

Abstract
Aims
Previous studies demonstrated direct correlation between CYP2C19 genotype and BMS‐823778 clearance in healthy volunteers. The objective of the present study was to develop a physiologically‐based pharmacokinetic (PBPK) model for BMS‐823778 and use the model to predict PK and drug–drug interaction (DDI) in virtual populations with multiple polymorphic genes.
Methods
The PBPK model was built and verified using existing clinical data. The verified model was simulated to predict PK of BMS‐823778 and significance of DDI with a strong CYP3A4 inhibitor in subjects with various CYP2C19 and UGT1A4 genotypes.
Results
The verified PBPK model of BMS‐823778 accurately recovered observed PK in different populations. In addition, the model was able to capture the exposure differences between subjects with different CYP2C19 genotypes. PK simulation indicated higher exposures of BMS‐823778 in CYP2C19 poor metabolizers who were also devoid of UGT1A4 activity, compared to those with normal UGT1A4 functionality. Moderate DDI with itraconazole was predicted in subjects with wild‐type CYP2C19 or UGT1A4. However, in subjects without CYP2C19 or UGT1A4 functionality, significant DDI was predicted when BMS‐823778 was coadministered with itraconazole.
Conclusions
A PBPK model was developed using clinical data that accurately predicted human PK in different population with various CYP2C19 phenotypes. Simulations with the verified PBPK model indicated that UGT1A4 was probably an important clearance pathway in CYP2C19 poor metabolizers. DDI with itraconazole is likely to be dependent on the genotypes of CYP2C19 and UGT1A4.
Keywords: 11β‐hydroxysteroid dehydrogenase, CYP2C19, drug–drug interaction, genetic polymorphism, physiologically based pharmacokinetic model (PBPK), UGT1A4
What is Already Known about this Subject
BMS‐823778 is a potent inhibitor of 11β‐HSD1; metabolism of BMS‐823778 is mediated mainly by polymorphic CYP2C19, with minor contribution from CYP3A4/5 and UGT1A4.
BMS‐823778 is safe and well tolerated in healthy volunteers following single and multiple ascending doses of BMS‐823778; genotypic analysis demonstrated direct correlation between CYP2C19 genotype and BMS‐823778 clearance in healthy volunteers.
UGT1A4 polymorphism did not impact significantly the PK of BMS‐823778 in CYP2C19 extensive and intermediate metabolizers; however, BMS‐823778 is likely to be affected by UGT1A4 polymorphism in subjects who are devoid of CYP2C19 activity.
What this Study Adds
A PBPK model of BMS‐823778 was developed using PK data from previous clinical studies; the model accurately captured observed PK in different populations and exposure differences between subjects with various CYP2C19 genotypes.
The model was used to simulate PK of BMS‐823778 and potential drug–drug interaction with a strong CYP3A4 inhibitor in subjects with various genetic polymorphism of CYP2C19 and UGT1A4.
This study demonstrated the utility of mechanistic PBPK modelling/simulation to yield crucial insights into the likelihood of changes in PK in scenarios in which clinical studies are practically challenging or not feasible.
Introduction
BMS‐823778 is a potent and selective inhibitor for http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2763 1, a microsomal enzyme that regulates tissue concentration of biologically active cortisol 2, 3, 4, 5 and a compelling target for the treatment of metabolic diseases including type‐2 diabetes, dyslipidaemia and obesity 6, 7, 8, 9. In a phase 1 combined single ascending‐dose (SAD) and multiple ascending‐dose (MAD) study in normal healthy volunteers, BMS‐823778 was well tolerated with no severe drug‐related adverse effects 10. In vitro reaction phenotyping studies of BMS‐823778, prompted by the large pharmacokinetic (PK) variability in SAD/MAD, demonstrated that the metabolism of BMS‐823778 was mainly mediated by polymorphic http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=262#1328, with minor contribution from http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1337 and http://www.genenames.org/cgi-bin/gene_symbol_report?hgnc_id=HGNC:12536 11.
CYP2C19 is one of the most important CYP enzymes responsible for the metabolism of many clinically used drugs 12, 13, 14. To date, >30 allelic variants of the human CYP2C19 gene have been identified, of which CYP2C19*2 and CYP2C19*3 are the most important as they account for the majority of the variants seen in many populations 15, 16. Both variants generate truncated enzymes with no enzyme activity, as opposed to CYP2C19*17 which is associated with increased enzyme expression and catalytic activity. Subjects with *1/*1 genotype are considered extensive metabolizers (EMs), subjects with *1/*2 or *1/*3 are considered intermediate metabolizers (IMs) and subjects with *2/*2, *2/*3 and *3/*3 are considered poor metabolizers (PMs) 15, 17. UGT1A4 is a polymorphic enzyme involved in the metabolism of several drugs including lamotrigine, tamoxifen and clozapine 18, 19. Substrate‐dependent enzyme activity of UGT1A4 has been reported with the most common genetic variants (*2 and *3) 20.
The impact of polymorphisms on the PK of BMS‐823778 was investigated in clinical studies in Chinese and Japanese subjects, as well as in human mass–balance study with healthy volunteers 10. A direct correlation between genetic variation of CYP2C19 and BMS‐823778 clearance was observed in all studies. In general, the clearance of BMS‐823778 was ~4–5 fold lower in CYP2C19 PMs compared to EMs. By contrast, genetic polymorphism of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=263#1338 and UGT1A4 did not seem to impact PK of BMS‐823778 in subjects who were CYP2C19 EMs or IMs. However, in a CYP2C19 PM subject, genetic polymorphism (*1/*2) of UGT1A4 appeared to further reduce the clearance of BMS‐823778 10. Because of limited number of subjects in the study, the impact of UGT1A4 or CYP3A5 polymorphisms on the PK of BMS‐823778 in subjects with predicted CYP2C19 PM phenotype was not fully characterized. In addition, drug–drug interaction (DDI) with inhibitors of CYP2C19, UGT1A4 or CYP3A4/5 is likely to be dependent on the subject's genotype, and thus DDI studies need to be carefully designed and may have practical limitations from subject recruitment perspective. Significant DDI with an inhibitor drug in subjects with certain genotypes can potentially lead to safety and tolerability issues, and poses challenges on drug development or even leads to program termination. Therefore, the ability to predict DDIs with inhibitors of metabolizing enzymes for BMS‐823778 or other drug candidates which are substrates of multiple polymorphic enzymes are highly desirable and essential in informing dosing with concomitant medications in clinical studies.
Physiologically‐based PK (PBPK) models are widely used in the pharmaceutical industry in various stages during drug discovery and development to enable decision making 21, 22, 23, 24. PBPK modelling and simulation integrate both system‐dependent and drug‐dependent parameters to quantitatively predict the time course of plasma concentration of a drug candidate in virtual populations 23, 25, and are very useful in assessing potential DDIs with coadministered drugs, impact of genetic polymorphism and ethnicity on systemic exposures, and drug exposures in special populations 26, 27, 28, 29. The current study describes the development of a PBPK model for BMS‐823778 incorporating pharmacogenetic information, and the simulations with the verified PBPK model to predict PK of BMS‐823778 and DDI with a strong CYP3A4 inhibitor in subjects with multiple polymorphic genes. The objective was to use BMS‐823778 as an example to further demonstrate the utility of mechanistic PBPK modelling/simulation to yield crucial insights into the likelihood of changes in PK in scenarios in which clinical studies are practically challenging or not feasible.
Methods
General model development workflow
The PBPK model of BMS‐823778 was built and verified with a population‐based simulator (Simcyp version 15; Certara L.P., Sheffield, UK). A hybrid bottom‐up and top‐down approach was used for model development. First, physicochemical properties and absorption, distribution, metabolism and excretion (ADME) parameters determined from in vitro experiments or from in silico prediction in Simcyp were used to construct the initial PBPK model. Comparative simulations leveraging available pharmacogenetic and PK data from the clinical studies in healthy male subjects (mainly Caucasian), Chinese and Japanese subjects with various CYP2C19 and UGT1A4 functionality were then performed to optimize the ADME parameters. For each simulation, 10 trials with 10 subjects in each trial were conducted to evaluate variability across study groups. Finally, the verified model was applied to predict PK in subjects with multiple polymorphic enzymes and extent of DDI when coadministered with a strong inhibitor of CYP3A4 (itraconazole, 200 mg solution, fasted, once daily).
Virtual populations
All clinical trial simulations of BMS‐823778 were performed with Simcyp virtual populations of Caucasian, Chinese and Japanese generated by matching age distribution and gender ratio to the actual study data. PBPK modelling of PK from human mass balance study was performed with Simcyp virtual Caucasian population since the majority of the subjects in the human ADME study were Caucasians (11 out of 14 subjects). Virtual populations with predicted CYP2C19 and UGT1A4 EM, IM and PM phenotypes were generated by setting frequency to 1 for a particular phenotype and 0 for the others in the demographic setting of each population. Intrinsic catalytic activity of CYP3A4, CYP2C19 and UGT1A4 per unit amount of enzyme were assumed to be the same in Caucasian, Chinese and Japanese. Enzyme expression levels of CYP2C19, CYP3A4 and UGT1A4 in Caucasian and Chinese were pre‐defined in Simcyp (version 15) and remained unchanged during simulation. Simcyp default CYP2C19 expression levels in Japanese population are 1 pmol mg–1 protein in liver and 0.57 pmol mg–1 protein in intestine. However, the PBPK model significantly over‐predicted plasma concentrations of BMS‐823778 in Japanese CYP2C19 EM or IM subjects with the default CYP2C19 expression levels. Thus CYP2C19 expression was adjusted to 8 pmol mg–1 in liver and 0.85 pmol mg–1 in intestine to match the observed PK. Mean enzyme activity of human liver microsomes with CYP2C19*1/*2 and *1/*3 genotypes toward the oxidation of BMS‐823778 was approximately half of those with CYP2C19*1*1 genotype 11, and thus the expression levels of CYP2C19 in the IM virtual population were re‐defined as half of these in EM subjects in both liver and intestine in order to reduce CYP2C19 activity. Assuming subjects with the predicted CYP2C19 PM phenotype have no enzyme activity, the expression levels were set at 0 pmol mg–1 in the PM subpopulation in both liver and intestine. Similar approach was applied to the virtual populations of UGT1A4 with PM phenotype.
Clinical PK studies for model development
PK data of BMS‐823778 for model development and verification were generated from three clinical studies in healthy volunteers, where subjects were genotyped for CYP2C19, CYP3A5 and UGT1A4 prior to or post‐treatment 10. These include a single dose human mass balance study in healthy male volunteers (11 White, two Asian and one Black); a single and multiple dose study in Chinese subjects; and a multiple dose study in Japanese subjects. The number of subjects and dose levels of BMS‐823778 in each study are summarized in Table 1. Prior to these studies, a combined phase 1 SAD/MAD study was conducted in healthy volunteers to evaluate PK and tolerability of BMS‐823778 10. However, genotyping was not performed in the study due to the lack of reaction phenotyping data prior to study initiation. Thus, PK from the SAD/MAD study was not used for model development.
Table 1.
Details of the clinical studies in healthy volunteers, Chinese and Japanese subjects
| Study no. | Population | Dosing Regimen | Dose (mg) | Formulation | Predicted CYP2C19 Phenotype (n) |
|---|---|---|---|---|---|
| 1 | Healthy volunteersa (male, age 19–34 years) | SD | 10 | Solution | EM (10), PM (4) |
| 2 | Chinese (male, age 20–55 years) | SD and QD × 12 days | 2, 15 | Capsule | EM (8), IM (17), PM (1) |
| 3 | Japanese (male, age 20–55 years) | QD × 14 days | 2, 12, 25 | Capsule | EM (6), IM (7), PM (4) |
n, number of subjects; SD, single dose; QD, once daily dose; EM, extensive metabolizers; PM, poor metabolizers; IM, intermediate metabolizers
11 Caucasians, two Asians and one African American
Input parameters for PBPK model development
Physicochemical properties and blood binding
The physicochemical and ADME parameters of BMS‐823778 are summarized in Table 2. BMS‐823778 is a weak base with an acid dissociation constant (pKa) of 3.5. It is bound to proteins in human plasma with a free fraction (fu) of 15%, determined with an equilibrium dialysis method. The blood/plasma partition ratio was determined to be 0.8.
Table 2.
Physicochemical and pharmacokinetic parameters of BMS‐823778 used for the physiologically based pharmacokinetic (PBPK) model
| Parameters | Values | Parameters | Values |
|---|---|---|---|
| MW (g mol –1 ) | 328 | CYP3A4 | Km = 62.77 μmol l–1 |
| Fraction of unbound (f u ) | 0.15 | Vmax = 0.35 (pmol min1 pmol protein–1) | |
| Blood/plasma | 0.8 | REF = 1 | |
| pKa | 3.5 | CYP2C19 | Km = 33.85 μmol l–1 |
| LogPo:w | 2.76 | Vmax = 1.48 (pmol/min/pmol protein) | |
| Absorption | ADAM model | REF = 8 | |
| P eff,man (10 –4 cm s –1 ) | 7.01 | UGT1A4 | CLint = 0.012 (μl min–1 mg–1 protein) |
| F uGut | 1 | CLR (L/h) | 0.007 |
| Formulation | solution | fa | 0.99 predicted in Simcyp |
| Distribution | Full PBPK | fg | 0.97–0.99 predicted in Simcyp |
| K p scalar | 1 | fh | 0.92–0.97 predicted in Simcyp |
| V ss (l kg –1 ) | 1.88 | F% | 0.90–0.96 predicted in Simcyp |
| Biliary clearance (l h –1 ) | 0.6 (100% re‐absorb) |
Tissue:plasma partition coefficients were taken directly from the rat tissue distribution study; Kp values are listed in Table S1 (Supporting Information). CLint, intrinsic clearance; CLR, renal clearance; F%, bioavailability; fa, fraction absorbed; fg, fraction escaped gut metabolism; fh, fraction escaped hepatic first metabolism; Fugut, unbound fraction in enterocytes; Km, Michaelis‐Menten constant; Kp, tissue to plasma partition coefficient; LogPo:w, partition coefficient between octanol and water; MW, molecular weight; Peff,man, effective permeability; pKa, negative logarithm of the acid dissociation constant; REF, relative expression factor; Vmax, maximum rate; Vss, volume of distribution
Absorption
Advanced dissolution, absorption and metabolism (ADAM) model was used to describe the absorption after oral dosing. In vitro permeability was determined in a Caco‐2 cell bi‐directional permeability assay (Supporting Information). Permeability coefficients (Pc) from basal‐to‐apical and from apical‐to‐basal were 5.26 × 10–5 and 5.24 × 10–5 cm/s, respectively, suggesting that BMS‐823778 is highly permeable and is likely not a substrate of efflux transporters. Effective permeability (Peff, 7.01 × 10–4 cm/s) of BMS‐823778 in humans was predicted with a Simcyp built‐in algorithm based on in vitro Caco‐2 permeability results. Because of rapid dissolution of the capsule formulation in simulated intestinal fluid, a solution formulation was used in the model.
Distribution
A full PBPK model was used to maximize the mechanistic structure. A perfusion‐limit liver model was used since BMS‐823778 does not appear to be a substrate of efflux or uptake transporters based on Caco‐2 bi‐directional permeability and hepatocyte uptake assay (Supporting Information). Tissue:plasma partition coefficients (Kp) in various organs including liver, kidney, spleen, adipose, bone, heart, gut, muscle and skin were directly taken from a rat tissue distribution study that was conducted with quantitative whole body autoradiography following a single dose of [14C]BMS‐823778 to Long–Evans rats (Supporting Information). Steady state volume of distribution (Vss) was predicted based on the individual input Kp values in the aforementioned tissues with a global Kp scalar of 1, using the Rodgers and Rowland method in Simcyp 30. The predicted Vss (1.88 l kg–1) was in the range of clinically observed values (Vss/F ranged from 1.6–2.1 l kg–1 in SAD/MAD study) 10.
Elimination
Renal clearance (CLR) of BMS‐823778 in various populations was directly measured in the clinical studies, and a mean observed value of 0.007 l h–1 was applied in the model. Biliary clearance (CLint,bile) was estimated based on biliary elimination of BMS‐823778 from a bile duct‐cannulated monkey study (Supporting Information) by assuming that % dose eliminated through biliary as intact drug (15% in monkey) was similar across species. CYP3A4/5 was a minor metabolic pathway in the clearance of BMS‐823778 11. The impact of CYP3A5 polymorphism on the PK of BMS‐823778 was not well understood. Thus, CYP3A5 pathway was not modelled alone but was rather combined into CYP3A4 pathway in the model. Michaelis–Menten enzyme kinetics of CYP2C19 and CYP3A4, determined with CYP supersomes, were used to describe metabolic clearance of BMS‐823778 in Simcyp. Intersystem relative expression factor (REF), a scaling factor to correct the difference between liver or intestine subcellular fraction and in vivo enzyme expression, was used to optimize the enzyme activity of CYP3A4 and CYP2C19 to match the existing clinical PK data. Enzyme activity of UGT1A4 could not be determined in vitro with UGT supersomes because of low turnover. CLint of UGT1A4 was estimated by Simcyp with a sensitivity analysis in the Chinese and Japanese subjects with predicted CYP2C19 PM phenotype by fixing CLint values of other pathways including CYP3A4, biliary and urine excretion. UGT1A4 was more sensitive to the total clearance of BMS‐823778 in the PM subjects and its enzyme activity was better optimized using PK data from CYP2C19 PM subjects. The difference on enzyme activity in relation to the predicted CYP2C19 and UGT1A4 phenotypes was reflected in the expression levels of each enzyme in both liver and intestine in the predefined subpopulation as described previously, rather than by changing CLint.
Model refinement through PK simulation in Caucasian, Chinese and Japanese populations
Primary model parameters to be optimized included CLint of UGT1A4, CLint,bile and REF of CYP3A4 and CYP2C19. As the first step in model development, a total of 10 trials with 10 subjects in each trial were simulated with the initial base model in CYP2C19 EM and PM Caucasian subjects (male only, age 19–34 years) after receiving a single oral dose of BMS‐823778 (10 mg). Plasma concentrations were simulated up to 504 h postdose and the simulated PK profiles were compared to the existing clinical data from human mass balance study to ensure that simulated PK profiles were comparable to the observed data. Since multiple pathways were involved in the clearance of BMS‐823778, fraction of metabolism/elimination (fm/fe) of CYP2C19, UGT1A4 and biliary pathways were estimated from the clinical data. These fm/fe values were critical in defining CL or REF of each clearance pathway. Given that simulated apparent clearance (CLT) of CYP2C19 EMs was 4–5‐fold high than PMs in different populations 10, 11, the fm through CYP2C19 metabolism was estimated to be 75–80%. In addition, UGT1A4 polymorphism increased exposure of BMS‐823778 by approximately 50% in a CYP2C19 PM subject, an fm 0.5 was assumed for UGT1A4 pathway in subjects who are devoid of CYP2C19 activity. Fraction of biliary elimination in humans was assumed to be same as in monkeys (0.15).
The optimized PBPK model was validated through simulations in virtual Chinese (male only, age 20–55 years) at 2 mg and 15 mg of BMS‐823778, and in virtual Japanese populations (male only, age 20–55 years) at 2, 12 and 25 mg of BMS‐823778. Simulations were conducted in populations with various predicted CYP2C19 phenotypes after single and multiple doses of BMS‐823778. An acceptance criterion was applied when evaluating acceptability of predictions to ensure that predicted values to be within 2‐fold of the observed values. The demographics of the subjects in virtual clinical trials in all populations were consistent with the actual clinical studies. To match the actual study design, the Japanese subjects received daily dose of BMS‐823778 for 14 days and plasma concentrations were simulated up to 168 h after the last dose. For the Chinese study, a custom trial design was implemented in Simcyp where PK samples were collected up to 120 h after the first dose, and the second dose was administered on day 6 followed with once daily dose of BMS‐823778 until day 17.
Prediction of BMS‐823778 PK in CYP2C19 PMs with different UGT1A4 genotypes
Following the development of BMS‐823778 PBPK model, BMS‐823778 plasma concentration–time profiles of BMS‐823778 was simulated in Caucasian CYP2C19 PMs (male only, age 20–55 years) carrying wild‐type (normal activity) or polymorphic UGT1A4 (without UGT1A4 activity) following multiple doses of BMS‐823778 at 10 mg (once every 3 days, total of 20 doses) for 70 days. Plasma concentrations were simulated up to 240 h post dose after the last dose of BMS‐823778. Minimum plasma concentration of BMS‐823778 was used to evaluate the time for reaching steady state in different subpopulations, and area under the curve [AUC(0–72 h)] of day 1 and day 60 were calculated to estimate the accumulation index.
Evaluation of DDI with CYP3A4 inhibitor in Caucasians with various polymorphic genes
Potential DDI with a CYP3A4 inhibitor (itraconazole, 200 mg solution, once daily) was evaluated through the simulation with the PBPK model. The primary metabolite of itraconazole, OH‐itraconazole, was also included as an inhibitor of CYP3A4. The inhibitor PBPK models of itraconazole and OH‐itraconazole in Simcyp compound library was used directly for DDI simulations and their primary model parameters are summarized in the Supporting Information. Plasma concentrations of BMS‐823778 in the presence or absence of itraconazole were simulated for 10 trials each containing 10 healthy Caucasian subjects (male only, age 20–55 years) with various predicted CYP2C19 phenotypes at different levels of UGT1A4 activity. For each simulation, itraconazole was administrated once daily for 11 days starting on day 1, with the dose level and dosing formulation all predefined in Simcyp. A single oral dose of 10 mg BMS‐823778 was administered concomitantly with the inhibitor on day 4. The AUC(INF) ratios with/without co‐medication were estimated to assess the extent of DDI.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 31, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 32.
Results
PBPK model performance in Caucasians following a single dose of BMS‐823778
The simulated and observed mean concentration–time profiles of BMS‐823778 following a single dose of 10 mg BMS‐823778 to CYP2C19 EM and PM Caucasians are illustrated in Figure 1. The PBPK model aligned well with the observed plasma concentration–time profiles. In addition, after reducing the UGT1A4 activity by 50%, the model was able to describe the PK profile in the subject (Caucasian) with the predicted CYP2C19 PM phenotype and UGT1A4*1/*2 genotype in which the exposure of BMS‐823778 was ~50% higher than typical CYP2C19 PMs (normal UGT1A4 activity) 11. The simulated and observed apparent clearance (CLT/F) values of BMS‐823778 in different subpopulations are summarized in Table 3. The ratios of predicted/observed CLT/F in CYP2C19 EM and PM subjects ranged from 0.88–1.06, well within the acceptance criteria (within 2‐fold between simulated and observed values).
Figure 1.
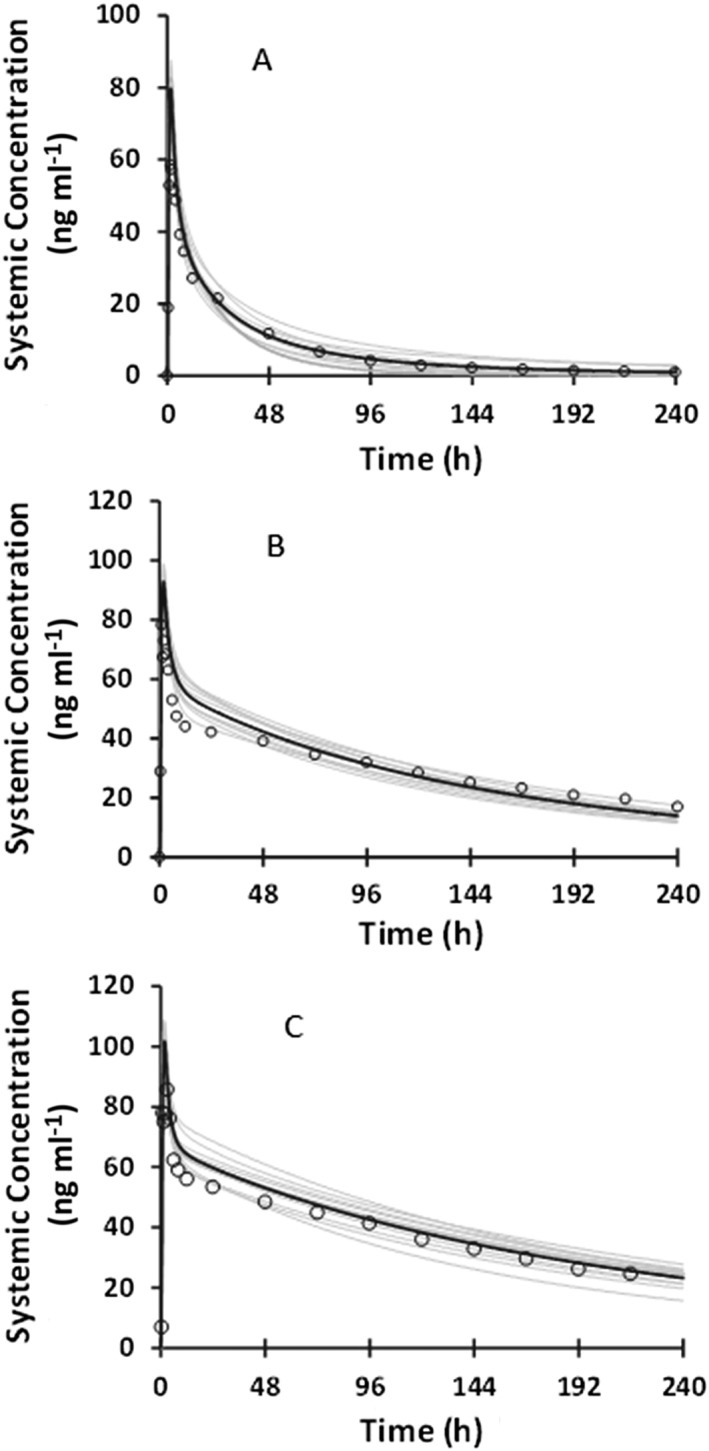
Comparison of observed and predicted plasma concentration–time profiles of BMS‐823778 following a single oral dose at 10 mg to healthy subjects with (A) CYP2C19 extensive metabolizers; (B) CYP2C19 poor metabolizers with UGT1A4*1/*1 genotype (normal activity); and (C) CYP2C19 poor metabolizers with UGT1A4*1/*2 genotype (simulated as 50% activity). Observed data: open circles; mean simulated profile: black line; individual simulated profile: grey line
Table 3.
Summary of observed and simulated apparent oral clearance (CLT/F) of BMS‐823778 in different populations with various predicted CYP2C19 phenotypes
| Study population | Predicted CYP2C19 phenotype | Observed CLT/Fc (ml min–1) | Simulated CLT/Fc (ml min–1) | Ratio of Predicted/ Observed |
|---|---|---|---|---|
| Healthy volunteers | EM | 89.2 | 78.9 | 0.88 |
| PMa | 14.7 | 15.4 | 1.05 | |
| PMb | 10.6 | 11.2 | 1.06 | |
| Chinese | EM | 52.4 | 51.3 | 0.98 |
| IM | 29.7 | 33.6 | 1.13 | |
| PM | 13.9 | 12.4 | 0.89 | |
| Japanese | EM | 50.9 | 58.5 | 1.15 |
| IM | 32.4 | 34.1 | 1.05 | |
| PM | 14.4 | 13.2 | 0.92 |
All CYP2C19 PM subjects in human mass balance study including the subject with UGT1A4 *1/*2 genotype
The CYP2C19 PM subject in human mass balance study with UGT1A4 *1/*2 genotype.
CLT/F values at 10, 15 and 12 mg for healthy volunteer, Chinese and Japanese, respectively, were used for comparison
EM, extensive metabolizers; PM, poor metabolizers; IM, intermediate metabolizers
PBPK model performance in Chinese and Japanese populations with different genotypes
The simulated and observed PK profiles of BMS‐823778 following single and multiple daily‐doses to healthy Chinese subjects are illustrated in Figure 2, and the predicted and observed CLT/F in Chinese subjects with various predicted CYP2C19 phenotypes are summarized in Table 3. The PBPK model described the observed concentration–time profiles at 15 mg BMS‐823778 both on day 1 and after the last dose. The ratios of predicted/observed geometric mean for the CLT/F ranged from 0.89–1.13 (Table 3). Exposure differences between various predicted CYP2C19 phenotypes were also accurately captured by the PBPK model in Chinese populations at 15 mg dose. At 2‐mg dose of BMS‐823778, the PBPK model over‐predicted the maximum plasma concentration and AUC (INF) in Chinese subjects after the first dose. However, the model was able to describe the exposure of BMS‐823778 at steady state after multiple daily‐doses of BMS‐823778.
Figure 2.
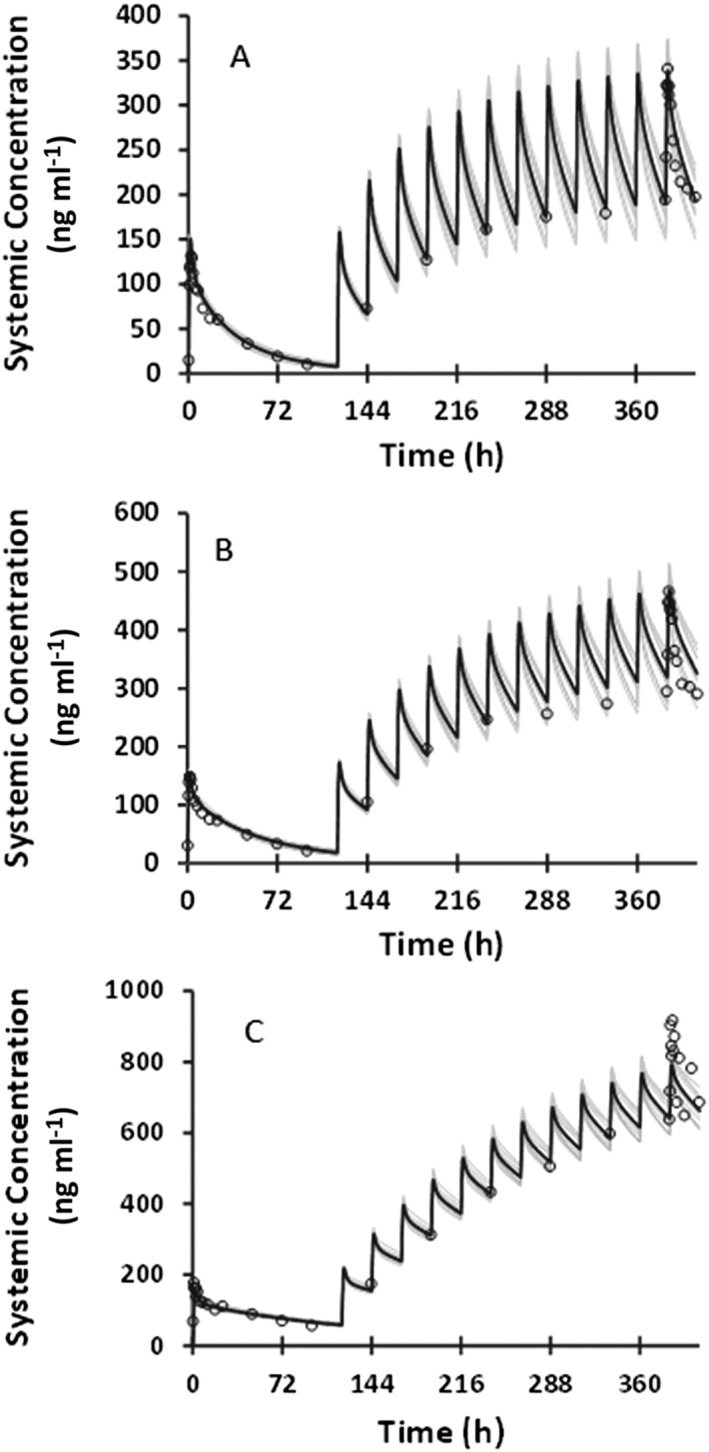
Comparison of observed and predicted plasma concentration–time profiles of BMS‐823778 following a single and multiple oral doses to healthy Chinese subjects with (A) CYP2C19 extensive metabolizers (15 mg); (B) CYP2C19 intermediate metabolizers (15 mg); (C) CYP2C19 poor metabolizers (15 mg). Observed data: open circles; mean simulated profile: black line; individual simulated profile: grey line
In the Japanese subjects, PK of BMS‐823778 was simulated following daily doses (2, 12 and 25 mg) for 14 days. The model reasonably recovered PK profiles in CYP2C19 PM subjects (Figure 3), but significantly over‐predicted plasma concentrations of BMS‐823778 in CYP2C19 EM or IM subjects with the Simcyp default CYP2C19 expression levels in both liver and intestine (Figure S1, Supporting Information). In order to describe the concentration–time profiles of BMS‐823778, the expression levels of CYP2C19 in both liver and intestine were adjusted by comparing the simulated PK profiles to the observed clinical data. It was found that using the similar abundance of CYP2C19 to those in Chinese population (8 pmol mg–1 protein in liver and 0.85 pmol mg–1 protein in intestine), the PBPK model accurately recovered observed clinical data from the Japanese CYP2C19 EM and PM subjects (Figure 3). Simulated CLT/F values from 10 trials were in good agreement with the observed values (Table 3). Similar to the Chinese subjects, the PBPK model was able to describe the PK profiles at 2 mg only after multiple dose of BMS‐823778. Day 1 exposure of BMS‐823778 at 2 mg was significantly over‐predicted by the model.
Figure 3.
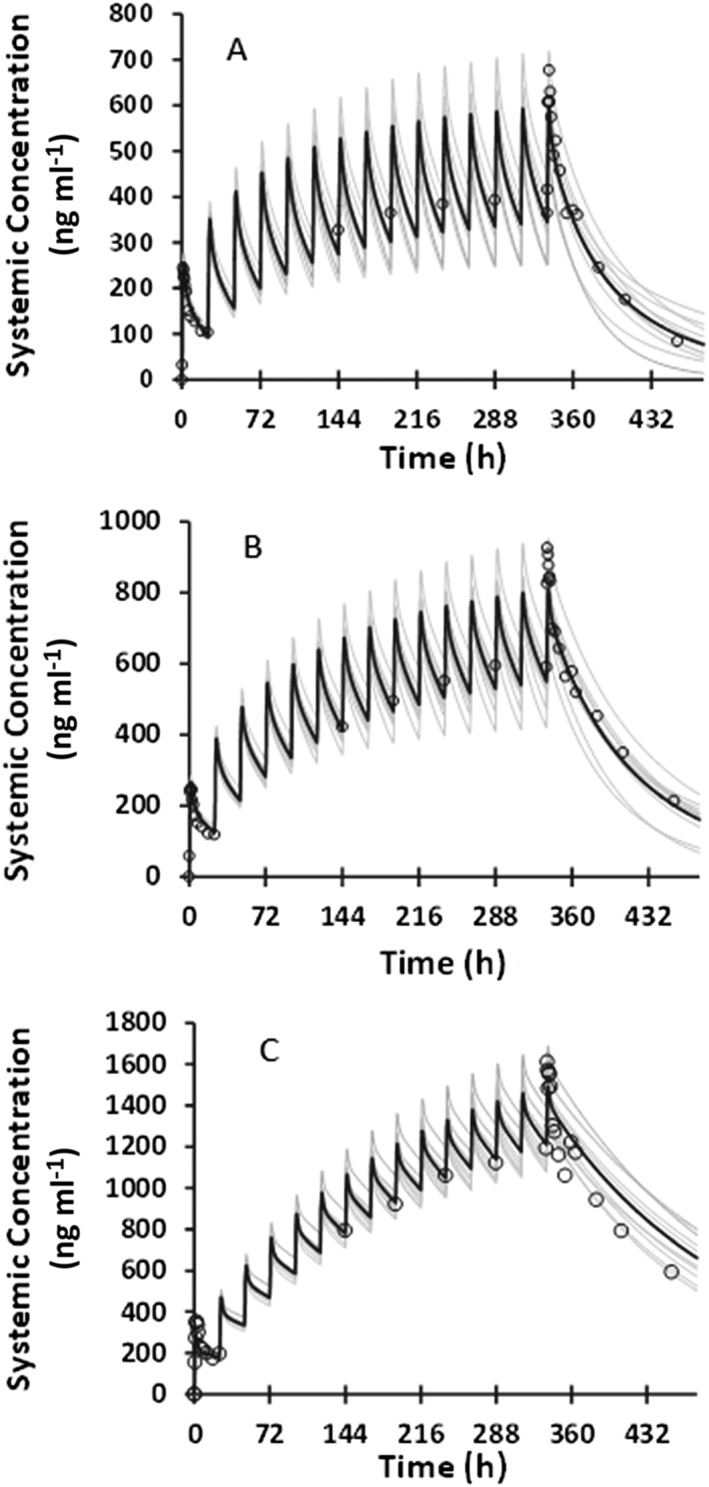
Comparison of observed and predicted plasma concentration–time profiles of BMS‐823778 following daily doses for 14 days to healthy Japanese subjects with (A) CYP2C19 extensive metabolizers (25 mg); (B) CYP2C19 intermediate metabolizers (25 mg); and (C) CYP2C19 poor metabolizers (25 mg). Observed data: open circles; mean simulated profile: black line; individual simulated profile: grey line
Prediction of BMS‐823778 PK in CYP2C19 PMs with different UGT1A4 functionality
The time course of BMS‐823778 plasma concentration after multiple doses was simulated with the verified PBPK model in CYP2C19 PMs with various UGT1A4 genotypes. Based on the previous clinical data, it was assumed that polymorphic UTG1A4 has reduced enzyme activity. The simulated concentration–time profiles following 10 mg of BMS‐823778 (once in 3 days, total of 20 doses) are illustrated in Figure 4, and PK parameters are summarized in Table 4. High exposure of BMS‐823778 was predicted in CYP2C19 PM subjects who were devoid of UGT1A4 functionality, ~1.8 fold higher at steady state than subjects with UGT1A4*1/*1 genotype (normal enzyme activity). Steady state was reached after 9 and 20 doses of BMS‐823778 in subjects with wild‐type or polymorphic UGT1A4 (assuming zero enzyme activity), respectively. Accumulation index of BMS‐823778 ranged from ~3.2 in subjects with normal UGT1A4 activity to 5.2 in subjects who were devoid of UGT1A4 functionality.
Figure 4.
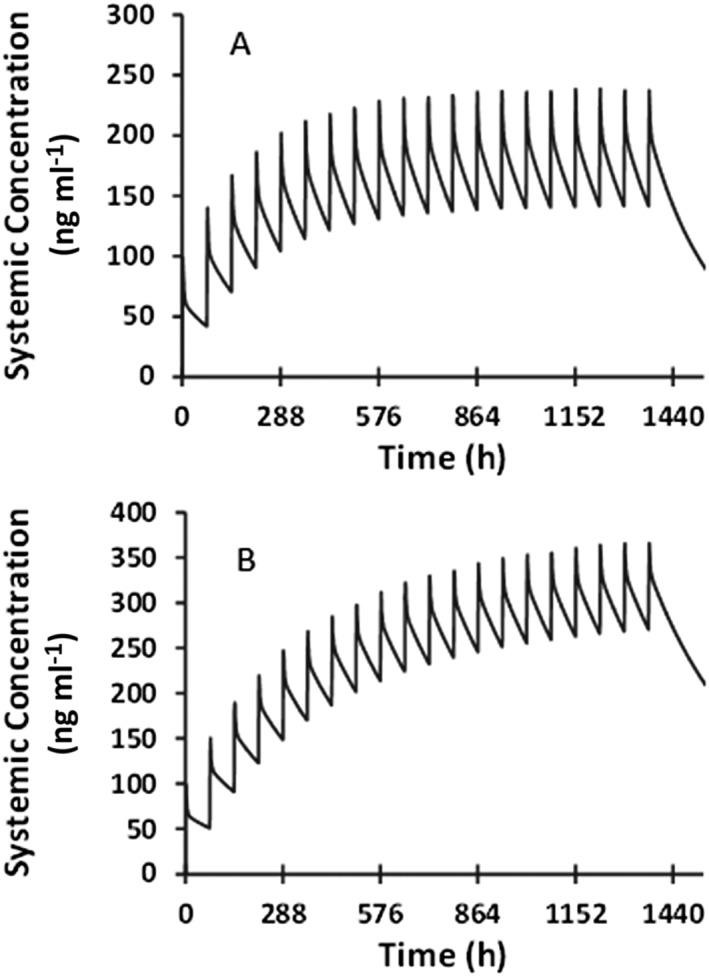
Simulated plasma concentration–time profiles of BMS‐823778 following oral doses (once every 3 days, total 20 doses) at 10 mg to CYP2C19 poor metabolizers with (A) UGT1A4 *1/*1 genotype (normal activity); and (B) polymorphic UGT1A4 (simulated with no enzyme activity)
Table 4.
Summary statistics of simulated BMS‐823778 pharmacokinetic with the physiologically based pharmacokinetic model following oral doses (once every 3 days) at 10 mg to CYP2C19 PM Caucasians with different UGT1A4 genotypes
| Treatment Group | Study Day | Cmax (ng ml–1) (CV%) | AUC(τ) (ng h ml–1) (CV%) | Half‐life (h) (CV%) | AI |
|---|---|---|---|---|---|
| UGT1A4*1/*1 (normal activity) | 1 | 98.8 (3.2) | 3776.6 (285.3) | NA | |
| 60 | 237.7 (22.8) | 12276.1 (1577.8) | 135.6 (16.8) | 3.2 | |
| Polymorphic UGT1A4 (no activity) | 1 | 99.3 (13.8) | 4242.7 (229.1) | NA | |
| 60 | 366.7 (25.3) | 21 948 (1849.3) | 235.7 (19.6) | 5.2 |
NA, not applicable; AI, accumulation index
T‐half on day 1 was not calculated because of short terminal phase.
Simulation of DDI with CYP3A4 inhibitor
The effect of itraconazole on the PK of BMS‐823778 was evaluated with the verified PBPK model in subjects with various predicted CYP2C19 phenotypes at different levels of UGT1A4 activity. Simulated exposures of itraconazole and OH‐itraconazole are summarized in Table S3 (Supporting Information) and were in the range of reported values 33, 34. As summarized in Table 5, itraconazole had little impact on BMS‐823778 exposure in CYP2C19 EMs, regardless of UGT1A4 activity. Moderate increase in AUC (1.9×) was observed when BMS‐823778 was coadministered with itraconazole to the CYP2C19 PM subjects with wild‐type UGT1A4 (normal enzyme activity). In subjects who were devoid of both CYP2C19 and UGT1A4 functionality, BMS‐823778 exposure was increased by 8.5‐fold when coadministered with itraconazole.
Table 5.
Pharmacokinetic simulation of BMS‐823778 with the physiologically based pharmacokinetic model following an oral dose with or without co‐administration of itraconazolea in CYP2C19 extensive metabolizer (EM) or poor metabolizer (PM) subjects, either with UGT1A4*1/*1 genotype (normal activity) or polymorphic UGT1A4 (no activity)
| Predicted CYP2C19 Phenotype | UGT1A4 Genotype | BMS‐823778 at 10 mg alone | With itraconazole | AUC Ratio | ||
|---|---|---|---|---|---|---|
| Cmax (ng ml–1) | AUC (INF) (ng h ml–1) | Cmax (ng ml–1) | AUC(INF) (ng h ml–1) | |||
| EM | UGT1A4*1/*1 | 85 | 1760 | 86 | 1940 | 1.1 |
| PM | UGT1A4*1/*1 | 98 | 7560 | 162 | 14 400 | 1.9 |
| EM | Polymorphic UGT1A4 | 86 | 1900 | 87 | 2100 | 1.1 |
| PM | Polymorphic UGT1A4 | 100 | 9260 | 101 | 78 700 | 8.5 |
OH‐itraconazole was included in the simulation; model parameters of itraconazole and OH‐itraconazole are included in the Supporting Information.
EM, extensive metabolizers; PM, poor metabolizers
Discussion
The aim of present work was to develop a mechanistic PBPK model for BMS‐823778, a substrate of multiple polymorphic enzymes. Pharmacogenetic studies have demonstrated a direct correlation between predicted CYP2C19 phenotype and in vivo clearance of BMS‐823778 10. However, the impact of polymorphism of UGT1A4 on the PK of BMS‐823778 has not been adequately characterized, and the significance of DDI in the presence of enzyme inhibitors in subjects with various genetic polymorphisms is unknown. The strategy of the present work was to use pharmacogenetic and PK data to develop and validate a PBPK model, and then use the model to predict PK profiles of BMS‐823778 as well as DDI potential with perpetrator drugs in subjects with various CYP2C19 and UGT1A4 functionality.
A base model with ADAM in absorption and full PBPK in distribution was constructed based on physicochemical properties, in vitro ADME parameters and rat tissue distribution data. In vivo clearance of BMS‐823778 was significantly underestimated with the in vitro enzyme kinetic values. Therefore, relative expression factors for CYP2C19 and CYP3A4 were introduced and subsequently optimized by comparing the simulated plasma concentration–time profiles to the observed clinical data. The refined model accurately recovered plasma concentration–time profiles of BMS‐823778 from the single dose human ADME study in healthy volunteers and the study in Chinese subjects with predicted CYP2C19 EM, IM or PM phenotypes, following single or multiple doses of BMS‐823778. Simulated apparent clearance CLT/F values in subjects with different predicted CYP2C19 phenotypes were consistent with the observed values from both studies. Furthermore, a 4–5‐fold difference in exposure between CYP2C19 EM and PM subjects was simulated for both Caucasian and Chinese populations which was closely matched with clinical results, demonstrating that the impact of CYP2C19 genetic polymorphism on the PK of BMS‐823778 was accurately captured by the model.
In Japanese subjects, however, even though the model reasonably described the time course of plasma concentration of BMS‐823778 in those with predicted CYP2C19 PM phenotype, it significantly over‐predicted the exposure in CYP2C19 EM or IM subjects. The discrepancy between simulated and observed PK in subjects with CYP2C19 activity indicated that Simcyp default enzyme expression for CYP2C19 might not represent the majority of Japanese population. The default liver expression of CYP2C19 in Simcyp was 14, 8 and 1 pmol mg–1 protein in Caucasian, Chinese and Japanese, respectively. It has been reported that CYP2C19 expression level in Japanese might be under‐estimated in Simcyp because of over‐prediction of omeprazole exposure in Japanese subjects by the Simcyp model 27. Subsequently the authors increased CYP2C19 liver expression (4.77 pmol mg–1 protein) in Simcyp virtual Japanese population to describe the in vivo clearance of omeprazole 27, 35. In the current study, the model was able to predict the PK in Japanese EM and IM subjects when both liver and intestine CYP2C19 expressions were adjusted to the same levels as in Chinese population. Overall, the PBPK model of BMS‐823778 could describe the exposure differences between the different ethnic groups with the adjusted enzyme abundance suggesting that the virtual population in Simcyp can be used to predict the clearance of CYP2C19 substrates when the enzyme expression levels are accurately defined.
The PBPK model over‐predicted day‐1 PK of BMS‐823778 at low doses in both Chinese and Japanese studies. This might be due to the nonlinear PK of BMS‐823778 after the first dose at low dose levels 10. In the SAD study, there was a greater than dose‐proportional increase in exposure in the dose range of 2–12 mg. However, when exposure has reached steady state after multiple daily‐doses of BMS‐823778, the exposure increased largely proportionally to dose even at low doses 10. The PBPK model was not able to capture the nonlinear PK because of unknown mechanism for the relatively low exposure of BMS‐823778 after the first dose at low dose levels.
Direct association between UGT1A4 genotype and phenotype has not been established. The impact of polymorphism on UGT1A4 enzyme activity appeared to be substrate‐dependent 20. PK from human mass balance study suggested that polymorphic UGT1A4 could have reduced enzyme activity for the metabolism of BMS‐823778 10. However, only one subject was identified with UGT1A4 genetic variation who was also a CYP2C19 PM, and the impact of UGT1A4 polymorphism needs to be tested further in the clinical study. The challenge would be to recruit enough CYP2C19 PMs carrying UGT1A4 allelic variants because of low frequency of these special populations. PBPK model has the advantage of using virtual populations with various genotypes for PK simulation to provide crucial insight in the PK of BMS‐823778 in rare populations which otherwise would be difficult to recruit. In the present study, the impact of UGT1A4 polymorphism on the clearance of BMS‐823778 in CYP2C19 PMs was investigated with the verified PBPK model. Assuming normal enzyme activity for wild‐type UGT1A4 and no activity for polymorphic UGT1A4, higher exposure, longer T‐half and greater accumulation at steady state was predicted in CYP2C19 PMs with polymorphic UGT1A4, comparing to those with wild‐type UGT1A4 (Table 4). The results suggested that UGT1A4 could be an important pathway in the clearance of BMS‐823778 in CYP2C19 PMs, retrospectively supporting the previous hypothesis on the reduced activity of polymorphic UGT1A4 towards the metabolism of BMS‐823778. In the current PBPK model, CLint of UGT1A4 pathway was estimated by fitting the model to the observed PK, instead of being characterized with UGT supersomes. Simulation with the PBPK model could be misleading if exposure increase in the subject with UGT1A4*1/*2 genotype resulted from intersubject variability rather than UGT1A4 activity change due to polymorphism. Further in vitro study is recommended to directly correlate the enzyme activity with different genotypes of UGT1A4 for the glucuronidation of BMS‐823778.
Clinical pharmacogenetic studies in subjects with various CYP2C19 genotypes demonstrated the important role of CYP2C19 in the clearance of BMS‐823778. The exposure difference between CYP2C19 EM and PM subjects provided a quantitative measure of the maximum DDI when BMS‐823778 is coadministered with a strong CYP2C19 inhibitor. CYP3A4 was a minor clearance pathway but its contribution could increase significantly in CYP2C19 PMs, particularly in subjects lacking functionality of both CYP2C19 and UGT1A4. Thus, DDI of BMS‐823778 with CYP3A4 inhibitors is probably dependent on the genotypes of CYP2C19 and UGT1A4. To estimate the DDI with a strong CYP3A4 inhibitor, PK of BMS‐823778 was simulated in the presence or absence of itraconazole in subjects with various CYP2C19 and UGT1A4 genotypes. Mild to moderate DDI was predicted when BMS‐823778 was co‐administered with itraconazole in subjects who have either CYP2C19 or UGT1A4 activity. By contrast, significant DDI with itraconazole was predicted in the CYP2C19 PM subjects with polymorphic UGT1A4 where CYP3A4 became a major clearance pathway for BMS‐823778.
Conclusions
We have developed a PBPK model for BMS‐823778 using clinical pharmacogenetic data that accurately predicted human PK following a single or multiple doses of BMS‐823778 to Caucasian, Chinese and Japanese populations. In addition, the PBPK model was able to describe the exposure differences in subjects with different predicated CYP2C19 phenotypes. Simulations with the verified PBPK model in subjects with various CYP2C19 and UGT1A4 activities indicated that UGT1A4 could be an important clearance pathway in CYP2C19 PMs. The extent of DDI with itraconazole was dependent on the genotypes of CYP2C19 and UGT1A4, and significant DDI with itraconazole was predicted in subjects who were devoid of both CYP2C19 and UGT1A4 functionality. The verified PBPK model can be simulated further to predict PK of BMS‐823778 in special populations, as well as PK in subjects with ultra‐metabolizer CYP2C19 *17 genotype if the enzyme activity for CYP2C19 *17 can be determined. Through this particular example, we have further demonstrated how a PBPK model could be developed with pharmacogenetics data and how the mechanistic PBPK model could be used to predict PK and extent of DDI in which clinical studies are practically challenging or not feasible.
Competing Interests
There are no competing interests to declare.
Supporting information
Table S1 Concentration of [14C]BMS‐823778‐derived radioactivity (ng equivalent g–1) in plasma and tissues of male Long–Evans rats following an oral dose of [14C]BMS‐823778 at 5 mg kg–1
Table S2 Parameters of itraconazole and OH‐itraconazole in the Simcyp physiologically based pharmacokinetic model
Table S3 Simulated pharmacokinetic parameters of itraconazole and OH‐itraconazole on day 1 and day 11
Figure S1 Simulated vs. observed pharmacokinetic of BMS‐823778 in Japanese CYP2C19 extensive metabolizer subjects at 2 and 12 mg with Simcyp default enzyme expression (1 pmol mg–1 protein in liver and 0.57 pmol mg–1 protein in intestine). Observed data: red circles; mean simulated profile: black line; individual simulated profile: coloured lines
Gong, J. , Iacono, L. , Iyer, R. A. , Humphreys, W. G. , and Zheng, M. (2018) Physiologically‐based pharmacokinetic modelling of a CYP2C19 substrate, BMS‐823778, utilizing pharmacogenetic data. Br J Clin Pharmacol, 84: 1335–1345. doi: 10.1111/bcp.13565.
References
- 1. Li J, Kennedy LJ, Wang H, Li JJ, Walker SJ, Hong Z, et al Optimization of 1,2,4‐triazolopyridines as inhibitors of human 11β‐hydroxysteroid dehydrogenase type 1 (11β‐HSD‐1). ACS Med Chem Lett 2014; 5: 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morentin Gutierrez P, Gyte A, deSchoolmeester J, Ceuppens P, Swales J, Stacey C, et al Continuous inhibition of 11beta‐hydroxysteroid dehydrogenase type I in adipose tissue leads to tachyphylaxis in humans and rats but not in mice. Br J Pharmacol 2015; 172: 4806–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morgan SA, McCabe EL, Gathercole LL, Hassan‐Smith ZK, Larner DP, Bujalska IJ, et al 11β‐HSD1 is the major regulator of the tissue‐specific effects of circulating glucocorticoid excess. Proc Natl Acad Sci 2014; 111: E2482–E2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morton NM. Obesity and corticosteroids: 11β‐hydroxysteroid type 1 as a cause and therapeutic target in metabolic disease. Mol Cell Endocrinol 2010; 316: 154–164. [DOI] [PubMed] [Google Scholar]
- 5. Dube S, Norby BJ, Pattan V, Carter RE, Basu A, Basu R. 11[beta]‐Hydroxysteroid dehydrogenase types 1 and 2 activity in subcutaneous adipose tissue in humans: implications in obesity and diabetes. J Clin Endocrinol Metab 2015; 100: E70–E76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, Yao W, et al The 11‐β‐hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 2010; 33: 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shah S, Hermanowski‐Vosatka A, Gibson K, Ruck RA, Jia G, Zhang J, et al Efficacy and safety of the selective 11beta‐HSD‐1 inhibitors MK‐0736 and MK‐0916 in overweight and obese patients with hypertension. J Am Soc Hypertens 2011; 5: 166–176. [DOI] [PubMed] [Google Scholar]
- 8. Shao S, Zhang X, Zhang M. Inhibition of 11β‐hydroxysteroid dehydrogenase type 1 ameliorates obesity‐related insulin resistance. Biochem Biophys Res Commun 2016; 478: 474–480. [DOI] [PubMed] [Google Scholar]
- 9. Stefan N, Ramsauer M, Jordan P, Nowotny B, Kantartzis K, Machann J, et al Inhibition of 11beta‐HSD1 with RO5093151 for non‐alcoholic fatty liver disease: a multicentre, randomised, double‐blind, placebo‐controlled trial. Lancet Diabetes Endocrinol 2014; 2: 406–416. [DOI] [PubMed] [Google Scholar]
- 10. Gong J, Iacono L, Hansen L. Clinical pharmacokinetics and the impact of genetic polymorphisms on a CYP2C19 substrate, BMS‐823778, in healthy subjects. Drug Metab Dispos 2018; 46: 316–325. [DOI] [PubMed] [Google Scholar]
- 11. Cheng Y, Wang L, Iacono L, Zhang D, Chen W, Gong J, et al Clinical significance of CYP2C19 polymorphisms on the metabolism and pharmacokinetics of 11β‐hydroxysteroid dehydrogenase type‐1 inhibitor BMS‐823778. Br J Clin Pharmacol 2018; 84: 130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang X‐L, Samant S, Lesko LJ, Schmidt S. Clinical pharmacokinetics and pharmacodynamics of Clopidogrel. Clin Pharmacokinet 2015; 54: 147–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirota T, Eguchi S, Ieiri I. Impact of genetic polymorphisms in CYP2C9 and CYP2C19 on the pharmacokinetics of clinically used drugs. Drug Metab Pharmacokinet 2013; 28: 28–37. [DOI] [PubMed] [Google Scholar]
- 14. Green B, Crauwels H, Kakuda TN, Vanveggel S, Brochot A. Evaluation of concomitant Antiretrovirals and CYP2C9/CYP2C19 polymorphisms on the pharmacokinetics of Etravirine. Clin Pharmacokinet 2016; 56: 1–12. [DOI] [PubMed] [Google Scholar]
- 15. Desta Z, Zhao X, Shin J‐G, Flockhart DA. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet 2002; 41: 913–958. [DOI] [PubMed] [Google Scholar]
- 16. McGraw J, Waller D. Cytochrome P450 variations in different ethnic populations. Expert Opin Drug Metab Toxicol 2012; 8: 371–382. [DOI] [PubMed] [Google Scholar]
- 17. Wojnowski L, Kamdem LK. Clinical implications of CYP3A polymorphisms. Expert Opin Drug Metab Toxicol 2006; 2: 171–182. [DOI] [PubMed] [Google Scholar]
- 18. Ehmer U, Vogel A, Schutte JK, Krone B, Manns MP, Strassburg CP. Variation of hepatic glucuronidation: novel functional polymorphisms of the UDP‐glucuronosyltransferase UGT1A4. Hepatology (Baltimore, Md) 2004; 39: 970–977. [DOI] [PubMed] [Google Scholar]
- 19. Reimers A, Sjursen W, Helde G, Brodtkorb E. Frequencies of UGT1A4*2 (P24T) and *3 (L48V) and their effects on serum concentrations of lamotrigine. Eur J Drug Metab Pharmacokinet 2016; 41: 149–155. [DOI] [PubMed] [Google Scholar]
- 20. Zhou J, Argikar UA, Remmel RP. Functional analysis of UGT1A4(P24T) and UGT1A4(L48V) variant enzymes. Pharmacogenomics 2011; 12: 1671–1679. [DOI] [PubMed] [Google Scholar]
- 21. Kostewicz ES, Aarons L, Bergstrand M, Bolger MB, Galetin A, Hatley O, et al PBPK models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci 2014; 57: 300–321. [DOI] [PubMed] [Google Scholar]
- 22. Huang SM, Abernethy DR, Wang Y, Zhao P, Zineh I. The utility of modeling and simulation in drug development and regulatory review. J Pharm Sci 2013; 102: 2912–2923. [DOI] [PubMed] [Google Scholar]
- 23. Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami‐Hodjegan A. The Simcyp® population‐based ADME simulator. Expert Opin Drug Metab Toxicol 2009; 5: 211–223. [DOI] [PubMed] [Google Scholar]
- 24. Rostami‐Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov 2007; 6: 140–148. [DOI] [PubMed] [Google Scholar]
- 25. Jamei M, Dickinson GL, Rostami‐Hodjegan A. A framework for assessing inter‐individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘bottom‐up’ vs ‘top‐down’ recognition of covariates. Drug Metab Pharmacokinet 2009; 24: 53–75. [DOI] [PubMed] [Google Scholar]
- 26. Duan P, Zhao P, Zhang L. Physiologically based pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug‐drug interactions (DDIs). Eur J Drug Metab Pharmacokinet 2017; 42: 689–705. [DOI] [PubMed] [Google Scholar]
- 27. Feng S, Cleary Y, Parrott N, Hu P, Weber C, Wang Y, et al Evaluating a physiologically based pharmacokinetic model for prediction of omeprazole clearance and assessing ethnic sensitivity in CYP2C19 metabolic pathway. Eur J Clin Pharmacol 2015; 71: 617–624. [DOI] [PubMed] [Google Scholar]
- 28. Steere B, Baker JAR, Hall SD, Guo Y. Prediction of in vivo clearance and associated variability of CYP2C19 substrates by genotypes in populations utilizing a pharmacogenetics‐based mechanistic model. Drug Metab Dispos 2015; 43: 870–883. [DOI] [PubMed] [Google Scholar]
- 29. Vieira MD, Kim MJ, Apparaju S, Sinha V, Zineh I, Huang SM, et al PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin Pharmacol Ther 2014; 95: 550–557. [DOI] [PubMed] [Google Scholar]
- 30. Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 2006; 95: 1238–1257. [DOI] [PubMed] [Google Scholar]
- 31. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al TThe IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hardin TC, Graybill JR, Fetchick R, Woestenborghs R, Rinaldi MG, Kuhn JG. Pharmacokinetics of itraconazole following oral administration to normal volunteers. Antimicrob Agents Chemother 1988; 32: 1310–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buchanan CM, Buchanan NL, Edgar KJ, Klein S, Little JL, Ramsey MG, et al Pharmacokinetics of itraconazole after intravenous and oral dosing of itraconazole‐cyclodextrin formulations. J Pharm Sci 2007; 96: 3100–3116. [DOI] [PubMed] [Google Scholar]
- 35. Inoue S, Howgate EM, Rowland‐Yeo K, Shimada T, Yamazaki H, Tucker GT, et al Prediction of in vivo drug clearance from in vitro data. II: potential inter‐ethnic differences. Xenobiotica 2006; 36: 499–513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Concentration of [14C]BMS‐823778‐derived radioactivity (ng equivalent g–1) in plasma and tissues of male Long–Evans rats following an oral dose of [14C]BMS‐823778 at 5 mg kg–1
Table S2 Parameters of itraconazole and OH‐itraconazole in the Simcyp physiologically based pharmacokinetic model
Table S3 Simulated pharmacokinetic parameters of itraconazole and OH‐itraconazole on day 1 and day 11
Figure S1 Simulated vs. observed pharmacokinetic of BMS‐823778 in Japanese CYP2C19 extensive metabolizer subjects at 2 and 12 mg with Simcyp default enzyme expression (1 pmol mg–1 protein in liver and 0.57 pmol mg–1 protein in intestine). Observed data: red circles; mean simulated profile: black line; individual simulated profile: coloured lines