

Abstract
Background and Purpose
Acid‐sensing ion channels (ASICs) are primary acid sensors in mammals, with the ASIC1b and ASIC3 subtypes being involved in peripheral nociception. The antiprotozoal drug diminazene is a moderately potent ASIC inhibitor, but its analgesic activity has not been assessed.
Experimental Approach
We determined the ASIC subtype selectivity of diminazene and the mechanism by which it inhibits ASICs using voltage‐clamp electrophysiology of Xenopus oocytes expressing ASICs 1–3. Its peripheral analgesic activity was then assessed relative to APETx2, an ASIC3 inhibitor, and morphine, in a Freund's complete adjuvant (FCA)‐induced rat model of inflammatory pain.
Key Results
Diminazene inhibited homomeric rat ASICs with IC50 values of ~200–800 nM, via an open channel and subtype‐dependent mechanism. In rats with FCA‐induced inflammatory pain in one hindpaw, diminazene and APETx2 evoked more potent peripheral antihyperalgesia than morphine, but the effect was partial for APETx2. APETx2 potentiated rat ASIC1b at concentrations 30‐fold to 100‐fold higher than the concentration inhibiting ASIC3, which may have implications for its use in in vivo experiments.
Conclusions and Implications
Diminazene and APETx2 are moderately potent ASIC inhibitors, both inducing peripheral antihyperalgesia in a rat model of chronic inflammatory pain. APETx2 has a more complex ASIC pharmacology, which must be considered when it is used as a supposedly selective ASIC3 inhibitor in vivo. Our use of outbred rats revealed responders and non‐responders when ASIC inhibition was used to alleviate inflammatory pain, which is aligned with the concept of number‐needed‐to‐treat in human clinical studies.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Abbreviations
- ASIC
acid‐sensing ion channel
- FCA
Freund's complete adjuvant
- i.pl.
intra‐plantar
- MES
2‐(N‐morpholino)ethanesulfonic acid
- PPT
paw pressure threshold
- RP
reverse phase
- RSA
rat serum albumin
- S‐D
Sprague–Dawley
Introduction
Over the last 20 years, many new drug targets have been identified in nociceptive‐signalling pathways, including a wide variety of neuronal ion channels (Bourinet et al., 2014; Waxman and Zamponi, 2014). There is a well‐established link between tissue acidosis and pain (Reeh and Steen, 1996), and low extracellular pH leads to the immediate excitation of nociceptors from many tissues (Steen et al., 1992; da Silva Serra et al., 2016). http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=118 strongly contribute to the low pH‐induced activation of sensory neurons (Krishtal and Pidoplichko, 1981; Deval et al., 2008) with several ASIC subtypes suggested as potential therapeutic targets in peripheral pain conditions (Deval et al., 2011; Diochot et al., 2012). In both rodents and humans, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=118#684, ASIC1b and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=118#686 are highly expressed in sensory neurons that innervate many tissues throughout the body including skin (Deval et al., 2008; Deval et al., 2011; da Silva Serra et al., 2016). Thus, they are appropriately placed to sense the acidosis associated with these conditions, and both their expression and function are substantially enhanced by inflammatory mediators (Voilley et al., 2001; Deval et al., 2008; Wang et al., 2013).
To date, the most convincing evidence for ASICs playing a role in nociception has come from their acute pharmacological modulation rather than genetic studies (Lin et al., 2015). Peripheral administration of both the non‐selective ASIC blockers (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2421 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=4116),and a suggested specific inhibitor of ASIC3 (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4135) evoked analgesia in rodent models of chemically induced pain, inflammatory pain and post‐operative pain (Ferreira et al., 1999; Dube et al., 2005; Karczewski et al., 2010; Deval et al., 2011; Izumi et al., 2012). pH‐dependent pain induced in humans by superficial injection or iontophoresis of acid was diminished by amiloride but not by http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=507 antagonists (Ugawa et al., 2002; Jones et al., 2004). However, these findings have recently been challenged, albeit using a substantially more intense acidosis (Schwarz et al., 2017).
Recently, the discovery of novel ASIC1 inhibitors from mamba snake venom (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=118 from Dendroaspis polylepis and Dendroaspis angusticeps) has suggested a key role for ASIC1b in peripheral pain in rodents (Diochot et al., 2012). Complementary to these studies using inhibitors, two ASIC activators, MitTx from the coral snake Micrurus tener (an ASIC1a, 1b and 3 activator), and the synthetic molecule http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=4203 (ASIC3 activator) both induce nocifensive behaviour when injected into the hindpaws of mice, effects that were ablated in ASIC1−/− or ASIC3−/− animals, respectively (Yu et al., 2010; Bohlen et al., 2011). More recently, a small conopeptide that selectively enhances ASIC3 activity was shown to markedly enhance acid‐induced muscle pain in wild‐type, but not ASIC3 knockout, mice (Reimers et al., 2017).
Given our reliance on pharmacological tools to study the role of ASICs in nociception, it is of utmost importance to understand the selectivity and mechanisms of action of the pharmacological agents used as fully as possible, in order to ensure meaningful data interpretation. Although the number of pharmacological tools available to study ASICs has increased greatly in the last 10 years, many of them are either non‐selective, not as selective as first thought, or insufficiently characterized (Cristofori‐Armstrong and Rash, 2017; Rash, 2017). Two examples of this are APETx2 and the veterinary diarylamidine drug, diminazene aceturate (hereafter, diminazene). APETx2, a 42‐residue sea anemone peptide, is the prototypical selective inhibitor of ASIC3‐containing channels (Diochot et al., 2004). However, APETx2 is pharmacologically promiscuous outside the ASIC family, with activity also at several voltage‐gated ion channels (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=579, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=572) (Blanchard et al., 2012; Peigneur et al., 2012; Jensen et al., 2014). Diminazene is an antiparasitic compound that potently inhibits ASICs with a suggested rank order of potency of 1b > 3 > 2a ≥ 1a (Chen et al., 2010). Given the expression of ASIC1b and ASIC3 in peripheral sensory neurons and their demonstrated role in pain, we hypothesized that diminazene would have peripheral analgesic activity.
The objective of this study was to use electrophysiology to determine the mechanism of inhibition and ASIC subtype selectivity of diminazene, followed by assessment of its peripheral antihyperalgesic efficacy in vivo, compared with that of APETx2, and the prototypic opioid receptor agonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627. We showed that diminazene has previously unreported, peripheral antihyperalgesic activity in a rat model of chronic inflammatory pain and that APETx2 has potent but partial antihyperalgesic activity that lost statistical significance when tested at higher doses. This latter finding may potentially be explained by previously unreported potentiating effects of APETx2 on ASIC1b. Furthermore, we observed substantial inter‐animal variability in the antihyperalgesic responses evoked in rats. The observation of responder and non‐responder animals parallels the heterogeneity of drug efficacy reported in humans and mice (Mogil, 1999; Moore et al., 2009).
Methods
Animals
All animal care and experimental procedures in this study were carried out in strict accordance with the recommendations in the Australian code of practice for the care and use of animals for scientific purposes (8th Ed., 2013). The protocol for Xenopus laevis studies was approved by the Anatomical Biosciences Animal Ethics Committee at The University of Queensland (UQ) (approval number: QBI/059/13/ARC/NHMRC). Frog recovery surgery was performed under anaesthesia (animals bathed in 1.3 mg·mL−1 of MS‐222), and all efforts were made to minimize suffering. The minimum time between surgeries on the same animal was 3 months, and on the final surgery (maximum of six), frogs were killed by decapitation under MS‐222 and ice anaesthesia. The protocol for rat pain behavioural studies was approved by the UQ Molecular Biosciences Animal Ethics Committee (approval number: CIPDD/007/14/NHMRC). At the completion of experiments, rats were sedated using 50% CO2 : O2 for ~30 s (until loss of muscle tone) then killed using 100% carbon dioxide. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Xenopus laevis oocyte electrophysiology
Two‐electrode voltage clamp was carried out using Xenopus oocytes as previously described (Cristofori‐Armstrong et al., 2015). cRNA encoding rat ASIC isoforms (1a, 1b, 2a and 3) was synthesized using an mMessage mMachine cRNA transcription kit and healthy stage V–VI oocytes injected with 4–100 ng cRNA per cell. All experiments were performed at room temperature (19–21°C) in ND96 solution (96 mM NaCl, 1.8 mM CaCl2, 2 mM KCl, 2 mM MgCl2, 5 mM HEPES, pH 7.45) containing 0.05% fatty acid free‐BSA (Sigma Aldrich) to prevent adsorption to plastic ware and tubing. Changes in extracellular pH were induced using a microperfusion system that allowed local, rapid exchange of solutions. HEPES was replaced by 2‐(N‐morpholino) ethanesulfonic acid (MES) to buffer solutions at pH <6.8. APETx2 was applied in the conditioning pH solution only, whereas diminazene was applied either in the conditioning pH only, the pH stimulus only or both as indicated in the results section. Current traces were normalized in pClamp10 prior to analysis of current rise/decay times (from 10 to 90% of maximum amplitude). Data fitting and statistical analyses were carried out using Prism 7.03 (GraphPad Software, San Diego, CA, USA). The Hill equation was fitted to normalized concentration–response curves to obtain the concentration of half‐maximal response (EC50 or IC50). Given the large size of oocytes and the physical limitations in solution exchange rates (our system uses a 25 μL chamber and 1 mL·min−1 flow rate), the data for ASIC current activation (rise times) is not representative of true activation rates as determined using excised patches (<5 to 20 ms) but is still sufficiently fast to observe channel‐dependent differences in global current kinetics (as illustrated in Figure 2).
Figure 2.
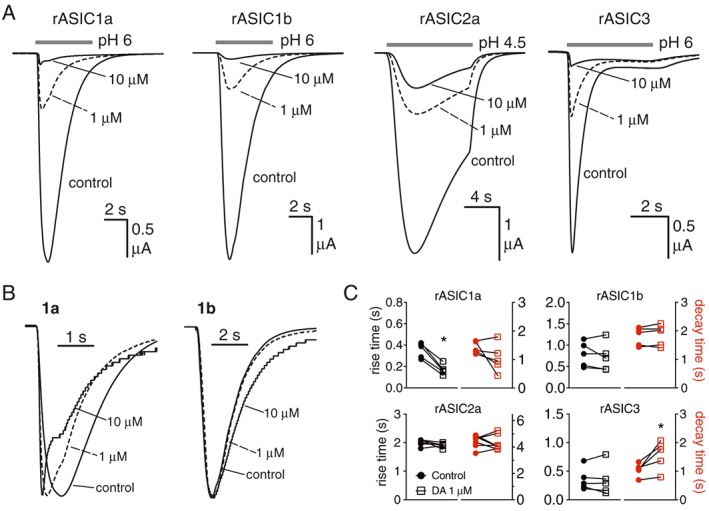
Diminazene (DA) affects the gating of ASIC1a but not other subtypes. (A) Current traces showing concentration‐dependent inhibitory effect of diminazene on rat ASIC subtypes when applied during low pH stimulation. (B) Same traces as in (A) scaled (i.e. normalized to peak current amplitude) to illustrate the significant effect of diminazene (1 and 10 μM) on the activation and desensitization kinetics of ASIC1a but not ASIC1b. Note the traces in the presence of 10 μM diminazene are stepped due to scaling up very small currents. (C) Quantitation of the effect of diminazene on the activation and desensitization of ASIC homomeric channels presented as rise and decay times (10–90%). ASIC1a, 1b and 3 n = 5; ASIC2a n = 7, * P < 0.05, significantly different from control: paired t‐test.
Inflammatory pain model
Adult male Sprague–Dawley (S‐D) rats weighing 200 to 225 g were purchased from the Animal Resources Centre (Perth, WA, Australia). Rats were housed in groups of 2–3 in individually ventilated cages in a temperature‐controlled room (23 ± 3°C) with a 12/12 h light–dark cycle. Environmental enrichment comprised placement of rodent hutches and rat chew sticks in all home cages. Standard rodent chow and water were available ad libitum. Animals were acclimatized in the animal holding facility for at least 3 days prior to initiation of experimentation, and all animals were handled daily from arrival to ensure minimal confounding stress effects.
A single intra‐plantar (i.pl.) injection of Freund's complete adjuvant [FCA, 1 mg·mL−1 of heat killed Mycobacterium tuberculosis (Sigma Aldrich, Castle Hill, NSW, Australia)] in a volume of 150 μL was given into the left hindpaw of male S‐D rats resulting in development of unilateral FCA‐induced inflammatory pain in the ipsilateral (injected) hindpaw that was confirmed by plethysmography and development of mechanical hyperalgesia as assessed using the Randall Selitto method of measuring paw pressure thresholds (PPT) (Stein et al., 1988).
FCA rats with fully developed mechanical hyperalgesia in the ipsilateral hindpaw (PPT values ≤80 g) were administered single i.pl. bolus doses of test or control drugs into this paw only in a volume of 100 μL. Specifically, rats received diminazene (1.9, 5.8, 19.4, 58.2, 194 and 582 nmol), 0.9% saline (vehicle for diminazene), APETx2 (0.03, 0.1, 1.1, 3.3 and 11 pmol), 0.05% rat serum albumin (RSA; vehicle for APETx2) or morphine [700 nmol (200 μg)]. Each rat received a maximum of three injections commencing on day ≥5 post‐i.pl and FCA treatment, with at least a 72 h wash‐out period between successive injections. PPTs were determined in the ipsilateral and contralateral hindpaws immediately prior to dosing and at various scheduled times for 3 h post‐dosing in individual animals. All paw pressure assessments were performed without knowledge of the treatments given (‘blinded’).
The areas under the change in PPT versus time curve (ΔPPT AUC) values for the ipsilateral hindpaws of individual FCA rats were determined as a measure of the extent and duration of the antihyperalgesic effect produced by single i.pl. bolus doses of diminazene and APETx2 relative to the positive control, morphine and the negative controls, 0.9% saline (vehicle) and 0.05% RSA (vehicle).
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as the mean (±SEM) PPT for each of the ipsilateral and contralateral hindpaws in each treatment group. ΔPPT values were calculated by subtracting pre‐dosing PPT values from post‐dosing PPT values for each drug and dose (thus accounting for variations in starting PPT), and any negative ΔPPT values were arbitrarily assigned a value of 0. For individual FCA rats, the extent and duration of action was determined by calculating the area under the ΔPPT versus time curve. Kruskal–Wallis ANOVA with Dunn's multiple comparison tests for respective individual comparisons was performed on the ΔPPT AUC values for groups of FCA rats administered single i.pl. bolus doses of each test compound or morphine relative to animals administered single bolus doses of vehicle (saline or 0.05% RSA). We re‐analysed the ΔPPT AUC data to determine responders and non‐responders using a threshold of two SDs (2SD) from the mean of the response to vehicle‐treated rats in each group. Responses that fell within this range were classified as non‐responders, whilst antihyperalgesic effects greater than 2SD higher than the mean response evoked by vehicle (thus outside the 95% confidence interval for the vehicle response) were classified as responders. The ΔPPT data take into account individual and time‐dependent variation in the response for each drug and at each dose, thus minimizing confounding factors. PPT is a reflexive measure, and the 2SD threshold has been applied to AUC data which is less prone to aberrant effects than peak reversal of hyperalgesia measures. For cases where n < 5 (as during the distinction of responder and non‐responder rats) because these data were collected blinded and the responder/non‐responder distinction only became apparent after un‐blinding and analysis of all data, we have not applied statistical analysis to data with n < 5, to comply with the recommendations of Curtis et al. (2015). Prism version 7.03 was used for all data and statistical analysis. The statistical significance criterion was set to P < 0.05.
Materials
Recombinant APETx2 was produced in‐house using an Escherichia coli expression system described previously (Anangi et al., 2012). The identity of this peptide was confirmed using matrix‐assisted laser desorption/ionization time‐of‐flight MS (observed monoisotopic m/z = 4558.8; theoretical monoisotopic m/z = 4558.9; model 4700 Proteomics Bioanalyser, Applied Biosystems, Foster City, CA, USA) and quantitated using reversed‐phase (RP) HPLC in comparison to a standard of synthetic APETx2 quantitated via amino acid analysis (Australian Proteome Analysis Facility, Macquarie Park, NSW, Australia). Diminazene aceturate was purchased from Sigma Aldrich (Castle Hill, NSW, Australia) and its purity confirmed using isocratic, analytical RP‐HPLC prior to use in experiments (flow rate 0.7 mL·min−1; Vydac 218TP54 column; solvent: acetonitrile : methanol : ammonium formate (pH 4.0, 20 mM) (10:10:80 v/v/v); detection at 254 nm with electrospray ionization MS for confirmation of mass). Morphine sulphate ampoules were purchased from Hospira (Melbourne, Victoria, Australia).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Diminazene is a potent, non‐selective, open‐channel inhibitor of ASICs
Electrophysiology was used to determine the mechanism of action and ASIC subtype selectivity of diminazene. Application of diminazene for ~50 s (the time between pH stimuli) in the conditioning solution only (pH 7.45) resulted in concentration‐dependent inhibition of ASIC1a, 1b and 3 with IC50 values in the low micromolar range (Figure 1A, B, D, G). Under these conditions, ASIC2a was resistant to inhibition by diminazene with only ~25% inhibition at the highest concentration tested (100 μM; Figure 1C). In contrast, all ASIC homomers were significantly more potently inhibited when diminazene was applied only during the pH stimulus (5 s for ASIC1a, 1b and 3 and 10 s for ASIC2a) (Figures 1 and 2A). Application of diminazene in both the conditioning and pH stimulus solutions did not significantly increase the potency of inhibition of any of the ASIC subtypes, compared to application in the low pH stimulus alone (Figure 1A–D, G). The fact that diminazene has greater ASIC inhibitory activity when applied during the low pH stimulus suggests that it functions via an open channel mechanism possibly by occlusion of the ion‐conducting pore. Application of diminazene only in the conditioning solution means that it is predominantly exposed to the resting (closed) state of the channel, whilst co‐application with the pH stimulus mostly exposes it to the open channel. To explore the possibility that diminazene acts via open pore block, that is, within the membrane electric field, we assessed its inhibitory activity when applied during a pH 6 stimulus at four different membrane potentials from −90 to 0 mV. Figure 1E shows that the inhibition of all rASIC homomers by diminazene (at both 100 nM and 1 μM) was strongly voltage‐dependent.
Figure 1.
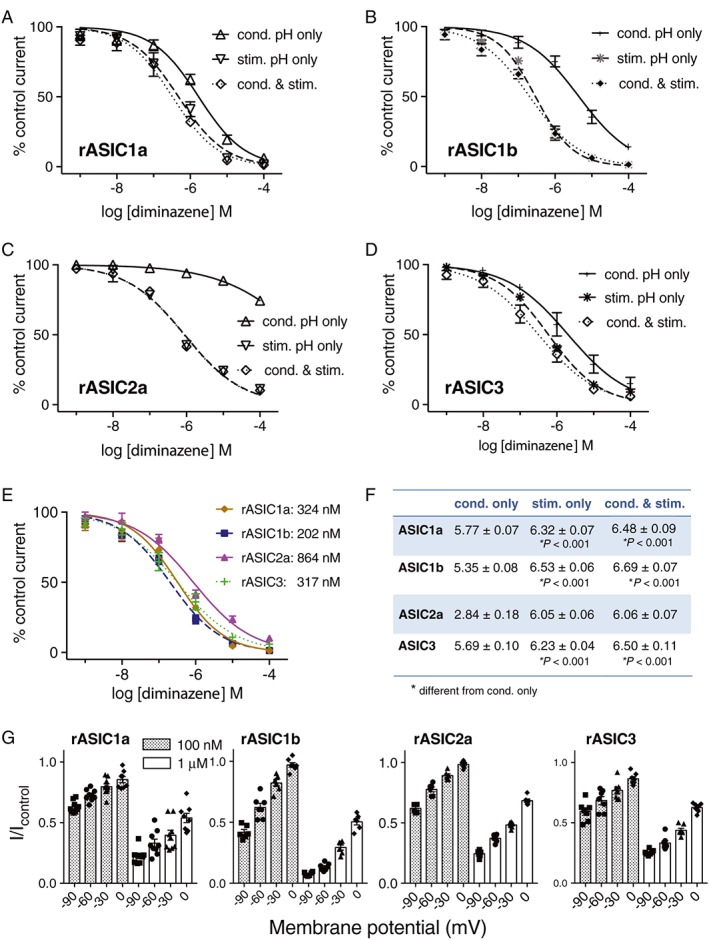
Diminazene inhibits ASICs via open channel block. (A–D) Concentration–effect curves comparing the inhibitory effect of diminazene on rat (r)ASIC channels expressed in Xenopus laevis oocytes when applied during conditioning alone (cond.), during the low pH stimulus alone (stim.) and during both conditioning and pH stimulus (cond. & stim.) (all n = 5 except ASIC3 cond. & stim. n = 8). (E) Voltage dependence of the inhibitory effect of diminazene (100 nM and 1 μM) applied during the pH 6 stimulus to rASIC1a (−90, −60 and −30 mV, n = 9; 0 mV, n = 8), rASIC1b (−90 and −30 mV, n = 6; −60 and 0 mV, n = 7), rASIC2a (n = 6) and rASIC3 (n = 7). (F) Concentration–effect curves for diminazene applied during both conditioning time and pH stimulus showing the subtype selectivity at ASIC1a (n = 5), 1b (n = 5), 2a (n = 5) and 3 (n = 8). (G) Summary of the pIC50 values for each condition and each channel subtype tested. Statistical comparison of logIC50 values was carried out using an extra sum‐of squares F test in Graphpad Prism 7 when fitting log[inhibitor] versus normalized response curves; statistical analysis of the rASIC2a IC50 was not possible due to lack of an accurate IC50 value from the incomplete concentration–effect curve.
The initial report by Chen et al. (2010) on the ASIC inhibitory activity of diminazene suggested a potency rank order of ASIC1b > 3 > 2a ≥ 1a, based on patch‐clamp analysis of ASICs heterologously expressed in CHO cells using a single concentration (3 μM) on all subtypes. Using X. laevis oocytes, full concentration–effect curves on all homomeric rat ASIC channels herein revealed that diminazene inhibits ASIC1b slightly more potently (IC50 202 nM) than ASIC1a and ASIC3 (IC50 ~320 nM) and approximately fourfold more potently than ASIC2a (IC50 864 nM) (Figure 1F and see Figure 1G for pIC50 ± SEM).
Co‐application of diminazene (1 and 10 μM) with the pH 6 stimulus appears to result in facilitation of ASIC1a gating kinetics characterized by a significant decrease in current rise time (Figure 2B, C) and an increased initial rate of current desensitization (Figure 2B) possibly resulting in faster overall desensitization, albeit not significant (Figure 2C). In contrast, diminazene had no effect on the activation or desensitization kinetics of the other ASIC subtypes studied except a minor increase in the decay time of ASIC3, which may be related to the sustained component of ASIC3 current (Figure 2C). The data shown in Figure 1 show that diminazene inhibits all of the ASIC subtypes via an open channel mechanism. However, the data in Figure 2 suggest a potential subtype‐dependent mechanism of action. For ASIC1b, ASIC2a and ASIC3, diminazene appears to behave as a simple open‐channel, pore blocker as it has no obvious effect on whole‐cell current kinetics. In contrast, diminazene may have a unique mixed mechanism of ASIC1a inhibition that involves enhancement of transition to the desensitized state, thus less time in the open state, and a reduced global current. Although this is consistent with the results of Chen et al. who observed a similar effect of diminazene on the kinetics of ASIC currents recorded from cultured hippocampal neurons (predominantly ASIC1a homomers) (Chen et al., 2010), this possibility requires further investigation using channel mutants and the rapid solution exchange afforded by macropatch electrophysiology.
Thus, diminazene is a non‐selective but moderately potent low MW inhibitor of ASICs. Given the potency of diminazene at inhibiting ASIC1b and ASIC3, both highly expressed in peripheral sensory neurons and implicated in peripheral nociception, we next assessed the potential analgesic activity of diminazene in a widely used rat model of chronic inflammatory pain.
Inter‐animal variability in the antihyperalgesic activity of diminazene and APETx2 in inflammatory pain: responders and non‐responders
Because ASICs are strongly linked to nociception in inflammatory conditions, we used a rat FCA model of unilateral inflammatory pain in the hindpaw to assess the in vivo antihyperalgesic efficacy of diminazene. As APETx2 has been used extensively to study the role of ASIC3 in inflammatory pain, we used this peptide and morphine for comparison. We allowed 5 days for chronic inflammatory pain to fully develop at which time the ipsilateral hindpaw showed mechanical hyperalgesia with baseline PPTs of ~50 g (Figures 3A and 4A). As expected, there was no hyperalgesia in the contralateral hindpaws, which had baseline PPTs of ~140 g for the diminazene group and ~130 g for the APETx2 group (Figures 3B and 4B). Thus, the degree of hyperalgesia was represented by a decrease in the ipsilateral PPTs of ~90 and ~80 g respectively.
Figure 3.
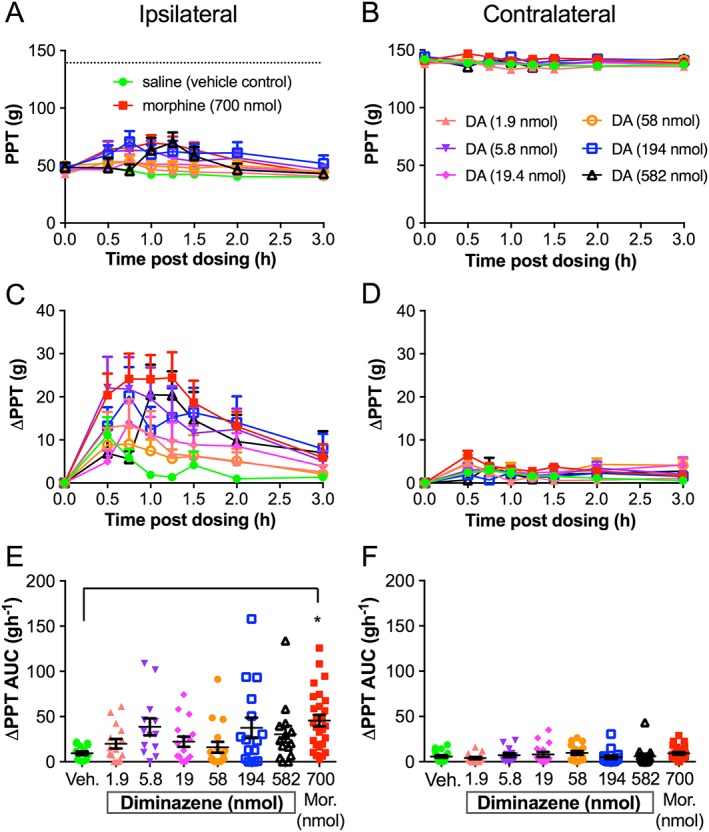
Diminazene (DA) has highly variable antihyperalgesic activity in the FCA rat model of unilateral inflammatory pain. Mean (±SEM) paw pressure thresholds (PPT values), mean (±SEM) change in paw pressure thresholds (ΔPPTs) and mean (±SEM) change in ΔPPT area under the curve values (ΔPPT AUCs) for the ipsilateral (A, C, E) and contralateral (B, D, F) hindpaws of FCA rats following administration of single 100 μL intra‐plantar bolus doses of saline (vehicle control) (n = 15), morphine (Mor) at 700 nmol (n = 27), diminazene at 1.9 nmol (n = 15), 5.8 nmol (n = 13), 19.4 nmol (n = 16), 58.2 nmol (n = 17), 194 nmol (n = 16) and 582 nmol (n = 14). PPTs were measured at time 0 (pre‐dosing) and at 30, 45, 60, 75, 90, 120 and 180 min post‐dosing. The dotted line in (A) indicates the PPT from the contralateral hindpaw. *P < 0.05, significantly different from vehicle; one‐way ANOVA.
Figure 4.
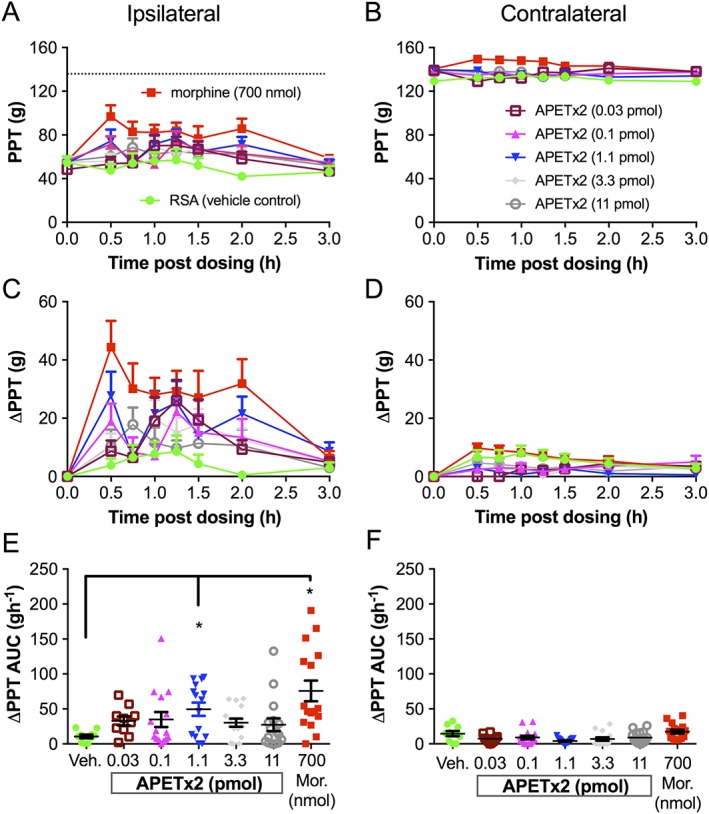
APETx2 has potent but highly variable antihyperalgesic activity in the FCA rat model of unilateral inflammatory pain. Mean (±SEM) paw pressure thresholds (PPT values), mean (±SEM) change in paw pressure thresholds (ΔPPTs) and mean (±SEM) change in PPT area under the curve (ΔPPT AUCs) for the ipsilateral (A, C, E) and contralateral (B, D, F) hindpaws of FCA rats following administration of single 100 μL intra‐plantar bolus doses of 0.05% RSA (vehicle control) (n = 10), morphine (Mor) at 700 nmol (n = 16), APETx2 at 0.03 pmol (n = 10), 0.1 pmol (n = 15), 1.1 pmol (n = 15), 3.3 pmol (n = 15) and 11 pmol (n = 16). PPTs were measured at time 0 (pre‐dosing) and at 30, 45, 60, 75, 90, 120 and 180 min post‐dosing. The dotted line in (A) indicates the PPT from the contralateral hindpaw. *P < 0.05, significantly different from vehicle; one‐way ANOVA.
The antihyperalgesic efficacy of diminazene relative to morphine and vehicle is shown in Figure 3, and that of APETx2 compared with morphine and vehicle is shown in Figure 4. The i.pl. injection of single bolus doses of diminazene and APETx2 resulted in modest antihyperalgesic effects (diminazene <30% and APETx2 ~35% reversal of hyperalgesia, Figures 3C and 4C) in the ipsilateral hindpaws, but there was considerable inter‐animal variability. Furthermore, the antihyperalgesic effects were not obviously dose‐dependent (Figures 3C, E and 4C, E). Closer inspection of the data revealed that the variability may be due to the presence of responder rats and those that are non‐responders to the drugs tested. much like the inter‐individual variability observed in human populations and between strains of mice for sensitivity to pain and responses to non‐opioid analgesics (Moore et al., 2009; Woolf, 2010). We therefore re‐assessed the ΔPPT AUC data after using a threshold of 2SD from the mean of the response to vehicle‐treated rats in each group. Responses that fell within this range were classified as non‐responders, whilst antihyperalgesic effects greater than 2SD higher than the mean response evoked by vehicle were classified as responders (Figures 5 and 6). On average, approximately 60–80% of rats were responders to morphine, whereas response rates to the two ASIC inhibitors were lower at 40 and 50% for diminazene and APETx2 respectively (see Tables 1 and 2).
Figure 5.
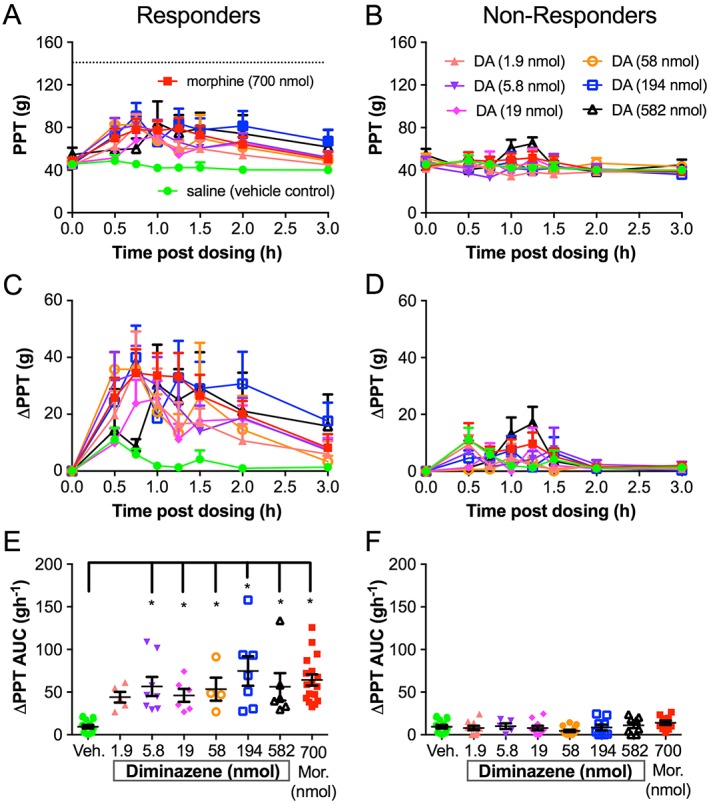
Diminazene (DA) has potent but heterogeneous antihyperalgesic activity in the FCA rat model of unilateral inflammatory pain. Rats were classified as responders/high responders (A, C, E) or non‐responders/low responders (B, D, F) based on examination of ΔPPT AUC values for the individual ipsilateral hindpaws. ΔPPT AUC values of responders given morphine (Mor) at 700 nmol (n = 17) or diminazene at 5.8 nmol (n = 8), 19.4 nmol (n = 6), 58.2 nmol (n = 4), 194 nmol (n = 7) and 582 nmol (n = 6) were significantly higher than the corresponding ΔPPT AUC values evoked by vehicle (n = 15) (*P < 0.05 one‐way ANOVA). Diminazene at 1.9 nmol (n = 5) was not significantly different from vehicle. There was no significant difference between ΔPPT AUC values of non‐responders/low responders given morphine (n = 10) or diminazene at 1.9 nmol (n = 10), 5.8 nmol (n = 5), 19.4 nmol (n = 10), 58.2 nmol (n = 13), 194 nmol (n = 9) and 582 nmol (n = 8) when compared to the ΔPPT AUC of vehicle. The dotted line in (A) indicates the PPT from the contralateral hindpaw.
Figure 6.
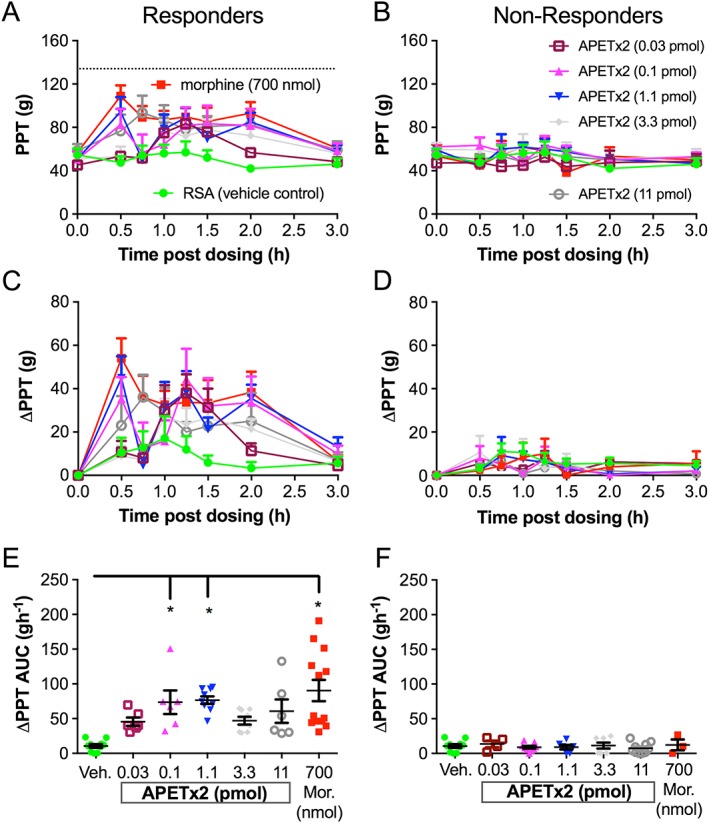
APETx2 has potent partial and heterogeneous antihyperalgesic activity in the FCA rat model of unilateral inflammatory pain. Rats were classified as responders/high responders (A, C, E) or non‐responders/low responders (B, D, F) based on examination of ΔPPT AUC values for the individual ipsilateral hindpaws. The ΔPPT AUC values of responders given morphine (Mor) at 700 nmol (n = 13) or APETx2 at 0.1 pmol (n = 6) and 1.1 pmol (n = 9) were significantly higher than the corresponding ΔPPT AUC values evoked by vehicle (n = 10). *P < 0.05, one‐way ANOVA. APETx2 at 0.03 pmol (n = 6), 3.3 pmol (n = 8) and at 11 pmol (n = 6) were not significantly different from vehicle. The dotted line in (A) indicates the PPT from the contralateral hindpaw.
Table 1.
Doses of diminazene or morphine classified as responders and non‐responders based upon the extent and duration of action (ΔPPT AUC values) for each FCA rat, compared to vehicle (saline)
| Drug | Dose (nmol) | Total number of FCA rats | Responders (R) | Non‐responders/low responders (NR/LR) | % R |
|---|---|---|---|---|---|
| Morphine | 700 | 27 | 17 | 10 | 63.0 |
| Diminazene | 1.9 | 15 | 5 | 10 | 33.3 |
| 5.8 | 13 | 8 | 5 | 61.5 | |
| 19.4 | 16 | 6 | 10 | 37.5 | |
| 58.2 | 17 | 4 | 13 | 23.5 | |
| 194 | 16 | 7 | 9 | 43.8 | |
| 582 | 14 | 6 | 8 | 42.9 | |
| Diminazene (totals) | – | 91 | 36 | 55 | 39.6 |
Table 2.
Doses of APETx2 or morphine classified as responders and non‐responders based upon the extent and duration of action (ΔPPT AUC values) for each FCA rat, compared to vehicle (0.05% RSA)
| Drug | Dose | Total number of FCA rats | Responders (R) | Non‐responders (NR) | % R |
|---|---|---|---|---|---|
| Morphine | 700 nmol | 16 | 13 | 3 | 81.3 |
| APETx2 | 0.03 pmol | 10 | 6 | 4 | 60.0 |
| 0.1 pmol | 15 | 6 | 9 | 40.0 | |
| 1.1 pmol | 15 | 9 | 6 | 60.0 | |
| 3.3 pmol | 15 | 8 | 7 | 53.3 | |
| 11 pmol | 16 | 6 | 10 | 37.5 | |
| APETx2 (totals) | – | 71 | 35 | 36 | 49.3 |
In responder rats, diminazene was potently and consistently antihyperalgesic for doses from 5.8 to 582 nmol per rat, resulting in a similar extent and duration of antihyperalgesia as morphine (700 nmol per rat) (Figure 5C, E). Neither morphine nor diminazene injected into the ipsilateral paw resulted in an analgesic effect in the contralateral paw (Figure 3B, D, F). Thus, diminazene appeared to be of similar efficacy but considerably more potent (~30‐fold) than morphine as an antihyperalgesic agent in the FCA rat model of inflammatory pain [the EC50 of i.pl. morphine in rats was ~175 nmol (50 μg) (Perrot et al., 1999; Rodrigues and Duarte, 2000)]. APETx2 also showed very potent and dose‐dependent antihyperalgesic activity in the inflamed hindpaw of responder rats; the 0.1 and 1.1 pmol i.pl. doses reached significance (compared to vehicle) and similar efficacy to morphine (700 nmol per rat) (i.e. ~50% reversal of the hyperalgesic decrease in PPT; Figure 6C). In contrast to the consistent antihyperalgesic effects observed for diminazene (once the response had plateaued), the antihyperalgesic efficacy of APETx2 was not statistically significant at 3.3 and 11 pmol (Figure 6E). Consistent with previous studies, our data show that APETx2 has potent antihyperalgesic effects in inflammatory pain in rats; however, by using a broad dose range (>2 log units), we revealed a complex antihyperalgesic profile for this peptide. In order to examine this aspect further, we carried out more experiments on the ASIC subtype selectivity of APETx2.
The ASIC3 inhibitor APETx2 also modulates the activity of ASIC1b and ASIC2a
Although widely used as an ASIC3 selective inhibitor, we and others have found that APETx2 inhibits several other ion channel families including voltage‐gated sodium channels (Blanchard et al., 2012; Peigneur et al., 2012) and hERG (Jensen et al., 2014), over a concentration range (i.e. low micromolar) that encompasses concentrations used to inhibit ASIC3 in several in vivo pain studies (Deval et al., 2008; Deval et al., 2011). Based on the complex analgesic effects of APETx2 that we observed in FCA‐induced inflammatory hyperalgesia here, and the potential involvement of ASIC1b in peripheral pain in rodents (Diochot et al., 2012), we reassessed the selectivity of APETx2 against all homomeric ASICs expressed in Xenopus oocytes.
When applied at pH 7.45 in the low micromolar range (1–10 μM), APETx2 robustly potentiated the activity of both homomeric ASIC1b and ASIC2a (Figure 7A–C) but not homomeric ASIC1a (Figure 7A, C). Analysis of the activation and desensitization kinetics of ASIC1b and ASIC2a in the absence and presence of APETx2 (10 μM) shows that it caused a significant increase in the current decay time (or desensitization) of rASIC1b with no apparent effect on the rise time (i.e. activation) of the whole‐cell current (Figure 7B, D). Conversely, the potentiating effect of APETx2 on rASIC2a seems to be accompanied by a significant decrease in the current rise time, with no effect on the decay time (Figure 7B, D). Regardless of the component of the whole‐cell ASIC1b or ASIC2a current affected by APETx2, the end result appears to be stabilization of the open state, thus allowing for increased ion flux and the observed current potentiation. The difference in the effects of APETx2 on decay time and rise time for ASIC1b and ASIC2a may be due to differences in its on‐ and off‐rate for the respective open states of each channel, with a faster on‐rate for ASIC2a, and slower off‐rate for ASIC1b. Effects of APETx2 on the pH dependence of activation and desensitization of these channels cannot be excluded and may also contribute to their potentiation by this peptide.
Figure 7.
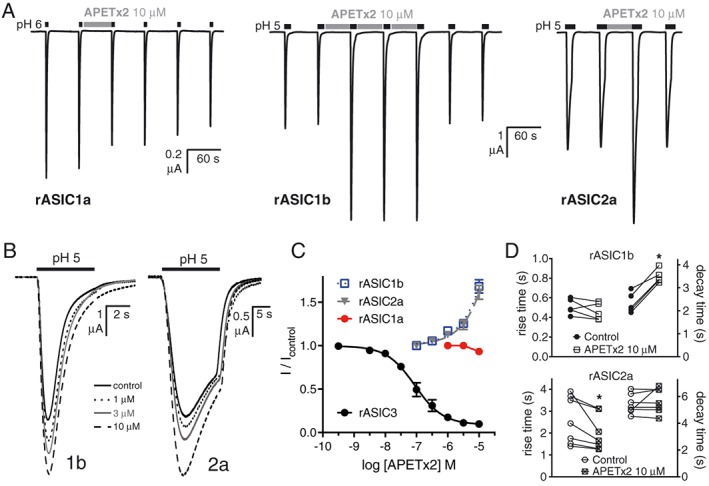
APETx2 potentiates the response of rASIC1b and rASIC2a (when applied during the conditioning pH) but not that of rASIC1a, expressed in Xenopus oocytes. (A) Traces showing the minor inhibitory effect on rASIC1a but rapid and reversible potentiating effect of APETx2 (10 μM) on rat ASIC1b and ASIC2a. (B) Traces showing the concentration‐dependent effect of APETx2 on ASIC1b and ASIC2a. (C) Concentration–response curves of APETx2 on homomeric rat ASIC1a (n = 5), ASIC1b (n = 5), ASIC2a (n = 5) and ASIC3 (n = 6). Curves were fitted using the log[inhibitor] versus normalized response equation in GraphPad 7.0. (D) Quantification of the effect of APETx2 on the activation and desensitization of rASIC1b (n = 5) and rASIC2a (n = 7) whole‐cell currents recorded from Xenopus oocytes [presented as rise and decay times (10–90%)], *P < 0.05, significantly different from control; paired t‐test.
Discussion
The present study revealed several new aspects of in vitro and in vivo ASIC pharmacology. We showed that diminazene inhibits ASICs in a non‐selective manner via an open‐channel mechanism, which has now been confirmed independently (Schmidt et al., 2017), and reduces mechanical hyperalgesia in a rat model of chronic inflammatory pain. We also showed that the antihyperalgesic activity of the ASIC3 inhibitory peptide APETx2 is very potent yet only partly efficacious at the doses tested. We report, for the first time, that APETx2 potentiates other ASIC homomers including ASIC1b, which is implicated in peripheral nociception in rodents. Together, our findings and previous studies by others suggest that potentiation of ASIC1b by APETx2 may counteract the antihyperalgesic effects produced by ASIC3 inhibition, resulting in the complex dose–response relationship we observed in vivo. Finally, by using outbred rats, we found that the effects of local ASIC inhibition resulted in incomplete relief of mechanical hyperalgesia and substantial between‐animal variability in antihyperalgesic responses.
Diminazene aceturate (trade name Berenil) has been used as a veterinary anti‐protozoal drug for decades (Riou and Benard, 1980). In 2010, diminazene was found to potently inhibit ASICs, but its selectivity and mechanism of inhibition were not fully characterized (Chen et al., 2010). We show that diminazene inhibits ASICs via open pore block, which is inconsistent with the proposal of Chen et al. (2010) that it binds to a hydrophobic groove on the outer surface of the channel, next to the acidic pocket some 30 Å above the level of the membrane, a suggestion that has so far not been experimentally validated. Confirming a pore block mechanism, the effect of diminazene is voltage dependent in a manner similar to the well‐characterized ASIC pore blocker amiloride (Dorofeeva et al., 2008) and consistent with its inhibitory effect on the related BLINaC channel (Wiemuth and Gründer, 2011). Our data (Fig. 2) also suggests a potential subtype‐dependent mechanism of action. For ASIC1b, ASIC2a and ASIC3, diminazene appears to behave as a simple open‐channel, pore blocker as it has no obvious effect on whole‐cell current kinetics. In contrast, diminazene may have a unique mixed mechanism of ASIC1a inhibition that involves enhancement of transition to the desensitized state, thus less time in the open state, and a reduced global current. Although this is consistent with the results of Chen et al. who observed a similar effect of diminazene on the kinetics of ASIC currents recorded from cultured hippocampal neurons (predominantly ASIC1a homomers) (Chen et al., 2010), this possibility requires further investigation using channel mutants and the rapid solution exchange afforded by macropatch electrophysiology.
Unfortunately, like many low MW drugs, diminazene has several other pharmacological actions including enhancement of the activity of ACE 2 (EC50 ~8 μM) (Kulemina and Ostrov, 2011) and in vitro and in vivo anti‐inflammatory effects in mice (pretreatment with 35 μM being effective in vitro) (Kuriakose et al., 2014). Nevertheless, the polypharmacology of diminazene (as currently known) appears to be substantially less complex than that of other agents used widely to study the biology of ASICs, such as amiloride and A‐317567. Furthermore, our data confirm that diminazene is more potent than other non‐selective ASIC inhibitors (Rash, 2017) and is ~25‐fold more potent at inhibiting ASICs 1 and 3 than potentiating ACE2, its next most potent activity. Thus, diminazene is a useful tool to study both biophysical and biological aspects of ASIC function. To this end, we demonstrated that diminazene has potent antihyperalgesic activity of comparable efficacy to similarly administered morphine. We hypothesize that this activity is related to its ability to inhibit ASICs in peripheral sensory afferents.
Although originally characterized as a potent and selective inhibitor of ASIC3 and ASIC3‐containing heteromeric channels, APETx2 was subsequently shown to inhibit the sensory neuron specific channel NaV1.8 and hERG in the low micromolar range (1–10 μM) (Blanchard et al., 2012; Jensen et al., 2014); Peigneur et al. (2012) suggest that APETx2 can even inhibit, at submicromolar concentrations, the NaV1.8 channels involved in inflammatory pain (Akopian et al., 1999; Payne et al., 2015). Thus, the activity of APETx2 on this channel has implications for the interpretation of inflammatory pain studies where it is used in high doses. In this study, we also showed that APETx2 is less selective within the ASIC family than previously reported, modulating both ASIC1b and ASIC2a homomers. Interestingly, as originally reported, ‘APETx2 (3 μM) blocked neither the rapid nor the slow component of the ASIC2a + 3 current (not shown)’, (Diochot et al., 2004). However, this does not exclude the possibility of minor potentiation of these heteromers by APETx2 at higher concentrations. Of particular relevance to pain studies, APETx2 robustly potentiated the activity of rASIC1b at concentrations 30‐fold to 100‐fold higher than it inhibits rASIC3 homomers, under the conditions used in vitro. This newly described activity has important implications for interpreting the results from pain studies in rodents.
Genetic knockout approaches to studying the role of ASICs in pain have so far not yielded very clear cut results. Staniland and McMahon (2009) noted the normal development of inflammatory pain (FCA‐induced) in mice lacking either ASIC1, ASIC2 or ASIC3, whereas Yen et al. (2009) did find a role for ASIC3 in the maintenance of sub‐acute phase inflammatory pain. Differences in these studies may be attributed to different methods of assessing pain, and/or the different KO mice used, which may have uniquely altered expression profiles of other nociceptive ion channels. Using a pharmacological approach, peripheral ASIC3 was first shown to be involved in inflammatory pain by Deval et al. (2008). Local administration of APETx2 or in vivo knockdown of ASIC3 (via intrathecal injection of siRNA) reduced thermal hyperalgesia induced in an FCA rat model of inflammatory pain (Deval et al., 2008). A second and independent study (Karczewski et al., 2010) demonstrated that APETx2 also had a peripheral antihyperalgesic effect in the FCA rat model. Despite all using an FCA rodent model of inflammatory pain, there were two main differences in the efficacy profile of APETx2 in our study when compared to these previous studies. Firstly, in contrast to the near complete alleviation of thermal and mechanical hyperalgesia by APETx2 reported by Deval et al., (2008) and Karczewski et al., (2010), respectively, we observed only partial reversal of hyperalgesia (~50%). Secondly, these anti‐hyperalgesic effects of APETx2 were observed at lower doses. These differences are likely to be due to differences in the study designs. Deval et al. used a single, high dose of APETx2 co‐administered with FCA and assessed thermal hyperalgesia 4 h later. In light of recent work showing that the immediate pain from bacterial infection (i.e. <12 h) is mediated primarily by nociceptor activation rather than an immune cell‐based inflammatory response (Chiu et al., 2013), the model used by Deval et al. may be more representative of acute pain than inflammatory pain. In contrast, Karczewski et al. tested three doses of APETx2 at 24 h post‐FCA induction (the two lower doses overlapping with those used in our study). In the present study, APETx2 was administered 5 days post‐FCA induction in a chronic phase of inflammation where mechanical hyperalgesia is fully developed (see Table 3 for study protocols). The greater antihyperalgesic efficacy observed for APETx2 in the previous studies was observed for high doses (10‐fold and 36‐fold higher than our highest dose) where APETx2 loses selectivity for ASIC3 and may synergistically engage another analgesic target, the NaV1.8 channels.
Table 3.
Summary of studies using APETx2 in FCA‐induced inflammatory pain models
| Study | Model | Measure of hyperalgesia | Concentration injected | Amount per paw (pmol) |
|---|---|---|---|---|
| Deval et al. (2008) | FCA, 4 h (male Wistar) | Thermal hyperalgesia ‐ paw withdrawal latency at 50°C | 20 μM (20 μL) | 400 |
| Karczewski et al. (2010) | FCA, 24 h (male S‐D) | Mechanical hyperalgesia, PPT | 0.022, 0.22 and 2.2 μM (50 μL) | 1.1, 11 and 110 |
| Current study | FCA, 5 days (male S‐D) | Mechanical hyperalgesia, PPT | 0.3–110 nM (100 μL) | 0.03–11 |
The other new observation in our study was the lack of a stable plateau in the dose‐dependent antihyperalgesia evoked by APETx2. Unlike the consistent level of relief observed for diminazene once a plateau effect was reached, APETx2 was not significantly analgesic at 3.3 and 11 pmol, although 11 pmol had a similar level of reversal of hyperalgesia, ~30%, to that observed by Karczewski et al. (2010) for the same dose. Interestingly, the 1.1 pmol dose had the greatest effect in our study, but no effect in the Karczewski study that employed a 24 h post‐FCA model that probably represents a developing inflammatory state such that immune cell‐mediated inflammation is only just beginning (Chiu et al., 2013). The 5 day model used in our study allows time for more profound sensitization of the nociceptive pathway to take place. Indeed, ASIC1a, 1b, 2b and 3 mRNA levels are highly up‐regulated (6‐fold to 15‐fold) in nociceptive neurons at 2 days post‐FCA‐induced inflammation in rats (Voilley et al., 2001). Thus, the ASIC expression levels are likely to be substantially different between the three time points assessed across these studies. This is consistent with the hypothesis that sensory neuron plasticity underlies the transition from acute to chronic pain states over several days (Reichling and Levine, 2009), a process shown to involve ASIC3, at least in a mouse model of fibromyalgia (Chen et al., 2014). Furthermore, the pH sensitivity and desensitization profiles of ASICs are modified by many different inflammatory mediators to enhance their function in inflammatory conditions. For example, lactate, 5‐HT, arachidonic acid, lysophosphatidylcholine, NO, prokineticin2 and ATP all potentiate the function of ASICs either directly (via decreasing desensitization or increasing activation) or indirectly (via phosphorylation) (Baron and Lingueglia, 2015; Marra et al., 2016). We suggest that the contribution of ASIC1 and ASIC3 to FCA‐induced hyperalgesia is likely to be more pronounced at day 5 post‐FCA, compared to the non‐inflamed (co‐injection with FCA) or early inflammation (24 h) conditions and thus may account for the substantial antihyperalgesic efficacy we observed at low doses of APETx2. It must be noted that nociception is highly complex ‐ >350 genes are involved (Mogil, 2012) ‐ and ASICs are not likely to be the sole mediators of inflammatory pain, consistent with our observation of partial reversal of hyperalgesia at the doses used.
Finally, the present study revealed substantial inter‐individual heterogeneity in the antihyperalgesic efficacy of ASIC inhibition. Inter‐subject variability in responsiveness to analgesic drugs is well documented in the clinical setting but is seldom taken into account in preclinical animal studies. There are many factors underlying the spectrum of sensitivity to analgesics, including genetics, sex, age and health status (Muralidharan and Smith, 2011; Smith and Muralidharan, 2012). All of these factors are equally applicable to preclinical pain studies in animals (Avsaroglu et al., 2007; Ji et al., 2007), with the addition of differing laboratory conditions and animal handling practices, which can contribute different levels of stress (Mogil, 2012; Mogil, 2017). Stress, for instance from handling or simple environment changes such as being placed in a different room, can induce substantial analgesic responses and is a confounding factor in pain studies that can result in wide variation in responses (Mogil et al., 2011; Mogil, 2017). In order to minimize such potential sources of variation, our protocol ensured that animals were handled daily during the acclimation period to minimize stress effects and that assays were carried out blinded to minimize unconscious bias during behavioural assessment. Thus, the wide variability in our behavioural results is consistent with the use of outbred rats, with a non‐homogeneous genetic background, and our data resemble the often marked inter‐individual variability in pain severity ratings and the doses of clinically available analgesic drugs needed for pain relief in patients with apparently similar pain states.
In conclusion, our experiments have revealed a new antihyperalgesic function for diminazene and further characterized the antihyperalgesic efficacy of APETx2 in a widely used rat model of chronic inflammatory pain. Using electrophysiological methods, we found that APETx2 not only inhibited ASIC3 but also potentiated agonist action at ASIC1b, at higher concentrations. This finding may be related to our observed lack of significant analgesic efficacy of APETx2 at higher doses and needs to be taken into account when selecting doses of APETx2 for administration in in vivo pain models. Indeed, the relevance of our observation that APETx2 enhances ASIC1b currents to its in vivo analgesic activity needs to be verified in further studies using APETx2 with modified ASIC1b/ASIC3 selectivity and assessment in ASIC3 and/or ASIC1b knockout mice. Finally, we highlight the need to take into account the wide variation in antihyperalgesic responses as a complicating factor in animal pain studies, akin to the widely differing human responsiveness to analgesics seen in the clinic. This feature emphasizes the need for open and thorough reporting of animal pain data (Muralidharan and Smith, 2011; Smith and Muralidharan, 2012). Efforts to recognize and understand the underlying causes of this source of variation in preclinical settings should help to improve translation of basic pain research from the laboratory to clinical practice.
Author contributions
Study conception and design were performed by L.D.R., G.F.K. and M.T.S. Peptide production was done by R.A. and B.C.‐A. Oocyte electrophysiology was carried out by N.J.S., B.C.‐A., L.D.R. Behavioural studies were performed by JYPL. Data were analysed by N.J.S., B.C.‐A., J.Y.P.L., L.D.R. and M.T.S. The MS was written by J.Y.P.L., M.T.S. and L.D.R, and all authors took part in editing the MS and giving final approval of the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was been supported by the Australian National Health and Medical Research Council via Project grant APP1067940 to L.D.R. and M.T.S., Project grant APP1063798 to G.F.K. and Principal Research Fellowship APP1044414 to G.F.K. We thank Prof John Wood for the rat ASIC1a, ASIC2a and ASIC3 clones and Prof Stefan Gründer for the rat ASIC1b clone. We thank Dr Nicholas Hamilton and Dr James Lefevre (Biomathematicians, UQ‐IMB) for helpful discussions on the statistical analysis of animal data.
Lee, J. Y. P. , Saez, N. J. , Cristofori‐Armstrong, B. , Anangi, R. , King, G. F. , Smith, M. T. , and Rash, L. D. (2018) Inhibition of acid‐sensing ion channels by diminazene and APETx2 evoke partial and highly variable antihyperalgesia in a rat model of inflammatory pain. British Journal of Pharmacology, 175: 2204–2218. doi: 10.1111/bph.14089.
Contributor Information
Maree T Smith, Email: maree.smith@uq.edu.au.
Lachlan D Rash, Email: l.rash@uq.edu.au.
References
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J et al (1999). The tetrodotoxin‐resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci 2: 541–548. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anangi R, Rash LD, Mobli M, King GF (2012). Functional expression in Escherichia coli of the disulfide‐rich sea anemone peptide APETx2, a potent blocker of acid‐sensing ion channel 3. Mar Drugs 10: 1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsaroglu H, van der Sar AS, van Lith HA, van Zutphen LF, Hellebrekers LJ (2007). Differences in response to anaesthetics and analgesics between inbred rat strains. Lab Anim 41: 337–344. [DOI] [PubMed] [Google Scholar]
- Baron A, Lingueglia E (2015). Pharmacology of acid‐sensing ion channels – physiological and therapeutical perspectives. Neuropharmacology 94: 19–35. [DOI] [PubMed] [Google Scholar]
- Blanchard MG, Rash LD, Kellenberger S (2012). Inhibition of voltage‐gated Na+ currents in sensory neurones by the sea anemone toxin APETx2. Br J Pharmacol 165: 2167–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen CJ, Chesler AT, Sharif‐Naeini R, Medzihradszky KF, Zhou S, King D et al (2011). A heteromeric Texas coral snake toxin targets acid‐sensing ion channels to produce pain. Nature 479: 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, Zamponi GW (2014). Calcium‐permeable ion channels in pain signaling. Physiol Rev 94: 81–140. [DOI] [PubMed] [Google Scholar]
- Chen X, Qiu L, Li M, Durrnagel S, Orser BA, Xiong ZG et al (2010). Diarylamidines: high potency inhibitors of acid‐sensing ion channels. Neuropharmacology 58: 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WN, Lee CH, Lin SH, Wong CW, Sun WH, Wood JN et al (2014). Roles of ASIC3, TRPV1, and NaV1.8 in the transition from acute to chronic pain in a mouse model of fibromyalgia. Mol Pain 10: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J et al (2013). Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501: 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofori‐Armstrong B, Rash LD (2017). Acid‐sensing ion channel (ASIC) structure and function: insights from spider, snake and sea anemone venoms. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2017.04.042. [DOI] [PubMed] [Google Scholar]
- Cristofori‐Armstrong B, Soh MS, Talwar S, Brown DL, Griffin JD, Dekan Z et al (2015). Xenopus borealis as an alternative source of oocytes for biophysical and pharmacological studies of neuronal ion channels. Sci Rep 5: 14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deval E, Noel J, Lay N, Alloui A, Diochot S, Friend V et al (2008). ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J 27: 3047–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deval E, Noel J, Gasull X, Delaunay A, Alloui A, Friend V et al (2011). Acid‐sensing ion channels in postoperative pain. J Neurosci 31: 6059–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diochot S, Baron A, Rash LD, Deval E, Escoubas P, Scarzello S et al (2004). A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid‐sensitive channel in sensory neurons. EMBO J 23: 1516–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diochot S, Baron A, Salinas M, Douguet D, Scarzello S, Dabert‐Gay AS et al (2012). Black mamba venom peptides target acid‐sensing ion channels to abolish pain. Nature 490: 552–555. [DOI] [PubMed] [Google Scholar]
- Dorofeeva NA, Barygin OI, Staruschenko A, Bolshakov KV, Magazanik LG (2008). Mechanisms of non‐steroid anti‐inflammatory drugs action on ASICs expressed in hippocampal interneurons. J Neurochem 106: 429–441. [DOI] [PubMed] [Google Scholar]
- Dube GR, Lehto SG, Breese NM, Baker SJ, Wang X, Matulenko MA et al (2005). Electrophysiological and in vivo characterization of A‐317567, a novel blocker of acid sensing ion channels. Pain 117: 88–96. [DOI] [PubMed] [Google Scholar]
- Ferreira J, Santos AR, Calixto JB (1999). Antinociception produced by systemic, spinal and supraspinal administration of amiloride in mice. Life Sci 65: 1059–1066. [DOI] [PubMed] [Google Scholar]
- Izumi M, Ikeuchi M, Ji Q, Tani T (2012). Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. J Biomed Sci 19: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JE, Cristofori‐Armstrong B, Anangi R, Rosengren KJ, Lau CH, Mobli M et al (2014). Understanding the molecular basis of toxin promiscuity: the analgesic sea anemone peptide APETx2 interacts with acid‐sensing ion channel 3 and hERG channels via overlapping pharmacophores. J Med Chem 57: 9195–9203. [DOI] [PubMed] [Google Scholar]
- Ji Y, Murphy AZ, Traub RJ (2007). Estrogen modulation of morphine analgesia of visceral pain in female rats is supraspinally and peripherally mediated. J Pain 8: 494–502. [DOI] [PubMed] [Google Scholar]
- Jones NG, Slater R, Cadiou H, McNaughton P, McMahon SB (2004). Acid‐induced pain and its modulation in humans. J Neurosci 24: 10974–10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski J, Spencer RH, Garsky VM, Liang A, Leitl MD, Cato MJ et al (2010). Reversal of acid‐induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br J Pharmacol 161: 950–960. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishtal OA, Pidoplichko VI (1981). A receptor for protons in the membrane of sensory neurons may participate in nociception. Neuroscience 6: 2599–2601. [DOI] [PubMed] [Google Scholar]
- Kulemina LV, Ostrov DA (2011). Prediction of off‐target effects on angiotensin‐converting enzyme 2. J Biomol Screen 16: 878–885. [DOI] [PubMed] [Google Scholar]
- Kuriakose S, Muleme H, Onyilagha C, Okeke E, Uzonna JE (2014). Diminazene aceturate (Berenil) modulates LPS induced pro‐inflammatory cytokine production by inhibiting phosphorylation of MAPKs and STAT proteins. Innate Immun 20: 760–773. [DOI] [PubMed] [Google Scholar]
- Lin SH, Sun WH, Chen CC (2015). Genetic exploration of the role of acid‐sensing ion channels. Neuropharmacology 94: 99–118. https://doi.org/10.1016/j.neuropharm.2014.12.011. [DOI] [PubMed] [Google Scholar]
- Marra S, Ferru‐Clement R, Breuil V, Delaunay A, Christin M, Friend V et al (2016). Non‐acidic activation of pain‐related acid‐sensing ion channel 3 by lipids. EMBO J 35: 414–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS (1999). The genetic mediation of individual differences in sensitivity to pain and its inhibition. Proc Natl Acad Sci U S A 96: 7744–7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS (2012). Pain genetics: past, present and future. Trends Genet 28: 258–266. [DOI] [PubMed] [Google Scholar]
- Mogil JS (2017). Laboratory environmental factors and pain behavior: the relevance of unknown unknowns to reproducibility and translation. Lab Anim (NY) 46: 136–141. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Sorge RE, LaCroix‐Fralish ML, Smith SB, Fortin A, Sotocinal SG et al (2011). Pain sensitivity and vasopressin analgesia are mediated by a gene‐sex‐environment interaction. Nat Neurosci 14: 1569–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ (2009). Pregabalin for acute and chronic pain in adults. Cochrane Database Syst Rev 3: CD007076. https://doi.org/10.1002/14651858.CD007076.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan A, Smith MT (2011). Pain, analgesia and genetics. J Pharm Pharmacol 63: 1387–1400. [DOI] [PubMed] [Google Scholar]
- Payne CE, Brown AR, Theile JW, Loucif AJ, Alexandrou AJ, Fuller MD et al (2015). A novel selective and orally bioavailable Nav 1.8 channel blocker, PF‐01247324, attenuates nociception and sensory neuron excitability. Br J Pharmacol 172: 2654–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peigneur S, Beress L, Moller C, Mari F, Forssmann WG, Tytgat J (2012). A natural point mutation changes both target selectivity and mechanism of action of sea anemone toxins. FASEB J 26: 5141–5151. [DOI] [PubMed] [Google Scholar]
- Perrot S, Guilbaud G, Kayser V (1999). Effects of intraplantar morphine on paw edema and pain‐related behaviour in a rat model of repeated acute inflammation. Pain 83: 249–257. [DOI] [PubMed] [Google Scholar]
- Rash LD (2017) Acid‐sensing ion channel pharmacology, Past, Present, and Future… In: Ion Channels Down Under, Geraghty D, Rash LD. (eds) Vol. 79, pp 35–66: Academic Press: Burlington, Massachusetts. [DOI] [PubMed] [Google Scholar]
- Reeh PW, Steen KH (1996). Tissue acidosis in nociception and pain. Prog Brain Res 113: 143–151. [DOI] [PubMed] [Google Scholar]
- Reichling DB, Levine JD (2009). Critical role of nociceptor plasticity in chronic pain. Trends Neurosci 32: 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimers C, Lee CH, Kalbacher H, Tian Y, Hung CH, Schmidt A et al (2017). Identification of a cono‐RFamide from the venom of Conus textile that targets ASIC3 and enhances muscle pain. Proc Natl Acad Sci U S A 114: E3507–E3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou G, Benard J (1980). Berenil induces the complete loss of kinetoblast DNA sequences in Trypanosoma equiperdum . Biochem Biophys Res Commun 96: 350–354. [DOI] [PubMed] [Google Scholar]
- Rodrigues AR, Duarte ID (2000). The peripheral antinociceptive effect induced by morphine is associated with ATP‐sensitive K+ channels. Br J Pharmacol 129: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Rossetti G, Joussen S, Grunder S (2017). Diminazene is a slow pore blocker of acid‐sensing ion channel 1a (ASIC1a). Mol Pharmacol 92: 665–675. [DOI] [PubMed] [Google Scholar]
- Schwarz MG, Namer B, Reeh PW, Fischer MJM (2017). TRPA1 and TRPV1 antagonists do not inhibit human acidosis‐induced pain. J Pain 18: 526–534. [DOI] [PubMed] [Google Scholar]
- da Silva Serra I, Husson Z, Bartlett JD, Smith ES (2016). Characterization of cutaneous and articular sensory neurons. Mol Pain 12 https://doi.org/10.1177/1744806916636387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MT, Muralidharan A (2012). Pharmacogenetics of pain and analgesia. Clin Genet 82: 321–330. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staniland AA, McMahon SB (2009). Mice lacking acid‐sensing ion channels (ASIC) 1 or 2, but not ASIC3, show increased pain behaviour in the formalin test. Eur J Pain 13: 554–563. [DOI] [PubMed] [Google Scholar]
- Steen KH, Reeh PW, Anton F, Handwerker HO (1992). Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro . J Neurosci 12: 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Herz A (1988). Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol Biochem Behav 31: 445–451. [DOI] [PubMed] [Google Scholar]
- Ugawa S, Ueda T, Ishida Y, Nishigaki M, Shibata Y, Shimada S (2002). Amiloride‐blockable acid‐sensing ion channels are leading acid sensors expressed in human nociceptors. J Clin Invest 110: 1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voilley N, de Weille J, Mamet J, Lazdunski M (2001). Nonsteroid anti‐inflammatory drugs inhibit both the activity and the inflammation‐induced expression of acid‐sensing ion channels in nociceptors. J Neurosci 21: 8026–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li WG, Yu Y, Xiao X, Cheng J, Zeng WZ et al (2013). Serotonin facilitates peripheral pain sensitivity in a manner that depends on the nonproton ligand sensing domain of ASIC3 channel. J Neurosci 33: 4265–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Zamponi GW (2014). Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci 17: 153–163. [DOI] [PubMed] [Google Scholar]
- Wiemuth D, Gründer S (2011). The pharmacological profile of brain liver intestine Na+ channel: inhibition by diarylamidines and activation by fenamates. Mol Pharmacol 80: 911–919. [DOI] [PubMed] [Google Scholar]
- Woolf CJ (2010). Overcoming obstacles to developing new analgesics. Nat Med 16: 1241–1247. [DOI] [PubMed] [Google Scholar]
- Yen YT, Tu PH, Chen CJ, Lin YW, Hsieh ST, Chen CC (2009). Role of acid‐sensing ion channel 3 in sub‐acute‐phase inflammation. Mol Pain 5: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Chen Z, Li WG, Cao H, Feng EG, Yu F et al (2010). A nonproton ligand sensor in the acid‐sensing ion channel. Neuron 68: 61–72. [DOI] [PubMed] [Google Scholar]