

Abstract
Aims
This study aimed to evaluate the effect of a strong CYP3A inducer, rifampin, on glasdegib pharmacokinetics in healthy volunteers.
Methods
In an open‐label, fixed‐sequence, two‐period Phase 1 study, subjects received a single 100‐mg oral dose of glasdegib alone or following once‐daily pre‐treatment with 600 mg rifampin. Glasdegib pharmacokinetics were calculated using a noncompartmental analysis.
Results
Twelve healthy male volunteers (3 whites, 5 blacks and 4 others) were enrolled in the study. Mean age, weight, height and body mass index was 37.8 years, 83.0 kg, 177.3 cm and 26.5 kg (m2) –1, respectively. When dosed alone, glasdegib geometric mean (% coefficient of variation) area under the plasma concentration–time curve from time zero to infinity (AUC inf) was 8145 ng × h ml−1 (23%) and maximum observed concentration (C max) was 703.2 ng ml−1 (19%). With rifampin, glasdegib AUC inf and C max decreased, with an adjusted geometric mean ratio (90% confidence interval) 29.66% (26.17–33.62) for AUC inf and 64.71% (57.21–73.19) for C max. Mean terminal half‐life decreased from 13.39 to 5.11 hours, geometric mean apparent oral clearance increased from 12.27 to 41.38 l h−1, whereas median time to C max remained similar (1.50 vs. 1.25 hours) in the presence of rifampin. All adverse events (n = 29) were mild in severity and resolved by the end of the study.
Conclusions
Co‐administration of rifampin expectedly decreased glasdegib AUC inf and C max by ~70% and ~35%, respectively. These results will help to formulate recommendations for dosing strategies in combination with CYP3A inducers in situations where co‐administration may be necessary. (http://clinicaltrials.gov identifier: NCT02430545).
Keywords: cytochrome P450, drug interaction, Phase I
What is Already Known about this Subject
Glasdegib is a potent and selective inhibitor of the G protein‐coupled receptor Smoothened, the key component of the Hedgehog signalling pathway involved in embryogenesis and potentially carcinogenesis.
Glasdegib inhibits the Hedgehog signalling pathway in vitro and has demonstrated anti‐tumour activity in patients with solid as well as haematopoietic tumours.
Glasdegib pharmacokinetics have been determined in healthy subjects in the absence and presence of ketoconazole, a strong inhibitor of CYP450 3A4/5, as well as in select cancer patients.
What this Study Adds
Glasdegib plasma exposure and maximum plasma concentration decreased by approximately 70% and 35%, respectively, in the presence of steady‐state rifampin in healthy volunteers.
Results of this study quantify the impact of a strong metabolic inducer on glasdegib pharmacokinetics and provide the extreme scenario for decrease in glasdegib exposure.
This information will assist in providing guidance in clinical studies for limiting the use of co‐medications that could decrease glasdegib exposure and potentially impact efficacy. These data will also allow for using in silico approaches to assess the impact of a moderate CYP3A inducer on glasdegib pharmacokinetics.
Introduction
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8201 (PF‐04449913) is a potent and selective inhibitor of the G protein‐coupled receptor http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=239, the key component of the Hedgehog signalling pathway, which is essential for embryogenic organ and tissue development 1, 2. Aberrant activation of the Hedgehog signalling pathway is thought to be linked to a number of human cancers 1, 3. Glasdegib inhibits the Hedgehog signalling pathway in vitro 4, 5 and has demonstrated potential anti‐tumour activity in vivo 6, 7, supporting the therapeutic potential of glasdegib in certain cancers.
The pharmacokinetics and safety of glasdegib following a single‐dose administration have been evaluated in healthy volunteers in three Phase 1 studies 8, 9, 10. Additional pharmacokinetics studies to determine the maximum tolerated dose (MTD) and recommended Phase 2 dose (RP2D) have been conducted in patients with advanced solid tumours 6 or select haematological malignancies 7 in the United States and Europe as well as in Japan 11. Glasdegib is currently under investigation in ongoing Phase 2 clinical trials for treatment of acute myeloid leukaemia and high‐risk myelodysplastic syndrome (http://clinicaltrials.gov identifiers, NCT01546038 and NCT01842646).
In vitro and ex vivo studies showed that glasdegib is primarily metabolised by cytochrome http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=263 3A4/5 9; hence, a strong inhibitor or inducer of CYP3A4/5 may impact glasdegib pharmacokinetics. A previous drug–drug interaction (DDI) study evaluating the effect of a strong CYP3A inhibitor found that co‐administration of ketoconazole increased glasdegib plasma exposure in healthy volunteers 8. The primary aim of this study was to investigate the effect of a strong inducer of CYP3A, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2765, on glasdegib pharmacokinetic parameters in healthy volunteers. Additionally, the safety and tolerability of a single dose of glasdegib administered alone or in the presence of steady‐state rifampin was evaluated.
Methods
Study design and population
This was a Phase 1, open‐label, fixed‐sequence, two‐period study of glasdegib in the absence or presence of rifampin, conducted in healthy volunteers at the Pfizer Clinical Research Unit (PCRU; New Haven, CT, USA).
Male and female subjects aged between 18 and 55 years with no clinically relevant abnormalities as identified by medical history, physical examination and clinical laboratory tests were eligible for the study. Other key inclusion criteria were body mass index (BMI) between 17.5 and 30.5 kg (m2) –1 and body weight >50 kg. Subjects were excluded if they were hypertensive (≥140/90 mm Hg) or had abnormal electrocardiogram results (ECG; corrected QT interval >450 ms or QRS interval >120 ms) at screening, had condition possibly affecting absorption, used tobacco‐ or nicotine‐containing products within 90 days of screening, consumed alcohol regularly (>14 drinks/week for males or >7 drinks/week for females), received an investigational drug within 30 days or 5 half‐lives (whichever was longer) preceding the first dose of study medication, or used prescription or non‐prescription drugs or dietary supplements within 7 days or 5 half‐lives (whichever was longer) preceding the first dose of study medication.
The primary endpoints of the study were plasma area under the plasma concentration–time curve (AUC) from time zero extrapolated to infinity time (AUC inf) and maximum observed plasma concentration (C max) for glasdegib; secondary endpoints included other pharmacokinetic parameters (AUC from time zero to the time of the last quantifiable concentration [AUC last], time to C max [T max], terminal half‐life [t 1/2], apparent oral clearance [CL/F] and apparent volume of distribution [Vz/F]) and safety.
The final protocol, any amendments and informed consent documentation were reviewed and approved by the institutional review board at the investigational centre. The study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and the International Conference on Harmonisation and Good Clinical Practice Guidelines, as well as any local regulatory requirements. All subjects participating in the study provided written informed consent. This study is registered at http://clinicaltrials.gov, identifier NCT02430545.
Procedures
Subjects were screened within 28 days prior to the first dose of glasdegib in Period 1. Each enrolled subject received 100 mg glasdegib alone (Treatment A) in Period 1, and 100 mg glasdegib after pre‐treatment with 600 mg rifampin (Treatment B) to induce CYP3A in Period 2, with a washout period of ≥11 days between the two doses of glasdegib to allow for glasdegib washout and adequate duration for maximal induction of rifampin (Table 1). The total study duration was approximately 17 days. Glasdegib 100 mg once daily is the clinical dosage of glasdegib that is under evaluation in Phase 2/3 trials, and was selected based on clinical activity, biomarker modulation, safety and tolerability data from previous studies in patients with haematologic malignancies and healthy volunteers 7, 8, 9, 11.
Table 1.
Study design
| Sequence | Period 1 (5 days) | Period 2 (12 days) |
|---|---|---|
| 1 (N = 12) | Treatment A | Treatment B |
| (Reference) | (Test) | |
| Glasdegib: 100 mg p.o., s.d., on Day 1 | Rifampin: 600 mg p.o., q.d. from Day −6 to Day 4 (11 days) | |
| Glasdegib: 100 mg p.o., s.d. on Day 1a |
Washout was 11 days between the two doses of glasdegib
p.o., oral dosing; q.d., once daily; s.d., single dose
On Day 0 in Period 1, subjects were admitted to the PCRU and remained in‐house for 5 days until collection of the 96‐h pharmacokinetic sample and safety assessments before discharge on Day 5. On Day 1 in Period 1, subjects received a single dose of 100 mg glasdegib administered orally after a 10‐h overnight fast, with fasting continuing for ≥4 hours post‐dose.
In Period 2, subjects were administered a 600 mg (2 × 300 mg capsules) oral dose of rifampin once daily, starting on Day −6 through to Day 4. On Day −6 and Days 0 through 4, rifampin was administered in the presence of PCRU staff, whereas on Days −5 to −1, rifampin was self‐administered at home. Subjects were instructed to fast 2 hours before and after rifampin dosing and to record date, time, dose and compliance with dosing instructions in the dosing diary. On Day 0 in Period 2, subjects were required to check back in the PCRU, where they remained until completion of the 96‐h pharmacokinetic sample and safety assessments on Day 5 before discharge. On Day 1 in Period 2, a single 100 mg dose of glasdegib was administered to subjects following a 10‐h overnight fast and immediately after the daily dose of rifampin. Throughout the study, including the washout period, subjects abstained from drinking alcohol and all concomitant treatments.
In both Periods 1 and 2, blood samples (2 ml) for assessing glasdegib pharmacokinetics were collected at pre‐dose and at 0.25, 0.5, 1, 1.5, 2, 4, 6, 10, 24, 48, 72 and 96 h following glasdegib administration.
Plasma samples were analysed within established stability for glasdegib concentrations at Covance Bioanalytical Services (Shanghai, China) using a validated liquid chromatography tandem mass spectrometric method that was selective for glasdegib. Calibration standard curves were linear over the quantitation range of 3.00 to 3000 ng ml−1 using a weighted (1/concentration2) linear least squares regression. Quality control (QC) samples at three concentration levels – low (9.00 ng ml−1), medium (100 ng ml−1) and high (2250 ng ml−1) – were included in each analytical run. Assay performance, as determined by calibration standard curve parameters, QC sample accuracy and precision, incurred sample reproducibility (ISR), and chromatographic data were considered acceptable during sample analysis. The coefficient of determination (r2) of the calibration standard curve across all analytical runs was ≥0.9916. Assay accuracy, expressed as percentage relative error (%RE) of the mean inter‐run QC sample concentration from the nominal QC concentration, ranged from −6.2% to 4.7% across all QC sample levels. Assay precision, expressed as percentage coefficient of variation (%CV) of the mean inter‐run QC sample concentration, was ≤5.3% for low‐, medium‐ and high‐concentration QC samples. For the ISR assessment, at least 10% of the pharmacokinetic samples were re‐assayed in singlicate, and all ISR sample concentrations were within an acceptable percent difference from their original concentrations.
The glasdegib pharmacokinetics in the absence or presence of rifampin were calculated for each subject using noncompartmental analysis of plasma concentration–time data. Samples with concentrations below the assay lower limit of quantitation (3.00 ng ml−1) were set to zero for analysis. The pharmacokinetic parameters were determined using Pfizer proprietary software (eNCA v2.2.4).
All subjects who received at least one dose of study medication were included in the safety analysis. The following assessments were performed during each treatment period: vital signs (blood pressure, pulse rate and body temperature) at screening, Day 1 and Day 5; safety laboratory tests (haematology, clinical chemistry and urinalysis) at screening, Day 0, Day 1, and Day 5; urine drug screening/breathalyser tests at screening and on Day 0; and a single 12‐lead ECG at screening, Day 0, Day 1, and Day 5. Physical examination was performed at screening, Day 0 and Day 5 of Period 1, and on Day 0 of Period 2. Subjects were questioned about prior/concomitant medication at each clinic visit. The safety and tolerability of glasdegib were assessed by monitoring adverse events (AEs) and serious AEs, graded according to the Medical Dictionary for Regulatory Activities (MedDRA) version 18.0.
Statistical analysis
A sample size of 12 subjects provided 90% confidence intervals (CIs) for the difference between glasdegib alone and in the presence of rifampin of ±0.124 and 0.167 on the natural log‐transformed AUC inf and C max, respectively, with 90% coverage probability. These calculations were based on the estimates for intra‐subject standard deviations of 0.135 and 0.182 for natural log‐transformed AUC inf and C max, respectively, as obtained from the mean of two previous clinical studies in healthy subjects 8, 10.
Natural log‐transformed AUC inf and C max for glasdegib were analysed using a mixed‐effect model with treatment as a fixed effect and subject as a random effect. Adjusted mean differences between the test (glasdegib at steady‐state rifampin) and reference (glasdegib alone) treatment and corresponding 90% CIs were obtained and exponentiated to provide estimates of the ratio of adjusted geometric mean ratio and 90% CI. Glasdegib pharmacokinetic parameters were summarised descriptively and individual subject and summary profile (median) of concentration–time data plotted by treatment. Matchbox plots were used to show the effect of rifampin on glasdegib AUC inf and C max.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 11, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 12, 13.
Results
A total of 12 healthy male volunteers were enrolled in the study. All subjects completed the study and were included in both the pharmacokinetic and safety analyses. For the 12 subjects, consisting of 3 whites, 5 blacks and 4 of other racial background, mean age, weight, height and BMI were 37.8 years, 83.0 kg, 177.3 cm and 26.5 kg (m2)−1, respectively (Table 2). No subjects received prior or concomitant drug or nondrug treatments.
Table 2.
Demographics and baseline characteristics of enrolled subjects
| Characteristic | All subjects (N = 12) |
|---|---|
| Sex, n (%) | |
| Male | 12 (100) |
| Age, years | |
| Mean (SD) | 37.8 (11.4) |
| Range | 24–52 |
| Race, n (%) | |
| White | 3 (25) |
| Black | 5 (42) |
| Other | 4 (33) |
| Weight, kg | |
| Mean (SD) | 83.0 (10.4) |
| Range | 65.5–99.7 |
| Height, cm | |
| Mean (SD) | 177.3 (7.6) |
| Range | 167–190 |
| BMI, kg m −2 | |
| Mean (SD) | 26.5 (3.5) |
| Range | 19.3–30.3 |
BMI, body mass index; SD, standard deviation
Median plasma concentration–time profiles following a single 100 mg oral dose of glasdegib administered alone and in the presence of rifampin in healthy volunteers are presented in Figure 1. The estimated pharmacokinetic parameters of glasdegib are summarised in Table 3. The geometric mean (geometric %CV) AUC inf and C max for glasdegib alone were 8145 ng × h ml−1 (23%) and 703.2 ng ml−1 (19%), respectively.
Figure 1.
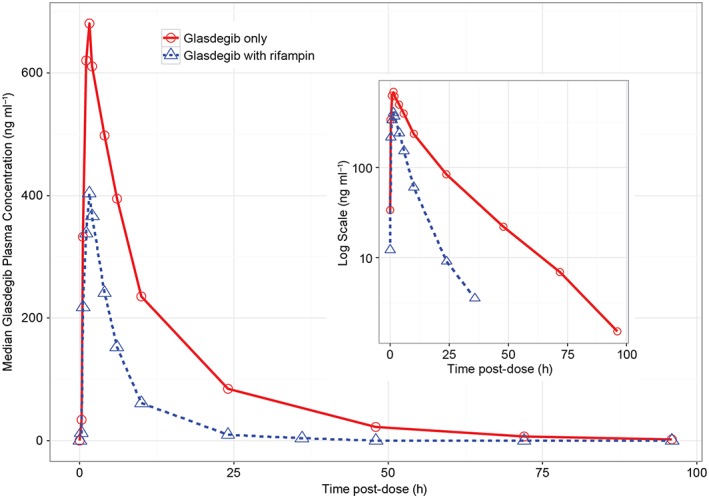
Linear median glasdegib plasma concentration–time profiles for glasdegib alone and in the presence of rifampin in healthy subjects; the inset shows the same data in a semi‐log scale
Table 3.
Glasdegib plasma pharmacokinetics
| Parameter a (unit) |
Glasdegib 100 mg s.d. + Rifampin 600 mg q.d. (Test)
(N = 12) |
Glasdegib 100 mg s.d.
(Reference) (N = 12) |
Mean Ratio b (90% CI) |
|---|---|---|---|
| AUC inf (ng × h ml −1 ) | 2416 (25) | 8145 (23) | 29.66 (26.17–33.62) |
| AUC last (ng × h ml −1 ) | 2385 (26) | 8051 (23) | 29.62 (26.17–33.54) |
| C max (ng ml −1 ) | 455.0 (26) | 703.2 (19) | 64.71 (57.21–73.19) |
| T max (h) | 1.25 (1.00–2.07) | 1.50 (1.00–4.05) | – |
| CL/F (l/h) | 41.38 (25) | 12.27 (23) | – |
| Vz/F (l) | 299.3 (23) | 232.7 (18) | – |
| t 1/2 (h) | 5.11 ± 1.06 | 13.39 ± 2.76 | – |
Geometric mean (geometric %CV) for all except median (range) for T max and arithmetic mean (±standard deviation) for t 1/2.
Adjusted geometric mean ratio for glasdegib in the presence vs. absence of rifampin and confidence interval expressed as percentages.
AUC inf, area under the plasma concentration–time curve from time zero extrapolated to infinity time; AUC last, area under the plasma–concentration curve from time zero to the time of the last quantifiable concentration; CI, confidence interval; C max, maximum observed concentration; CL/F, apparent oral clearance; %CV, percent coefficient of variation; q.d., once daily; s.d., single dose; T max, time to maximum observed concentration; t 1/2, terminal half‐life; Vz/F, apparent volume of distribution
When glasdegib was administered following pre‐treatment with rifampin, glasdegib exposure (i.e., AUCi nf) and peak exposure (i.e., C max) decreased in all 12 subjects (Figure 2). The mean ratio (glasdegib in the presence of rifampin/glasdegib alone) of adjusted geometric mean (90% CI) AUC inf and C max was 29.66% (26.17–33.62) and 64.71% (57.21–73.19), respectively. Co‐administration of rifampin resulted in approximately 70% and 35% decreases in AUC inf and C max, respectively (Table 3). Inter‐individual variability for glasdegib exposure was similar for both treatments, with geometric %CV ranging from 23% to 25% for AUC inf and 19% to 26% for C max.
Figure 2.
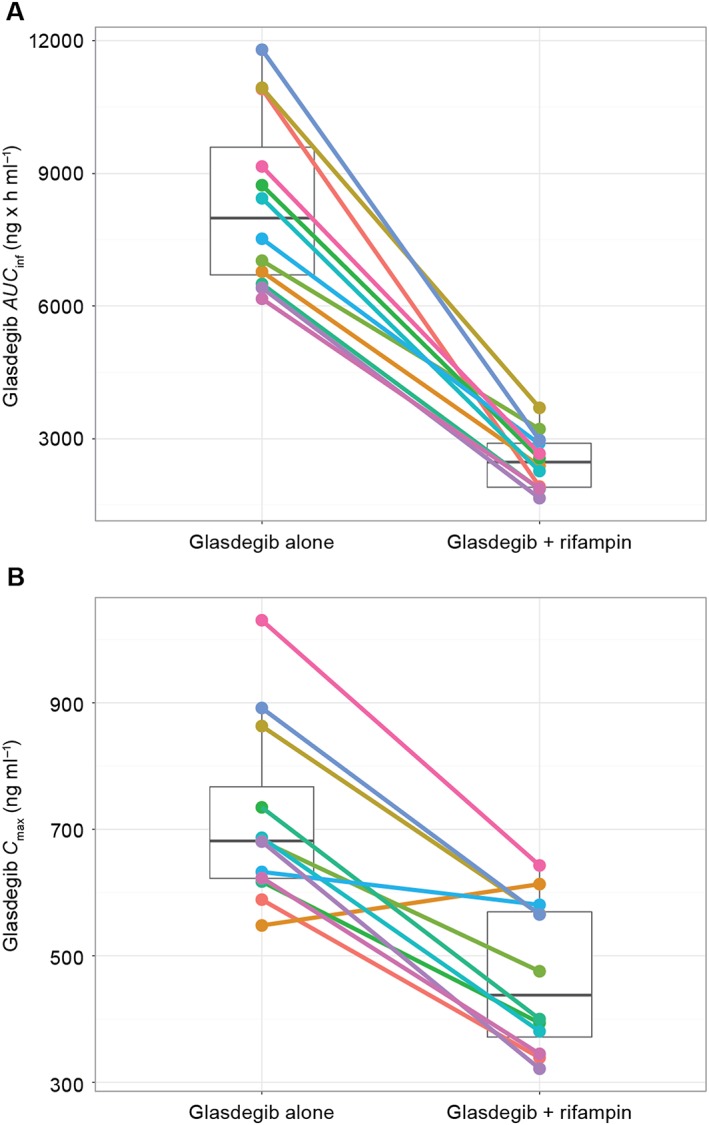
Matchstick plots for glasdegib (A) AUC inf and (B) C max when administered alone vs. in the presence of rifampin. Closed circles represent individual subject values; the top and bottom of the overlaid box plots represent 75% and 25% percentiles, respectively, and the middle bar represents the geometric mean of the observed data
Median T max for glasdegib was comparable between glasdegib in the presence of steady‐state rifampin and glasdegib alone (1.25 vs. 1.50 hours), but mean t 1/2 decreased from 13.39 to 5.11 hours with co‐administration of rifampin. Glasdegib geometric mean CL/F increased from 12.27 l h−1 in the absence of rifampin to 41.38 l h−1 in the presence of rifampin.
A total of 29 treatment‐emergent, all‐causality AEs were reported: one AE with 100 mg glasdegib alone; 16 with 600 mg rifampin alone; and 12 AEs with glasdegib co‐administered with rifampin (Table 4). Of 29 AEs, 24 were considered treatment‐related by the investigator. The most common AE (76%; n = 22/29), reported with rifampin alone or rifampin plus glasdegib, was chromaturia, which is a known side effect of rifampin treatment. All AEs were mild in severity and resolved by the end of the study.
Table 4.
Treatment‐emergent adverse events
| No. subjects with adverse event, n, all‐causality (n, treatment‐related) |
Glasdegib 100 mg
(N = 12) |
Rifampin 600 mg q.d. (N = 12) | Glasdegib 100 mg + Rifampin 600 mg q.d. (N = 12) |
|---|---|---|---|
| Any | 1 (0) | 16 (13) | 12 (11) |
| Gastrointestinal disorders | 0 | 2 (0) | 0 |
| Abdominal discomfort | 0 | 1 (0) | 0 |
| Aphthous stomatitis | 0 | 1 (0) | 0 |
| Metabolic and nutrition disorders | 0 | 1 (1) | 2 (1) |
| Decreased appetite | 0 | 0 | 1 (0) |
| Increased appetite | 0 | 1 (1) | 1 (1) |
| Renal and urinary disorders | 0 | 12 (12) | 10 (10) |
| Chromaturia | 0 | 12 (12) | 10 (10) |
| Skin and subcutaneous tissue disorders | 1 (0) | 1 (0) | 0 |
| Acne | 1 (0) | 1 (0) | 0 |
q.d., once daily
Glasdegib administered alone or in the presence of steady‐state rifampin was safe and well tolerated in healthy subjects evaluated in the current study.
Discussion
A human mass balance study showed glasdegib to be metabolised primarily by hepatic CYP3A4, and to a lesser extent, by uridine diphosphoglucuronosyltransferase 1A9 9. The involvement of CYP3A4 in the glasdegib metabolism was supported by the result of the previous DDI study, in which a known strong inhibitor of CYP3A, ketoconazole, increased glasdegib AUC inf and C max by 140% and 40%, respectively, in healthy volunteers 8. In the current DDI study, the co‐administration of rifampin, a potent CYP3A inducer, resulted in approximately 70% and 35% decreases in glasdegib AUC inf and C max, respectively, providing further evidence for the role of CYP3A in the metabolism of glasdegib. These results provide the limit of decrease in glasdegib exposures that could be expected with strong CYP3A4 induction when glasdegib is administered to patients with cancer. The pharmacokinetic data from the first‐in‐human dose escalation study indicated that the steady‐state pharmacokinetics of glasdegib could be predicted from single‐dose pharmacokinetics 7. Therefore, the results of this single‐dose study can be extrapolated to the multiple‐dosing setting in patients with cancer.
All subjects had a measurable decrease in AUC inf and 11 out of 12 subjects had a decrease in C max in the presence of rifampin (Figure 2). The presence of rifampin did not impact the variability in the pharmacokinetic parameters (%CV in Table 3), suggesting that CYP3A expression may not be the driver for inter‐individual variability in glasdegib pharmacokinetics. This observation is consistent with the results from the study assessing the effect of a strong CYP3A inhibitor 8.
The presence of rifampin, in addition to its effect on AUC inf and C max, increased the apparent oral clearance of glasdegib (by ~240%), with a smaller increase in the apparent volume of distribution (by ~30%). The T max of glasdegib was not affected by rifampin (1.5 vs. 1.25 hours); however, the t 1/2 was reduced by ~60% in the presence of rifampin.
Assuming a well‐stirred model for hepatic drug clearance, the observed changes in the pharmacokinetic parameters can be considered as follows. According to the well‐stirred model, the hepatic clearance is determined by liver blood flow, fraction unbound in blood and the intrinsic clearance of the liver 14. Intrinsic clearance mainly reflects enzymatic processes in the liver and can be altered during induction and inhibition of enzyme systems, such as treatment with rifampin, which induces CYP3A and, thereby increasing the intrinsic ability of the liver to clear glasdegib. This is reflected in the change in the apparent oral clearance observed in the study. Treatment with a metabolic inducer, such as rifampin, is not expected to affect either liver blood flow or the fraction unbound in blood. An increase in intrinsic clearance of a drug due to metabolic induction could result in decreased oral bioavailability (assuming complete absorption and no gut wall metabolism) because of the increased first‐pass effect. The fraction of drug escaping the first‐pass effect, and thus making it to the systemic circulation (FH), is also a function of hepatic blood flow, fraction unbound in blood and the intrinsic clearance 14. However, in this case, there is an inverse relationship between FH and intrinsic clearance such that an increase in intrinsic clearance would result in a larger first‐pass effect (FH decrease).
The smaller decrease observed in C max in the presence of metabolic induction, when compared with the large drop in AUC inf, suggests that, although the first‐pass effect might have some impact on the oral bioavailability of glasdegib, it does not play a major role. This conclusion is further supported by the small increase observed in the apparent volume of distribution. The increase in Vz/F from 233 to 299 L in the presence of rifampin, is most likely due to the reduction in oral bioavailability because of the decrease in FH, and does not reflect an increase in the distribution of the drug. Further studies, such as an absolute bioavailability study, may provide more insight into the mechanism for the observed effects.
The mean t 1/2 of glasdegib was reduced to 5.11 hours in the presence of a metabolic inducer, compared with 13.39 hours when glasdegib was dosed in the absence of an inducer. The t 1/2 of drug is dependent on the apparent volume of distribution (directly proportional) and the apparent oral clearance (inversely proportional). Therefore, the decrease in the half‐life parameter can be explained by the large increase in CL/F compared with the small increase in Vz/F, thereby resulting in an overall decrease in the terminal half‐life of glasdegib. This indicates that glasdegib will be washed out of the systemic circulation in a shorter time in the condition of metabolic induction of CYP3A, resulting in lower plasma exposure.
In‐vitro in the transfected MDCKII‐MDR1 cell system, glasdegib was shown to be a substrate of P‐glycoprotein (P‐gp). However, this efflux mechanism is expected to have minimal effect for the following reasons: (a) the absolute oral bioavailability of a 100 mg oral dose of glasdegib was high (77.12%), indicating that most of the drug reaches the systemic circulation and passive absorption is the predominant process; (b) there were dose proportional increases in glasdegib exposure (AUC and C max) observed in humans over the dose range of 5 to 600 mg following both a single dose and multiple dosing 7; and (c) the changes in glasdegib exposures observed with a strong CYP3A4 inhibitor (ketoconazole) and with a strong CYP3A4/pan‐inducer (rifampin) are in line with what would be expected based on inhibition/induction of CYP3A4. Therefore, while glasdegib was shown to be a substrate in‐vitro in a transfected system, in humans based on the observed clinical data, P‐gp does not appear to play a role in either absorption or elimination of glasdegib and hence is not considered relevant to the decrease in exposures due to rifampin treatment.
The current study also confirmed the safety and tolerability of glasdegib in healthy volunteers since there were no serious AEs, AE‐related treatment discontinuations or AE‐related dose reductions or temporary discontinuations.
In conclusion, this study estimated the decrease in glasdegib plasma exposure and maximum observed plasma concentration in the presence of a strong CYP3A inducer in healthy volunteers. These results provided the maximum reduction in exposure expected in cancer patients for 100 mg glasdegib once daily when co‐administered with a CYP3A inducer. These results will help to formulate recommendations for dosing strategies in combination with CYP3A inducers in situations where co‐administration may be necessary. For example, having an estimate of the maximal effect allows for use of in silico tools, such as Simcyp® population‐based absorption, distribution, metabolism and excretion simulator (Simcyp® Ltd, Sheffield, UK), to assess the effect of a moderate inducer of CYP3A and provide guidance for use with glasdegib dosing, although avoidance of the use of strong CYP3A inducers is recommended. The study further confirmed the safety and tolerability of glasdegib in healthy volunteers.
Competing Interests
All authors are employees of Pfizer and M.N.S., B.H. and R.R.L. own stock in Pfizer.
This study was sponsored by Pfizer . Medical writing support was provided by Mariko Nagashima, PhD, of Engage Scientific Solutions (Southport, CT, USA) and was funded by Pfizer.
Contributors
M.N.S. and B.H. were involved in conception or design of the work. M.N.S., B.H. and H.W. were involved in acquisition, analysis or interpretation of data. All authors revised the article critically for important intellectual content and approved the final version.
Shaik, M. N. , Hee, B. , Wei, H. , and LaBadie, R. R. (2018) Evaluation of the effect of rifampin on the pharmacokinetics of the Smoothened inhibitor glasdegib in healthy volunteers. Br J Clin Pharmacol, 84: 1346–1353. doi: 10.1111/bcp.13568.
References
- 1. Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev 2008; 22: 2454–2472. [DOI] [PubMed] [Google Scholar]
- 2. Lee RTH, Zhao Z, Ingham PW. Hedgehog signalling. Development 2016; 143: 367–372. [DOI] [PubMed] [Google Scholar]
- 3. Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel) 2016; 8: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Munchhof MJ, Li Q, Shavnya A, Borzillo GV, Boyden TL, Jones CS, et al Discovery of PF‐04449913, a potent and orally bioavailable inhibitor of smoothened. ACS Med Chem Lett 2012; 3: 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giordani G, Barraco M, Giangrande A, Martinelli G, Guadagnuolo V, Simonetti G, et al The human smoothened inhibitor PF‐04449913 induces exit from quiescence and loss of multipotent drosophila hematopoietic progenitor cells. Oncotarget 2016; 7: 55313–55327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wagner AJ, Messersmith WA, Shaik MN, Li S, Zheng X, McLachlan KR, et al A phase I study of PF‐04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2015; 21: 1044–1051. [DOI] [PubMed] [Google Scholar]
- 7. Martinelli G, Oehler VG, Papayannidis C, Courtney R, Shaik MN, Zhang X, et al Treatment with PF‐04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study. Lancet Haematol 2015; 2: e339–e346. [DOI] [PubMed] [Google Scholar]
- 8. Shaik MN, LaBadie RR, Rudin D, Levin WJ. Evaluation of the effect of food and ketoconazole on the pharmacokinetics of the smoothened inhibitor PF‐04449913 in healthy volunteers. Cancer Chemother Pharmacol 2014; 74: 411–418. [DOI] [PubMed] [Google Scholar]
- 9. Lam JL, Vaz A, Hee B, Liang Y, Yang X, Shaik MN. Metabolism, excretion and pharmacokinetics of [14C]glasdegib (PF‐04449913) in healthy volunteers following oral administration. Xenobiotica 2017: Jan 3; 47: 1–13. [DOI] [PubMed] [Google Scholar]
- 10. Giri N, Lam LH, LaBadie RR, Hee B, Liang Y, Woolfson A , et al Effect of a proton pump inhibitor on the plasma pharmacokinetics of the smoothened (SMO) inhibitor glasdegib (PF‐04449913). Proceedings of the The American Assocation of Pharmaceutical Scientists (AAPS) Annual Meeting; Nov 13–17; Denver, CO.
- 11. Minami Y, Minami H, Miyamoto T, Yoshimoto G, Kobayashi Y, Munakata W, et al Phase I study of glasdegib (PF‐04449913), an oral smoothened inhibitor, in Japanese patients with select hematologic malignancies. Cancer Sci 2017; 108: 1628–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The concise guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The concise guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilkinson GR, Shand DG. Commentary: a physiological approach to hepatic drug clearance. Clin Pharmacol Ther 1975; 18: 377–390. [DOI] [PubMed] [Google Scholar]