

Abstract
Background and Purpose
Pharmacotherapy for pain currently involves trial and error. A previous study on inherited erythromelalgia (a genetic model of neuropathic pain due to mutations in the sodium channel, Nav1.7) used genomics, structural modelling and biophysical and pharmacological analyses to guide pharmacotherapy and showed that carbamazepine normalizes voltage dependence of activation of the Nav1.7‐S241T mutant channel, reducing pain in patients carrying this mutation. However, whether this approach is applicable to other Nav channel mutants is still unknown.
Experimental Approach
We used structural modelling, patch clamp and multi‐electrode array (MEA) recording to assess the effects of carbamazepine on Nav1.7‐I234T mutant channels and on the firing of dorsal root ganglion (DRG) sensory neurons expressing these mutant channels.
Key Results
In a reverse engineering approach, structural modelling showed that the I234T mutation is located in atomic proximity to the carbamazepine‐responsive S241T mutation and that activation of Nav1.7‐I234T mutant channels, from patients who are known to respond to carbamazepine, is partly normalized with a clinically relevant concentration (30 μM) of carbamazepine. There was significantly higher firing in intact sensory neurons expressing Nav1.7‐I234T channels, compared with neurons expressing the normal channels (Nav1.7‐WT). Pre‐incubation with 30 μM carbamazepine also significantly reduced the firing of intact DRG sensory neurons expressing Nav1.7‐I234T channels. Although the expected use‐dependent inhibition of Nav1.7‐WT channels by carbamazepine was confirmed, carbamazepine did not enhance use‐dependent inhibition of Nav1.7‐I234T mutant channels.
Conclusion and Implications
These results support the utility of a pharmacogenomic approach to treatment of pain in patients carrying sodium channel variants.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Abbreviations
- DRG
dorsal root ganglion
- IEM
inherited erythromelalgia
- MEA
multi‐electrode array
Introduction
Pharmacogenomics has, as a major goal, the matching of specific medications with particular patients on the basis of their genetic background. The voltage gated http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=584 sets the gain on dorsal root ganglion (DRG) sensory neurons (Waxman, 2006) and plays a major role in the pathophysiology of pain in humans (Dib‐Hajj et al., 2013). Over the past decade, it has become clear that point mutations that hyperpolarize the voltage dependence of activation of Nav1.7 channels cause hereditary pain disorders characterized by severe distal limb pain (Cummins et al., 2004; Dib‐Hajj et al., 2005; Faber et al., 2012; Hoeijmakers et al., 2012; Huang et al., 2014). Pain in most of inherited erythromelalgia (IEM) patients is not relieved by pharmacotherapy. However, an IEM family carrying the NaV1.7‐V400M mutation was found to be responsive to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5339 which normalized activation of the mutant channel, shifting its activation in a depolarizing direction towards wild‐type (WT) values (Fischer et al., 2009). Yang et al. capitalized on this finding, using the V400M mutation as a seed to predict, by structural modelling, that another Nav1.7 mutation, S241T, should also be responsive to treatment with carbamazepine, confirming responsiveness of DRG sensory neurons carrying S241T mutant channel in vitro (Yang et al., 2012). In a double‐blind, placebo‐controlled follow‐up clinical study, we showed that treatment of patients carrying the S241T mutation with carbamazepine produced substantial pain relief (Geha et al., 2016). However, whether this pharmacogenomic approach is applicable to other Nav1.7 channel mutations has not been established.
Here, we followed up a report of pain relief by carbamazepine in a patient with severe pain due to the Nav1.7 mutation I234T (Meijer et al., 2014). This patient reported warmth‐induced pain attacks, similar to the vast majority of IEM patients carrying Nav1.7 mutations. We have used a reverse engineering approach to show that the NaV1.7‐I234T mutation is located in proximity to the carbamazepine‐responsive V400M and S241T mutations within the folded channel protein, and revealed that carbamazepine partly normalized hyperpolarized activation of the mutant channel and attenuated physiologically relevant thermally induced hyperexcitability of DRG sensory neurons expressing Nav1.7‐I234T channels. Our demonstration of carbamazepine‐induced restoration of channel activation and neuronal excitability towards WT values supports the notion that a pharmacogenomic approach to pain treatment is an achievable objective.
Methods
Structural modelling
Structural modelling was carried out as described in our previous reports (Yang et al., 2012) (Yang et al., 2013). Sequence alignment was performed using Clustal Omega and refined manually (Yarov‐Yarovoy et al., 2012). Individual models for the four transmembrane domains of human hNav1.7 channel were generated with GPCR‐ITASSER (Roy et al., 2011) (Zhang et al., 2015) by using templates including bacterial sodium channels (Protein Data Bank ID code 3RVY) (Payandeh et al., 2011). Four individual domains were assembled in a clockwise order viewed from the extracellular side (Dudley et al., 2000) (Li et al., 2001). The four domain complex structural model was refined by fragment‐guided molecular dynamics simulation (Zhang et al., 2011). PyMol (Schrödinger, New York, NY, USA) was used to generate figures from the models.
Plasmid preparation and HEK293 cell transfection
A channel construct encoding eGFP with a ‘StopGo’ 2A linker (Atkins et al., 2007) in‐frame with the N‐terminus of human Nav1.7‐WT channel that has been rendered resistant to tetrodotoxin (TTX‐R) by the Tyr362Ser substitution has been previously described (Huang et al., 2014) (Yang et al., 2016). The I234T mutation was created using a site‐directed mutagenesis kit (Agilent). These TTX‐R derivatives of hNav1.7 were used to establish HEK293 cell lines stably expressing Nav1.7‐I234T or Nav1.7‐WT. HEK293 cells were maintained in DMEM/F‐12 supplemented with 10% FBS (HyClone) in an incubator with 5% CO2 at 37°C. HEK293 cells were seeded on glass coverslips coated with poly‐l‐lysine (BD Biosciences, San Jose, CA, USA) in a 24‐well plate.
Voltage clamp recording of HEK293 cells
Whole‐cell voltage clamp recordings were obtained from HEK293 cells expressing WT or mutant Nav1.7 channels using an EPC‐10 amplifier and the PatchMaster program (v 53; HEKA Elektronik, Holliston, MA, USA) at room temperature. The extracellular solution contained the following (in mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 20 dextrose and 10 HEPES, pH = 7.3 with NaOH (320 mOsm adjusted with dextrose). The pipette solution contained the following (in mM): 140 Cs‐fluoride, 10 NaCl, 1 EGTA, 10 HEPES, 20 dextrose, pH = 7.3 with CsOH (310 mOsm adjusted with dextrose). Patch pipettes had a resistance of 1–2 MΩ when filled with pipette solution. Series resistance and prediction compensation (80–90%) was applied to reduce voltage errors. The recorded currents were digitized at a rate of 50 kHz after passing through a low‐pass Bessel filter setting of 10 kHz. After achieving whole‐cell configuration, a 5 min equilibration period was applied before starting the recording.
Voltage clamp recording data were analysed using Fitmaster (HEKA Elektronik) and OriginPro (Microcal Software, Northampton, MA, USA). For activation curves to be generated, cells were held at −120 mV and stepped to potentials of −80 to 40 mV in 5 mV increments for 100 ms. Peak inward currents were extracted by Origin automatically and fitted with a Boltzmann function (BoltzIV) to determine the voltage at half‐activation (V1/2), activation curve slope at half‐activation (Z) and reversal potential (ENa) for each successful recording. Conductance was calculated as G = I/(Vm − ENa), normalized by the maximum conductance value and fitted with Boltzmann equation (Cummins et al., 2004; Dib‐Hajj et al., 2005). To examine the use‐dependent effect of carbamazepine on the WT and I234T channels, we depolarized HEK293 cells expressing either Nav1.7‐WT or Nav1.7‐I234T mutant channels to −10 mV from a −120 mV holding potential at a frequency of 5, 10 and 20 Hz, with pre‐incubation with DMSO or 30 μM carbamazepine (Choi et al., 2009; Estacion et al., 2010).
Isolation and transfection of primary DRG sensory neurons
All animal care and experimental studies were approved by the Veterans Administration Connecticut Healthcare System Institutional Animal Care and Use Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Dorsal root ganglia (DRGs) were harvested from 4‐ to 6‐week‐old female and male Sprague Dawley rats and dissociated as described previously (Dib‐Hajj et al., 2009b) (Huang et al., 2013). Briefly, DRGs were dissociated with a 20 min incubation in 1.5 mg·mL−1 collagenase A (Roche, Indianapolis, IN, USA) and 0.6 mM EDTA, followed by an 18 min incubation in 1.5 mg·mL−1 collagenase D (Roche), 0.6 mM EDTA and 30 U·mL−1 papain (Worthington Biochemical, Lakewood, NJ, USA). DRGs were then centrifuged and triturated in 0.5 mL of DRG media containing 1.5 mg·mL−1 BSA (low endotoxin) and 1.5 mg·mL−1 trypsin inhibitor (Sigma, St. Louis, MO, USA). After trituration, 2 mL of DRG media was added to the cell suspension, which was filtered with a 70 μm nylon mesh cell strainer (Becton Dickinson, Franklin Lakes, NJ, USA). The mesh was washed twice with 2 mL of DRG media. Neurons were then transfected with Nav1.7‐I234T or Nav1.7‐WT using a Nucleofector IIS (Lonza, Basel, Switzerland) and Amaxa Basic Neuron SCN Nucleofector Kit (VSPI‐1003).
Multi‐electrode array (MEA) recording
MEA experiments were carried out according to our recently developed protocol (Yang et al., 2016). Briefly, dissociated neurons were maintained at 37°C in a 5% CO2 incubator for 3 days before MEA recordings. Action potential activity in these neurons was assessed using a multi‐well MEA system (Maestro, Axion Biosystems, Atlanta, GA, USA). A 12‐well recording plate was used, embedded with a total of 768 electrodes. For each experiment, three wells (with ~192 available electrodes for recording) were used to assess neurons expressing Nav1.7‐WT or Nav1.7‐I234T mutant channels. For potential variations to be minimized during the recording, neurons expressing WT or I234T constructs, derived from DRG sensory neurons pooled from two rats on the same day by the same investigator, were plated in the same 12‐well MEA recording plate. Additionally, we always analyse sister cultures prepared in parallel by the same technician, in a head‐to‐head comparison. For experiments with carbamazepine, neurons were pretreated with either carbamazepine (30 μM) or DMSO for more than 30 min in its normal culture medium as described previously (Yang et al., 2012). Measurement of the mean firing frequency and the number of active electrodes with DMSO or carbamazepine treatment was made from the same culture, providing an assessment on the efficacy of carbamazepine on the same neurons.
The investigator was blinded to the identity of the constructs expressed in the DRG sensory neurons during the recording. A spike detection criterion of >6 standard deviations above background signals was used to separate action potential spikes from noise. We defined active electrodes as registering >1 recorded spike over a 200 s period (Yang et al., 2016). MEA data were analysed using Axion Integrated Studio AxIS2.1 (Axion Biosystems) and NeuroExplorer (Nex Technologies, Madison, AL, USA).
For assessment of the effects of carbamazepine on DRG sensory neurons expressing either Nav1.7‐WT or Nav1.7‐I234T mutant channels under different temperatures, the precise temperature control of the MEA system was utilized, which enables continuous monitoring of neuronal firing during temperature ramps. DRG sensory neurons expressing either Nav1.7‐WT or Nav1.7‐I234T channels were plated on the same MEA plate for temperature ramp study and assessed by an investigator blinded to the identity of the constructs. Three different temperatures (33, 37 and 40°C) were used for the study, and each temperature was maintained for 7–10 min to allow analysis of steady‐state neuronal firing at each temperature.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are presented as mean ± SEM. Unless otherwise noted, statistical significance was determined using Student's t‐test. Use‐dependent inhibition analysis was done using two‐factor ANOVA with replication and Bonferroni post hoc test. We accepted statistical significance when P < 0.05.
Materials
All chemicals used in this study, including carbamazepine, were supplied by Sigma‐Aldrich.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
The I234T mutation is located in atomic proximity to the carbamazepine‐responsive S241T mutation within the folded sodium channel structure
Using the carbamazepine‐responsive V400M Nav1.7 mutation as a seed, we previously observed that the S241T mutation, located in proximity to V400M in the channel folded structure, responds to carbamazepine (Yang et al., 2012). We hypothesized that this atomic proximity of mutations within the channel 3D structure might be an indicator of their responsiveness to carbamazepine, which was confirmed at the channel level (by voltage clamp) and at the cell level (by current‐clamp) in vitro (Yang et al., 2012), and in patients with IEM carrying this mutation (Geha et al., 2016). This finding encouraged us to hypothesize that structural modelling might identify additional candidate mutations of Nav1.7 channels for further biophysical and pharmacological analysis. Among existing disease‐causing IEM mutations of Nav1.7 channels, we identified the I234T mutation as a primary candidate using this approach (Figure 1A). As shown in the structural model of the Nav1.7 channel, I234 is located in the same S4–S5 linker as S241 (Figure 1B). Upon closer examination, we found that the side chain of I234 was located within ~6.5 Å of S241, pointing towards the S6 helix with the same orientation of S241 (Figure 1C). Extrapolating from our previous demonstration that the proximity to V400M, a carbamazepine‐responsive mutation, identified S241T as a potential carbamazepine‐responsive mutation, here we hypothesized that the proximity of I234 to S241 and the orientation of their side chains also serves as a potential indicator of responsiveness to carbamazepine.
Figure 1.
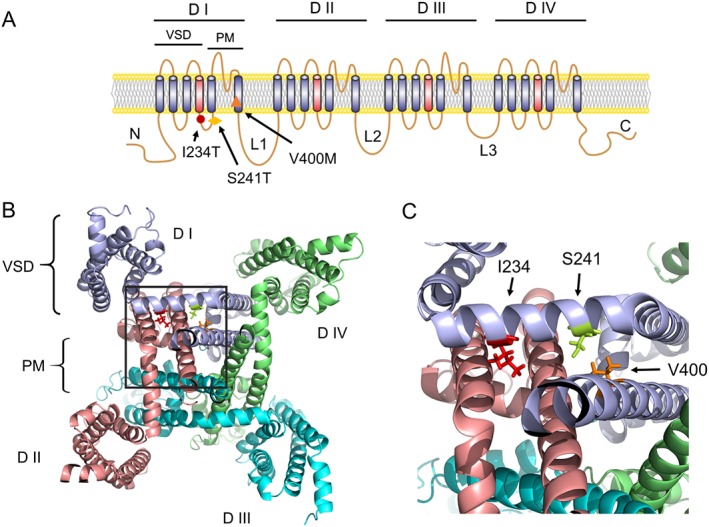
Structural modelling of the I234T of human Nav1.7 channels. (A) Diagram of the human Nav1.7 channel topology showing the location of I234T, S241T and V400M, all carbamazepine‐responsive mutations, in the domain I (D I). (B) Cytosolic view of the 3D structural model of human Nav1.7 transmembrane domains. (C) Close‐up cytosolic view of the boxed area of panel (B). I234, S241 and V400 are shown as stick model. VSD, voltage‐sensing domain; PM, pore module. L1/2/3, intracellular loop 1/2/3.
Carbamazepine partly corrects hyperpolarized activation of Nav1.7‐I234T mutant channel
To determine whether carbamazepine could also normalize the biophysical properties of the Nav1.7‐I234T mutant channel as it did for Nav1.7‐S241T channels, we studied the voltage dependence of activation of Nav1.7‐I234T channels using voltage clamp recording. Compared with Nav1.7‐WT, the Nav1.7‐I234T channel displays a remarkable ~18 mV hyperpolarizing shift in the V1/2 of activation (Ahn et al., 2010), making the mutant channel easier to open in response to small depolarizing stimuli. To determine whether carbamazepine corrected this hyperpolarizing shift of channel activation, we pre‐incubated the Nav1.7‐I234T cells with either DMSO or a clinically relevant concentration (30 μM) of carbamazepine for at least 30 min or longer before recording (Fischer et al., 2009; Yang et al., 2012). Carbamazepine or DMSO was maintained in the external bath solution during the recording. Representative current traces from neurons expressing Nav1.7‐WT or Nav1.7‐I234T channels are shown in Figure 2A, B. Notably, pre‐incubation with 30 μM carbamazepine produced a significant depolarizing shift (~6 mV) in the V1/2 of activation compared with that with DMSO (I234T with DMSO: −41.48 ± 0.7 mV, n = 11; I234T with carbamazepine: −35.69 ± 1.29 mV, n = 12; P < 0.05, Student's t‐test; Figure 2C). This type of partial correction of V1/2 of activation was seen in Nav1.7‐S241T and Nav1.7‐V400M channels pretreated with carbamazepine (Fischer et al., 2009; Yang et al., 2012). I234T channels respond to carbamazepine in a similar manner to S241T channels, suggesting that a partial correction of activationV1/2 may confer carbamazepine responsiveness to DRG sensory neurons expressing this mutant channel.
Figure 2.
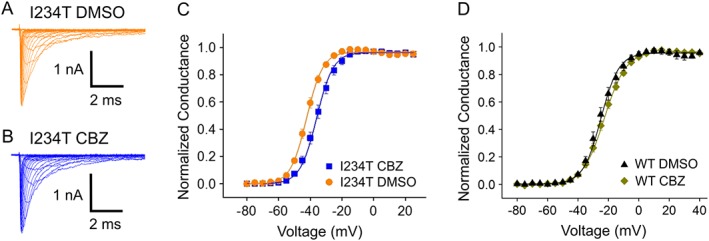
Carbamazepine depolarizes the voltage dependence of activation of the Nav1.7‐I234T mutant channel. (A, B) Representative current traces recorded from HEK293 cells expressing Nav1.7‐I234T mutant channels with either (A) DMSO or (B) carbamazepine (CBZ) pre‐incubation. (C) The voltage dependence of activation curves of Nav1.7‐I234T mutant channels treated with DMSO or carbamazepine were plotted and fitted with Boltzman equation. A depolarizing shift of activation of ~6 mV was observed when Nav1.7‐I234T mutant channels was pre‐incubated with carbamazepine. (D) The voltage dependence of activation curves of Nav1.7‐WT channels treated with DMSO or carbamazepine were plotted and fitted with Boltzman equation. No significant difference was found between the curves from Nav1.7‐WT channels incubated with DMSO and those incubated with carbamazepine.
To determine whether carbamazepine might have a similar effect on the Nav1.7‐WT channel, we studied the activation of WT Nav1.7 channels with or without carbamazepine pretreatment. Interestingly, we found that carbamazepine does not shift the activation of WT Nav1.7 (Nav1.7‐WT DMSO: −25.2 ± 1.3 mV, n = 8; Nav1.7‐WT carbamazepine: −23.0 ± 1.2 mV, n = 9; P > 0.05) (Figure 2D), suggesting that the carbamazepine effect on channel activation is mutant‐specific.
We also assessed whether carbamazepine affects fast inactivation of the Nav1.7‐I234T mutant channel. As found previously for the V400M and S241T mutant channels, carbamazepine did not change the fast inactivation of I234T mutant channels (I234T DMSO: −75.4 ± 1.2 mV; I234T carbamazepine: −74.5 ± 1.3 mV, P > 0.05).
Nav1.7‐I234T mutation increases the firing of intact DRG sensory neurons across a physiological temperature range
The hyperpolarizing shift of activation of Nav1.7‐I234T channels suggests that the expression of this mutation may cause hyperexcitability of DRG sensory neurons. To assess the firing properties of DRG sensory neurons expressing Nav1.7‐I234T mutant channels, we used MEA recording, which provides a non‐invasive, high‐throughput, extracellular recording approach that can monitor the firing of intact neurons, at three physiological temperatures: skin temperature (33°C), core body temperature (37°C), and non‐noxious warmth (40°C) (Yang et al., 2016).
We observed a marked difference in response to increasing temperature between DRG sensory neurons expressing Nav1.7‐WT channels and Nav1.7‐I234T mutant channels. Firing frequencies, which are represented by colours as a heatmap (Figure 3A–F), were clearly different between DRG neurons expressing Nav1.7‐WT and Nav1.7‐I234T channels. The firing level of DRG sensory neurons expressing Nav1.7‐WT channels was fairly low across all three temperatures. In contrast, the expression of I234T mutant channels significantly increased the firing of DRG sensory neurons at all three temperatures (Figure 3G).
Figure 3.
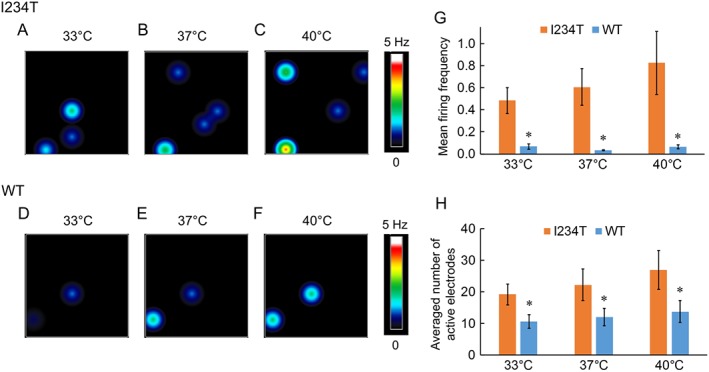
DRG sensory neurons expressing Nav1.7‐I234T mutant channels display increased firing. (A–F) Heatmap of representative MEA recordings from DRG sensory neurons expressing Nav1.7‐I234T (A–C) or WT channels (D–F). The firing frequency of each active electrode is colour‐coded: white/red represents high firing frequency; blue/black represents low firing frequency. Each circle represents an active electrode within an 8 × 8 electrode array. (G) Mean firing frequency of neurons expressing Nav1.7‐WT channels and Nav1.7‐I234T mutant channels at 33, 37 and 40°C. *P <0.05, significantly different from WT; I234T, n = 4 experiments with eight rats; WT, n = 5 experiments with 10 rats. (H) Averaged number of active electrodes from neurons expressing Nav1.7‐WT or Nav1.7‐I234T channels at these three temperatures. *P <0.05, significantly different from WT.
We found that the number of active electrodes for DRG sensory neurons expressing Nav1.7‐WT channels was also lower than for those expressing Nav1.7 I234T mutant channels at each experimental temperature (Figure 3H). Although neurons expressing I234T mutant channel exhibited a trend for increased firing frequency and number of active electrodes with increased temperature, this difference did not reach statistical significance. Taken together, our data indicate that expression of Nav1.7‐I234T mutant channels causes hyperexcitability of DRG sensory neurons by increasing the firing frequency and the number of active neurons.
Carbamazepine reduces the firing of intact DRG sensory neurons expressing Nav1.7‐I234T mutant channels across a physiological temperature range
To determine whether carbamazepine has an inhibitory effect on the firing of DRG sensory neurons expressing Nav1.7‐I234T channels, we assayed the firing of DRG sensory neurons with or without pre‐incubation with a clinically relevant concentration of carbamazepine (30 μM) using MEAs at 33, 37 and 40°C. We found that carbamazepine markedly attenuated the firing of DRG sensory neurons expressing I234T mutant channels (Figure 4A–F) in terms of both mean firing frequency and average number of active electrodes across all three temperatures (Figure 4G,H).
Figure 4.
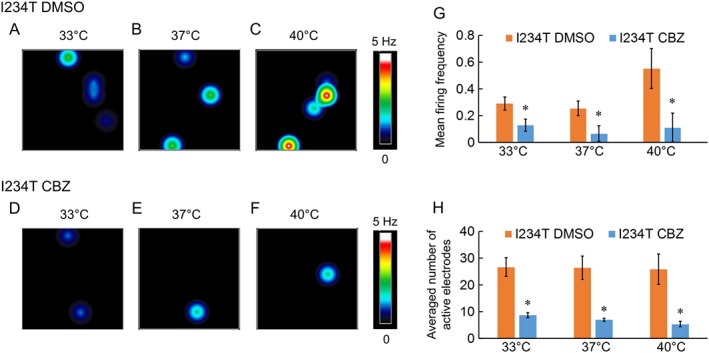
Carbamazepine reduces the firing of DRG sensory neurons expressing Nav1.7‐I234T mutant channels. (A–F) Heatmap of a representative MEA recording of DRG sensory neurons expressing Nav1.7‐I234T with DMSO or carbamazepine (CBZ) pre‐incubation. Carbamazepine produces a pronounced reduction in the number of active electrodes and mean firing frequency. (G) mean firing frequency of neurons expressing Nav1.7‐I234T channels with DMSO or carbamazepine pre‐incubation at all three temperatures (33, 37 and 40°C). *P <0.05, significantly different from DMSO; n = 6 experiments with 12 rats. (H) Averaged number of active electrodes from neurons expressing Nav1.7‐I234T channels with DMSO or carbamazepine pre‐incubation at all three temperatures (33, 37 and 40°C). *P <0.05, significantly different from DMSO.
These results indicate that a clinically relevant concentration of carbamazepine indeed inhibited firing of DRG sensory neurons expressing Nav1.7‐I234T channels, by reducing both the number of active neurons and the firing frequency of neurons across a physiological temperature range.
Carbamazepine did not significantly reduce the firing of intact DRG sensory neurons expressing Nav1.7‐WT channels
To assess whether the inhibitory effect of carbamazepine on the firing of DRG sensory neurons expressing Na1.7‐I234T mutant channels is specific, we performed similar MEA experiments on DRG sensory neurons expressing Nav1.7‐WT channels. We found that carbamazepine had no significant effect on the firing of DRG sensory neurons expressing Nav1.7‐WT channels, compared with DMSO (Figure 5A–G). Additionally, the number of active electrodes remained relatively unchanged with DMSO or carbamazepine treatment, for all conditions (Figure 5H). These data suggest that the main effect of carbamazepine on DRG sensory neurons expressing Nav1.7‐I234T mutant channels was mutant‐specific.
Figure 5.
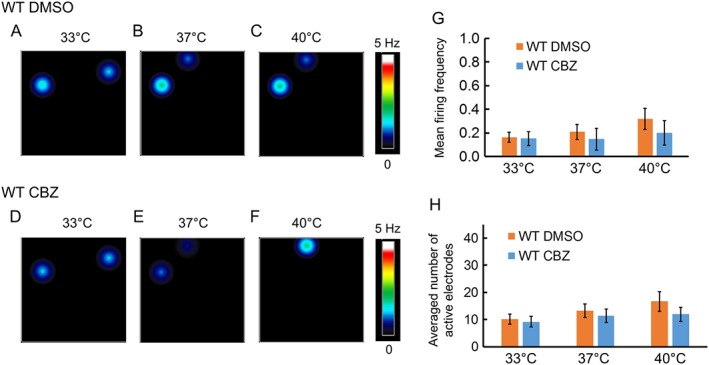
Carbamazepine does not reduce the firing of DRG sensory neurons expressing Nav1.7‐WT channels significantly. (A–F) Heatmap of a representative MEA recording of DRG sensory neurons expressing Nav1.7‐WT channels with DMSO or carbamazepine (CBZ) pre‐incubation. Carbamazepine did not significantly affect the firing frequency of these neurons. (G) Mean firing frequency of neurons expressing Nav1.7‐WT channels with pre‐incubation with DMSO or carbamazepine, at all three temperatures (33, 37 and 40°C). n = 5 experiments with 10 rats. (H) Averaged number of active electrodes from neurons expressing Nav1.7‐WT channels with DMSO or carbamazepine pre‐incubation at all three temperatures (33, 37 and 40°C).
The effect of carbamazepine is not mediated via use‐dependent inhibition of the channel peak current
Local anaesthetics, anticonvulsants (e.g. carbamazepine) and tricyclic antidepressants exhibit use‐dependent inhibition against voltage‐gated sodium channels (Tanelian and Brose, 1991; Kuo et al., 1997; Kuo, 1998; Yang et al., 2010). In the case of V400M, S241T and I234T channels, we found that carbamazepine acts via a novel mechanism by depolarizing activation of mutant channels (Fischer et al., 2009; Yang et al., 2012). Here, we wanted to further determine whether carbamazepine might have a classical use‐dependent inhibitory effect on the Nav1.7‐I234T mutant channel. To examine the use‐dependent effect of carbamazepine on the WT and I234T channels, we depolarized HEK293 cells expressing either Nav1.7‐WT or Nav1.7‐I234T mutant channels to −10 mV from a −120 mV holding potential at a frequency of 20 Hz with the pre‐incubation of DMSO or 30 μM carbamazepine.
We found that 30 μM carbamazepine indeed produced a notable use‐dependent inhibition of Nav1.7‐WT channels, compared with DMSO control at 20 Hz frequency (Figure 6A, B). For Nav1.7‐WT channels, use‐dependent current amplitude at the 30th pulse was reduced to a significantly greater extent after carbamazepine than after DMSO (Figure 6E). On the other hand, Nav1.7‐I234T channels displayed a stronger use‐dependent fall‐off with DMSO pre‐incubation, similar to that with 30 μM carbamazepine pre‐incubation (Figure 6C, D, F). Additionally, we studied use‐dependent inhibition of Nav1.7‐I234T channels at other frequencies (5 and 10 Hz) and found that carbamazepine did not affect use‐dependent inhibition at these frequencies either (at 5 Hz, I234T DMSO: 91.2 ± 1.3%; I234T carbamazepine: 91.1 ± 1.1%; n = 9, P > 0.05. At 10 Hz, I234T DMSO: 84.3 ± 1.9%; I234T carbamazepine: 84.7 ± 2.0%; n = 8, P > 0.05). Taken together, our data demonstrate that a clinically relevant concentration of carbamazepine produces a use‐dependent inhibition of the Nav1.7‐WT channel but not of the Nav1.7‐I234T mutant channel, suggesting that use‐dependent current reduction is not likely to be a contributing factor for carbamazepine‐mediated effects on the firing of DRG sensory neurons expressing Nav1.7‐I234T mutant channels.
Figure 6.
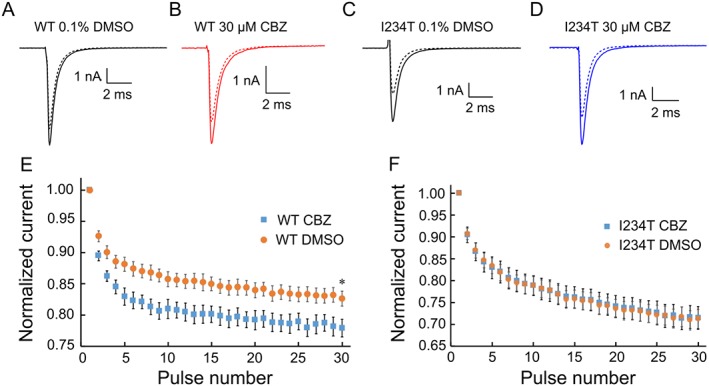
The effect of carbamazepine is not mediated via a use‐dependent inhibition of Nav1.7‐I234T mutant channels. The use‐dependent block, defined as the ratio of the peak from the 30th pulse normalized to the peak of the first pulse, at a frequency of 20 Hz recording, is determined for each cell. (A, B) Representative first and last (30th) current traces recorded from HEK293 cells expressing Nav1.7‐WT channels with either (A) DMSO or (B) carbamazepine (CBZ) pre‐incubation. (C, D) Representative first and last (30th) current traces recorded from HEK293 cells expressing Nav1.7‐I234T mutant channels with either DMSO (C) or carbamazepine (D) pre‐incubation. (E, F) Use‐dependent block curves of Nav1.7‐WT and Nav1.7‐I234T mutant channels at a frequency of 20 Hz recording were plotted. (E) Use‐dependent fall‐off of peak Nav1.7‐WT current with the pre‐incubation of DMSO or 30 μM carbamazepine.*P <0.05, significantly different from DMSO; n=9. (F) Use‐dependent fall‐off of peak Nav1.7‐I234T current with the pre‐incubation of DMSO or 30 μM carbamazepine.
Discussion
IEM, a painful disorder caused by gain‐of‐function mutations in Nav1.7 channels, provides a genetic model of neuropathic pain (Drenth and Waxman, 2007; Dib‐Hajj et al., 2009a; Dib‐Hajj et al., 2013). Pharmacotherapy of IEM remains generally ineffective, and the mainstay of treatment at this time is identification and avoidance of triggers of pain episodes, combined with cooling of the distal extremities when episodes occur. However, sodium channel blockers have been used with some success in certain patients. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2623 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2629 have been reported to be efficacious in some patients, although their effects are transient (Harty et al., 2006). In a child with another Nav1.7 mutation, treatment with mexiletine was effective for 1 month before termination due to concerns about side effects (Choi et al., 2009).
The selection of a pharmacological therapy for most of these IEM patients has been thus far based on trial and error. Building upon the observation that carbamazepine normalizes activation in the V400M IEM mutant Nav1.7 channel and that carbamazepine monotherapy produces pain relief in patients carrying this mutation (Fischer et al., 2009), a pharmacogenomic approach aided by structural modelling and in vitro functional assays (Yang et al., 2012) predicted the efficacy of treatment of patients carrying the S241T mutation of Nav1.7 channels (Geha et al., 2016), suggesting that pharmacogenomically guided therapy for pain might be applied more broadly.
Initially identified in a 5‐year‐old British girl with episodes of severe intermittent pain, the I234T mutation substitutes a highly conserved isoleucine 234 by threonine at the N‐terminus of the linker between S4 and S5 in domain I (D1/S4–S5) of the Nav1.7 channel. When studied by voltage clamp, this mutation hyperpolarizes activation by ~18 mV (Ahn et al., 2010). A trial of carbamazepine in this patient was terminated because of side effects (Ahn et al., 2010). A second patient of French‐Canadian ancestry carrying the same mutation, also with a history of severe intermittent pain, was subsequently reported (Meijer et al., 2014) (Kim et al., 2015). Episodes of severe pain, associated with sweating and erythema of the lower limbs and hands, lasting 30–60 s, recurred multiple times per hour. In response to treatment with carbamazepine, at a therapeutic dose (30 mg·kg−1) with adequate blood levels, the episodes became less frequent and less intense (Meijer et al., 2014). This case represents the third mutation of IEM for which patients respond to treatment with carbamazepine and has provided the opportunity to test our approach using structural modelling and in vitro pharmacological analysis.
As a comparator, we have studied the Nav1.7‐F1449V mutation from a family that has been anecdotally reported to be non‐responsive to carbamazepine. We reported that carbamazepine does not shift the activation of Nav1.7‐F1449V channels, nor does carbamazepine reduce the firing of DRG sensory neurons expressing these channels (Yang et al., 2012). We would note, however, that anecdotal reporting of lack of clinical efficacy should be interpreted with caution. Ethical considerations preclude a formal clinical study on patients to test a drug that has been predicted not to be efficacious based on in vitro pharmacology.
A dose–response correlation for carbamazepine was obtained in our previous study on the V400M Nav1.7 mutation (Fischer et al., 2009), and 30 μM was chosen as a clinically relevant concentration to use in in vitro studies on the V400M (Fischer et al., 2009), S241T (Yang et al., 2012) and I234T channels (this current study). This concentration is consistent with therapeutic ranges of carbamazepine suggested in previous reports (Tomson et al., 1980; O'Dougherty et al., 1987; Breton et al., 2005). More importantly, we recently studied the effect of carbamazepine in patients with IEM and observed a clinical response (reduction in pain) with serum concentrations of carbamazepine of ~3.6–6.0 mg·L−1, corresponding to 15.2–25.4 μM (Geha et al., 2016).
In our previous study, we used mutant cycle analysis to study the coupling between S241T and V400M because these two residues are located 159 amino acids away from each other, in the S4–5 linker and S6 transmembrane regions of domain I of the channel. The conventional view, which the S4–5 linker may exert an effect on the S6 transmembrane segment (Payandeh et al., 2011), prompted an empirical assessment of energetic coupling. By contrast, I234 and S241 are located within the same helix, so that mutant cycle analysis is not needed.
In the current study, we found that DRG sensory neurons expressing Nav1.7‐WT channels do not display a notable temperature‐related response, when studied via MEA recording. This phenomenon has been observed in our previous studies (Yang et al., 2016). We suggest that other sensory neurons in the culture that do not normally respond to temperature may mask the effect of temperature‐sensitive neurons (Yang et al., 2016).
A characteristic action of carbamazepine on mutant Nav1.7 channels, identified from carbamazepine‐responsive individuals with IEM, is that it shifts the voltage dependence of activation in the direction of WT channels. We found that in all three mutants (V400M, S241T and I234T), incubation with carbamazepine for at least 30 min is required, to affect the channel activation. We hypothesize that the I234T, S241T or V400M mutations may have an unusual allosteric effect on the channel and that carbamazepine may act on these mutant channels via an action as a chaperone that ‘corrects’ the folding of the mutant channels in a similar manner to the rescue of folding defective proteins by binding to their inhibitors, which may require additional time (Yang et al., 2012).
In the present study, voltage clamp experiments showed that pre‐incubation of cells expressing the Nav1.7‐I234T mutant channel with clinically relevant concentrations of carbamazepine (30 μM) shifted the V1/2 of activation of the Nav1.7‐I234T mutant channel ~6 mV in a depolarizing direction, thereby partly returning activation to its normal voltage‐dependence. The magnitude of this shift is comparable with that observed when similar treatment was applied to cells expressing the V400M (Fischer et al., 2009) and S241T (Yang et al., 2012) IEM mutations. Carbamazepine treatment of DRG sensory neurons expressing these mutant channels reduced hyperexcitability (Yang et al., 2012; Geha et al., 2016) (and this study). Importantly, the magnitude of carbamazepine‐mediated reduced excitability of DRG sensory neurons expressing Nav1.7‐I234T channels was substantially greater than the effect of carbamazepine on DRG sensory neurons expressing Nav1.7‐WT channels. These findings argue that pain relief in these patients is due, at least in part, to an effect of carbamazepine on the responsive mutant Nav1.7 channels that reduces peripheral barrage rather than a non‐specific effect on sodium channels.
The mode of action of clinically relevant concentrations of carbamazepine, which induced a depolarizing shift of activation, returning its voltage dependence towards that of WT channels (Fischer et al., 2009; Yang et al., 2012), is distinct from its widely accepted activity as a use‐dependent inhibitor of sodium channels (Tanelian and Brose, 1991; Kuo et al., 1997; Kuo, 1998; Yang et al., 2010). Indeed, we confirmed here the use‐dependent blockade of WT Nav1.7 channels by carbamazepine. By contrast, I234T mutant channels displayed a use‐dependent fall‐off in the current which was not enhanced by treatment with carbamazepine. These data suggest that the classical blockade mechanism by carbamazepine (use‐dependent inhibition of hyperactive channels) does not play a detectable role in reducing the firing of DRG sensory neurons expressing Nav1.7‐I234T mutant channels.
One possible explanation for the effect of carbamazepine on specific Nav1.7 mutations is that it acts as a chemical chaperone, in a manner analogous to the effect of carbamazepine, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2624 and mexiletine on folding‐defective http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=578 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=582 sodium channels (Valdivia et al., 2004; Tan et al., 2006; Rusconi et al., 2007; Rusconi et al., 2009) which rescue channel activity. However, unlike these previous studies, carbamazepine has an effect after about 30 min on cells expressing the Nav1.7 mutant channel, rather than the ~24 h incubation that was used in the Nav1.1 and Nav1.5 mutant channels. The novel mode of action of carbamazepine on I234T, S241T and V400M channels, normalizing voltage dependence of activation, is also distinct from its effect in rescuing current density of folding‐defective channels. For these reasons, we suggest that the effect of carbamazepine on the Nav1.7 mutant channels involves a mechanism, other than those involved with folding‐defective Nav1.1 and Nav1.5 channels or reflects a channel‐specific response to treatment.
Examination of carbamazepine classic binding using the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=579 channel model suggests that carbamazepine interacts with three key residues of the Nav1.2 channel (Leu1465 of DIII‐S6, Phe1764 of DIV‐S6 and Tyr1771 of DIV‐S6) (Lipkind and Fozzard, 2010). The corresponding residues in the Nav1.7 channel are Leu1438 of DIII‐S6, Phe1737 of DIV‐S6 and Tyr1744 of DIV‐S6 respectively. In the structural model of Nav1.7 channels, these residues face the ion‐conducting pathway, just as in the Nav1.2 channel model. Given the orientations and locations of these residues, no obvious interactions between these residues and the V400M, S241T or I234T mutants could be found. We hypothesize that the classic interaction of carbamazepine with the three residues in the ion‐conducting pathway reflects its traditional mechanism of action via high affinity binding to the inactivated state of sodium channels, while the I234T mutation, like V400M and S241T, may allosterically affect the binding of carbamazepine to the channel.
In conclusion, we show here that structural modelling and in vitro functional assays demonstrate that the voltage dependence of Nav1.7‐I234T channels, from patients with IEM who responded to treatment with carbamazepine, is shifted to more depolarized potentials, closer to the value of WT channels, by clinically relevant concentrations of this agent. Carbamazepine treatment of DRG sensory neurons expressing the Nav1.7‐I234T mutant channel led to significant reduction in mutant‐induced hyperexcitability in response to a physiologically relevant thermal stimulus, in agreement with amelioration of pain in patients carrying the I234T mutation. Our data also support the conclusion that the efficacy of carbamazepine in patients with the V400M, S241T and I234T mutations is due to a direct action on the mutant Nav1.7 channels in primary afferents, rather than a pan‐sodium channel effect of this drug. These data more generally support a pharmacogenomic approach, guided by structural modelling and functional assessment in vitro, to treatment of neuropathic pain, and suggest that the carbamazepine‐induced depolarizing shift of voltage dependence activation might provide a predictor of efficacy in patients carrying Nav1.7 channel variants.
Author contributions
Y.Y., S.D.D.‐H. and S.G.W. designed research; Y.Y., T.A. and L.C. performed research; Y.Y., T.A., L.C., P.E., S.D.D.‐H. and S.G.W. analysed data; Y.Y., P.E., S.D.D.‐H. and S.G.W. wrote the paper with inputs from T.A. and L.C.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs, and by the Kenneth Rainin Foundation; Y.Y. is supported by Regenerative Medicine Research Fund (RMRF) of CT Innovations. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralysed Veterans of America with Yale University. We thank Dr. Mark Estacion, Dr. Jianying Huang, Dr. Chongyang Han and Dr. Andrew Tan for valuable comments. We thank Lawrence Macala, Palak Shah, Fadia Dib‐Hajj and Peng Zhao for technical support.
Yang, Y. , Adi, T. , Effraim, P. R. , Chen, L. , Dib‐Hajj, S. D. , and Waxman, S. G. (2018) Reverse pharmacogenomics: carbamazepine normalizes activation and attenuates thermal hyperexcitability of sensory neurons due to Nav1.7 mutation I234T. British Journal of Pharmacology, 175: 2261–2271. doi: 10.1111/bph.13935.
References
- Ahn HS, Dib‐Hajj SD, Cox JJ, Tyrrell L, Elmslie FV, Clarke AA et al. (2010). A new Nav1.7 sodium channel mutation I234T in a child with severe pain. Eur J Pain 14: 944–950. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins JF, Wills NM, Loughran G, Wu CY, Parsawar K, Ryan MD et al. (2007). A case for “StopGo”: reprogramming translation to augment codon meaning of GGN by promoting unconventional termination (Stop) after addition of glycine and then allowing continued translation (Go). RNA 13: 803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton H, Cociglio M, Bressolle F, Peyriere H, Blayac JP, Hillaire‐Buys D (2005). Liquid chromatography‐electrospray mass spectrometry determination of carbamazepine, oxcarbazepine and eight of their metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 828: 80–90. [DOI] [PubMed] [Google Scholar]
- Choi JS, Zhang L, Dib‐Hajj SD, Han C, Tyrrell L, Lin Z et al. (2009). Mexiletine‐responsive erythromelalgia due to a new Na(v)1.7 mutation showing use‐dependent current fall‐off. Exp Neurol 216: 383–389. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Dib‐Hajj SD, Waxman SG (2004). Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci 24: 8232–8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Black JA, Waxman SG (2009a). Voltage‐gated sodium channels: therapeutic targets for pain. Pain Med 10: 1260–1269. [DOI] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Choi JS, Macala LJ, Tyrrell L, Black JA, Cummins TR et al. (2009b). Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc 4: 1118–1126. [DOI] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L et al. (2005). Gain‐of‐function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 128: 1847–1854. [DOI] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Yang Y, Black JA, Waxman SG (2013). The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14: 49–62. [DOI] [PubMed] [Google Scholar]
- Drenth JP, Waxman SG (2007). Mutations in sodium‐channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest 117: 3603–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley SC Jr, Chang N, Hall J, Lipkind G, Fozzard HA, French RJ (2000). mu‐conotoxin GIIIA interactions with the voltage‐gated Na(+) channel predict a clockwise arrangement of the domains. J Gen Physiol 116: 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, Waxman SG, Dib‐Hajj SD (2010). Effects of ranolazine on wild‐type and mutant hNav1.7 channels and on DRG neuron excitability. Mol Pain 6: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS et al. (2012). Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 71: 26–39. [DOI] [PubMed] [Google Scholar]
- Fischer TZ, Gilmore ES, Estacion M, Eastman E, Taylor S, Melanson M et al. (2009). A novel Nav1.7 mutation producing carbamazepine‐responsive erythromelalgia. Ann Neurol 65: 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geha P, Yang Y, Estacion M, Schulman BR, Tokuno H, Apkarian AV et al. (2016). Pharmacotherapy for pain in a family with inherited erythromelalgia guided by genomic analysis and functional profiling. JAMA Neurol 73: 659–667. [DOI] [PubMed] [Google Scholar]
- Harty TP, Dib‐Hajj SD, Tyrrell L, Blackman R, Hisama FM, Rose JB et al. (2006). Na(V)1.7 mutant A863P in erythromelalgia: effects of altered activation and steady‐state inactivation on excitability of nociceptive dorsal root ganglion neurons. J Neurosci 26: 12566–12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JG, Han C, Merkies IS, Macala LJ, Lauria G, Gerrits MM et al. (2012). Small nerve fibres, small hands and small feet: a new syndrome of pain, dysautonomia and acromesomelia in a kindred with a novel Nav1.7 mutation. Brain 135: 345–358. [DOI] [PubMed] [Google Scholar]
- Huang J, Yang Y, Dib‐Hajj SD, van Es M, Zhao P, Salomon J et al. (2014). Depolarized inactivation overcomes impaired activation to produce DRG neuron hyperexcitability in a Nav1.7 mutation in a patient with distal limb pain. J Neurosci 34: 12328–12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Yang Y, Zhao P, Gerrits MM, Hoeijmakers JG, Bekelaar K et al. (2013). Small‐fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J Neurosci 33: 14087–14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DT, Rossignol E, Najem K, Ospina LH (2015). Bilateral congenital corneal anesthesia in a patient with SCN9A mutation, confirmed primary erythromelalgia, and paroxysmal extreme pain disorder. J AAPOS 19: 478–479. [DOI] [PubMed] [Google Scholar]
- Kuo CC (1998). A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels. Mol Pharmacol 54: 712–721. [PubMed] [Google Scholar]
- Kuo CC, Chen RS, Lu L, Chen RC (1997). Carbamazepine inhibition of neuronal Na+ currents: quantitative distinction from phenytoin and possible therapeutic implications. Mol Pharmacol 51: 1077–1083. [DOI] [PubMed] [Google Scholar]
- Li RA, Ennis IL, French RJ, Dudley SC Jr, Tomaselli GF, Marban E (2001). Clockwise domain arrangement of the sodium channel revealed by (mu)‐conotoxin (GIIIA) docking orientation. J Biol Chem 276: 11072–11077. [DOI] [PubMed] [Google Scholar]
- Lipkind GM, Fozzard HA (2010). Molecular model of anticonvulsant drug binding to the voltage‐gated sodium channel inner pore. Mol Pharmacol 78: 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer IA, Vanasse M, Nizard S, Robitaille Y, Rossignol E (2014). An atypical case of SCN9A mutation presenting with global motor delay and a severe pain disorder. Muscle Nerve 49: 134–138. [DOI] [PubMed] [Google Scholar]
- O'Dougherty M, Wright FS, Cox S, Walson P (1987). Carbamazepine plasma concentration. Relationship to cognitive impairment. Arch Neurol 44: 863–867. [DOI] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N, Catterall WA (2011). The crystal structure of a voltage‐gated sodium channel. Nature 475: 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Xu D, Poisson J, Zhang Y (2011). A protocol for computer‐based protein structure and function prediction. J Vis Exp : e3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi R, Combi R, Cestele S, Grioni D, Franceschetti S, Dalpra L et al. (2009). A rescuable folding defective Nav1.1 (SCN1A) sodium channel mutant causes GEFS+: common mechanism in Nav1.1 related epilepsies? Hum Mutat 30: E747–E760. [DOI] [PubMed] [Google Scholar]
- Rusconi R, Scalmani P, Cassulini RR, Giunti G, Gambardella A, Franceschetti S et al. (2007). Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J Neurosci 27: 11037–11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan BH, Valdivia CR, Song C, Makielski JC (2006). Partial expression defect for the SCN5A missense mutation G1406R depends on splice variant background Q1077 and rescue by mexiletine. Am J Physiol Heart Circ Physiol 291: H1822–H1828. [DOI] [PubMed] [Google Scholar]
- Tanelian DL, Brose WG (1991). Neuropathic pain can be relieved by drugs that are use‐dependent sodium channel blockers: lidocaine, carbamazepine, and mexiletine. Anesthesiology 74: 949–951. [DOI] [PubMed] [Google Scholar]
- Tomson T, Tybring G, Bertilsson L, Ekbom K, Rane A (1980). Carbamazepine therapy in trigeminal neuralgia: clinical effects in relation to plasma concentration. Arch Neurol 37: 699–703. [DOI] [PubMed] [Google Scholar]
- Valdivia CR, Tester DJ, Rok BA, Porter CB, Munger TM, Jahangir A et al. (2004). A trafficking defective, Brugada syndrome‐causing SCN5A mutation rescued by drugs. Cardiovasc Res 62: 53–62. [DOI] [PubMed] [Google Scholar]
- Waxman SG (2006). Neurobiology: a channel sets the gain on pain. Nature 444: 831–832. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dib‐Hajj SD, Zhang J, Zhang Y, Tyrrell L, Estacion M et al. (2012). Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Na(V)1.7 mutant channel. Nat Commun 3: 1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Estacion M, Dib‐Hajj SD, Waxman SG (2013). Molecular architecture of a sodium channel S6 helix: radial tuning of the voltage‐gated sodium channel 1.7 activation gate. J Biol Chem 288: 13741–13747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Huang J, Mis MA, Estacion M, Macala L, Shah P et al. (2016). Nav1.7‐A1632G mutation from a family with inherited erythromelalgia: enhanced firing of dorsal root ganglia neurons evoked by thermal stimuli. J Neurosci 36: 7511–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YC, Huang CS, Kuo CC (2010). Lidocaine, carbamazepine, and imipramine have partially overlapping binding sites and additive inhibitory effect on neuronal Na+ channels. Anesthesiology 113: 160–174. [DOI] [PubMed] [Google Scholar]
- Yarov‐Yarovoy V, DeCaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D et al. (2012). Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci U S A 109: E93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liang Y, Zhang Y (2011). Atomic‐level protein structure refinement using fragment‐guided molecular dynamics conformation sampling. Structure 19: 1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang J, Jang R, Zhang Y (2015). GPCR‐I‐TASSER: a hybrid approach to G protein‐coupled receptor structure modeling and the application to the human genome. Structure 23: 1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]