

Abstract
Aims
Gemcitabine (2′,2′‐difluoro‐2′‐deoxycytidine; dFdC) is a prodrug that has to be phosphorylated within the tumour cell to become active. Intracellularly formed gemcitabine diphosphate (dFdCDP) and triphosphate (dFdCTP) are considered responsible for the antineoplastic effects of gemcitabine. However, a major part of gemcitabine is converted into 2′,2′‐difluoro‐2′‐deoxyuridine (dFdU) by deamination. In the cell, dFdU can also be phosphorylated to its monophosphate (dFdUMP), diphosphate (dFdUDP) and triphosphate (dFdUTP). In vitro data suggest that these dFdU nucleotides might also contribute to the antitumour effects, although little is known about their intracellular pharmacokinetics (PK). Therefore, the objective of the present study was to gain insight into the intracellular PK of all dFdC and dFdU nucleotides formed during gemcitabine treatment.
Methods
Peripheral blood mononuclear cell (PBMC) samples were collected from 38 patients receiving gemcitabine, at multiple time points after infusion. Gemcitabine, dFdU and their nucleotides were quantified in PBMCs. In addition, gemcitabine and dFdU plasma concentrations were monitored. The individual PK parameters in plasma and in PBMCs were determined.
Results
Both in plasma and in PBMCs, dFdU was present in higher concentrations than gemcitabine [mean intracellular area under the concentration–time curve from time zero to 24 h (AUC0–24 h) 1650 vs. 95 μM*h]. However, the dFdUMP, dFdUDP and dFdUTP concentrations in PBMCs were much lower than the dFdCDP and dFdCTP concentrations. The mean AUC0–24 h for dFdUTP was 312 μM*h vs. 2640 μM*h for dFdCTP.
Conclusions
The study provides the first complete picture of all nucleotides that are formed intracellularly during gemcitabine treatment. Low intracellular dFdU nucleotide concentrations were found, which calls into question the relevance of these nucleotides for the cytotoxic effects of gemcitabine.
Keywords: 2′,2′‐difluorodeoxycytidine triphosphate (dFdCTP); 2′,2′‐difluorodeoxyuridine triphosphate (dFdUTP); gemcitabine; nucleotides; peripheral blood mononuclear cells (PBMCs); pharmacokinetics
What is Already Known about this Subject
Gemcitabine is a prodrug; two intracellularly formed gemcitabine nucleotides are considered responsible for the antitumour effect.
A major part of gemcitabine is converted into 2′,2′‐difluorodeoxyuridine (dFdU).
In vitro data suggest that intracellularly formed dFdU nucleotides also contribute to the antitumour effect, although little is known about their formation in patients.
What this Study Adds
This study provides the first complete picture of all nucleotides that are formed intracellularly during gemcitabine treatment.
Despite the high dFdU plasma concentrations, the intracellular dFdU nucleotide concentrations were low – much lower than the gemcitabine nucleotide concentrations.
This finding calls into question the relevance of dFdU nucleotides for the cytotoxic effects of gemcitabine.
Introduction
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4793 (2′,2′‐difluoro‐2′‐deoxycytidine; dFdC) is an important anticancer drug in the treatment of a variety of solid tumours, including advanced pancreatic, bladder, breast, ovarian and nonsmall cell lung cancer. It is a deoxycytidine analogue in which the two hydrogen atoms at the 2′‐position of the deoxyribose moiety are replaced by fluorine.
To become active, gemcitabine has to enter the tumour cell, where it has to be activated by phosphorylation. The intracellular metabolism of gemcitabine is illustrated in Figure 1. Cellular uptake of this agent is largely mediated by human equilibrative nucleoside transporter (hENT) 1 and, to a lesser extent, also by hENT2 and the human concentrative nucleoside transporters (hCNT) 1 and 3 1, 2. Once inside the cell, gemcitabine is phosphorylated in the cytoplasm by deoxycytidine kinase (dCK) to its monophosphate (dFdCMP) and then by pyrimidine nucleoside monophosphate kinase (UMP‐CMP kinase) to gemcitabine diphosphate (dFdCDP). The enzyme responsible for the final phosphorylation step, the phosphorylation of dFdCDP to gemcitabine triphosphate (dFdCTP) is unclear, although nucleoside diphosphate kinase may carry out this role 3.
Figure 1.
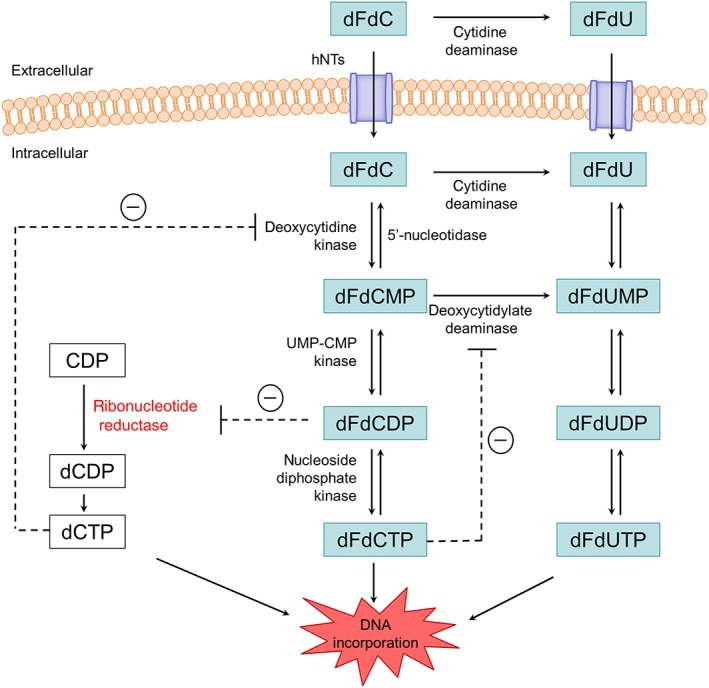
Gemcitabine metabolism and mechanisms of action. dFdC, 2′,2′‐difluorodeoxycytidine (gemcitabine); dFdCMP, 2′,2′‐difluorodeoxycytidine 5′‐monophosphate; dFdCDP, 2′,2′‐difluorodeoxycytidine 5′‐diphosphate; dFdCTP, 2′,2′‐difluorodeoxycytidine 5′‐triphosphate; dFdU, 2′,2′‐difluorodeoxyuridine; dFdUMP, 2′,2′‐difluorodeoxyuridine 5′‐monophosphate; dFdUDP, 2′,2′‐difluorodeoxyuridine 5′‐diphosphate; dFdUTP, 2′,2′‐difluorodeoxyuridine 5′‐triphosphate; dCMP, deoxycytidine monophosphate; dCDP, deoxycytidine diphosphate; dCTP, deoxycytidine triphosphate; hNTs, human nucleoside transporters; UMP‐CMP kinase, pyrimidine nucleoside monophosphate kinase
Intracellularly formed dFdCDP and dFdCTP are considered responsible for the cytotoxic effects of gemcitabine. dFdCTP competes with deoxycytidine triphosphate (dCTP) for incorporation into DNA. When dFdCTP is incorporated into DNA, this interferes with the DNA synthesis and triggers apoptosis 4, 5, 6. An additional mechanism of action of gemcitabine is self‐potentiation. dFdCDP inhibits the enzyme http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2630, which catalyses the reduction of ribonucleotides to deoxyribonucleotides 7. By inhibiting this enzyme, the intracellular deoxyribonucleotide triphosphate (dNTP) pool is depleted. This increases the dFdCTP/dCTP ratio, which favours the incorporation of dFdCTP into the DNA. As dCK activity is also regulated by dCTP, reducing the dNTP pool also promotes gemcitabine phosphorylation 8.
A major mechanism for the elimination of gemcitabine is deamination to 2′,2′‐difluoro‐2′‐deoxyuridine (dFdU) by cytidine deaminase (CDA) (Figure 1). This deamination takes place in the liver, in the blood compartment, and within normal and tumour cells 9, 10. dFdU can also be taken up by cells – for instance, by hCNT1 11. In the cell, dFdU can be phosphorylated to its monophosphate (dFdUMP), diphosphate (dFdUDP) and triphosphate (dFdUTP) 11. Alternatively, dFdCMP can be deaminated to dFdUMP, by deoxycytidylate deaminase, and then further phosphorylated 9, 10. In vitro studies have demonstrated that dFdUTP is incorporated into both DNA and RNA, and that the extent of incorporation is correlated with the cytotoxicity of dFdU 11. This suggests that dFdUTP incorporation might also contribute to the cytotoxicity of gemcitabine. In addition, recent studies have indicated that dFdUMP, which is structurally similar to deoxyuridine monophosphate (dUMP), inhibits the enzyme thymidylate synthase in tumour cell lines. This might also contribute to the cytotoxic effect of gemcitabine 12.
Measurement of the intracellular concentrations of the active metabolites during gemcitabine treatment provides useful information that might help to optimize dosing regimens for gemcitabine. Therefore, numerous studies have examined the intracellular pharmacokinetics (PK) of dFdCTP in patients treated with gemcitabine 13, 14, 15, 16, 17, 18, 19, 20, 21. However, much less is known about the intracellular PK of dFdCDP, which also contributes to the activity of gemcitabine. In addition, very little is known about the intracellular PK of the dFdU nucleotides (i.e. dFdUMP, dFdUDP and dFdUTP) during gemcitabine treatment, although in vitro data suggest that these dFdU nucleotides might also contribute to the cytotoxic effect of gemcitabine 11. The objective of the present study was to gain insight into the intracellular concentration–time course of all dFdC and dFdU nucleotides during treatment with gemcitabine. To this end, the intracellular concentrations of gemcitabine, dFdU and all nucleotides that could be expected (i.e. dFdCMP, dFdCDP dFdCTP, dFdUMP, dFdUDP and dFdUTP) were monitored in peripheral blood mononuclear cells (PBMCs) obtained at several time points during and after gemcitabine treatment.
Methods
Study design and treatment schedule
The intracellular PK of gemcitabine, dFdU and their nucleotides was examined in two patient cohorts. Cohort A consisted of 12 female patients who were participating in a phase I study in advanced breast cancer 22, exploring the safety, PK profile and the preliminary antitumour activity of the combination of gemcitabine and lapatinib. Gemcitabine was given as a 30‐min intravenous infusion at a dose of 1000 mg m–2 on days 1, 8 and 15 of a 28‐day treatment cycle. Patients received lapatinib once daily according to a dose escalation schedule. PBMC samples were collected on day 1 of the first treatment cycle prior to gemcitabine infusion, at the end of the infusion, and 2 h and 24 h after the start of the infusion. On the same day, plasma samples were collected to monitor gemcitabine and dFdU plasma concentrations prior to, at the end of, and at 1, 2, 4, 8 and 24 h after start of the infusion.
Cohort B consisted of 26 patients with advanced solid tumours who were participating in a phase I study investigating the safety, PK and preliminary antitumour activity of the combination of gemcitabine, sorafenib and carboplatin. Gemcitabine was given as a 30‐min intravenous infusion of 500 mg m–2 on days 1 and 8 of a 21‐day treatment cycle. Patients received sorafenib once or twice daily according to a dose escalation schedule. Carboplatin [area under the plasma concentration–time curve (AUC) 2 mg*min ml–1 or 3 mg*min ml–1) was administered as a 30‐min intravenous infusion on day 1 of every cycle. The gemcitabine infusion was started 90 min after the start of the carboplatin infusion. PBMC samples were collected on day 1 of the first treatment cycle prior to the infusion, at the end of the infusion, and 2 h and 22 h after the start of the infusion. Plasma samples were collected prior to, at the end of, and at 2, 4, 6 and 22 h after the start of the gemcitabine infusion.
The studies were approved by the medical ethics committee of our institute and were conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent before enrolment.
Quantification of gemcitabine and dFdU in plasma
To monitor the gemcitabine and dFdU plasma concentrations, 3 ml of blood was collected at the defined time points. Samples were collected in heparinized tubes containing 500 μg of the CDA inhibitor tetrahydrouridine (Calbiochem, La Jolla, CA, USA) and were immediately centrifuged at 4°C for 5 min at 1500 × g. Plasma was collected and stored at −20°C until analysis. Gemcitabine and dFdU were quantified in plasma by a validated liquid chromatography tandem‐mass spectrometry (LC–MS/MS) assay, as previously described by our group 23, with some minor adjustments. In brief, 13C15N2‐labelled isotopes of the analytes were used as internal standards and added to 200 μl of plasma. A solid‐phase extraction (SPE) was performed using OASIS HLB 30 mg SPE cartridges (Waters Corporation, Milford, MA, USA). The SPE cartridges were conditioned with 500 μl methanol and 500 μl water. The plasma sample was then loaded onto the cartridge, followed by 500 μl water. After drying the cartridge for 2 min with air, the analytes were eluted with 400 μl methanol. Methanol was evaporated under a gentle stream of nitrogen at 40°C and the residue was redissolved in 100 μl of reconstitution solvent [1 mM ammonium acetate (pH 6.8)/acetonitrile (97:3, v/v)]. Chromatographic separations were performed using a Synergi Hydro‐RP column (150 mm × 4.6 mm ID, particle size 5 μm; Phenomenex, Torrance, CA, USA) kept at 30°C. The mobile phase consisted of 1 mM ammonium acetate (pH 6.8)/acetonitrile (94:6, v/v) and was delivered isocratically at a flow rate of 0.2 ml min–1. Detection of the analytes was performed on an API3000 triple quadrupole mass spectrometer equipped with an electrospray ionization probe (Sciex, Framingham, MA, USA). Gemcitabine was detected in positive ion mode, and dFdU in negative ion mode. The validated concentration ranges in plasma were 0.5–1000 ng ml–1 (1.90–3799 nM) for gemcitabine and 50–10 000 ng ml–1 (189–37 853 nM) for dFdU. The accuracies for gemcitabine and dFdU in plasma were within ±16.3% at the lower limit of quantification (LLOQ) and within ±10.7% at the other levels tested. The precisions were <12.2% at all concentration levels.
Quantification of gemcitabine, dFdU and their nucleotides in PBMCs
Previous studies performed by our group demonstrated that, if samples were processed on ice‐water, the intracellular dFdC and dFdU nucleotide concentrations in PBMCs remained stable for at least 3 h and were not influenced by the addition of a CDA inhibitor (tetrahydrouridine) or deoxycytidylate deaminase inhibitor (zebularine) 24. Based on these data, PBMCs were isolated and processed on ice‐water, as described previously 25, without addition of an enzyme inhibitor. In brief, 15 ml of blood was collected in heparinized tubes and PBMCs were isolated immediately using cold Ficoll‐Paque PLUS density gradient (GE Healthcare, Pittsburgh, PA, USA). The collected PBMCs were resuspended in 100 μl cold phosphate‐buffered saline (PBS), resulting in a homogeneous cell suspension with a total volume of approximately 130 μl. A 10 μl aliquot of this cell suspension was used for the analysis of protein concentrations using the Bio‐Rad Protein Assay 26, which was corrected for interference by residual haemoglobin, as previously described by our group 27. A 100 μl aliquot of the cell suspension was used for the quantification of gemcitabine, dFdU and their monophosphates, diphosphates and triphosphates. To this end, cells were lysed by the addition of 100 μl 1.2 M perchloric acid (HClO4), followed by extensive vortex mixing. After centrifugation (5 min, 1500 × g, at 4°C), the supernatant (PBMC lysate) was collected and stored at −70°C until analysis. Directly prior to LC–MS/MS analysis, the PBMC lysates were spiked with internal standard: a mixture of 13C15N2‐labelled gemcitabine and dFdU nucleotides, which were synthesized in‐house 24.
The concentrations of gemcitabine, dFdCMP, dFdCDP, dFdCTP, dFdU, dFdUMP, dFdUDP and dFdUTP in the PBMC lysates were determined using the validated simultaneous LC–MS/MS assay previously described by our group 24, with some minor adjustments. In brief, chromatographic separation of all eight compounds was obtained using a porous graphitic carbon (Hypercarb) column (100 mm × 2.1 mm ID, 5 μm particles, Thermo Fisher Scientific Inc., Waltham, MA, USA) kept at 45°C. Elution was performed using a gradient from 0 to 25 mM ammonium bicarbonate in acetonitrile–water (15:85, v/v). As previously described, the chromatographic separation was only reproducible when the redox state of the porous graphitic carbon was carefully maintained and the column was properly cleaned 24, 28. Therefore, before each analytical run the column was treated for 30 min with preconditioning buffer (consisting of 1 mM ammonium acetate in acetonitrile/water (15:85, v/v) with 0.05% hydrogen peroxide and with the pH adjusted to 4 with glacial acetic acid); between each analytical injection, 100 μl diluted formic acid (10% v/v, in water) was injected, and after each analytical run the column was back‐flushed with about 20 column volumes of tetrahydrofuran 24.
Detection was performed on an API4000 triple quadrupole mass spectrometer equipped with an electrospray ionization probe operating in the positive ionization mode (Sciex). Calibration ranges in PBMC lysate were 2.91–289.1 nM for dFdC; 22.1–2197 nM for dFdCMP; 46.9–4655 nM for dFdCDP; 39.2–3888 nM for dFdCTP; 49.0–4864 nM for dFdU; 20.8–2063 nM for dFdUMP; 48.9–4847 nM for dFdUDP; and 46.7–4630 nM for dFdUTP. Accuracies were within ±19.4% at the LLOQ and within ±14.4% at the other levels tested. The precisions were <19.7% at the LLOQ and <14.9% at the other levels tested.
The analytical results, expressed as nM in PBMC lysate, were multiplied with the lysate sample volume to obtain the absolute nucleoside and nucleotide amounts in a sample. These amounts were then divided by the number of cells in the sample, to obtain the nucleoside and nucleotide amounts per million PBMCs. Instead of performing a direct cell count, which requires immediate analysis of each sample, the number of cells was derived from the amount of protein in the sample, as samples could be stored for longer periods prior to analysis of the protein concentrations. In addition, we assessed the correlation between the cellular protein content and the cell number. To this end, a cell count was performed for 27 PBMC samples prior to cell lysis. The mean PBMC concentration in the cell suspensions in PBS was 53 × 106 PBMCs ml–1 [coefficient of variation (CV) = 81%]. The mean amount of protein in the cell suspensions (corrected for haemoglobin) was 6.6 mg ml–1 (CV = 60%). Thus, the samples contained, on average, 0.12 mg protein per 106 PBMCs.
In order to compare the intracellular concentrations with the plasma concentrations, the intracellular concentrations were also converted to the nucleoside and nucleotide amounts per volume unit (μM). To this end, the number of PBMCs in each sample was derived from the amount of protein, and an average cell volume of 282.9 fl was assumed (as determined by Simiele et al. 29).
PK and statistical analysis
The individual PK parameters in plasma and in PBMCs were derived using noncompartmental analysis, using validated scripts in the software package R (version 3.1.2). Sufficient plasma samples were collected from each patient, to determine the plasma half‐lives (t½) of dFdC and dFdU and to extrapolate the areas under the plasma concentration‐time curves to infinity (AUCinf). The terminal plasma t½ of dFdC and dFdU were calculated based on the last three data points.
The number of PBMC samples collected per patient was limited, to minimize the burden of blood sampling (as a larger blood volume of 15 ml was required per time point, to isolate sufficient PBMCs) and because of the practical feasibility (each sample had to be processed immediately after collection owing to limited stability of the analytes). As only four PBMC samples were collected per patient, it was not possible to determine the intracellular t½. Instead, the percentage of the maximum observed concentration (Cmax) that was still present 22 h or 24 h after gemcitabine infusion was reported. Despite the limited number of PBMC sample times, the intracellular AUC from time zero up to the last sample time (AUC0–t) was calculated.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 30, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 31.
Results
Plasma PK of gemcitabine and dFdU
The mean plasma concentration–time curves for gemcitabine and dFdU are shown in Figure 2, and the results of the noncompartmental PK analysis in plasma are shown in Table 1. As expected, gemcitabine plasma concentrations were highest at the end of the infusion. The highest dFdU plasma concentration was also measured at this time in the majority of patients, indicating that gemcitabine is converted rapidly to dFdU.
Figure 2.
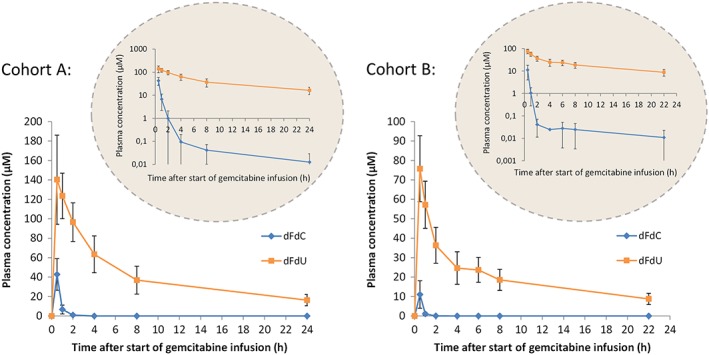
Plasma concentrations of gemcitabine (dFdC) and 2′,2′‐difluorodeoxyuridine (dFdU) for patient cohorts A and B, receiving gemcitabine 1000 mg m–2 and 500 mg m–2, respectively. The data are shown as mean values (symbols) with standard deviations (error bars). Inset: semi‐logarithmic presentation
Table 1.
Summary statistics for the pharmacokinetic parameters of gemcitabine (2′,2′‐difluorodeoxycytidine; dFdC) and its metabolite 2′,2′‐difluorodeoxyuridine (dFdU) in plasma. The data are shown as mean values (range)
| dFdC | dFdU | |
|---|---|---|
| Cohort A (receiving gemcitabine 1000 mg m –2 ) | ||
| n | 12 | 12 |
| C max (μM) | 42.8 (16.7–81.7) | 147 (99.9–224) |
| t max (h) | 0.5 (0.5–0.5) | 0.8 (0.5–2) |
| t ½ , β (h) | 7.32 (3.43–19.5) | 11.2 (8.65–13.2) |
| CL (l h –1 m –2 ) | 153 (66.7–341) | – |
| Percentage of C max still present after 24 h (%) | 0.027 (<LLOQ – 0.14) | 12 (6.2–24) |
| AUC 0–24 h (μM • h) | 28.8 (11.1–56.9) | 999 (673–1595) |
| AUC 0–inf (μM • h) | 28.9 (11.1–57.0) | 1265 (869–2010) |
| Cohort B (receiving gemcitabine 500 mg m –2 ) | ||
| n | 26 | 26 |
| C max (μM) | 11.1 (0.802–29.3) | 75.8 (43.5–115) |
| t max (h) | 0.5 (0.5–0.5) | 0.52 (0.5–1) |
| t½ , β (h) | 10.5 (4.16–42.8) | 13.9 (8.78–24.5) |
| CL (l h –1 m –2) | 324 (109–873) | – |
| Percentage of C max still present after 22 h (%) | 0.089 (LLOQ – 0.39) | 12 (5.6–22) |
| AUC 0–22 h (μM • h) | 6.75 (0.535–17.3) | 478 (289–748) |
| AUC 0–inf (μM • h) | 7.89 (2.18–17.5) | 662 (364–1060) |
AUC0–t, area under the concentration–time curve from time zero up to the time point of the last quantifiable data point; AUCinf, the area under the concentration–time curve extrapolated to infinity, using the terminal elimination constant; CL, plasma clearance; Cmax, the maximum observed concentration; LLOQ, lower limit of quantification; n, the number of patients for whom pharmacokinetics were evaluable; t½,β, terminal elimination half‐life; tmax, the time to reach the maximum observed concentration
Patients in cohort A received gemcitabine doses twice as high as patients in cohort B. However, the maximum observed gemcitabine plasma concentrations and AUCs were almost four times higher for cohort A than for cohort B. For the dFdU plasma concentrations, by contrast, a dose‐proportional increase was observed for cohort A compared with cohort B. We had no clear explanation for the differences found between the two patient cohorts. One possibility was that the comedication had had some influence. However, previous combination studies with lapatinib 22, 32, sorafenib 33 and carboplatin 34 showed no clinically relevant influence of concomitant use of these drugs on the plasma PK of gemcitabine.
In all patients, the dFdU levels measured in plasma were substantially higher than the gemcitabine levels. In addition, dFdU remained in the plasma for longer. Two hours after the start of the infusion, gemcitabine was only present at very low concentrations, and 24 h after the start of the infusion gemcitabine had almost completely disappeared, whereas the dFdU concentrations were, on average, still 12% of the Cmax. The dFdU plasma exposure was, therefore, much higher than the gemcitabine plasma exposure in both patient cohorts.
Although gemcitabine concentrations were low within 2 h after the infusion, in most patients very low gemcitabine plasma concentrations (<0.06 μM) were measurable up to 24 h after infusion. This is more apparent when the data are displayed on a semi‐logarithmic scale (Figure 2, inset). Plasma concentrations of dFdU peaked at the end of infusion, followed by a rapid distribution phase and a longer elimination phase. The plasma t½ values reported here refer to the terminal elimination phase and were based on the last three sampling time points.
Intracellular PK of gemcitabine, dFdU and their nucleotides
The mean intracellular concentration–time curves of gemcitabine, dFdU and their nucleotides are shown in Figure 3, and the results of the noncompartmental analysis of the intracellular PK data are presented in Table 2. The highest intracellular gemcitabine and dFdU concentrations were measured at the end of the infusion. The intracellular gemcitabine and dFdU concentrations increased dose‐proportionately: the Cmax values for gemcitabine and dFdU were twice as high in cohort A as in cohort B. However, the mean intracellular AUCs of gemcitabine and dFdU were 4 and 3 times higher, respectively, in cohort A than in cohort B.
Figure 3.
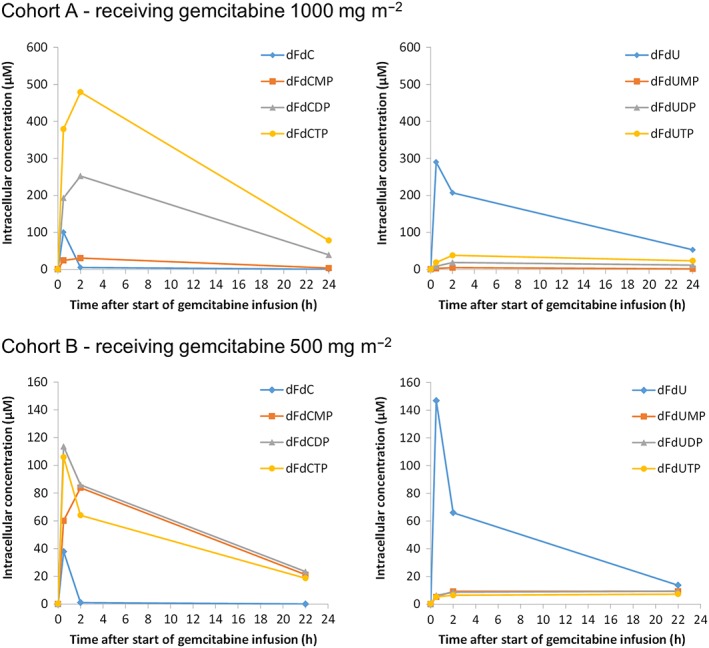
Mean intracellular concentrations of gemcitabine, 2′,2′‐difluorodeoxyuridine (dFdU) and their nucleotides in peripheral blood mononuclear cells. dFdC, 2′,2′‐difluorodeoxycytidine (gemcitabine); dFdCMP, 2′,2′‐difluorodeoxycytidine 5′‐monophosphate; dFdCDP, 2′,2′‐difluorodeoxycytidine 5′‐diphosphate; dFdCTP, 2′,2′‐difluorodeoxycytidine 5′‐triphosphate; dFdU, 2′,2′‐difluorodeoxyuridine; dFdUMP, 2′,2′‐difluorodeoxyuridine 5′‐monophosphate; dFdUDP, 2′,2′‐difluorodeoxyuridine 5′‐diphosphate; dFdUTP 2′,2′‐difluorodeoxyuridine 5′‐triphosphate
Table 2.
Summary statistics for the pharmacokinetic parameters of gemcitabine, 2′,2′‐difluorodeoxyuridine (dFdU) and their nucleotides in peripheral blood mononuclear cells. The data are shown as mean values (range)
| dFdC | dFdCMP | dFdCDP | dFdCTP | dFdU | dFdUMP | dFdUDP | dFdUTP | |
|---|---|---|---|---|---|---|---|---|
| Cohort A (receiving gemcitabine 1000 mg m –2 ) | ||||||||
| n | 12 | 12 | 12 | 12 | 12 | 12 | 12 | 12 |
| C max (μM) | 100 (21.7–482) | 30.9 (5.12–67.7) | 253 (75.0–461) | 497 (194–1060) | 330 (107–1120) | 4.53 (1.39–7.99) | 18.5 (5.78–32.0) | 38.5 (9.73–87.9) |
| t max (h) | 0.5 (0.5) | 1.4 (0.5–2) | 1.5 (0.5–2) | 1.5 (0.5–2) | 0.9 (0.5–2) | 1.8 (0.5–2) | 5.4 (0.5–24) | 5.4 (0.5–24) |
| Percentage of C max still present after 24 h (%) | (<LLOQ) | 14 (<LLOQ –34) | 17 (<LLOQ – 42) | 16 (<LLOQ –40) | 22 (<LLOQ – 75) | 53 (<LLOQ – 83) | 67 (<LLOQ – 100) | 64 (<LLOQ – 100) |
| AUC 0–24 h (μM • h) | 195 (57.6–514) | 411 (68.5–873) | 3480 (1080–6650) | 6580 (2830–15 700) | 3200 (848–9890) | 77.0 (31.7–171) | 342 (117–674) | 695 (230–1620) |
| Cohort B (receiving gemcitabine 500 mg m –2 ) | ||||||||
| n | 26 | 26 | 26 | 26 | 26 | 26 | 26 | 26 |
| C max (μM) | 37.8 (4.80–245) | 90.8 (3.63–440) | 128 (10.1–401) | 96.1 (<LLOQ – 440) | 158 (35.5–798) | 10.7 (<LLOQ – 48.5) | 10.9 (<LLOQ – 28.4) | 7.97 (<LLOQ – 29.0) |
| t max (h) | 0.5 (0.5) | 1.4 (0.5–2) | 1.2 (0.5–2) | 1.7 (0.5–22) | 0.8 (0.5–2) | 11 (0.5–22) | 16 (0.5–22) | 12 (0.5–22) |
| Percentage of C max still present after 22 h (%) | (<LLOQ) | 25 (<LLOQ – 46) | 23 (<LLOQ – 46) | 43 (<LLOQ – 100) | 16 (<LLOQ – 96) | 85 (<LLOQ – 100) | 87 (<LLOQ – 100) | 92 (<LLOQ – 100) |
| AUC 0–22 h (μM • h) | 48.5 (15.6–252) | 1170 (55.8–5800) | 1300 (138–3100) | 824 (54.9–3030) | 937 (289–5350) | 194 (23.5–938) | 193 (24.8–500) | 135 (23.5–452) |
AUC0–t, the area under the concentration‐time curve from time zero up to the time point of the last quantifiable data point; Cmax, the maximum observed concentration; dFdC, 2′,2′‐difluorodeoxycytidine (gemcitabine); dFdCMP, gemcitabine monophosphate; dFdCDP, gemcitabine diphosphate; dFdCTP, gemcitabine triphosphate; dFdUMP, dFdU monophosphate; dFdUDP, dFdU diphosphate; dFdUTP, dFdU triphosphate; n, the number of evaluable patients; tmax, the time to reach the maximum observed concentration
The gemcitabine and dFdU levels measured in PBMCs were both higher than the levels measured in plasma. The Cmax values for gemcitabine and dFdU in PBMCs were, on average, 2–3 times higher than the Cmax values measured in plasma.
In PBMCs, as well as in plasma, dFdU was present at higher concentrations than gemcitabine. However, the intracellular dFdU nucleotide concentrations (i.e. dFdUMP, dFdUDP and dFdUTP) were relatively low (<88 μM) – much lower than the dFdC nucleotide concentrations that were measured (up to 1060 μM). In 8% of the patients, the dFdU nucleotide concentrations were below the LLOQ of the assay at all time points.
The ratios between the monophosphates, diphosphates and triphosphates differed between the two patient cohorts. In cohort A, dFdCTP showed the highest abundance, followed by dFdCDP. For dFdCMP, dFdUMP, dFdUDP and dFdUTP, only low concentrations were found (<88 μM). For cohort B, the dFdCMP, dFdCDP and dFdCTP concentrations were approximately equally high. The concentrations of the dFdU nucleotides were also low for this patient cohort (<49 μM). We had no clear explanation for the differences between the two patient cohorts. It could not be excluded that the comedication had had an influence on the intracellular nucleotide accumulation. However, to the best of our knowledge, the influences of carboplatin, sorafenib and lapatinib on the intracellular accumulation of dFdCTP have never been studied 21.
Two hours after the start of the infusion, very low gemcitabine concentrations were found in PBMCs. However, the dFdC nucleotides remained present in the cells for much longer. Twenty‐four hours after the start of the infusion, substantial intracellular dFdC nucleotide concentrations were still found in most patients. dFdU also remained present in the cells for an extended period.
High interindividual variability was observed for the intracellular Cmax and AUC of gemcitabine, dFdU and the dFdC nucleotides. Although some interpatient variability was also seen for the dFdU nucleotide concentrations, these concentrations were systematically low in all patients.
Discussion
The plasma PK data found in the present study were in line with previously published data on the plasma PK of gemcitabine and dFdU 14, 19, 35, 36. In addition, the intracellular PK results for dFdCTP were in line with those in previous reports 35, 36, 37, 38. However, data on the intracellular PK of the other dFdC and dFdU nucleotides in patients treated with gemcitabine were lacking. The present study provides the first complete picture of the PK of all nucleotides that are formed intracellularly during gemcitabine treatment.
Although dFdU was traditionally believed to be an inactive metabolite, in vitro studies demonstrated that it has cytotoxic activity in tumour cell lines 11, 12, 39. The half‐maximal inhibitory concentration (IC50) for dFdU for gemcitabine‐sensitive tumour cell lines is highly variable, but was found to be at least 1000–2000‐fold higher than for gemcitabine 11, 12, 39. However, Veltkamp et al. showed that the cytotoxicity of dFdU was dependent on the duration of drug exposure 11. The present study demonstrated that, during gemcitabine treatment, the concentrations of dFdU were well above those of gemcitabine, both in the plasma and in PBMCs. In addition, dFdU had a longer t½ than gemcitabine. Therefore, both in the plasma and in PBMCs, the exposure to dFdU exceeded the exposure to the parent drug, gemcitabine.
Despite the high intracellular dFdU concentrations that were measured in PBMCs during gemcitabine treatment, the intracellular dFdUMP, dFdUDP and dFdUTP concentrations were much lower than the intracellular dFdCDP and dFdCTP concentrations.
In the present study, PBMCs were used to investigate the intracellular PK of the gemcitabine and dFdU nucleotides. PBMCs are commonly used as a surrogate matrix to study the intracellular PK of nucleoside analogues. They represent the intracellular ‘activation machinery’ and, unlike tumour cells, can be obtained at multiple time points after drug administration. Nevertheless, it should be kept in mind that the concentrations of nucleotides measured in PBMCs might differ from those that would be found in tumour cells – for instance, because enzymes involved in the synthesis of nucleotides might have a different activity in PBMCs than in tumour cells. As depicted in Figure 1, dFdU nucleotides can be formed via two pathways: (i) by direct phosphorylation of dFdU and (ii) through the conversion of dFdCMP to dFdUMP by deoxycytidylate deaminase. Direct phosphorylation of dFdU is most likely to be mediated by dCK and the mitochondrial thymidine kinase (TK) 2. However, compared with gemcitabine, dFdU has a much lower affinity for dCK and a higher affinity for TK2 40, 41. The S‐phase‐specific enzyme TK1 was not involved in the phosphorylation of dFdU, as demonstrated by Veltkamp et al. 11. Unlike TK1, TK2 and dCK are not cell cycle dependent. However, their activity in PBMCs might be different than in tumour cells. Eriksson et al. 42 showed that the in vitro activity of dCK was higher in (non‐activated) PBMCs than in solid tumour tissue. The activity of TK2, by contrast, was lower in (non‐activated) PBMCs than in solid tumour tissue 42. These differences might implicate that dFdU nucleotide concentrations measured in PBMCs are lower than those that would be found in tumour cells.
The low dFdU nucleotide concentrations might also be related to some of the feedback mechanisms that have previously been described for gemcitabine. For instance, the activity of deoxycytidylate deaminase, the enzyme which is responsible for the conversion of dFdCMP to dFdUMP, is directly inhibited by dFdCTP (Figure 1). Moreover, lowering of the dCTP pool (an important effect of gemcitabine) is associated with a decrease in the deoxycytidylate deaminase enzyme activity 9. Previous in vitro studies with arabinosylcytosine (ara‐C), another cytidine analogue, demonstrated that deamination via deoxycytidylate deaminase was the predominant pathway in the formation of arabinosyluracil triphosphate (ara‐UTP) 43. Inhibition of deoxycytidylate deaminase suppressed ara‐UTP formation from ara‐C, whereas inhibition of CDA did not perturb the formation of ara‐UTP from ara‐C 43. As the intracellular metabolism of gemcitabine resembles the metabolism of ara‐C, deamination via deoxycytidylate deaminase might also be the most important pathway for the formation of dFdU nucleotides. Intracellular feedback mechanisms which inhibit this enzyme might therefore play a substantial role in limiting the degree of deamination of gemcitabine nucleotides.
The question is whether sufficient dFdU is phosphorylated to make a contribution to the cytotoxic effect of gemcitabine. To answer this question, the dFdU nucleotide concentrations found in the present study can be compared with the concentrations that have been found in in vitro studies.
Recent cell line studies indicated that dFdUMP, which is structurally similar to deoxyuridine monophosphate (dUMP), might contribute to the cytotoxic effect of gemcitabine by direct inhibition of the enzyme thymidylate synthase 12. Honeywell et al. demonstrated that, although dFdUMP was a 10 000‐fold less potent inhibitor than 5‐fluorodeoxyuridine monophosphate (the active metabolite of 5‐fluorouracil), inhibition of the enzyme did result in increased dUMP mis‐incorporation into the DNA 12. However, the dFdUMP concentrations measured in PBMCs in the present study were relatively low compared with those that were needed to inhibit thymidylate synthase in the cell line study by Honeywell et al. (inhibitory constant = 130 μM) 12. This suggests that thymidylate synthase inhibition by dFdUMP does not play a major role, at least not in PBMCs.
Incorporation of dFdUTP into the DNA and RNA, however, might play a role. In the study by Veltkamp et al., incorporation of dFdUTP into both the DNA and the RNA has been found after cells were incubated for 24 h with relatively low concentrations (0.5 μM) of gemcitabine or dFdU 11. The intracellular dFdUTP concentrations measured in the incubated cells were also relatively low compared with our data in patients. This suggests that dFdU incorporation can also be expected in our patient population. In the study by Veltkamp et al., a strong correlation was found between the extent of dFdU incorporation and the cytotoxicity of dFdU 11. Further studies are warranted to elucidate the importance of the incorporation of dFdU into the DNA for the cytotoxicity of gemcitabine in patients.
It is also interesting to consider the present intracellular PK data in the light of previous clinical reports investigating the relationship between the enzyme activity of CDA and the clinical outcome of gemcitabine therapy. Several studies have shown that patients with low CDA activity displayed more toxicity 44 and better efficacy of gemcitabine‐containing therapy 45, 46. Conversely, Serdjebi et al. showed that pancreatic cancer patients with high CDA activity were five times more likely to have progressive disease following gemcitabine therapy than patients with normal or low CDA activity 47. The present intracellular PK data support the hypothesis that high dFdU plasma concentrations play a limited role in the cytotoxic effects of gemcitabine therapy, as a result of limited intracellular activation by the formation of dFdU nucleotides. If this hypothesis can be confirmed in other tissues, a pharmacological basis might be provided for why differences in CDA activity, by modifying the dFdC/dFdU ratio, have a substantial impact on the efficacy and toxicity of gemcitabine therapy.
In conclusion, the present study provides the first complete picture of all nucleotides that are formed intracellularly during gemcitabine treatment. Although dFdU was present at higher concentrations than gemcitabine in PBMCs, low intracellular dFdUMP, dFdUDP and dFdUTP concentrations were found. This calls into question the relevance of these dFdU nucleotides for the cytotoxic effects of gemcitabine.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
Contributors
E.J.B.D. was responsible for the study concept and design, data collection, data analysis and interpretation, and the writing of the manuscript. A.D.R.H., H.R., J.H.M.S. and J.H.B. had final approval of the manuscript.
Derissen, E. J. B. , Huitema, A. D. R. , Rosing, H. , Schellens, J. H. M. , and Beijnen, J. H. (2018) Intracellular pharmacokinetics of gemcitabine, its deaminated metabolite 2′,2′‐difluorodeoxyuridine and their nucleotides. Br J Clin Pharmacol, 84: 1279–1289. doi: 10.1111/bcp.13557.
References
- 1. Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, et al Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 1998; 58: 4349–4357. [PubMed] [Google Scholar]
- 2. Ritzel MW, Ng AM, Yao SY, Graham K, Loewen SK, Smith KM, et al Recent molecular advances in studies of the concentrative Na+‐dependent nucleoside transporter (CNT) family: identification and characterization of novel human and mouse proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides. Mol Membr Biol 2001; 18: 65–72. [DOI] [PubMed] [Google Scholar]
- 3. Wong A, Soo RA, Yong W‐P, Innocenti F. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab Rev 2009; 41: 77–88. [DOI] [PubMed] [Google Scholar]
- 4. Gandhi V, Legha J, Chen F, Hertel LW, Plunkett W. Excision of 2′,2′‐difluorodeoxycytidine (gemcitabine) monophosphate residues from DNA. Cancer Res 1996; 56: 4453–4459. [PubMed] [Google Scholar]
- 5. Huang P, Chubb S, Hertel L, Grindey G, Plunkett W. Action of 2′,2′‐difluorodeoxycytidine on DNA synthesis. Cancer Res 1991; 51: 6110–6117. [PubMed] [Google Scholar]
- 6. Mini E. Cellular pharmacology of gemcitabine. Ann Oncol 2006; 17: 7–12. [DOI] [PubMed] [Google Scholar]
- 7. Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, et al Inhibition of ribonucleotide reduction in CCRF‐CEM cells by 2′,2′‐difluorodeoxycytidine. Mol Pharmacol 1990; 38: 567–572. [PubMed] [Google Scholar]
- 8. de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol 2014; 741: 8–16. [DOI] [PubMed] [Google Scholar]
- 9. Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, et al Cellular elimination of 2′,2′‐difluorodeoxycytidine 5′‐triphosphate: a mechanism of self‐potentiation. Cancer Res 1992; 52: 533–539. [PubMed] [Google Scholar]
- 10. Xu Y‐Z, Plunkett W. Modulation of deoxycytidylate deaminase in intact human leukemia cells. Action of 2′,2′‐difluorodeoxycytidine. Biochem Pharmacol 1992; 44: 1819–1827. [DOI] [PubMed] [Google Scholar]
- 11. Veltkamp SA, Pluim D, van Eijndhoven MAJ, Bolijn MJ, Ong FHG, Govindarajan R, et al New insights into the pharmacology and cytotoxicity of gemcitabine and 2′,2′‐difluorodeoxyuridine. Mol Cancer Ther 2008; 7: 2415–2425. [DOI] [PubMed] [Google Scholar]
- 12. Honeywell RJ, Ruiz Van Haperen VWT, Veerman G, Smid K, Peters GJ. Inhibition of thymidylate synthase by 2′,2′‐difluoro‐2′‐deoxycytidine (Gemcitabine) and its metabolite 2′,2′‐difluoro‐2′‐deoxyuridine. Int J Biochem Cell Biol 2015; 60: 73–81. [DOI] [PubMed] [Google Scholar]
- 13. Grunewald R, Kantarjian H, Keating MJ, Abbruzzese J, Tarassoff P, Plunkett W. Pharmacologically directed design of the dose rate and schedule of 2′,2′‐difluorodeoxycytidine (Gemcitabine) administration in leukemia. Cancer Res 1990; 50: 6823–6826. [PubMed] [Google Scholar]
- 14. Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, et al A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol 1991; 9: 491–498. [DOI] [PubMed] [Google Scholar]
- 15. Grunewald R, Abbruzzese JL, Tarassoff P, Plunkett W. Saturation of 2′,2′‐difluorodeoxycytidine 5′‐triphosphate accumulation by mononuclear cells during a phase I trial of gemcitabine. Cancer Chemother Pharmacol 1991; 27: 258–262. [DOI] [PubMed] [Google Scholar]
- 16. Grunewald R, Kantarjian H, Du M, Faucher K, Tarassoff P, Plunkett W. Gemcitabine in leukemia: a phase I clinical, plasma, and cellular pharmacology study. J Clin Oncol 1992; 10: 406–413. [DOI] [PubMed] [Google Scholar]
- 17. Patel SR, Gandhi V, Jenkins J, Papadopolous N, Burgess MA, Plager C, et al Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol 2001; 19: 3483–3489. [DOI] [PubMed] [Google Scholar]
- 18. Gandhi V, Plunkett W, Du M, Ayres M, Estey EH. Prolonged infusion of gemcitabine: clinical and pharmacodynamic studies during a phase I trial in relapsed acute myelogenous leukemia. J Clin Oncol 2002; 20: 665–673. [DOI] [PubMed] [Google Scholar]
- 19. Cattel L, Airoldi M, Delprino L, Passera R, Milla P, Pedani F. Pharmacokinetic evaluation of gemcitabine and 2′,2′‐difluorodeoxycytidine‐5′‐triphosphate after prolonged infusion in patients affected by different solid tumors. Ann Oncol 2006; 17 ((Suppl. 5)): v142–v147. [DOI] [PubMed] [Google Scholar]
- 20. Grimison P, Galettis P, Manners S, Jelinek M, Metharom E, de Souza PL, et al Randomized crossover study evaluating the effect of gemcitabine infusion dose rate: evidence of auto‐induction of gemcitabine accumulation. J Clin Oncol 2007; 25: 5704–5709. [DOI] [PubMed] [Google Scholar]
- 21. Rizzuto I, Ghazaly E, Peters GJ. Pharmacological factors affecting accumulation of gemcitabine's active metabolite, gemcitabine triphosphate. Pharmacogenomics 2017; 18: 911–925. [DOI] [PubMed] [Google Scholar]
- 22. van der Noll R, Smit WM, Wymenga ANM, Boss DS, Grob M, Huitema ADR, et al Phase I and pharmacological trial of lapatinib in combination with gemcitabine in patients with advanced breast cancer. Invest New Drugs 2015; 33: 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vainchtein LD, Rosing H, Thijssen B, Schellens JHM, Beijnen JH. Validated assay for the simultaneous determination of the anti‐cancer agent gemcitabine and its metabolite 2′,2′‐difluorodeoxyuridine in human plasma by high‐performance liquid chromatography with tandem mass spectrometry. Rapid Commun Mass Spectrom 2007; 21: 2312–2322. [DOI] [PubMed] [Google Scholar]
- 24. Jansen RS, Rosing H, Schellens JHM, Beijnen JH. Simultaneous quantification of 2′,2′‐difluorodeoxycytidine and 2′,2′‐difluorodeoxyuridine nucleosides and nucleotides in white blood cells using porous graphitic carbon chromatography coupled with tandem mass spectrometry. Rapid Commun Mass Spectrom 2009; 23: 3040–3050. [DOI] [PubMed] [Google Scholar]
- 25. Veltkamp SA, Hillebrand MJX, Rosing H, Jansen RS, Wickremsinhe ER, Perkins EJ, et al Quantitative analysis of gemcitabine triphosphate in human peripheral blood mononuclear cells using weak anion‐exchange liquid chromatography coupled with tandem mass spectrometry. J Mass Spectrom 2006; 41: 1633–1642. [DOI] [PubMed] [Google Scholar]
- 26. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 1976; 72: 248–254. [DOI] [PubMed] [Google Scholar]
- 27. Pluim D, Jacobs BAW, Krähenbühl MD, Ruijter AEM, Beijnen JH, Schellens JHM. Correction of peripheral blood mononuclear cell cytosolic protein for hemoglobin contamination. Anal Bioanal Chem 2013; 405: 2391–2395. [DOI] [PubMed] [Google Scholar]
- 28. Jansen RS, Rosing H, Schellens JHM, Beijnen JH. Retention studies of 2′,2′‐difluorodeoxycytidine and 2′,2′‐difluorodeoxyuridine nucleosides and nucleotides on porous graphitic carbon: development of a liquid chromatography‐tandem mass spectrometry method. J Chromatogr A 2009; 1216: 3168–3174. [DOI] [PubMed] [Google Scholar]
- 29. Simiele M, D'Avolio A, Baietto L, Siccardi M, Sciandra M, Agati S, et al Evaluation of the mean corpuscular volume of peripheral blood mononuclear cells of HIV patients by a coulter counter to determine intracellular drug concentrations. Antimicrob Agents Chemother 2011; 55: 2976–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cerbone L, Sternberg CN, Sengeløv L, Agerbaek M, Van Herpen C, Marreaud S, et al Results from a phase I study of lapatinib with gemcitabine and cisplatin in advanced or metastatic bladder cancer: EORTC Trial 30061. Oncology 2016; 90: 21–28. [DOI] [PubMed] [Google Scholar]
- 33. Schultheis B, Kummer G, Zeth M, Brendel E, Xia C, Kornacker M, et al Phase IB study of sorafenib in combination with gemcitabine and cisplatin in patients with refractory solid tumors. Cancer Chemother Pharmacol 2012; 69: 333–339. [DOI] [PubMed] [Google Scholar]
- 34. Carmichael J, Allerheiligen S, Walling J. A phase I study of gemcitabine and carboplatin in non‐small cell lung cancer. Semin Oncol 1996; 23: 55–59. [PubMed] [Google Scholar]
- 35. Kroep JR, Giaccone G, Voorn DA, Smit EF, Beijnen JH, Rosing H, et al Gemcitabine and paclitaxel: pharmacokinetic and pharmacodynamic interactions in patients with non‐small‐cell lung cancer. J Clin Oncol 1999; 17: 2190–2197. [DOI] [PubMed] [Google Scholar]
- 36. Soo RA, Wang LZ, Tham LS, Yong WP, Boyer M, Lim HL, et al A multicentre randomised phase II study of carboplatin in combination with gemcitabine at standard rate or fixed dose rate infusion in patients with advanced stage non‐small‐cell lung cancer. Ann Oncol 2006; 17: 1128–1133. [DOI] [PubMed] [Google Scholar]
- 37. van Moorsel CJ, Kroep JR, Pinedo HM, Veerman G, Voorn DA, Postmus PE, et al Pharmacokinetic schedule finding study of the combination of gemcitabine and cisplatin in patients with solid tumors. Ann Oncol 1999; 10: 441–448. [DOI] [PubMed] [Google Scholar]
- 38. Tempero M, Plunkett W, Ruiz van Haperen VW, Hainsworth JD, Hochster H, Abbruzzese JL, et al Randomized phase II comparison of dose‐intense gemcitabine: thirty‐minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J Clin Oncol 2003; 21: 3402–3408. [DOI] [PubMed] [Google Scholar]
- 39. Ruiz van Haperen VW, Veerman G, Eriksson S, Boven E, Stegmann AP, Hermsen M, et al Development and molecular characterization of a 2′,2′‐difluorodeoxycytidine‐resistant variant of the human ovarian carcinoma cell line A2780. Cancer Res 1994; 54: 4138–4143. [PubMed] [Google Scholar]
- 40. Hodge LS, Taub ME, Tracy TS. The deaminated metabolite of gemcitabine, 2′,2′‐difluorodeoxyuridine, modulates the rate of gemcitabine transport and intracellular phosphorylation via deoxycytidine kinase. Drug Metab Dispos 2011; 39: 2013–2016. [DOI] [PubMed] [Google Scholar]
- 41. Wang L, Munch‐Petersen B, Herrström Sjöberg A, Hellman U, Bergman T, Jörnvall H, et al Human thymidine kinase 2: molecular cloning and characterisation of the enzyme activity with antiviral and cytostatic nucleoside substrates. FEBS Lett 1999; 443: 170–174. [DOI] [PubMed] [Google Scholar]
- 42. Eriksson S, Arnér E, Spasokoukotskaja T, Wang L, Karlsson A, Brosjö O, et al Properties and levels of deoxynucleoside kinases in normal and tumor cells: implications for chemotherapy. Adv Enzyme Regul 1994; 34: 13–25. [DOI] [PubMed] [Google Scholar]
- 43. Gandhi V, Xu YZ, Estey E. Accumulation of arabinosyluracil 5′‐triphosphate during arabinosylcytosine therapy in circulating blasts of patients with acute myelogenous leukemia. Clin Cancer Res 1998; 4: 1719–1726. [PubMed] [Google Scholar]
- 44. Ciccolini J, Dahan L, André N, Evrard A, Duluc M, Blesius A, et al Cytidine deaminase residual activity in serum is a predictive marker of early severe toxicities in adults after gemcitabine‐based chemotherapies. J Clin Oncol 2010; 28: 160–165. [DOI] [PubMed] [Google Scholar]
- 45. Tibaldi C, Giovannetti E, Vasile E, Mey V, Laan AC, Nannizzi S, et al Correlation of CDA, ERCC1, and XPD polymorphisms with response and survival in gemcitabine/cisplatin‐treated advanced non‐small cell lung cancer patients. Clin Cancer Res 2008; 14: 1797–1803. [DOI] [PubMed] [Google Scholar]
- 46. Tibaldi C, Giovannetti E, Tiseo M, Leon LG, D'incecco A, Loosekoot N, et al Correlation of cytidine deaminase polymorphisms and activity with clinical outcome in gemcitabine‐/platinum‐treated advanced non‐small‐cell lung cancer patients. Ann Oncol 2012; 23: 670–677. [DOI] [PubMed] [Google Scholar]
- 47. Serdjebi C, Seitz J‐F, Ciccolini J, Duluc M, Norguet E, Fina F, et al Rapid deaminator status is associated with poor clinical outcome in pancreatic cancer patients treated with a gemcitabine‐based regimen. Pharmacogenomics 2013; 14: 1047–1051. [DOI] [PubMed] [Google Scholar]