRED. The final model was also assessed using a visual predictive check (VPC) and nonparametric bootstrap analysis. In VPC analysis, a total of 1000 hypothetical data sets were simulated by random sampling using the NONMEM program. The 50th percentile (median) and 90% prediction interval of the simulated concentrations were plotted using the observed concentrations. In the bootstrap analysis, a total of 1000 replication data sets were generated by random sampling using the Perl‐speaks‐NONMEM version 4.7.0 program 29. The estimates of each population pharmacokinetic parameter obtained using the final model were compared with the medians and 95% prediction intervals using the bootstrap analysis.
Model‐based simulation
The AUC from 0 to 12 h at steady state (AUC0–12) as well as the CL/F of apixaban based on the final model were calculated using a Monte Carlo simulation to determine the impact of Ccr and genetic polymorphisms on apixaban exposure. Two hundred pharmacokinetic profiles were simulated for a patient with various Ccr values (30, 60, 90, and 120 ml min−1) and genetic polymorphisms, using the NONMEM program. The oral dosing schedules were set to 5 mg twice daily. The AUC0–12 of apixaban were calculated as its dose divided by the CL/F.
Statistical analysis
Data are expressed as the median value unless otherwise indicated. For multiple comparisons against a control group, significant differences were evaluated using the Kruskal–Wallis test, followed by Dunn's post hoc test using Prism6 software (GraphPad Software, San Diego, CA, USA). Allele frequencies of polymorphisms were evaluated according to the Hardy–Weinberg equilibrium using the χ2 test. A probability value of less than 0.05 was considered statistically significant.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org,the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 30, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 31, 32.
Results
Patient characteristics and genotype frequencies
No patients were treated with combination of apixaban and the CYP3A4 and/or P‐gp inhibitors/inducers except amiodarone, verapamil and rifampicin. Because only one patient was treated with rifampicin, this patient was excluded from this analysis. As a result, a total of 276 observations from 81 patients were included in analyses and characteristics of patients are summarized in Table 1. Blood samples were drawn during hospitalization for 23 inpatients, while they were drawn during hospital visit for 32 outpatients. For 26 patients, blood samples were drawn during both hospitalization and hospital visit. Blood samples were drawn at three time points for 48 of 49 inpatients. For only one patient, blood samples were drawn at two time points (trough sampling and sampling at approximately 10 h after last apixaban dose). Twenty‐three patients were treated with combinations of apixaban and the CYP3A4 and/or P‐glycoprotein inhibitors amiodarone (17 patients) and verapamil (6 patients). Frequencies of the CYP3A5, ABCB1 and ABCG2 genotypes are shown in Table 2. All allele frequencies in this study were comparable to the Hardy–Weinberg equilibrium.
Table 1.
Clinical characteristics and demographics of patients with atrial fibrillationa
| Number of patients | 81 |
| Number of measurements | 276 |
| Sex (male/female) | 61/20 |
| Age (years) | 68.1 (40.5–84.9) |
| Body weight (kg) | 65.0 (41.0–92.2) |
| Dosage of apixaban (mg day −1 ) | 10 (5–20) |
| Plasma concentration of apixaban (ng ml −1 ) | 157.9 (15.6–673.6) |
| Serum creatinine (mg dl −1 ) | 0.88 (0.41–1.34) |
| Creatinine clearance (ml min −1 ) | 69.8 (30.6–145.5) |
| Aspartate amino transferase (IU l −1 ) | 23 (13–97) |
| Alanine amino transferase (IU l −1 ) | 19 (5–115) |
| Number of patients treated with CYP3A4 and/or P‐glycoprotein inhibitors | |
| Amiodarone | 17 |
| Verapamil | 6 |
| Number of patients with co‐morbidities | |
| Hypertension | 36 |
| Diabetes mellitus | 34 |
| Heart failure | 20 |
| Dyslipidemia | 18 |
Data are presented as the number or median with the range in parentheses.
Table 2.
Allele frequencies of ABCB1, ABCG2 and CYP3A5 polymorphisms in Japanese patients
| Genotype | Number of patients | Frequency | Allele frequency a |
|---|---|---|---|
| ABCB1 1236C>T | 0.451 | ||
| C/C | 24 | 0.296 | |
| C/T | 41 | 0.506 | |
| T/T | 16 | 0.198 | |
| ABCB1 2677G>T/A | 0.556 | ||
| G/G | 20 | 0.247 | |
| G/T | 11 | 0.136 | |
| G/A | 21 | 0.259 | |
| A/A | 16 | 0.198 | |
| T/A | 11 | 0.136 | |
| T/T | 2 | 0.024 | |
| ABCB1 3435C>T | 0.407 | ||
| C/C | 31 | 0.383 | |
| C/T | 34 | 0.420 | |
| T/T | 16 | 0.197 | |
| ABCG2 421C>A | 0.315 | ||
| C/C | 39 | 0.482 | |
| C/A | 33 | 0.407 | |
| A/A | 9 | 0.111 | |
| CYP3A5 *3 | 0.765 | ||
| *1/*1 | 4 | 0.049 | |
| *1/*3 | 30 | 0.370 | |
| *3/*3 | 47 | 0.581 |
All allele frequencies were comparable to the Hardy–Weinberg equilibrium.
Population pharmacokinetic model
To develop the population pharmacokinetic model of apixaban, the effect of Ccr on apixaban pharmacokinetics was first examined. The OBJ value with the basic model was calculated to be 2671.80. The OBJ values with covariate models including Ccr with Eqs. (6) and (7) were calculated to be 2625.89 and 2627.80, respectively, suggesting that these OBJ values were significantly smaller than those obtained using the basic model. Therefore, the covariate model including Ccr with Eq. (6) was considered to be the most suitable to characterize the relationship between CLR/F and Ccr.
The results of the covariate analyses are summarized in Table 3. A forward inclusion method revealed that Ccr significantly affected the CLR/F of apixaban, and that CYP3A5*3 and ABCG2 421A/A genotypes had a significant impact on the CLNR/F of apixaban. A backward elimination method did not exclude these covariates. Other covariate models including AST, ALT, body weight, age of patients, three ABCB1 genotypes, co‐morbidities (hypertension, diabetes mellitus, heart failure, dyslipidemia), and concomitant CYP3A4 and/or P‐glycoprotein inhibitors (amiodarone and verapamil) did not affect the CLR/F or CLNR/F of apixaban. Additionally, no covariate models affected the Vd/F of apixaban. The population pharmacokinetic parameter estimates of apixaban in Japanese AF patients are shown in Table 4. The final population pharmacokinetic model for CL/F was as follows:
| (11) |
where the dichotomous parameter CYP3A5 is equal to 1 if patients had the CYP3A5*1/*3 or *3/*3 genotype, otherwise it was set to 0, and the dichotomous parameter ABCG2 is equal to 1 if patients had the ABCG2 421A/A genotype, otherwise it was set to 0. A nonlinear relationship between CL/F of apixaban and Ccr was observed. The population mean of CL/F for a typical patient (Ccr value of 70 ml min−1) with the CYP3A5*1/*1 and ABCG2 421C/C or C/A genotypes was estimated to be 3.06 l h−1. When Ccr values were set to the typical value, the population mean of CL/F was 1.52 times higher in patients with the CYP3A5*1/*1 genotype compared with patients with the CYP3A5*1/*3 or *3/*3 genotype, while the population mean of CL/F was 1.49 times higher in patients with the ABCG2 421C/C or C/A genotype compared with patients with the ABCG2 421A/A genotype. The CL/F value for patients with both CYP3A5*1/*3 or *3/*3 and ABCG2 421A/A genotypes was decreased further, compared with patients with both the CYP3A5*1/*1 and the ABCG2 421C/C or C/A genotype (Figure 1). Inter‐individual variabilities for CL/F and Vd/F were 26.6% and 56.6%, respectively. The covariance between inter‐individual variability for CL/F and that for Vd/F was 11.6%, and the correlation coefficient between individual CL/F and Vd/F was 0.770. The intra‐individual variability was 34.0% (Table 4).
Table 3.
Summary of the tested covariate effects on OBJs
| Tested covariates | ΔOBJ (1st selection) a | ΔOBJ (2nd selection) | ΔOBJ (3rd selection) |
|---|---|---|---|
| Effect on CL R /F | |||
| ABCB1 1236 T/T | −2.72 | ||
| ABCG2 421A/A | −3.93 | −4.24 | 1.17 |
| CYP3A5*3 | −3.73 | ||
| Amiodarone | −b | ||
| Effect on CL NR /F | |||
| ABCB1 1236 T/T | −4.82 | −3.31 | |
| ABCG2 421A/A | −8.99 | −6.80 | Included |
| CYP3A5*3 | −9.12 | Included | Included |
| Amiodarone | −7.09 | −b | |
| Effect on Vd/F | |||
| BW with Eq. (8) | 0.01 | ||
| BW with Eq. (9) | 1.96 |
The difference from the OBJ value was calculated using the covariate model including Ccr with Eq. (6) (OBJ value, 2625.89), and it is expressed to two decimal places.
Calculation of the OBJ value was not conducted.
Table 4.
Population pharmacokinetic parameter estimates for apixaban in AF patientsa
| Parameters | Original data | 1000 Bootstrap sample data | |||
|---|---|---|---|---|---|
| Mean | 95% CI | Median | 2.5th–97.5th percentiles | ||
|
|
|||||
| θ1 (l h–1) | 1.53 | 1.39−1.67 | 1.49 | 1.16−1.81 | |
| θ4 | 0.700 | 0.471−0.929 | 0.714 | 0.340−0.976 | |
| θ5 b | 0.312 | 0.273−0.351 | 0.342 | 0.124−0.737 | |
| θ6 c | 0.341 | 0.160−0.522 | 0.478 | 0.08−0.861 | |
| Vd/F (l) = θ2 | 24.7 | 15.8−33.6 | 25.0 | 19.1−43.7 | |
| ka (h–1) = θ3 | 0.42 fixed | − | 0.42 fixed | − | |
| Inter‐ and intra‐individual variabilities d , e | |||||
| η1 (%) | 26.6 (21.5) | 18.7−34.5 | 25.9 | 16.4−34.4 | |
| η2 (%) | 56.6 (35.0) | 8.8−104 | 57.3 | 21.3−103 | |
| ε (%) | 34.0 (12.0) | 28.0−40.0 | 33.6 | 28.3−38.7 | |
The ka was set to the literature value 13.
If patients had the CYP3A5*1/*3 or *3/*3 genotype, then the dichotomous parameter CYP3A5 was equal to 1, otherwise it was set to 0.
If patients had the ABCG2 421A/A genotype, then the dichotomous parameter ABCG2 was equal to 1, otherwise it was set to 0.
The η 1, η 2 and ε values denote the inter‐individual variabilities for CL/F, V/F and the intra‐individual variability, respectively.
Data are presented as the mean with the shrinkage (%) in parentheses.
Figure 1.
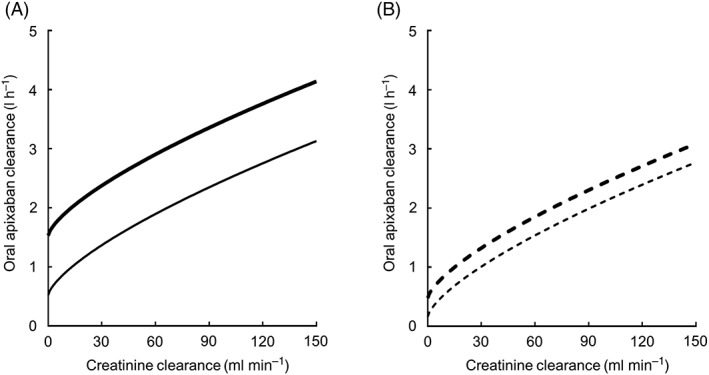
Correlation between the population mean estimates of both oral apixaban and creatinine clearance in the final model. In panel A, the thick and thin lines indicate the population mean estimates for a typical patient with the CYP3A5*1/*1 and ABCG2 421C/C or C/A genotype, and for a typical patient with the CYP3A5*1/*1 and ABCG2 421A/A genotypes, respectively. In panel B, the thick dotted and thin dotted lines indicate the population mean estimates for a typical patient with the CYP3A5*1/*3 or *3/*3 and ABCG2 421C/C or C/A genotype, and for a typical patient with the CYP3A5*1/*3 or *3/*3 and ABCG2 421A/A genotypes, respectively
Model evaluation
The goodness‐of‐fit plots for the final model are shown in Figure 2. PRED was shown to correlate reasonably with OBS, and IPRED was shown to correlate well with OBS. No systematic deviation was observed in the relationship between CWRES and time after last dose or PRED. The final model was also assessed by estimating the population pharmacokinetic parameters from 1000 bootstrap resamplings. The median values of population pharmacokinetic parameters calculated from the bootstrapping samples were generally similar to the population estimates in the final model (Table 4). The final model was further evaluated using VPC analysis. As shown in Figure 3, VPC analysis generally resulted in a reasonable predictability of the final model. Additionally, the VPC of apixaban concentrations showed that the model fit was similar for inpatients and outpatients (Figure S1).
Figure 2.
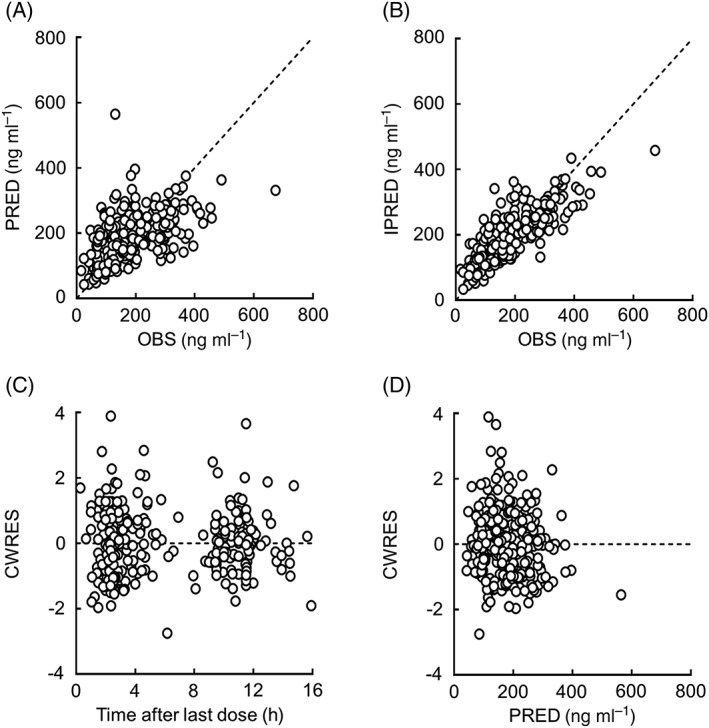
Goodness‐of‐fit plots for the final model of apixaban. The relationship between the observed concentrations (OBS) and population predictions (PRED), and individual predictions (IPRED), are shown in panels A and B, respectively. The relationship between conditional weighted residuals (CWRES) and the time after last dose, and PRED are shown in panels C and D, respectively. Open circles indicate the observed values. Each dotted line shows a line of identity
Figure 3.
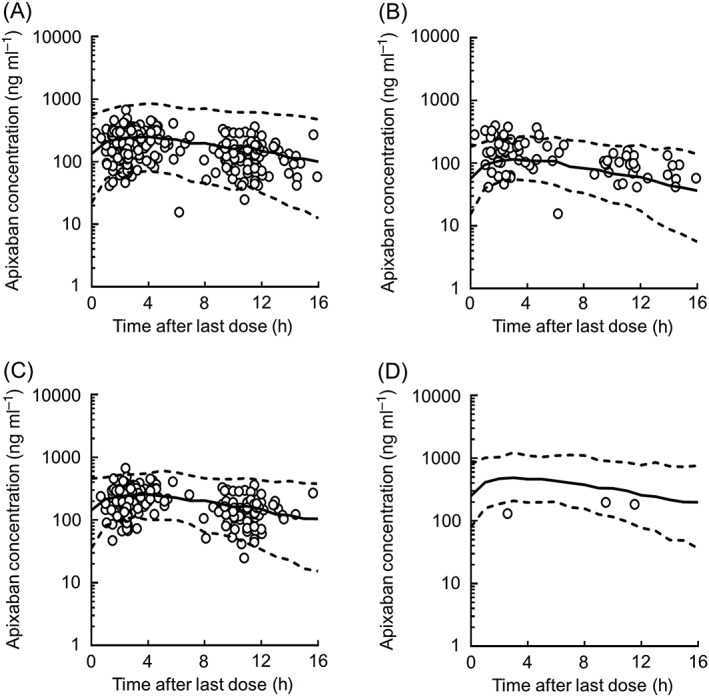
Visual predictive checks of apixaban concentrations with data obtained from the final model. Open circles show the observed concentrations in all patients (A), patients administered twice daily a dose of 5 mg day−1 (B), 10 mg day−1 (C) and 20 mg day−1 (D). The top dotted, middle solid, and bottom dotted lines are shown as the 95th, 50th, and 5th percentiles, respectively, as calculated from 1000 simulated data sets
Model‐based simulation
To simulate the AUC0–12 of apixaban at steady state using the final population pharmacokinetic parameters, data sets were divided into four groups according to the combination of the ABCG2 and CYP3A5 genotypes in hypothetical patients, as follows: patients with the CYP3A5*1/*1 and ABCG2 421C/C or C/A genotype (group A); patients with the CYP3A5*1/*1 and ABCG2 421A/A genotypes (group B); patients with the CYP3A5*1/*3 or *3/*3 and the ABCG2 421C/C or C/A genotype (group C); and patients with CYP3A5*1/*3 or *3/*3 and ABCG2 421A/A genotypes (group D). As shown in Figure 4, the median predicted AUC0–12 in group A decreased from 2104 to 1329 ng h ml−1 when the Ccr values were changed from 30 to 120 ml min−1. The median predicted AUC0–12 in group A was significantly lower compared with that in groups B, C and D independent of the Ccr values (P < 0.001).
Figure 4.
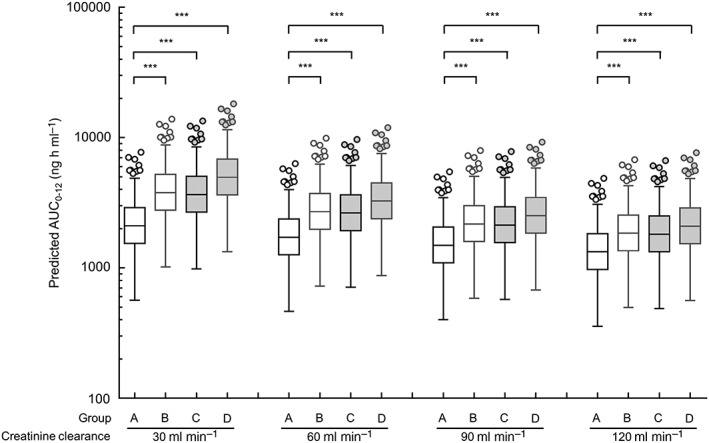
Simulations of AUC from time 0 to 12 h (AUC0−12) for apixaban in the 200 replication data sets in a patient administered a typical dose of 5 mg twice daily. These simulations are conducted using the final model. The box‐and‐whisker plots are presented according to the Tukey style. Open circles show the outliers. Four groups consist of patients with the CYP3A5*1/*1 and ABCG2 421C/C or C/A genotype (group A), patients with the CYP3A5*1/*1 and ABCG2 421A/A genotypes (group B), patients with the CYP3A5*1/*3 or *3/*3 and ABCG2 421C/C or C/A genotype (group C), and patients with the CYP3A5*1/*3 or *3/*3 and ABCG2 421A/A genotypes (group D). *** P < 0.001 by the Kruskal–Wallis test, followed by Dunn's multiple comparison test
Discussion
We recently reported that trough concentrations of apixaban were significantly higher in patients with the ABCG2 421A/A genotype than in patients with the ABCG2 421C/C or C/A genotype, and that trough concentrations of apixaban were significantly higher in patients with the CYP3A5*1/*3 or *3/*3 genotype compared with patients with the CYP3A5*1/*1 genotype. Additionally, although apixaban is known to be a substrate of ABCB1, no ABCB1 polymorphism altered its trough concentration 24. In this study, we estimated the population pharmacokinetic parameters of apixaban in Japanese AF patients and examined the impact of polymorphisms in ABCB1, ABCG2, CYP3A5, and clinical laboratory data on its pharmacokinetic parameters. It was reported that the CLR of apixaban in healthy subjects administered intravenously was approximately 34% of total clearance 33. Therefore, its CL/F was described as the sum of CLR/F and CLNR/F to examine the effect of Ccr on the CLR/F of apixaban. Our data indicated the nonlinear relationship between CL/F and Ccr. A few population pharmacokinetic studies of apixaban showed that the relationship between Ccr and CLR/F of apixaban was described using a linear 12, E max 13, or power 25 model. Thus, our model is considered to be compatible with these models. In a subsequent covariate analysis, only the Ccr was identified as a significant covariate for CLR/F, while the ABCG2 421A/A and CYP3A5*3 genotypes were identified as significant covariates for CLNR/F (Figure 1, Table 3). As shown in Figures 2 and 3 and in Table 4, bootstrap and VPC analyses indicated that the final model provided a robust and unbiased fit to the data. Our results indicated for the first time the impact of the genetic polymorphisms of ABCG2 and CYP3A5 on pharmacokinetic parameters of apixaban.
The CL/F value of apixaban in this study was smaller than values for a typical patient or healthy subject in previous population pharmacokinetic analyses (4.29–4.53 l h−1), while the Vd/F value of apixaban (24.7 l) in this study was comparable to that of previous population pharmacokinetic analyses (22.9–43.9 l) 12, 13, 25. Recently, it was reported that the CL/F value in the Asian race was lower than that in non‐Asian races 24. This racial difference in the CL/F value of apixaban may be explained by the higher allele frequency of ABCG2 421C>A in East Asians (30–60%) compared with Caucasians and African‐Americans (5–10%) 34. The CL/F value in Japanese AF patients in our study is slightly lower than that in healthy male Japanese subjects 35. Therefore, the racial and pathological differences among AF patients or subjects were considered to affect the pharmacokinetics of apixaban.
It was reported that renal clearance of apixaban in healthy Japanese subjects after intravenous administration ranged from 15.2 to 17.8 ml min−1, which corresponds to 22.2–33.2% of its 35. Apixaban exhibits 87% protein binding in plasma 36, and renal clearance corrected by the unbound apixaban concentration is estimated to range from 116.9 to 136.9 ml min−1, which is close to normal glomerular filtration rate of nearly 100 ml min−1. These results suggest that ABCG2 protein may contribute to the hepatic and/or intestinal secretion of apixaban rather than its renal excretion. Further studies are required to clarify the relative contribution of ABCG2 and CYP3A5 to hepatic, intestinal and renal clearances of apixaban.
There is little information concerning the relationship between the pharmacokinetics of non‐vitamin K anticoagulants and co‐morbidities in AF patients. It was reported that heart failure significantly affected the CL/F of dabigatran in AF patients, although patients with heart failure showed a 6.7% decreased CL/F 37. In the present study, co‐morbidities did not affect the apixaban pharmacokinetics. Thus, the influence of co‐morbidities on pharmacokinetics of non‐vitamin K anticoagulants in AF patients may be considered to be negligible.
The concomitant CYP3A4 and/or P‐glycoprotein inhibitors (amiodarone and verapamil) had no effects on the CL/F of apixaban in this study (Table 3). Drug–drug interactions between apixaban and inhibitors (ketoconazole and diltiazem) or inducers (rifampicin) of both CYP3A4 and P‐glycoprotein were observed in healthy subjects 33, 38, and a previous population pharmacokinetic analysis indicated that strong/moderate CYP3A4 and P‐glycoprotein inhibitors decreased the CL/F of apixaban by 20.3% 25. In the present study, all patients treated with apixaban and amiodarone had the CYP3A5*1/*3 or *3/*3 genotype, and five of six patients treated with apixaban and verapamil also had these genotypes. Thus, we expected that it would be difficult to separately evaluate the effect of amiodarone, verapamil and the CYP3A5*3 genotype on the pharmacokinetic parameters of apixaban.
There is no information concerning the exposure–response study for AF patients. Recent exposure–response studies for patients undergoing orthopaedic surgery have shown that an increase in the daily AUC at steady state of 1000 ng h ml−1 would increase the odds ratio for bleeding by 0.118 and decrease the odds ratio for venous thromboembolism (VTE) by 0.0499 12. The present simulation study showed that the median of the predicted steady‐state apixaban AUC0–12 had large variability using the final model (Figure 4). For example, the predicted AUC0–12 of apixaban at steady state was calculated to range from 463 to 3254 ng h ml−1 when a patient with Ccr of 60 ml min−1 took apixaban at a typical dose of 5 mg twice daily. The predicted AUC0–12 of apixaban in a patient with a Ccr of 30, 90 or 120 ml min−1 was similarly variable compared with a patient with a Ccr of 60 ml min−1. Therefore, the ABCG2 and CYP3A5 genotypes as well as apixaban concentrations can predict the rate of bleeding and VTE events because apixaban exhibits considerable inter‐individual variabilities for these events. Further exposure–response studies in AF patients are needed to clarify the effect of the ABCG2 and CYP3A5 genotypes as well as apixaban concentrations on the rate of bleeding and VTE events.
A limitation of this study is the small number of patients with CYP3A5*1/*1 or ABCG2 421A/A genotype. It was reported that the false positive rate (type 1 error inflation) of covariate effects tended to be high when the frequency of covariate was less than 20% 39. In the present study, the frequency of the ABCG2 421A/A and CYP3A5*1/*1 genotypes were only 11.1% and 4.9%, respectively (Table 1). The covariate effects of these genotypes on apixaban pharmacokinetics may be poorly estimated, although our population pharmacokinetic and pharmacogenomics model was validated (Table 4). The population pharmacokinetics and pharmacogenomics of apixaban in a larger number of Japanese population will need to be examined in the future.
In conclusion, our results suggest that the ABCG2 421A/A and CYP3A5*3 genotypes and renal function are significant predictors of apixaban pharmacokinetics. These findings may provide useful information for individualized apixaban pharmacotherapy to prevent the risk of adverse reactions.
Competing Interests
There are no competing interests to declare.
This study was supported in part by JSPS KAKENHI Grant Number 15K18938, the Japan Research Foundation for Clinical Pharmacology, and the Uehara Memorial Foundation. We thank the Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Contributors
S.U., D.H., M.H., T.T. and T.K. were involved in the conception and design of the study. S.U., D.H., Y.K., R.F., C.T., T.Y., M.H., T.T. and T.K. were involved in the analysis and/or interpretation of data. D.H., Y.T., T.O., H.I. and S.O. were involved in the acquisition of data. S.U. drafted the manuscript. D.H., M.H., T.T. and T.K. revised the manuscript critically for important intellectual content. All authors gave approval of the final version of the manuscript to be published.
Supporting information
Figure S1 Visual predictive checks of apixaban concentrations with data obtained from the final model. Open circles show the observed concentrations in inpatients (A) and outpatients (B). The top dotted, middle solid, and bottom dotted lines are shown as the 95th, 50th, and 5th percentiles, respectively, as calculated from 1000 simulated data sets
Ueshima, S. , Hira, D. , Kimura, Y. , Fujii, R. , Tomitsuka, C. , Yamane, T. , Tabuchi, Y. , Ozawa, T. , Itoh, H. , Ohno, S. , Horie, M. , Terada, T. , and Katsura, T. (2018) Population pharmacokinetics and pharmacogenomics of apixaban in Japanese adult patients with atrial fibrillation. Br J Clin Pharmacol, 84: 1301–1312. doi: 10.1111/bcp.13561.
References
- 1. Pinto DJ, Orwat MJ, Koch S, Rossi KA, Alexander RS, Smallwood A, et al Discovery of 1‐(4‐methoxyphenyl)‐7‐oxo‐6‐(4‐(2‐oxopiperidin‐1‐yl)phenyl)‐4,5,6,7‐tetrahydro‐1H‐pyrazolo[3,4‐c]pyridine‐3‐carboxamide (apixaban, BMS‐562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem 2007; 50: 5339–5356. [DOI] [PubMed] [Google Scholar]
- 2. Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S, et al Apixaban in patients with atrial fibrillation. N Engl J Med 2011; 364: 806–817. [DOI] [PubMed] [Google Scholar]
- 3. Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek EM, Hanna M, et al Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365: 981–992. [DOI] [PubMed] [Google Scholar]
- 4. Easton JD, Lopes RD, Bahit MC, Wojdyla DM, Granger CB, Wallentin L, et al Apixaban compared with warfarin in patients with atrial fibrillation and previous stroke or transient ischaemic attack: a subgroup analysis of the ARISTOTLE trial. Lancet Neurol 2012; 11: 503–511. [DOI] [PubMed] [Google Scholar]
- 5. Hu PT, Lopes RD, Stevens SR, Wallentin L, Thomas L, Alexander JH, et al Efficacy and safety of apixaban compared with warfarin in patients with atrial fibrillation and peripheral artery disease: insights from the ARISTOTLE Trial. J Am Heart Assoc 2017; 6: e004699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weitz JI, Semchuk W, Turpie AG, Fisher WD, Kong C, Ciaccia A, et al Trends in prescribing oral anticoagulants in Canada, 2008–2014. Clin Ther 2015; 37: 2506–2514. [DOI] [PubMed] [Google Scholar]
- 7. Hogg K, Bahl B, Latrous M, Scaffidi Argentina S, Thompson J, Chatha AA, et al Time trends in intracranial bleeding associated with direct oral anticoagulants: a 5‐year cohort study. CMAJ Open 2015; 3: E432–E437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Toyoda K, Arihiro S, Todo K, Yamagami H, Kimura K, Furui E, et al Trends in oral anticoagulant choice for acute stroke patients with nonvalvular atrial fibrillation in Japan: the SAMURAI‐NVAF study. Int J Stroke 2015; 10: 836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hylek EM, Skates SJ, Sheehan MA, Singer DE. An analysis of the lowest effective intensity of prophylactic anticoagulation for patients with nonrheumatic atrial fibrillation. N Engl J Med 1996; 335: 540–546. [DOI] [PubMed] [Google Scholar]
- 10. Yasaka M, Minematsu K, Yamaguchi T. Optimal intensity of international normalized ratio in warfarin therapy for secondary prevention of stroke in patients with non‐valvular atrial fibrillation. Intern Med 2001; 40: 1183–1188. [DOI] [PubMed] [Google Scholar]
- 11. Gong IY, Kim RB. Importance of pharmacokinetic profile and variability as determinants of dose and response to dabigatran, rivaroxaban, and apixaban. Can J Cardiol 2013; 29: S24–S33. [DOI] [PubMed] [Google Scholar]
- 12. Leil TA, Feng Y, Zhang L, Paccaly A, Mohan P, Pfister M. Quantification of apixaban's therapeutic utility in prevention of venous thromboembolism: selection of phase III trial dose. Clin Pharmacol Ther 2010; 88: 375–382. [DOI] [PubMed] [Google Scholar]
- 13. Leil TA, Frost C, Wang X, Pfister M, LaCreta F. Model‐based exposure–response analysis of apixaban to quantify bleeding risk in special populations of subjects undergoing orthopedic surgery. CPT Pharmacometrics Syst Pharmacol 2014; 3: e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berger J, Balfour DC 3rd, Biskupiak JE, Dunn J, Liotta DB, Merli GJ, et al Research to real‐world application: experts weigh in on the underuse of anticoagulants. Am J Manag Care 2014; 20: S312–S318. [PubMed] [Google Scholar]
- 15. Zhang D, He K, Raghavan N, Wang L, Mitroka J, Maxwell BD, et al Comparative metabolism of 14C‐labeled apixaban in mice, rats, rabbits, dogs, and humans. Drug Metab Dispos 2009; 37: 1738–1748. [DOI] [PubMed] [Google Scholar]
- 16. Wang L, Zhang D, Raghavan N, Yao M, Ma L, Frost CE, et al In vitro assessment of metabolic drug–drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos 2010; 38: 448–458. [DOI] [PubMed] [Google Scholar]
- 17. Zhang D, He K, Herbst JJ, Kolb J, Shou W, Wang L, et al Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab Dispos 2013; 41: 827–835. [DOI] [PubMed] [Google Scholar]
- 18. Takahashi H, Wilkinson GR, Nutescu EA, Morita T, Ritchie MD, Scordo MG, et al Different contributions of polymorphisms in VKORC1 and CYP2C9 to intra‐ and inter‐population differences in maintenance dose of warfarin in Japanese, Caucasians and African‐Americans. Pharmacogenet Genomics 2006; 16: 101–110. [DOI] [PubMed] [Google Scholar]
- 19. Pirmohamed M, Burnside G, Eriksson N, Jorgensen AL, Toh CH, Nicholson T, et al A randomized trial of genotype‐guided dosing of warfarin. N Engl J Med 2013; 369: 2294–2303. [DOI] [PubMed] [Google Scholar]
- 20. Goulding R, Dawes D, Price M, Wilkie S, Dawes M. Genotype‐guided drug prescribing: a systematic review and meta‐analysis of randomized control trials. Br J Clin Pharmacol 2015; 80: 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paré G, Eriksson N, Lehr T, Connolly S, Eikelboom J, Ezekowitz MD, et al Genetic determinants of dabigatran plasma levels and their relation to bleeding. Circulation 2013; 127: 1404–1412. [DOI] [PubMed] [Google Scholar]
- 22. Vandell AG, Lee J, Shi M, Rubets I, Brown KS, Walker JR. An integrated pharmacokinetic/pharmacogenomic analysis of ABCB1 and SLCO1B1 polymorphisms on edoxaban exposure. Pharmacogenomics J 2016; 18: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gouin‐Thibault I, Delavenne X, Blanchard A, Siguret V, Salem JE, Narjoz C, et al Interindividual variability in dabigatran and rivaroxaban exposure: contribution of ABCB1 genetic polymorphisms and interaction with clarithromycin. J Thromb Haemost 2017; 15: 273–283. [DOI] [PubMed] [Google Scholar]
- 24. Ueshima S, Hira D, Fujii R, Kimura Y, Tomitsuka C, Yamane T, et al Impact of ABCB1, ABCG2, and CYP3A5 polymorphisms on plasma trough concentrations of apixaban in Japanese patients with atrial fibrillation. Pharmacogenet Genomics 2017; 27: 329–336. [DOI] [PubMed] [Google Scholar]
- 25. Byon W, Sweeney K, Frost C, Boyd RA. Population pharmacokinetics, pharmacodynamics, and exploratory exposure–response analyses of apixaban in subjects treated for venous thromboembolism. CPT Pharmacometrics Syst Pharmacol 2017; 6: 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976; 16: 31–41. [DOI] [PubMed] [Google Scholar]
- 27. Wang X, Tirucherai G, Marbury TC, Wang J, Chang M, Zhang D, et al Pharmacokinetics, pharmacodynamics, and safety of apixaban in subjects with end‐stage renal disease on hemodialysis. J Clin Pharmacol 2016; 56: 628–636. [DOI] [PubMed] [Google Scholar]
- 28. Chang M, Yu Z, Shenker A, Wang J, Pursley J, Byon W, et al Effect of renal impairment on the pharmacokinetics, pharmacodynamics, and safety of apixaban. J Clin Pharmacol 2016; 56: 637–645. [DOI] [PubMed] [Google Scholar]
- 29. Lindbom L, Pihlgren P, Jonsson EN. PsN‐Toolkit – a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 2005; 79: 241–257. [DOI] [PubMed] [Google Scholar]
- 30. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acid Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, et al The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 2017; 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vakkalagadda B, Frost C, Byon W, Boyd RA, Wang J, Zhang D, et al Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa. Am J Cardiovasc Drugs 2016; 16: 119–127. [DOI] [PubMed] [Google Scholar]
- 34. Hira D, Terada T. BCRP/ABCG2 and high‐alert medications: biochemical, pharmacokinetic, pharmacogenetic, and clinical implications. Biochem Pharmacol 2018; 147: 201–210. [DOI] [PubMed] [Google Scholar]
- 35. Yamahira N, Frost C, Fukase H, Yu Z, Wang J, Pursley J, et al Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of apixaban in healthy Japanese male subjects. Int J Clin Pharmacol Ther 2014; 52: 564–573. [DOI] [PubMed] [Google Scholar]
- 36. Eriksson BI, Quinlan DJ, Weitz JI. Comparative pharmacodynamics and pharmacokinetics of oral direct thrombin and factor Xa inhibitors in development. Clin Pharmacokinet 2009; 48: 1–22. [DOI] [PubMed] [Google Scholar]
- 37. Liesenfeld KH, Lehr T, Dansirikul C, Reilly PA, Connolly SJ, Ezekowitz MD, et al Population pharmacokinetic analysis of the oral thrombin inhibitor dabigatran etexilate in patients with non‐valvular atrial fibrillation from the RE‐LY trial. J Thromb Haemost 2011; 9: 2168–2175. [DOI] [PubMed] [Google Scholar]
- 38. Frost CE, Byon W, Song Y, Wang J, Schuster AE, Boyd RA, et al Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Br J Clin Pharmacol 2015; 79: 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lagishetty CV, Duffull SB. Evaluation of approaches to deal with low‐frequency nuisance covariates in population pharmacokinetic analyses. AAPS J 2015; 17: 1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Visual predictive checks of apixaban concentrations with data obtained from the final model. Open circles show the observed concentrations in inpatients (A) and outpatients (B). The top dotted, middle solid, and bottom dotted lines are shown as the 95th, 50th, and 5th percentiles, respectively, as calculated from 1000 simulated data sets