

Abstract
Voltage‐gated calcium channels (VGCCs) play important roles in physiological functions including the modulation of neurotransmitter release, neuronal network activities, intracellular signalling pathways and gene expression. Some pathological conditions, including nerve injuries, can cause the dysregulation of VGCCs and their subunits. This in turn can lead to a functional maladaptation of VGCCs and their subunits, which can contribute to the development of disorders such as pain sensations. This review has summarized recent findings related to maladaptive changes in the dysregulated VGCC α2δ1 subunit (Cavα2δ1) with a focus on exploring the mechanisms underlying the contribution of Cavα2δ1 to pain signal transduction. At least under neuropathic pain conditions, the dysregulated Cavα2δ1 can modulate VGCC functions as well as other plasticity changes. The latter includes abnormal excitatory synaptogenesis resulting from its interactions with injury‐induced extracellular matrix glycoprotein molecule thrombospondins, which is independent of the VGCC functions. Blocking Cavα2δ1 with gabapentinoids can reverse neuropathic pain significantly with relatively mild side effects, but only in a small population of neuropathic pain patients due to reasons yet to be explored. There are emerging data suggesting that early preventive treatment with gabapentinoids can prevent aberrant excitatory synapse formation and the development of chronic pain. If these findings are confirmed clinically, this could be an attractive approach for neuropathic pain management.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Abbreviations
- DRG
dorsal root ganglion
- HVA
high voltage‐activated
- LVA
low voltage‐activated
- TSP4
thrombospondin‐4
- VGCC
voltage‐gated calcium channels
Introduction
Under physiological conditions, nociceptive sensations primarily serve as a protective mechanism for our body. A stimulus detected by sensory nerve endings evokes action potentials that propagate along the primary afferent nerve fibres up to the soma of dorsal root ganglion (DRG) neurons and then through their central axons to the first relay in the dorsal spinal cord. As a sensory information processing hub referring periphery sensory information to the brain, the spinal dorsal horn sensory circuitry is critically involved in processing sensory information locally and sending it through ascending tracts to the higher CNS for further processing. The brain then sends descending signals to the spinal cord through descending pathways to modulate sensory and motor neuron circuitry activities. However, under pathological conditions, damage to the nervous system can induce plasticity changes in the sensory pathway so that its activation thresholds are lowered. As a consequence, exaggerated pain perception can occur in the absence of painful stimuli or with mild painful stimulation. This kind of abnormal sensation gives rise to painful phenomena including ‘allodynia’ (i.e. painful perception to an innocuous stimulus) and ‘hyperalgesia’ (exacerbated pain perception to a mildly noxious stimulus), collectively called pain states here. Among the factors that play critical roles in pain development after nerve injuries (neuropathic pain states), voltage‐gated calcium channels (http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=80) contribute to this process through the modulation of important pathophysiological functions. These include the release of excitatory neurotransmitters (Lee, 2013), calcium‐dependent enzyme activation (Park and Luo, 2010), effects on gene expression (Perret and Luo, 2009; Park and Luo, 2010; Wheeler et al., 2012) and other short‐ and long‐term plasticity changes in the spinal dorsal horn (Russo and Hounsgaard, 1994; Naka et al., 2013; Youn et al., 2013; Liu and Zhou, 2015). In addition, injury‐induced dysregulation of VGCC subunits, such as α2δ1 (Cavα2δ), may also contribute to pain signal transduction by modulating additional neural plasticity such as abnormal synaptogenesis (Eroglu et al., 2009; Park et al., 2016). These changes can alter the activities of the neural network that fundamentally contribute to the cellular underpinning of chronic pain development.
Voltage‐gated calcium channels and accessory subunits
So far, 10 VGCC subtypes have been identified that are divided broadly into two categories based on their thresholds of activation by membrane depolarization: high voltage‐activated (HVA) and low voltage‐activated (LVA) channels (Benarroch, 2010; Simms and Zamponi, 2014; Zamponi et al., 2015). The HVA channel family can be further sub‐divided, based on their pharmacological and functional characteristics, into L‐, N‐, P/Q‐ and R‐types, and the LVA channels are mainly T‐type VGCCs. Structurally, all VGCCs contain a pore‐forming α1 subunit that determines their main biophysical properties. There are three major families of α1 subunits (Cav1, Cav2 and Cav3), which can undergo alternative splicing in a tissue‐, age‐ and pathology‐dependent manner (Lipscombe et al., 2013). In addition, the HVA channels are heteromultimers with three auxiliary subunits, Cavβ, Cavα2δ and Cavγ. Four Cavβ subunits (Cavβ1–β4), four Cavα2δ subunits (Cavα2δ−4) and eight Cavγ subunits (Cavγ1–8) have been identified. Unlike the HVA channels that require co‐assembly with auxiliary calcium channel subunits to function, the LVA channels appear as α1 subunit monomers that can be functionally regulated by the auxiliary subunits (Dubel et al., 2004). However, other studies reported that auxiliary subunits of calcium channels have little or no modulatory effect on LVA channel currents (Perez‐Reyes, 2003; Turner and Zamponi, 2014).
The functions and details of the Cavα1, Cavβ and Cavγ subunits are reviewed and summarized in other chapters in this issue and recent reviews (Simms and Zamponi, 2014; Zamponi et al., 2015; Campiglio and Flucher, 2015). In addition, the structural feature, regulation and function of different types of VGCCs and their implications in neurological diseases including pain, migraine, epilepsy, cerebellar ataxia, Parkinson disease and hypertension have been the subjects of numerous recent reviews (Perret and Luo, 2009; Park and Luo, 2010; Cain and Snutch, 2011; Vink and Alewood, 2012; Simms and Zamponi, 2014; Zamponi et al., 2015; Zamponi, 2016). In this review, we focus on recent findings related to maladaptive and dysregulation of Cavα2δ and its potential contribution to pain development, as well as prospective views of blocking Cavα2δ in neuropathic pain development and management.
Cav α 2 δ subunits
Each Cavα2δ subunit consists of the Cavα2 and Cavδ peptides that are encoded by the same gene, post‐translationally cleaved and then linked by disulfide‐bounds (Ellis et al., 1988). Site‐directed mutagenesis data have indicated that a single intermolecular disulfide bound between cysteine residue 404 in the von Willebrand factor A (VWA) domain of Cavα2 and cysteine residue 1047 in the extracellular domain of Cavδ is critical for the structural and functional integrity of the Cavα2δ protein (Calderon‐Rivera et al., 2012). Recently, data from cryo‐electron microscopy reveal that the primary sequence of Cavα2δ contains four tandem cache domains and one VWA domain that are intertwined (Wu et al., 2016). In addition, there are four disulfide bounds between the Cavα2 and Cavδ peptides and two within the Cavδ peptide (Wu et al., 2016). In combination with the site‐directed mutagenesis data, it is likely that each of these disulfide bounds is mutually critical in stabilizing the calcium channel complex and for its functions. Data from biochemical characterization of Cavα2δ proteins support the view that Cavα2δ is attached to the extracellular leaflet of the plasma membrane via a glycophosphatidylinositol anchor (Davies et al., 2010), which seems to be supported by structural data revealed by cryo‐electron microscopy (Wu et al., 2016). The known functions of Cavα2δ subunits include promoting and stabilizing cell surface expression and modulating functions of VGCC (Dolphin, 2012; Dolphin, 2013).
Calcium channel Cav α 2 δ 1 subunit as a therapeutic target in pain management
Pharmacological and biochemical studies
Data from biochemical studies have indicated that gabapentinoids, including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7560 (Neurontin; Pfizer) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5484 (Lyrica; Pfizer), bind to both Cavα2δ1 and Cavα2δ2 subunit proteins (Gee et al., 1996; Gong et al., 2001; Marais et al., 2001b; Li et al., 2011; Kukkar et al., 2013; Verma et al., 2014; Landmark et al., 2015). Gabapentinoids are widely used in management of neuropathic pain, including that derived from diabetic neuropathy, nerve injuries, drugs and radiation‐induced neuropathies, as well as post‐herpetic and trigeminal neuralglia (Johnson and Rice, 2014; Moore et al., 2014; Zamponi et al., 2015). However, only a significant up‐regulation of Cavα2δ1, but not Cavα2δ2, has been observed in DRG/spinal cord of neuropathic pain models (Luo et al., 2001; 2002; Valder et al., 2003; Bauer et al., 2009), supporting the concept that the anti‐hyperalgesic properties of gabapentinoids are mediated by their binding to the Cavα2δ1 subunit. This is confirmed by point mutation of the binding site for gabapentinoids on Cavα2δ1 in a transgenic mouse line, which diminishes Cavα2δ1 binding and the anti‐hyperalgesic properties of gabapentinoids (Field et al., 2006).
Genetic studies
One way to confirm that Cavα2δ1 has a role in pain processing is to use Cavα2δ1 knockout mice to generate pain research models, which certainly come with limitations, or correlate abnormal sensation deficits with Cavα2δ1 genetic defects in patients. Table 1 summarizes findings of the studies done in Cavα2δ genetic‐knockout mouse models or from patients with genetic defects regarding a potential role for each Cavα2δ subunit in pain signal transduction. Even though Cavα2δ1 knockout is conceived as embryonic lethal (Simms and Zamponi, 2014), it has been reported that mice with a targeted deletion of Cavα2δ1 (Fuller‐Bicer et al., 2009) display normal sensitivity to thermal stimulation but reduced sensitivity to mechanical and cold stimuli that correlates with reduced responses of dorsal horn wide dynamic range neurons to stimuli (Fuller‐Bicer et al., 2009; Patel et al., 2013). In addition, the onset time of mechanical hypersensitivity is delayed, and the anti‐hyperalgesic activity of gabapentinoids after peripheral nerve injury is absent in this line of mice (Patel et al., 2013). These findings confirm that the induction of Cavα2δ1 is critical for the initial development of neuropathic pain states after nerve injury (Boroujerdi et al., 2008) and that Cavα2δ1 does indeed mediate the anti‐hyperalgesic properties of gabapentinoids (Field et al., 2006), as reported previously. Peripherally, general Cavα2δ1 knockout results in a significant reduction in calcium entry per action potential, and in the duration and frequency of action potentials in DRG sensory neurons (Margas et al., 2016). However, linking a direct causal role of Cavα2δ1 to these electrophysiological and behavioural changes in this mouse line is difficult because we cannot rule out the contributions from other compensatory factors and/or developmental changes due to general embryonic knockout of Cavα2δ1. It is conceivable that general knockout of Cavα2δ1 can affect a wide range of important organ development and physiological functions due to the importance of Cavα2δ1 in VGCC trafficking, membrane expression and in modulating different functions of VGCCs (Hendrich et al., 2008; Cassidy et al., 2014). With the application of Cre‐induced recombination technology, sensory neuron‐type specific deletion of Cavα2δ1 (Park et al., 2016) allows us to study the contribution of Cavα2δ1 up‐regulation in a subpopulation of sensory neurons to modality‐specific processing of neuropathic pain states.
Table 1.
Genetic studies regarding the role of Cavα2δ subunits in pain signal transduction
| Cavα2δ isoform | Genetic modification | Sensory phenotype | Reference |
|---|---|---|---|
| Cavα2δ1 | General (embryonic) knockout in mice | ↓ Mechanical/cold sensitivity | Fuller‐Bicer et al., 2009 |
| Delayed mechanical hypersensitivity onset after nerve injury | Patel et al., 2013 | ||
| Natural aberrations/mutations in patients | Not reported | Vergult et al., 2015 | |
| Hino‐Fukuyo et al., 2015 | |||
| Cavα2δ2 | Spontaneous mutations in mice | Not reported | Barclay et al., 2001; Brodbeck et al., 2002 |
| Donato et al., 2006 | |||
| Brill et al., 2004 | |||
| Targeted disruption in mice | Ivanov et al., 2004 | ||
| Homozygous mutations in patients | Edvardson et al., 2013 | ||
| Pippucci et al., 2013 | |||
| Cavα2δ3 | Spontaneous mutations in Drosophila | ↓ Acute heat pain sensitivity | Neely et al., 2010 |
| Deletion by homologous recombination in mice | Delayed inflammatory thermal hyperalgesia | ||
| Single‐nucleotide polymorphisms in patients | ↓ Low‐back pain sensitivity | ||
| Splice site mutation | Not reported | Iossifov et al., 2012 | |
| Cavα2δ4 | Spontaneous homozygous frame shift mutation in mice | Not reported | Wycisk et al., 2006a |
| Homozygous nucleotide substitution in patients | Wycisk et al., 2006b |
Genomic aberration of the CACNA2D1 gene encoding Cavα2δ1 has been identified in total blood DNA of three patients with epilepsy and intellectual disability, supporting a potential contribution of Cavα2δ1 to these clinical phenotypes (Vergult et al., 2015). Similarly, another study has identified mutations in Cavα2δ1 from peripheral blood DNA of patients with early‐onset epileptic encephalopathy (West syndrome) (Hino‐Fukuyo et al., 2015). Unfortunately, abnormal behavioural sensitivity has not been reported or tested in these patients. However, Cavα2δ1 over‐expression in mouse neuronal tissues has been reported to cause brain epileptiform activity associated with behavioural arrests (Faria et al., 2017) and spinal synaptic hyperexcitability associated with behavioural hypersensitivity to mechanical/thermal/cold stimuli (Li et al., 2006; Zhou and Luo, 2013; Zhou and Luo, 2015). These data support a role for Cavα2δ1 in mediating the neural circuitry hyperexcitability associated with both epileptic and pain phenotypes.
Since gabapentinoids bind to both Cavα2δ1 and Cavα2δ2 subunits and show antiepileptic, anxiolytic and antihyperalgesic efficacy in preclinical studies, a recent study was well‐designed to distinguish the role of Cavα2δ1 and Cavα2δ2 in the anticonvulsant activity of gabapentinoids. Mutations of the binding site for gabapentinoids in Cavα2δ1, but not in Cavα2δ2, result in a significant reduction in the anticonvulsant efficacy of pregabalin compared with wild‐type controls and the Cavα2δ2 mutants (Lotarski et al., 2014). Similarly, these mouse mutants were used to reveal the association of drug binding to Cavα2δ1, but not Cavα2δ2, with the anxiolytic‐like activity of gabapentinoids (Lotarski et al., 2011). Together with other findings indicating a role for Cavα2δ1 in pain signal transduction and in mediating the antihyperalgesic activity of gabapentinoids, findings from these genetic studies support the involvement of Cavα2δ1 in mediating pain signal transduction, epileptic and anxiolytic activities as well as the inhibitory effects of gabapentinoids on these disorders.
Genetic studies on other Cav α 2 δ isoforms
Naturally occurring mutations in the CACNA2D2 gene encoding Cavα2δ2 in ducky (Barclay et al., 2001; Brodbeck et al., 2002), ducky2j (Donato et al., 2006) and entla (Brill et al., 2004) mice as well as targeted Cavα2δ2 knockout (Ivanov et al., 2004) result in reduced calcium current densities, gabapentinoid binding sites and ataxia and epileptic phenotypes. Two recent clinical studies have identified different homozygous mutations in the CACNA2D2 gene that correlate with epileptic encephalopathy (Edvardson et al., 2013; Pippucci et al., 2013), supporting the possibility that Cavα2δ2 may play a role in mediating epileptic activities. Even though abnormalities in both Cavα2δ1 and Cavα2δ2 correlate with the development of epileptic activity, gabapentinoids' anti‐seizure effect is likely to be mediated through their binding to Cavα2δ1, but not Cavα2δ2, because the anti‐seizure efficacy of pregabalin is reduced in mice with a gabapentinoid binding site mutation in Cavα2δ1, but not in Cavα2δ2 (Lotarski et al., 2014). Further mechanistic studies regarding the relative contribution of Cavα2δ1 and Cavα2δ2 to brain neural circuitry hyperexcitability in a cell type‐specific manner may provide answers to this discrepancy. Abnormal nociceptive sensitivity was not reported in these studies (Table 1).
A genome‐wide screen study in drosophila has identified the CACNA2D3 gene encoding Cavα2δ3 as an evolutionarily conserved pain gene mediating thermal hyperalgesia (Neely et al., 2010). Deletion of Cavα2δ3 with homologous recombination in mice results in deficits in acute heat pain sensitivity and delayed thermal hyperalgesia induced by inflammation (Neely et al., 2010). In addition, single‐nucleotide polymorphisms within or close to the human CACNA2D3 gene are shown to be associated with reduced thermal pain sensitivity in healthy volunteers, and with chronic low‐back pain in patients (Neely et al., 2010). Data from functional imaging in Cavα2δ3 mutant mice indicate that thermal pain signal transmission from the thalamus to higher pain centres is impaired (Neely et al., 2010). Since Cavα2δ3 is mainly expressed in the brain, and it is not a binding site for gabapentinoids (Marais et al., 2001a), these findings support a role for Cavα2δ3 in transmitting pain signals in the brain through a mechanism that could be insensitive to gabapentinoids. Interestingly, sensory (noxious thermal and tactile) stimulation can trigger strong cross‐activation of brain regions related to hearing, vision and olfaction in Cavα2δ3 mutant mice (Neely et al., 2010), supporting a role for Cavα2δ3 in maintaining normal sensory signal distinction in the brain. In addition, splice site mutation of the human CACNA2D3 gene might be involved in autism spectrum disorders (Iossifov et al., 2012). However, sensory testing is not reported in these studies (Table 1).
As Cavα2δ4 is the major Cavα2δ subunit expressed in mouse retina cells (Knoflach et al., 2013), mutations in the CACNA2D4 gene encoding Cavα2δ4 result in loss of retina signalling, abnormal synapse morphology and dysfunction in mouse rods and cones (Wycisk et al., 2006a), as well as autosomal recessive cone dystrophy associated with night blindness in patients (Wycisk et al., 2006b). Abnormal nociceptive sensitivity was not reported or tested in these studies (Table 1).
Potential mechanisms underlying Cav α 2 δ 1 dysregulation in pain signal transduction and the anti‐hyperalgesic effects of gabapentinoids
Cavα2δ1 expression is up‐regulated in DRG (and trigeminal ganglia) and spinal cord in some neuropathic pain models derived from mechanical nerve injury, spinal cord injury, diabetic or chemical neuropathies that correlate with the onset and maintenance of neuropathic pain states (Luo et al., 2001; 2002; Newton et al., 2001; Yusaf et al., 2001; Li et al., 2004; 2014b; Boroujerdi et al., 2008; 2011). The Cavα2δ1 ligand gabapentinoids can reverse neuropathic pain states in these animal models (Hwang and Yaksh, 1997; Luo et al., 2002; Boroujerdi et al., 2011; Li et al., 2014b). In addition, transgenic mice overexpressing Cavα2δ1 in neuronal tissues, including sensory and spinal cord neurons, display neuropathic pain states such as tactile allodynia and thermal hyperalgesia, in the absence of any nerve injury, that can be blocked by gabapentinoids (Li et al., 2006). Together, these findings support the view that injury‐induced Cavα2δ1 up‐regulation may play a critical role in neuropathic pain development.
Data from detailed mechanistic investigations indicate that nerve injury‐induced Cavα2δ1 expression occurs predominantly in DRG sensory neurons, then undergoes axonal translocation to presynaptic terminals in spinal dorsal horn (Figure 1), since the axonal transport can be interrupted by cutting the dorsal root connecting injured DRG and dorsal spinal cord through dorsal rhyzotomy (Li et al., 2004). Subsequently, this axonal transport of an elevated expression of Cavα2δ1 from DRG neurons to dorsal spinal cord was independently confirmed. Data from electron microscopy show directly that injury‐induced Cavα2δ1 in DRG is indeed trafficking to pre‐synaptic terminals of sensory fibres in dorsal spinal cord, and this process can be blocked by gabapentinoids (prebabalin) (Bauer et al., 2009). Thus, injury‐induced increased expression of Cavα2δ1 probably regulates sensory pathway sensitivity peripherally and centrally.
Figure 1.
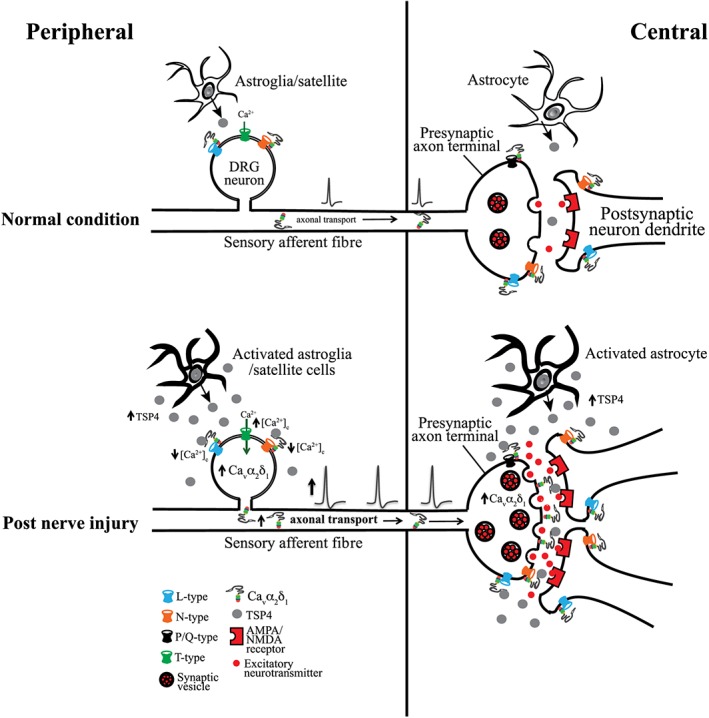
Cartoon illustration of injury‐induced peripheral and central plasticity contributing to sensory sensitization and pain states. Nerve injury induces an up‐regulation of Cavα2δ1 in DRG that undergoes axonal transport to the presynaptic terminal of injured sensory fibres in the dorsal spinal cord. Nerve injury also leads to increased TSP4 expression and secretion from activated DRG neurons/non‐neuronal cells and spinal cord astrocytes. Peripherally, the elevated TSP4/Cavα2δ1 can interact to modulate VGCC currents and intracellular calcium levels, which may cause sensory neuron/afferent activation and sensitization of presynaptic terminals. Centrally, elevated TSP4/Cavα2δ1 can interact to promote presynaptic excitatory neurotransmitter release and excitatory synapse formation, which together can lead to activation of postsynaptic dorsal horn neurons and central sensitization.
At both the peripheral and central levels, it is possible that different Cavα2δ1 isoforms play distinct roles in mediating pain states. Early reports have indicated that peripheral nerve injury induces the up‐regulation of distinct forms of Cavα2δ1 in DRG and spinal cord that correlates with the development of neuropathic pain states (Luo et al., 2001). In addition, injury‐induced DRG Cavα2δ1 isoforms differ from that detected in other tissues such as spinal cord, brain and skeletal muscle as they have unique migration rates in Western blots and different glycosylation patterns (Luo, 2000; Luo et al., 2001). These findings suggest that Cavα2δ1 may have tissue‐specific functions and injury‐induced Cavα2δ1 up‐regulation may modulate pain processing in a tissue type‐specific manner. Recently, it was reported that DRG Cavα2δ1 isoforms are indeed derived from alternative splicing (Lana et al., 2014). In addition, a minor splice variant (ΔA + BΔC) of Cavα2δ1 is preferentially up‐regulated in small DRG neurons after spinal nerve ligation injury, which also enhances calcium currents as it is the major splice variant of DRG Cavα2δ1, but has significantly reduced affinity for gabapentin (Lana et al., 2014). It is speculated that differential expression of specifically spliced Cavα2δ1 variants in neuropathic pain patients may account for the variations in efficacy of pain relief by gabapentinoids observed clinically (Lana et al., 2014).
At the peripheral level, dysregulated Cavα2δ1 can regulate DRG neuron activities through the modulation of calcium channel activities (Li et al., 2006; D'Arco et al., 2015). At the spinal cord level, it can increase presynaptic excitatory input into superficial and deep dorsal horn that regulates dorsal horn neuron excitabilities (Nguyen et al., 2009; Zhou and Luo, 2013, 2015). Since different VGCC blockers have distinct inhibitory effects on Cavα2δ1‐mediated nociception, it is likely that Cavα2δ1 dysregulation contributes to pain processing at least partially through modulation of pathways associated with specific VGCCs (Chang et al., 2015).
Data from in vitro studies indicate that Cavα2δ1 can increase cell‐surface and synaptic expression of VGCCs, including the P/Q‐type (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=532) (Hendrich et al., 2008; Hoppa et al., 2012) and N‐type (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=533) (Cassidy et al., 2014) calcium channels. Calcium entry through these channels predominantly drives synaptic transmission and initiates fast release of classical neurotransmitters (Catterall, 2011; Campiglio and Flucher, 2015). Mechanistically, it has been shown that increased transient Cavα2δ1 expression in cultured rat DRG neurons leads to increased membrane expression of N‐type (Cav2.2) VGCC. This plasticity couples to a prolonged Ca2+ signal evoked by membrane depolarization and activity‐dependent slowing of axonal mitochondrial transport, both of which can be inhibited by treatments with N‐type VGCC blockers. These findings suggest that increased Cavα2δ1 expression modulates sensory neuron responses to stimulation through the regulation of mitochondrial calcium buffering, which is mediated by N‐type VGCCs (D'Arco et al., 2015). Interestingly, chronic treatment with gabapentinoids can reduce the densities of Cavα2δ1 and Cav2 (Cav2.1 and Cav2.2) subunits in plasma membrane and inhibit excitatory presynaptic transmission (Hendrich et al., 2008, 2012; Bauer et al., 2009, 2010; Cassidy et al., 2014), presumably by interfering with Cavα2δ1 trafficking process through disrupting the recycling of Cavα2δ1 to the plasma membrane (Tran‐Van‐Minh and Dolphin, 2010). This is supported by in vitro findings that the membrane surface expression of Cav2.2 channels is reduced and become insensitive to gabapentin blockade when co‐expressed with a Cavα2δ1 mutant lacking the binding site for gabapentinoids (Cassidy et al., 2014). Cavα2δ1 can also enhance the expression of T‐type calcium channels in plasma membrane without changing their electrophysiological properties (Dubel et al., 2004). However, Cavα2δ1 has also been found to have little or no modulatory effect on T‐type channel currents (Perez‐Reyes, 2003; Turner and Zamponi, 2014), and gabapentin failed to affect VGCC trafficking or function in cultured hippocampal neurons (Brown and Randall, 2005; Hoppa et al., 2012).
Recent data from biochemical studies suggest that an elevation in Cavα2δ1 promotes presynaptic neurotransmitter release by modulating presynaptic VGCC abundance and Ca2+ influx‐driven exocytosis. While the former can be explained by the ability of Cavα2δ1 to promote VGCC trafficking, the latter is believed to be mediated through an extracellular metal ion‐dependent adhesion site within the VWA domain of Cavα2δ1 (Hoppa et al., 2012; Korber and Kuner, 2016). These findings provide mechanistic insights, revealing how Cavα2δ plays a significant pathological role in modulating the release of numerous neurotransmitters, including pain‐inducing excitatory neurotransmitters and peptides at the spinal cord level. This can be one of the primary targets of gabapentinoids in pain modulation.
While alterations in VGCC trafficking may lead to changes in VGCC functions, and subsequently changes in sensory neuron excitability or presynaptic neurotransmitter release, it is unable to explain the fast actions of gabapentinoids in reversing pain states in animal models, which occur within 1 h of administration (Hwang and Yaksh, 1997; Luo et al., 2002). Recently, it was reported that Cavα2δ3 is required for the rapid induction and sustained maintenance of presynaptic homeostatic potentiation, a form of neuroplasticity in which the presynaptic neurotransmitter release apparatus is modulated in response to postsynaptic perturbations that may occur in neurological disorders (Wang et al., 2016). It appears that extracellular Cavα2δ3 residing in the presynaptic release sites of the synaptic cleft serves as a transsynaptic homeostatic signalling molecule that relays retrograde information to presynaptic cytoplasm to modulate active zone activities, presumably through interactions with other proteins. This plasticity seems to be independent of its ability to regulate VGCC abundance (Wang et al., 2016). If this plasticity and its sensitivity to blockade by gabapentinoids are confirmed in neuropathic pain conditions, it may underlie a new mechanism through which gabapentinoids reverse neuropathic pain states so promptly. Identification of Cavα2δ3 as a pain gene (Neely et al., 2010) (Table 1) supports this notion. However, there is no evidence so far indicating that Cavα2δ3 is a binding site for gabapentinoids, or is up‐regulated after nerve injuries. In fact, Cavα2δ3 mRNA is significantly reduced in injured DRG after nerve injury (Bauer et al., 2009). More detail studies may help to distinguish the relative contribution of Cavα2δ1 and Cavα2δ3 to pain signal transduction and the anti‐hyperalgesic actions of gabapentinoids.
In addition, it has been reported that gabapentin can block the interactions between Cavα2δ1 and synaptogenic thrombospondin‐4 (TSP4) (Park et al., 2016), which is also up‐regulated in DRG and spinal cord after peripheral nerve injury (Kim et al., 2012; Pan et al., 2016). This leads to diminished dorsal horn neuron hypersensitivity and behavioural hypersensitivity within 1 h (Park et al., 2016). This fast action of gabapentin may provide another possible explanation about the fast anti‐hyperalgesic actions of gabapentinoids.
Current states of gabapentinoids in the management and prevention of neuropathic pain states
Gabapentinoids provide pain relief mainly in a pathophysiological state‐dependent manner; they are only effective in alleviating pathological pain sensations but do not affect acute physiological noxious sensations (Field et al., 1997b; Stanfa et al., 1997; Dickenson et al., 2005). These findings suggest that gabapentinoids most likely work on a sensitized sensory pathway related to Cavα2δ1 up‐regulation. This may explain why gabapentinoids only provide significant pain relief in about one third of neuropathic pain patients (Moore et al., 2014), since it is conceivable that pathological changes induced by Cavα2δ1 up‐regulation may not be present in all neuropathic pain patients non‐selectively. This is supported by data from animal model studies indicating that gabapentin is only effective in reversing neuropathic pain states associated with detectable Cavα2δ1 up‐regulation in dorsal spinal cord and/or DRG (Luo et al., 2002). Gabapentinoids have relatively mild side effects, which include dizziness, somnolence and ataxia. The latter is believed to be mediated through an effect on calcium channel trafficking (Jun et al., 1999; Mark et al., 2011) and by binding to Cavα2δ2 (Barclay et al., 2001; Ivanov et al., 2004).
Based on evidence from randomized clinical trials (RCTs), expert panels from the Neuropathic Pain Special Interest Group of the International Association for the Study of Pain (Dworkin et al., 2007), European Federation of Neurological Societies (Attal et al., 2006) and Canadian Pain Society (Moulin et al., 2007) recommended, almost simultaneously, that gabapentinoids should be one of the first line medication classes for neuropathic pain management in 2006–2007. Recent updated reviews for using gabapentinoids in neuropathic pain management are summarized in Table 2. Depending on the aetiology of a neuropathic pain condition, the conclusions from these reviews still confirm that gabapentinoids can be used as first‐line medications or are recommended for the management of neuropathic pain derived from certain aetiologies (Dworkin et al., 2010; Vargas‐Espinosa et al., 2012; Hershman et al., 2014; Finnerup et al., 2015; Loh et al., 2016). Findings from these reviews also point out the need for more studies for dose‐standardization, to assess their adverse effects and their suitability for neuropathic pain derived from other aetiologies such as cancer, chemotherapy‐induced peripheral neuropathies, and combination therapies, so that better drug efficacy can be achieved and/or their side effects reduced (Dworkin et al., 2010; Chaparro et al., 2012; Vargas‐Espinosa et al., 2012; Hershman et al., 2014; Guan et al., 2016).
Table 2.
Recommendation of gabapentinoids for neuropathic pain management
| Review criteria/strength | Neuropathic pain‐inducing condition | Recommendation | Reference |
|---|---|---|---|
| Randomized clinical trials (RCTs)/systemic reviews/pooled analysis/retrospective study | Post‐herpetic neuralgia | Type A recommendation from consistent, good quality patient‐oriented evidence | Vargas‐Espinosa et al., 2012 |
| More studies are needed for standardizing doses and assessing adverse effects | |||
| RCTs ≥1 week, combination pharmacotherapy, neuropathic pain in cancer patients | Cancer | Further studies are needed | Guan et al., 2016 |
| Double‐blind, RCTs comparing two or more drug combinations to placebo and/or ≥1 other comparator | Varies | More studies are needed for two‐drug combinations including comparisons with placebo and both single‐agent components in neuropathic pain treatments | Chaparro et al., 2012 |
| Review established guidelines (Dworkin et al., Pain 2007) and resent RCT studies for development of future recommendations | Varies | Single medication therapy: First‐line medication | Dworkin et al., 2010 |
| Combination therapy: additional studies are needed to develop specific combination therapies. | |||
| Recent RCTs | Chemotherapy‐induced peripheral neuropathies (CIPN) | Reasonable to try for selective CIPN pain patients who should be informed about limited scientific evidence, potential benefits, harms and costs. | Hershman et al., 2014 |
| Expert opinion on clinical experience, side effect profile, effectiveness in other neuropathic pain conditions, other relevant factors | Spinal cord injury | First line therapy | Loh et al., 2016 |
| Update of evidence‐based recommendations/systemic review/double‐blind RCTs, including unpublished clinical trials, and meta‐analysis used Numbers needed to treat for 50% pain relief and assessed publication bias | Varies | First line therapy | Finnerup et al., 2015 |
Even though most animal model studies confirm that gabapentinoids are effective at reversing pain states, clinical data for the efficacy of gabapentinoids in acute and persistent pain relief are not consistent. While some studies show that gabapentinoids are effective in pain relief after surgeries, this comes at the expense of increased adverse effects (Buvanendran et al., 2010; Engelman and Cateloy, 2011; Clarke et al., 2012; Eipe et al., 2015; Mishriky et al., 2015). Other studies do not show similar benefits of gabapentinoid treatment (Lunn et al., 2015; Kharasch and Eisenach, 2016; Martinez et al., 2017a,b). This could be due to the fact that for various reasons negative data from animal model studies are often not getting published, but that is less likely to be the case for most clinical trials. These discrepancies in clinical studies has lead to the suggestion that more studies in different surgical models and specific patient populations are needed to explore the benefits of a surgical model‐specific application of gabapentinoids (Fletcher and Martinez, 2015).
One way to optimize the pharmacological benefits and reduce the adverse effects of gabapentinoids in pain management is to lower the dose of gabapentinoids by using them as a combination treatment. A few studies have addressed this option with inconsistent outcomes, as summarized in the reviews presented in Table 2. Alternatively, pre‐emptive treatment with gabapentinoids has been shown to be efficacious in blocking post‐operative pain development in patients (Field et al., 1997a; Clivatti et al., 2009; Mardani‐Kivi et al., 2013; Ravindran, 2014; Hwang et al., 2015). However, few studies have addressed the underlying mechanism of the preventive effects of gabapentinoids in chronic pain development. In another study reported in this issue, we tested the hypothesis that early low‐dose gabapentin treatment can prevent injury‐induced neuropathic pain development by inhibiting abnormal excitatory synaptogenesis between sensory and spinal cord neurons (Yu et al., 2017).
Synaptogenic effects of Cav α 2 δ 1/TSP4 in chronic pain processing
At the dorsal spinal cord level, the interactions of the up‐regulated Cavα2δ1 with TSP4 post injury can lead to aberrant excitatory synaptogenesis that is highly likely to contribute to the development of chronic neuropathic pain, as illustrated in Figure 1 and is supported by the following findings from animal studies. Peripheral nerve injury induces an up‐regulation of both Cavα2δ1 and TSP4 in the DRG and spinal cord that precedes the onset and correlates with the duration of neuropathic pain states (Luo et al., 2001; 2002; Newton et al., 2001; Li et al., 2004, 2014a; Kim et al., 2012; Pan et al., 2015). Blocking injury‐induced Cavα2δ1 up‐regulation (Boroujerdi et al., 2008; Park et al., 2016) or genetic ablation of TSP4 (Kim et al., 2012) prevents the onset and development of injury‐induced pain states. Activation of a Cavα2δ1/TSP4 dependent pathway at the spinal cord level leads to sensitization of spinal dorsal horn neurons through enhanced presynaptic excitatory neurotransmission and aberrant excitatory synapse formation that are known to contribute to neuropathic pain development after peripheral nerve injuries (Zhou and Luo, 2013; 2015; Park et al., 2016). Treatments that block injury‐induced pain states, such as genetic ablation of either Cavα2δ1 or TSP4 alone, also block injury‐induced aberrant excitatory synaptogenesis (Park et al., 2016). Furthermore, nerve injury‐induced TSP4 seems to affect VGCC subtypes in sensory neurons differently, resulting in decreased HVA and increased LVA calcium currents, by interacting with Cavα2δ1 (Pan et al., 2016). Together, these findings strongly support a critical role for injury‐induced Cavα2δ1/TSP4 in chronic neuropathic pain processing, mediated through the promotion of aberrant excitatory synapse formation and presynaptic neurotransmission in the dorsal spinal cord. The exact location of Cavα2δ1/TSP4 interactions in vivo remains to be established since data from a recent in vitro co‐transfection study suggest that Cavα2δ1/TSP4 interactions occur intracellularly (Lana et al., 2016).
New prospects of targeting Cav α 2 δ 1 for neuropathic pain treatment and prevention
The translational value of these findings suggest that blocking injury‐induced synapse formation may lead to the prevention or reversal of neuropathic pain states post‐nerve injuries. While genetic ablation or biochemical knockdown of Cavα2δ1 or TSP4 are powerful tools in mechanistic research with animal models, these approaches are not practical currently for preventing/treating neuropathic pain states in patients. Blocking injury‐induced abnormal synapse formation by small molecule drugs can be an attractive approach in preventing/managing neuropathic pain development after nerve injuries. Since gabapentinoids can block the Cavα2δ1/TSP4‐induced aberrant excitatory synaptogenesis and development of pain states (Park et al., 2016), using these drugs to block abnormal synapse formation could be a novel approach for chronic neuropathic pain management. Unfortunately, recent in vitro data indicate that late treatments, starting after the initiation of synapse formation, with gabapentinoids cannot reverse already formed excitatory synapses (Eroglu et al., 2009; Yu et al., 2017). Thus, the acute effects of gabapentinoids in blocking neuropathic pain states are unlikely to be due to reversing abnormal synapse formation.
Can early gabapentinoid treatment prevent abnormal synapse formation and pain state development? To address this question, we have tested the effects of gabapentin in synaptogenesis between cultured DRG sensory neurons and spinal cord neurons in vitro and in TSP4 injected mice in vivo (Yu et al., 2017). Our data indicate that pre‐emptive gabapentin treatment at a low‐dose can prevent TSP4‐induced excitatory synapse formation in vitro. However, delayed gabapentin treatment for 2 days after TSP4 addition, when excitatory synapses have been formed, has no anti‐synaptogenic effects. These findings were confirmed in vivo by analysing synapse formation in the dorsal spinal cord of mice 4 days after bolus intrathecal TSP4 injection, which is known to cause pain‐like behavioural hypersensitivity (Kim et al., 2012; Park et al., 2016). Early daily treatment with a sub‐anti‐hyperalgesic dose of gabapentin blocks TSP4‐induced behavioural hypersensitivity and in vivo excitatory synapse formation. However, delayed gabapentin treatment starting 2 days later is not effective in blocking TSP4‐induced pain states and excitatory synapse formation in vivo (Yu et al., 2017). These findings confirm that early blocking of the activation of the TSP4/Cavα2δ1‐dependent pathway by a low dose of gabapentin may prevent aberrant excitatory synapse formation in the spinal cord resulting from TSP4/Cavα2δ1 up‐regulation, such as in the case post‐peripheral nerve injury. Clinical validation of these results may lead to this approach being used to prevent the development of pain states.
Conclusions
Maladaptive changes in the expression and functions of the VGCC Cavα2δ1 subunit post injuries in the peripheral and central nervous systems can contribute to chronic pain development through pathological changes associated with or independent of VGCC functions. Gabapentinoids, by acting as Cavα2δ1 ligands, can reverse pain states significantly in a subpopulation of neuropathic pain patients through the various potential mechanisms discussed, some of which remain to be validated. Emerging data suggest that early preventive treatment with gabapentinoids can prevent aberrant excitatory synapse formation and pain state development, which, if confirmed clinically, could be an attractive approach for preventing neuropathic pain development.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported in part by NIH grants NS064341 and DE021847 to Z.D.L.
Gong, N. , Park, J. , and Luo, Z. D. (2018) Injury‐induced maladaptation and dysregulation of calcium channel α2δ subunit proteins and its contribution to neuropathic pain development. British Journal of Pharmacology, 175: 2231–2243. doi: 10.1111/bph.13930.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attal N, Cruccu G, Haanpaa M, Hansson P, Jensen TS, Nurmikko T et al. (2006). EFNS guidelines on pharmacological treatment of neuropathic pain. Eur J Neurol 13: 1153–1169. [DOI] [PubMed] [Google Scholar]
- Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J et al. (2001). Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci 21: 6095–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CS, Nieto‐Rostro M, Rahman W, Tran‐Van‐Minh A, Ferron L, Douglas L et al. (2009). The increased trafficking of the calcium channel subunit alpha2delta‐1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J Neurosci 29: 4076–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CS, Rahman W, Tran‐van‐Minh A, Lujan R, Dickenson AH, Dolphin AC (2010). The anti‐allodynic alpha(2)delta ligand pregabalin inhibits the trafficking of the calcium channel alpha(2)delta‐1 subunit to presynaptic terminals in vivo. Biochem Soc Trans 38: 525–528. [DOI] [PubMed] [Google Scholar]
- Benarroch EE (2010). Neuronal voltage‐gated calcium channels: brief overview of their function and clinical implications in neurology. Neurology 74: 1310–1315. [DOI] [PubMed] [Google Scholar]
- Boroujerdi A, Kim HK, Lyu YS, Kim DS, Figueroa KW, Chung JM et al. (2008). Injury discharges regulate calcium channel alpha‐2‐delta‐1 subunit upregulation in the dorsal horn that contributes to initiation of neuropathic pain. Pain 139: 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroujerdi A, Zeng J, Sharp K, Kim D, Steward O, Luo ZD (2011). Calcium channel alpha‐2‐delta‐1 protein upregulation in dorsal spinal cord mediates spinal cord injury‐induced neuropathic pain states. Pain 152: 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill J, Klocke R, Paul D, Boison D, Gouder N, Klugbauer N et al. (2004). entla, a novel epileptic and ataxic Cacna2d2 mutant of the mouse. J Biol Chem 279: 7322–7330. [DOI] [PubMed] [Google Scholar]
- Brodbeck J, Davies A, Courtney JM, Meir A, Balaguero N, Canti C et al. (2002). The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated alpha 2 delta‐2 protein with abnormal function. J Biol Chem 277: 7684–7693. [DOI] [PubMed] [Google Scholar]
- Brown JT, Randall A (2005). Gabapentin fails to alter P/Q‐type Ca2+ channel‐mediated synaptic transmission in the hippocampus in vitro. Synapse 55: 262–269. [DOI] [PubMed] [Google Scholar]
- Buvanendran A, Kroin JS, Della Valle CJ, Kari M, Moric M, Tuman KJ (2010). Perioperative oral pregabalin reduces chronic pain after total knee arthroplasty: a prospective, randomized, controlled trial. Anesth Analg 110: 199–207. [DOI] [PubMed] [Google Scholar]
- Cain SM, Snutch TP (2011). Voltage‐gated calcium channels and disease. Biofactors 37: 197–205. [DOI] [PubMed] [Google Scholar]
- Calderon‐Rivera A, Andrade A, Hernandez‐Hernandez O, Gonzalez‐Ramirez R, Sandoval A, Rivera M et al. (2012). Identification of a disulfide bridge essential for structure and function of the voltage‐gated Ca(2+) channel alpha(2)delta‐1 auxiliary subunit. Cell Calcium 51: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campiglio M, Flucher BE (2015). The role of auxiliary subunits for the functional diversity of voltage‐gated calcium channels. J Cell Physiol 230: 2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy JS, Ferron L, Kadurin I, Pratt WS, Dolphin AC (2014). Functional exofacially tagged N‐type calcium channels elucidate the interaction with auxiliary α2δ‐1 subunits. Proc Natl Acad Sci U S A 111: 8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2011). Voltage‐gated calcium channels. Cold Spring Harb Perspect Biol 3: a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E, Chen X, Kim M, Gong N, Bhatia S, Luo ZD (2015). Differential effects of voltage‐gated calcium channel blockers on calcium channel alpha‐2‐delta‐1 subunit protein‐mediated nociception. Eur J Pain 19: 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaparro LE, Wiffen PJ, Moore RA, Gilron I (2012). Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database Syst Rev 7: CD008943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke H, Bonin RP, Orser BA, Englesakis M, Wijeysundera DN, Katz J (2012). The prevention of chronic postsurgical pain using gabapentin and pregabalin: a combined systematic review and meta‐analysis. Anesth Analg 115: 428–442. [DOI] [PubMed] [Google Scholar]
- Clivatti J, Sakata RK, Issy AM (2009). Review of the use of gabapentin in the control of postoperative pain. Rev Bras Anestesiol 59: 87–98. [DOI] [PubMed] [Google Scholar]
- D'Arco M, Margas W, Cassidy JS, Dolphin AC (2015). The upregulation of alpha2delta‐1 subunit modulates activity‐dependent Ca2+ signals in sensory neurons. J Neurosci 35: 5891–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A, Kadurin I, Alvarez‐Laviada A, Douglas L, Nieto‐Rostro M, Bauer CS et al. (2010). The alpha2delta subunits of voltage‐gated calcium channels form GPI‐anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci U S A 107: 1654–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickenson AH, Bee LA, Suzuki R (2005). Pains, gains, and midbrains. Proc Natl Acad Sci U S A 102: 17885–17886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2012). Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat Rev Neurosci 13: 542–555. [DOI] [PubMed] [Google Scholar]
- Dolphin AC (2013). The alpha2delta subunits of voltage‐gated calcium channels. Biochim Biophys Acta 1828: 1541–1549. [DOI] [PubMed] [Google Scholar]
- Donato R, Page KM, Koch D, Nieto‐Rostro M, Foucault I, Davies A et al. (2006). The ducky(2J) mutation in Cacna2d2 results in reduced spontaneous Purkinje cell activity and altered gene expression. J Neurosci 26: 12576–12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubel SJ, Altier C, Chaumont S, Lory P, Bourinet E, Nargeot J (2004). Plasma membrane expression of T‐type calcium channel alpha(1) subunits is modulated by high voltage‐activated auxiliary subunits. J Biol Chem 279: 29263–29269. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, O'Connor AB, Audette J, Baron R, Gourlay GK, Haanpaa ML et al. (2010). Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc 85: S3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin RH, O'Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS et al. (2007). Pharmacologic management of neuropathic pain: evidence‐based recommendations. Pain 132: 237–251. [DOI] [PubMed] [Google Scholar]
- Edvardson S, Oz S, Abulhijaa FA, Taher FB, Shaag A, Zenvirt S et al. (2013). Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. J Med Genet 50: 118–123. [DOI] [PubMed] [Google Scholar]
- Eipe N, Penning J, Yazdi F, Mallick R, Turner L, Ahmadzai N et al. (2015). Perioperative use of pregabalin for acute pain‐a systematic review and meta‐analysis. Pain 156: 1284–1300. [DOI] [PubMed] [Google Scholar]
- Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT et al. (1988). Sequence and expression of mRNAs encoding the alpha 1 and alpha 2 subunits of a DHP‐sensitive calcium channel. Science 241: 1661–1664. [DOI] [PubMed] [Google Scholar]
- Engelman E, Cateloy F (2011). Efficacy and safety of perioperative pregabalin for post‐operative pain: a meta‐analysis of randomized‐controlled trials. Acta Anaesthesiol Scand 55: 927–943. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E et al. (2009). Gabapentin receptor alpha2delta‐1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139: 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria LC, Gu F, Parada I, Barres B, Luo ZD, Prince DA (2017). Epileptiform activity and behavioral arrests in mice overexpressing the calcium channel subunit alpha2delta‐1. Neurobiol Dis 102: 70–80. [DOI] [PubMed] [Google Scholar]
- Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ et al. (2006). Identification of the {alpha}2‐{delta}‐1 subunit of voltage‐dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci USA 103: 17537–17542 Epub 12006 Nov 17536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MJ, Holloman EF, McCleary S, Hughes J, Singh L (1997a). Evaluation of gabapentin and S‐(+)‐3‐isobutylgaba in a rat model of postoperative pain. J Pharmacol Exp Ther 282: 1242–1246. [PubMed] [Google Scholar]
- Field MJ, Oles RJ, Lewis AS, McCleary S, Hughes J, Singh L (1997b). Gabapentin (neurontin) and S‐(+)‐3‐isobutylgaba represent a novel class of selective antihyperalgesic agents. Brit J Pharmacol 121: 1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH et al. (2015). Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurol 14: 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher D, Martinez V (2015). Should we use gabapentin for postoperative pain control? Pain 156: 2402–2403. [DOI] [PubMed] [Google Scholar]
- Fuller‐Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N et al. (2009). Targeted disruption of the voltage‐dependent calcium channel alpha2/delta‐1‐subunit. Am J Physiol Heart Circ Physiol 297: H117–H124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN (1996). The novel anticonvulsant drug, gabapentin (neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem 271: 5768–5776. [DOI] [PubMed] [Google Scholar]
- Gong HC, Hang J, Kohler W, Li L, Su TZ (2001). Tissue‐specific expression and gabapentin‐binding properties of calcium channel alpha2delta subunit subtypes. J Membr Biol 184: 35–43. [DOI] [PubMed] [Google Scholar]
- Guan J, Tanaka S, Kawakami K (2016). Anticonvulsants or antidepressants in combination pharmacotherapy for treatment of neuropathic pain in cancer patients: a systematic review and meta‐analysis. Clin J Pain 32: 719–725. [DOI] [PubMed] [Google Scholar]
- Hendrich J, Bauer CS, Dolphin AC (2012). Chronic pregabalin inhibits synaptic transmission between rat dorsal root ganglion and dorsal horn neurons in culture. Channels (Austin) 6: 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich J, Van Minh AT, Heblich F, Nieto‐Rostro M, Watschinger K, Striessnig J et al. (2008). Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci U S A 105: 3628–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershman DL, Lacchetti C, Dworkin RH, Lavoie Smith EM, Bleeker J, Cavaletti G et al. (2014). Prevention and management of chemotherapy‐induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 32: 1941–1967. [DOI] [PubMed] [Google Scholar]
- Hino‐Fukuyo N, Kikuchi A, Arai‐Ichinoi N, Niihori T, Sato R, Suzuki T et al. (2015). Genomic analysis identifies candidate pathogenic variants in 9 of 18 patients with unexplained West syndrome. Hum Genet 134: 649–658. [DOI] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA (2012). alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature 486: 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JH, Yaksh TL (1997). Effect of subarachnoid gabapentin on tactile‐evoked allodynia in a surgically induced neuropathic pain model in the rat. Reg Anesth 22: 249–256. [DOI] [PubMed] [Google Scholar]
- Hwang SH, Park IJ, Cho YJ, Jeong YM, Kang JM (2015). The efficacy of gabapentin/pregabalin in improving pain after tonsillectomy: a meta‐analysis. Laryngoscope. https://doi.org/10.1002/lary.25636. [DOI] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74: 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov SV, Ward JM, Tessarollo L, McAreavey D, Sachdev V, Fananapazir L et al. (2004). Animal model – cerebellar ataxia, seizures, premature death, and cardiac abnormalities in mice with targeted disruption of the Cacna2d2 gene. Am J Pathol 165: 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RW, Rice AS (2014). Clinical practice. Postherpetic neuralgia. N Engl J Med 371: 1526–1533. [DOI] [PubMed] [Google Scholar]
- Jun K, Piedras‐Rentería ES, Smith SM, Wheeler DB, Lee SB, Lee TG et al. (1999). Ablation of P/Q‐type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)‐subunit. Proc Natl Acad Sci U S A 96: 15245–15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharasch ED, Eisenach JC (2016). wherefore gabapentinoids?: was there rush too soon to judgment? Anesthesiology 124: 10–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Li KW, Boroujerdi A, Peter Yu Y, Zhou CY, Deng P et al. (2012). Thrombospondin‐4 contributes to spinal sensitization and neuropathic pain states. J Neurosci 32: 8977–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoflach D, Kerov V, Sartori SB, Obermair GJ, Schmuckermair C, Liu X et al. (2013). Cav1.4 IT mouse as model for vision impairment in human congenital stationary night blindness type 2. Channels 7: 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber C, Kuner T (2016). Molecular machines regulating the release probability of synaptic vesicles at the active zone. Front Synaptic Neurosci 8: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukkar A, Bali A, Singh N, Jaggi AS (2013). Implications and mechanism of action of gabapentin in neuropathic pain. Arch Pharm Res 36: 237–251. [DOI] [PubMed] [Google Scholar]
- Lana B, Page KM, Kadurin I, Ho S, Nieto‐Rostro M, Dolphin AC (2016). Thrombospondin‐4 reduces binding affinity of [(3)H]‐gabapentin to calcium‐channel α2δ‐1‐subunit but does not interact with α2δ‐1 on the cell‐surface when co‐expressed. Sci Rep 6: 24531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana B, Schlick B, Martin S, Pratt WS, Page KM, Goncalves L et al. (2014). Differential upregulation in DRG neurons of an alpha2delta‐1 splice variant with a lower affinity for gabapentin after peripheral sensory nerve injury. Pain 155: 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmark CJ, Beiske G, Baftiu A, Burns ML, Johannessen SI (2015). Experience from therapeutic drug monitoring and gender aspects of gabapentin and pregabalin in clinical practice. Seizure‐Eur J Epilep 28: 88–91. [DOI] [PubMed] [Google Scholar]
- Lee S (2013). Pharmacological inhibition of voltage‐gated Ca(2+) channels for chronic pain relief. Curr Neuropharmacol 11: 606–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CY, Song YH, Higuera ES, Luo ZD (2004). Spinal dorsal horn calcium channel alpha2delta‐1 subunit upregulation contributes to peripheral nerve injury‐induced tactile allodynia. J Neurosci 24: 8494–8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CY, Zhang XL, Matthews EA, Li KW, Kurwa A, Boroujerdi A et al. (2006). Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain 125: 20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KW, Kim DS, Zaucke F, Luo ZD (2014a). Trigeminal nerve injury‐induced thrombospondin‐4 up‐regulation contributes to orofacial neuropathic pain states in a rat model. Eur J Pain 18: 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KW, Yu YP, Zhou C, Kim DS, Lin B, Sharp K et al. (2014b). Calcium channel alpha2delta1 proteins mediate trigeminal neuropathic pain states associated with aberrant excitatory synaptogenesis. J Biol Chem 289: 7025–7037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Taylor CP, Weber M, Piechan J, Prior F, Bian F et al. (2011). Pregabalin is a potent and selective ligand for alpha(2)delta‐1 and alpha(2)delta‐2 calcium channel subunits. Eur J Pharmacol 667: 80–90. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Andrade A, Allen SE (2013). Alternative splicing: functional diversity among voltage‐gated calcium channels and behavioral consequences. Biochim Biophys Acta 1828: 1522–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XG, Zhou LJ (2015). Long‐term potentiation at spinal C‐fiber synapses: a target for pathological pain. Curr Pharm Des 21: 895–905. [DOI] [PubMed] [Google Scholar]
- Loh E, Guy SD, Mehta S, Moulin DE, Bryce TN, Middleton JW et al. (2016). The CanPain SCI clinical practice guidelines for rehabilitation management of neuropathic pain after spinal cord: introduction, methodology and recommendation overview. Spinal Cord 54 (Suppl 1): S1–S6. [DOI] [PubMed] [Google Scholar]
- Lotarski S, Hain H, Peterson J, Galvin S, Strenkowski B, Donevan S et al. (2014). Anticonvulsant activity of pregabalin in the maximal electroshock‐induced seizure assay in alpha2delta1 (R217A) and alpha2delta2 (R279A) mouse mutants. Epilepsy Res 108: 833–842. [DOI] [PubMed] [Google Scholar]
- Lotarski SM, Donevan S, El‐Kattan A, Osgood S, Poe J, Taylor CP et al. (2011). Anxiolytic‐like activity of pregabalin in the Vogel conflict test in alpha2delta‐1 (R217A) and alpha2delta‐2 (R279A) mouse mutants. J Pharmacol Exp Ther 338: 615–621. [DOI] [PubMed] [Google Scholar]
- Lunn TH, Husted H, Laursen MB, Hansen LT, Kehlet H (2015). Analgesic and sedative effects of perioperative gabapentin in total knee arthroplasty: a randomized, double‐blind, placebo‐controlled dose‐finding study. Pain 156: 2438–2448. [DOI] [PubMed] [Google Scholar]
- Luo ZD (2000). Rat dorsal root ganglia express distinctive forms of the alpha2 calcium channel subunit. Neuroreport 11: 3449–3452. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI et al. (2002). Injury type‐specific calcium channel alpha 2 delta‐1 subunit up‐regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J Pharmacol Exp Ther 303: 1199–1205. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME et al. (2001). Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve‐injured rats. J Neurosci 21: 1868–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais E, Klugbauer N, Hofmann F (2001a). Calcium channel alpha2delta subunits‐structure and gabapentin binding. Mol Pharmacol 59: 1243–1248. [DOI] [PubMed] [Google Scholar]
- Marais E, Klugbauer N, Hofmann F (2001b). Calcium channel alpha(2)delta subunits – Structure and gabapentin binding. Mol Pharmacol 59: 1243–1248. [DOI] [PubMed] [Google Scholar]
- Mardani‐Kivi M, Mobarakeh MK, Keyhani S, Motlagh KH, Ekhtiari KS (2013). Is gabapentin effective on pain management after arthroscopic anterior cruciate ligament reconstruction? A triple blinded randomized controlled trial. Arch Bone Jt Surg 1: 18–22. [PMC free article] [PubMed] [Google Scholar]
- Margas W, Ferron L, Nieto‐Rostro M, Schwartz A, Dolphin AC (2016). Effect of knockout of alpha2delta‐1 on action potentials in mouse sensory neurons. Philos Trans R Soc Lond B Biol Sci 371: 20150430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark MD, Maejima T, Kuckelsberg D, Yoo JW, Hyde RA, Shah V et al. (2011). Delayed postnatal loss of P/Q‐type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1A mutations. J Neurosci 31: 4311–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez V, Carles M, Marret E, Beloeil H, Regional Anaesthesia PCotFSoAIC (2017a). Perioperative use of gabapentinoids in France. Anaesth Crit Care Pain Med. In Press. [DOI] [PubMed] [Google Scholar]
- Martinez V, Pichard X, Fletcher D (2017b). Perioperative pregabalin administration does not prevent chronic postoperative pain. Systematic review with a meta‐analysis of randomized trials. Pain 158: 775–783. [DOI] [PubMed] [Google Scholar]
- Mishriky BM, Waldron NH, Habib AS (2015). Impact of pregabalin on acute and persistent postoperative pain: a systematic review and meta‐analysis. Br J Anaesth 114: 10–31. [DOI] [PubMed] [Google Scholar]
- Moore RA, Wiffen PJ, Derry S, Toelle T, Rice AS (2014). Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev 4: CD007938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulin DE, Clark AJ, Gilron I, Ware MA, Watson CP, Sessle BJ et al. (2007). Pharmacological management of chronic neuropathic pain ‐ consensus statement and guidelines from the Canadian Pain Society. Pain Res Manag 12: 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka A, Gruber‐Schoffnegger D, Sandkuhler J (2013). Non‐Hebbian plasticity at C‐fiber synapses in rat spinal cord lamina I neurons. Pain 154: 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely GG, Hess A, Costigan M, Keene AC, Goulas S, Langeslag M et al. (2010). A genome‐wide Drosophila screen for heat nociception identifies alpha2delta3 as an evolutionarily conserved pain gene. Cell 143: 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton RA, Bingham S, Case PC, Sanger GJ, Lawson SN (2001). Dorsal root ganglion neurons show increased expression of the calcium channel alpha2delta‐1 subunit following partial sciatic nerve injury. Brain Res Mol Brain Res 95: 1–8. [DOI] [PubMed] [Google Scholar]
- Nguyen D, Deng P, Matthews EA, Kim DS, Feng G, Dickenson AH et al. (2009). Enhanced pre‐synaptic glutamate release in deep‐dorsal horn contributes to calcium channel alpha‐2‐delta‐1 protein‐mediated spinal sensitization and behavioral hypersensitivity. Mol Pain 5: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Guo Y, Wu HE, Park J, Trinh VN, Luo ZD et al. (2016). Thrombospondin‐4 divergently regulates voltage‐gated Ca2+ channel subtypes in sensory neurons after nerve injury. Pain 157: 2068–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Yu H, Park J, Yu YP, Luo ZD, Hogan QH (2015). Painful nerve injury upregulates thrombospondin‐4 expression in dorsal root ganglia. J Neurosci Res 93: 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Luo ZD (2010). Calcium channel functions in pain processing. Channels (Austin) 4: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Yu YP, Zhou CY, Li KW, Wang D, Chang E et al. (2016). Central mechanisms mediating thrombospondin‐4‐induced pain states. J Biol Chem 291: 13335–13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Bauer CS, Nieto‐Rostro M, Margas W, Ferron L, Chaggar K et al. (2013). alpha2delta‐1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. J Neurosci 33: 16412–16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E (2003). Molecular physiology of low‐voltage‐activated t‐type calcium channels. Physiol Rev 83: 117–161. [DOI] [PubMed] [Google Scholar]
- Perret D, Luo ZD (2009). Targeting voltage‐gated calcium channels for neuropathic pain management. Neurotherapeutics 6: 679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippucci T, Parmeggiani A, Palombo F, Maresca A, Angius A, Crisponi L et al. (2013). A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS ONE [Electronic Resource] 8: e82154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran D (2014). Chronic postsurgical pain: prevention and management. J Pain Palliat Care Pharmacother 28: 51–53. [DOI] [PubMed] [Google Scholar]
- Russo RE, Hounsgaard J (1994). Short‐term plasticity in turtle dorsal horn neurons mediated by L‐type Ca2+ channels. Neuroscience 61: 191–197. [DOI] [PubMed] [Google Scholar]
- Simms BA, Zamponi GW (2014). Neuronal voltage‐gated calcium channels: structure, function, and dysfunction. Neuron 82: 24–45. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfa LC, Singh L, Williams RG, Dickenson AH (1997). Gabapentin, ineffective in normal rats, markedly reduces C‐fibre evoked responses after inflammation. Neuroreport 8: 587–590. [DOI] [PubMed] [Google Scholar]
- Tran‐Van‐Minh A, Dolphin AC (2010). The alpha2delta ligand gabapentin inhibits the Rab11‐dependent recycling of the calcium channel subunit alpha2delta‐2. J Neurosci 30: 12856–12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RW, Zamponi GW (2014). T‐type channels buddy up. Pflugers Arch 466: 661–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valder CR, Liu JJ, Song YH, Luo ZD (2003). Coupling gene chip analyses and rat genetic variances in identifying potential target genes that may contribute to neuropathic allodynia development. J Neurochem 87: 560–573. [DOI] [PubMed] [Google Scholar]
- Vargas‐Espinosa ML, Sanmarti‐Garcia G, Vazquez‐Delgado E, Gay‐Escoda C (2012). Antiepileptic drugs for the treatment of neuropathic pain: a systematic review. Med Oral Patol Oral Cir Bucal 17: e786–e793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergult S, Dheedene A, Meurs A, Faes F, Isidor B, Janssens S et al. (2015). Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur J Hum Genet 23: 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma V, Singh N, Jaggi AS (2014). Pregabalin in neuropathic pain: evidences and possible mechanisms. Current Neuropharmacol 12: 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink S, Alewood PF (2012). Targeting voltage‐gated calcium channels: developments in peptide and small‐molecule inhibitors for the treatment of neuropathic pain. Br J Pharmacol 167: 970–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Jones RT, Whippen JM, Davis GW (2016). α2δ‐3 is required for rapid Transsynaptic homeostatic signaling. Cell Rep 16: 2875–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P et al. (2012). Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB‐dependent gene expression. Cell 149: 1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M et al. (2016). Structure of the voltage‐gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature 537: 191–196. [DOI] [PubMed] [Google Scholar]
- Wycisk KA, Budde B, Feil S, Skosyrski S, Buzzi F, Neidhardt J et al. (2006a). Structural and functional abnormalities of retinal ribbon synapses due to Cacna2d4 mutation. Invest Ophthalmol Vis Sci 47: 3523–3530. [DOI] [PubMed] [Google Scholar]
- Wycisk KA, Zeitz C, Feil S, Wittmer M, Forster U, Neidhardt J et al. (2006b). Mutation in the auxiliary calcium‐channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet 79: 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn DH, Gerber G, Sather WA (2013). Ionotropic glutamate receptors and voltage‐gated Ca(2)(+) channels in long‐term potentiation of spinal dorsal horn synapses and pain hypersensitivity. Neural Plast 2013: 654257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YP, Kweon TD, Vo B, Gong N, Luo ZD (2017). Thrombospondin 4 acts on pre‐synaptic calcium channel alpha‐2‐delta‐1 subunits to induce synaptogenesis between sensory and spinal cord neurons through a T‐type calcium channel pathway that is blocked by early, low‐dose gabapentin treatments. in revision.
- Yusaf SP, Goodman J, Gonzalez IM, Bramwell S, Pinnock RD, Dixon AK et al. (2001). Streptozocin‐induced neuropathy is associated with altered expression of voltage‐gated calcium channel subunit mRNAs in rat dorsal root ganglion neurones. Biochem Biophys Res Commun 289: 402–406. [DOI] [PubMed] [Google Scholar]
- Zamponi GW (2016). Targeting voltage‐gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov 15: 19–34. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A, Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67: 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Luo ZD (2013). Electrophysiological characterization of spinal neuron sensitization by elevated calcium channel alpha‐2‐delta‐1 subunit protein. Eur J Pain 18: 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Luo ZD (2015). Nerve injury‐induced calcium channel alpha‐2‐delta‐1 protein dysregulation leads to increased pre‐synaptic excitatory input into deep dorsal horn neurons and neuropathic allodynia. Eur J Pain 19: 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]