

Abstract
Background and Purpose
Endocannabinoids play an important role in modulating spinal nociceptive signalling, crucial for the development of pain. The cannabinoid CB1 receptor and the TRPV1 cation channel are both activated by the endocannabinoid anandamide, a product of biosynthesis from the endogenous lipid precursor N‐arachidonoylphosphatidylethanolamine (20:4‐NAPE). Here, we report CB1 receptor‐ and TRPV1‐mediated effects of 20:4‐NAPE on spinal synaptic transmission in control and inflammatory conditions.
Experimental Approach
Spontaneous (sEPSCs) and dorsal root stimulation‐evoked (eEPSCs) excitatory postsynaptic currents from superficial dorsal horn neurons in rat spinal cord slices were assessed. Peripheral inflammation was induced by carrageenan. Anandamide concentration was assessed by mass spectrometry.
Key Results
Application of 20:4‐NAPE increased anandamide concentration in vitro. 20:4‐NAPE (20 μM) decreased sEPSCs frequency and eEPSCs amplitude in control and inflammatory conditions. The inhibitory effect of 20:4‐NAPE was sensitive to CB1 receptor antagonist PF514273 (0.2 μM) in both conditions, but to the TRPV1 antagonist SB366791 (10 μM) only after inflammation. After inflammation, 20:4‐NAPE increased sEPSCs frequency in the presence of PF514273 and this increase was blocked by SB366791.
Conclusions and Implications
While 20:4‐NAPE treatment inhibited the excitatory synaptic transmission in both naive and inflammatory conditions, peripheral inflammation altered the underlying mechanisms. Our data indicate that 20:4‐NAPE application induced mainly CB1 receptor‐mediated inhibitory effects in naive animals while TRPV1‐mediated mechanisms were also involved after inflammation. Increasing anandamide levels for analgesic purposes by applying substrate for its local synthesis may be more effective than systemic anandamide application or inhibition of its degradation.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=56 |
| Enzymes b |
| http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=273#1398 |
| Voltage‐gated ion channels c |
| http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=507 |
| Ligand‐gated ion channels d |
| http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=75 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,dAlexander et al., 2015a,b,c,d).
Abbreviations
- 20:4‐NAPE
N‐arachidonoylphosphatidylethanolamine
- DRG
dorsal root ganglion
- NAPE‐PLD
N‐acyl phosphatidylethanolamine phospholipase D
- PF514273
2‐(2‐chlorophenyl)‐3‐(4‐chlorophenyl)‐7‐(2,2‐difluoropropyl)‐6,7‐dihydro‐2H‐pyrazolo[3,4‐f][1,4]oxazepin‐8(5H)‐one
- PWL
paw withdrawal latency
Introduction
Modulation of synaptic transmission in the spinal dorsal horn is pivotal in nociceptive signalling. An important part in this process is mediated by the TRPV1 cation channel and the Gi/o protein‐coupled cannabinoid CB1 receptors (Katona and Freund, 2008; Nagy et al., 2014; Sousa‐Valente et al., 2014a). However, our current understanding does not allow us to realise fully the analgesic potential of controlling spinal signalling through these two pathways.
Spinal TRPV1 channels are expressed predominantly by the central branches of nociceptive small‐ and medium‐sized dorsal root ganglion (DRG) neurons (Caterina et al., 1997). Activation of these channels has an excitatory effect through increasing the transmitter release from the terminals of DRG neurons, but may depress evoked currents (Baccei et al., 2003). CB1 receptors are expressed in various structures including inhibitory neurons, astrocytes and central terminals of nociceptive small to medium size DRG neurons (Ahluwalia et al., 2000; Alkaitis et al., 2010; Veress et al., 2013). Activation of both presynaptic CB1 receptors and those on inhibitory interneurons leads to reduced transmitter release from the respective neurons (Morisset and Urban, 2001; Pernia‐Andrade et al., 2009).
Several endogenous agents including N‐arachidonoylethanolamine (anandamide) activate both TRPV1 channels and CB1 receptors (Devane et al., 1992; Zygmunt et al., 1999; Tognetto et al., 2001; Ahluwalia et al., 2003). Importantly, sub‐populations of DRG neurons as well as spinal cord cells are able to synthesize or degrade anandamide (Carrier et al., 2004; van der Stelt et al., 2005; Vellani et al., 2008; Varga et al., 2014). Anandamide synthesis occurs through many metabolic pathways either in a Ca2+‐insensitive or Ca2+‐sensitive manner (Ueda et al., 2013). N‐arachidonoylphosphatidylethanolamine (20:4‐NAPE) constitutes the precursor for anandamide synthesis in all pathways (Wang and Ueda, 2009; Snider et al., 2010; Ueda et al., 2013). We have shown recently that application of 20:4‐NAPE to cultured DRG neurons results in anandamide production in a concentration and temperature‐dependent manner (Varga et al., 2014).
Painful peripheral pathologies including tissue inflammation alter the expression and/or activity of both TRPV1 channels and CB1 receptors (Richardson et al., 1998; Amaya et al., 2003, 2006; Luo et al., 2004; Kanai et al., 2005; Spicarova and Palecek, 2009; Spicarova et al., 2011; Kwon et al., 2014). Inflammation also induces changes in anandamide levels in the spinal cord (Buczynski et al., 2010; Costa et al., 2010) and the expression of enzymes synthesizing and hydrolysing anandamide, in DRG neurons (Lever et al., 2009; Sousa‐Valente et al., 2017). Here, for the first time, instead of ‘flooding’ the entire preparation by exogenous anandamide, we studied how providing substrate (20:4‐NAPE) for anandamide‐synthesizing pathways in the spinal cord affected nociceptive spinal synaptic transmission and what role TRPV1 channels and CB1 receptors played in that process, under naive and inflammatory conditions.
Methods
Animals
All animal care and experimental procedures were in accordance with local Institutional Animal Care and Use Committees and consistent with the guidelines of the International Association for the Study of Pain, the UK Animals (Scientific Procedures) Act (1986) and EU Directive 2010/63/EU for animal experiments. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Altogether, 49 male Wistar rats (Institute of Physiology CAS, Czech Republic) of postnatal days (P19–P23) were used in this study. Animals were maintained under temperature (22 ± 2°C) and light‐controlled (12 h light/dark cycle) conditions with free access to food and water.
Spinal cord slice preparation
Male Wistar rats (P19–P23) were anaesthetised with isoflurane (3%), the lumbar spinal cords were removed and immersed in oxygenated, ice‐cold, dissection solution containing (in mM) 95 NaCl, 1.8 KCl, 7 MgSO4, 0.5 CaCl2, 1.2 KH2PO4, 26 NaHCO3, 25 D‐glucose and 50 sucrose. Animals were killed by subsequent medulla interruption and exsanguination. Each spinal cord was fixed to a vibratome stage (VT 1000S, Leica, Germany) using cyanoacrylate glue in a groove between two agar blocks. Acute transverse slices 300–350 μm thick were cut from L4–L5 segments, incubated in the dissection solution for 30 min at 33°C, stored in a recording solution at room temperature and allowed to recover for 1 h before the electrophysiological experiments. The recording solution contained (in mM) 127 NaCl, 1.8 KCl, 1.2 KH2PO4, 2.4 CaCl2, 1.3 MgSO4, 26 NaHCO3, and 25 D‐glucose. For the actual measurement, slices were transferred into a recording chamber that was perfused continuously with recording solution (room temperature (21–24°C)) at a rate of ~2 mL·min−1. All extracellular solutions were saturated with carbogen (95% O2, 5% CO2) during the whole process.
Patch‐clamp recording
Altogether, sEPSCs were recorded from 98 and eEPSCs from 79 superficial dorsal horn neurons. Individual neurons were visualized using a differential interference contrast microscope (DM LFSA, Leica, Germany) equipped with a 63 × 0.90 water‐immersion objective and an infrared‐sensitive camera (KP‐200P, Hitachi, Japan) with a standard TV/video monitor (Hitachi VM‐172, Japan). Patch pipettes were pulled from borosilicate glass tubing when filled with intracellular solution; they had resistances of 3.5–6.0 MΩ. The intracellular pipette solution contained (in mM) 125 gluconic acid lactone, 15 CsCl, 10 EGTA, 10 HEPES, 1 CaCl2, 2 MgATP, and 0.5 NaGTP and was adjusted to pH 7.2 with CsOH. Voltage‐clamp recordings in the whole cell configuration were performed with an AxoPatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) at room temperature (21–24°C). Whole cell responses were low‐pass filtered at 2 kHz and digitally sampled at 10 kHz. The series resistance of the recorded neurons was routinely compensated by 80% and was monitored during the whole experiment. AMPA receptor‐mediated spontaneous excitatory postsynaptic currents (sEPSCs) were recorded from superficial dorsal horn neurons in laminae I and II(outer), clamped at −70 mV in the presence of 10 μM bicuculline and 5 μM strychnine. To evoke EPSCs, a dorsal root was stimulated with a suction electrode using a constant current isolated stimulator (Digitimer, DS3, Hertfordshire, UK). Test pulses of 0.5 ms duration and an intensity ranging between 20 and 350 μA were applied at a frequency of 0.033 Hz. The intensity of the stimulation was adjusted to evoke stable EPSCs. Application of each drug lasted for 4 min period (recording solution, 20:4‐NAPE, capsaicin, co‐application 20:4‐NAPE and PF514273, co‐application 20:4‐NAPE and SB366791) or 6 min antagonist pretreatment (PF514273, SB366791, co‐application of PF514273 with SB366791). Neurons with capsaicin‐ sensitive primary afferent input were identified by increase of sEPSC frequency (>20%) following capsaicin (0.2 μM) application at the end of the experimental protocol. Capsaicin was applied in 87% of the recorded neurons and 92% of these responded with sEPSC frequency increase. The software package pCLAMP version 10.0 (Axon Instruments, CA, USA) was used for data acquisition and subsequent off‐line analysis. Cells with a series resistance >20 MΩ were excluded from the analysis.
Anandamide release experiments
Spinal cord slices from five animals were prepared in the same way as for the electrophysiological experiments. For each of the five experiments, 18 acute spinal cord slices were used. Slices were put into plastic safe‐lock tube with 200 μL of the recording solution, saturated with carbogen (95% O2–5% CO2) during the whole process. Incubation of 10 min was used before the whole volume of the solution (sample) was extracted and immediately frozen for later mass spectrometry analysis. The solution in the tube was immediately replaced with another solution sample. In each experiment, eight samples were taken after 10 min of incubation: 1 (recording solution), 2 (recording solution), 3 (20:4‐NAPE, 20 μM), 4 (recording solution), 5 (20:4‐NAPE, 100 μM), 6 (recording solution), 7 (20:4‐NAPE, 200 μM), 8 (recording solution). Additional samples with only 20:4‐NAPE, 20–100‐200 μM without the slices were also prepared and analysed.
The collected samples were analysed for the presence of anandamide with mass spectrometry. For calibration purposes solutions of anandamide and 20:4‐NAPE were used. Anandamide content was determined by reversed‐phase high‐performance chromatography using Agilent 1100 LC system (Agilent, Palo Alto, CA, USA) consisting of a degasser, a binary pump, an autosampler, and an thermostatic column compartment. Chromatographic separation was carried out in a Kinetex 2.6u PFP, 100A column (100 × 2.1 mm I.D., Phenomenex, Torrence, CA, USA). The sample (10 μL) was injected into the column and eluted with a gradient consisting of (A) water‐formic acid 100:0.1 v/v and (B) acetontrile‐formic acid 100:0.085 v/v (flow rate 0.35 mL·min−1 and temperature 40°C). The gradient started at A/B 80:20 for 5 min, reaching 100% B after 10 min. For the next 5 min the elution was isocratic at 100% B. Elution was monitored by an ion‐trap mass spectrometer (Agilent LC‐MSD Trap XCT‐Ultra; Agilent, Palo Alto, CA, USA). Atmospheric pressure ionization‐electrospray ionization (API‐ESI) positive mode ion‐trap mass spectrometry at MRM (multiple reaction monitoring) mode was used with transition of m/z 348.1 > 287.1 for anandamide (retention time 11.2 min) when monitored mass range was 100–400 m/z. Operating conditions: drying gas (N2), 12 l.min−1; drying gas temperature, 350°C; nebulizer pressure, 30 ψ (207 kPa). The areas of the anandamide peak were measured. The results from each experiment (peak areas of anandamide were normalized to the production of anandamide after the 200 μM 20:4‐NAPE application (100%).
Peripheral inflammation
Peripheral inflammation was induced in a group of animals 24 h before the spinal cord slice preparation was made. Under isoflurane (3%) anaesthesia, both hind paws were injected subcutaneously by a 3% mixture of carrageenan (50 μL) in a physiological saline solution. The animals were left to recover in their home cages. This carrageenan injection in peripheral tissue is a thoroughly characterized and established animal model of inflammatory pain (Ren and Dubner, 1999). Naive animals were used as controls.
Behavioural testing
The animals used in the model of peripheral inflammation were tested for responsiveness to thermal stimuli before and 24 h after the model induction. Paw withdrawal latencies (PWLs) to radiant heat stimuli were determined for both hind paws using Plantar Test 37370 apparatus (Ugo Basile, Italy). The rats were placed under non‐binding, clear plastic cages on a 3 mm thick glass plate and left to adapt at least for 20 min. The radiant heat was applied to the plantar surface of each hind paw until a deliberate escape movement of the paw was detected by the Plantar Test apparatus. The PWLs were tested four times for each hind paw with at least 5 min intervals between the trials. Results from each hind paw were averaged. Baseline withdrawal latencies were determined in all animals before any experimental procedure.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Some data were normalized as a percentage of the control values (100%). Results are shown as means ± SEM. For offline analysis of the recorded sEPSCs data, segments of 2 min duration were used for each experimental condition. Only sEPSCs with amplitudes 5 pA or greater (which corresponded to at least twice the noise level) were included in the frequency analysis. In the case of amplitude analysis, the same sEPSCs events were used. Statistics were calculated using SigmaStat 3.5 (Systat Software, CA, USA). A Kolmogorov–Smirnov test was used to evaluate statistical significance for cumulative data. One‐way ANOVA or one‐way ANOVA repeated measures followed by a post hoc test (Student–Newman–Keuls) or paired t‐test was used for data with normal distribution and non‐parametric rank test or RM on ranks was used where appropriate for statistical comparisons. P‐value <0.05 was considered statistically significant. Detailed information is given in the figure legends.
Materials
All chemicals used for extracellular and intracellular solutions were of analytical grade and purchased from Sigma Aldrich (St. Louis, MO, USA) and Tocris Bioscience (Bristol, UK). Capsaicin, SB366791 and PF514273 (Tocris Bioscience) and anandamide (Avanti Polar Lipids, Alabaster, AL, USA) were dissolved in DMSO (Sigma Aldrich), which had a concentration of < 0.1% in the final solution. 20:4‐NAPE (Avanti Polar Lipids) was dissolved in chloroform, which had a concentration of < 0.1% in the final solution. The concentration of the selective TRPV1 channel antagonist SB366791 (10 μM) used here was based on our previous studies (Spicarova et al., 2014a; Spicarova and Palecek, 2009) and its selectivity pA2 = 7.71 (Gunthorpe et al., 2004). The concentration of the highly selective CB1 receptor antagonist PF514273 (0.2 μM) was determined by considering Ki ‐ 1 nM (Dow et al., 2009) and the needed diffusion through the spinal cord slice. 20:4‐NAPE was applied in the recording solution in concentration (20 μM) based on our preliminary results and previous experiments performed on DRG cultures (Varga et al., 2014). Carrageenan for induction of inflammation was purchased from Sigma Aldrich.
Results
Application of 20:4‐NAPE increased anandamide concentration in spinal cord slices
To verify the production of anandamide from 20:4‐NAPE in our preparation, mass spectrometry was used to analyse anandamide content after application of different concentrations of 20:4‐NAPE (20, 100 and 200 μM) on spinal cord slices in vitro. Under the control conditions with extracellular solution only, the average anandamide concentration in the solution was very low (7067 ± 4532 of peak area). Anandamide concentration increased gradually with increasing concentration of 20:4‐NAPE application (20 μM: 48 324 ± 27 502; 100 μM: 103 310 ± 38 179; 200 μM: 298 004 ± 139 867 anandamide peak area). To reduce the differences between the individual experiments and facilitate statistical analysis, these results were normalised against the content found for the highest concentration of 20:4‐NAPE (200 μM) used (set to 100%) (Figure 1). There was no anandamide detected in the samples where 20:4‐NAPE was present without the slices. These results indicate that 20:4‐NAPE (20 μM) application to the spinal cord slices led to increased anandamide concentration in the slice.
Figure 1.
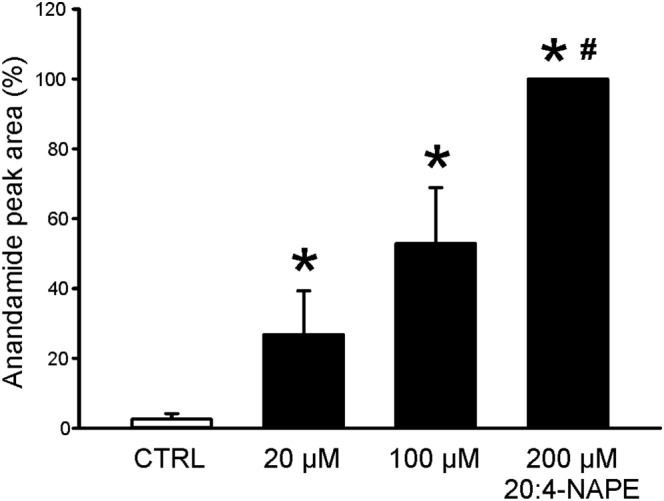
Anandamide concentration after 20:4‐NAPE application to spinal cord slices. Three different concentrations of 20:4‐NAPE (20, 100 and 200 μM) were applied to spinal cord slices. Increasing content of anandamide was detected in the extracellular solution after 20:4‐NAPE application, in a concentration dependent manner. *P < 0.05, significantly different from control, # P < 0.05, significantly different from 20 and 100 μM 20:4‐NAPE; repeated measures ANOVA on ranks followed by Student–Newman–Keuls test; n = 5).
Application of 20:4‐NAPE reduced both spontaneous and evoked activity of spinal dorsal horn neurons
The role of 20:4‐NAPE in nociceptive synaptic transmission was investigated using recordings of spontaneous EPSCs (sEPSCs) and dorsal root stimulation‐evoked EPSCs (eEPSCs) from neurons in laminae I and II(outer) of the dorsal horn. Overall, the mean control frequency of sEPSCs recorded in neurons from slices prepared from naive animals was 1.09 ± 0.14 Hz (n = 42). Application of 20:4‐NAPE (20 μM, 4 min) clearly decreased sEPSC frequency in 12 of the 13 recorded neurons (Figure 2A–C) when data was averaged from all neurons in this group. The average amplitude of the sEPSCs was also reduced significantly after the 20:4‐NAPE application from 24.9 ± 2.5 pA to 21.4 ± 1.4 pA (n = 13, P < 0.05, paired t‐test). However, the decrease of the amplitude (>15%) was present in only four cells out of 13 neurons and the cumulative distribution of sEPSC amplitudes did not show significant change after 20:4‐NAPE (Figure 2D).
Figure 2.
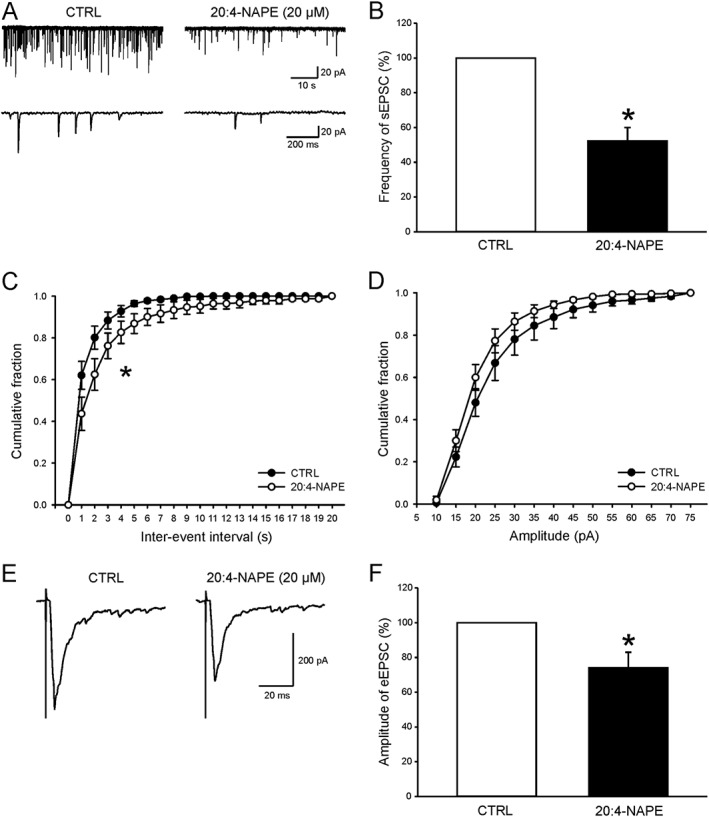
Inhibitory effect of 20:4‐NAPE application on excitatory postsynaptic currents in spinal cord slices from naive animals. (A) An example of native recording of spontaneous EPSCs from one superficial dorsal horn neuron before (CTRL) and during 20:4‐NAPE (20 μM) application. (B) Application of 20:4‐NAPE (20 μM) robustly decreased the average frequency of sEPSCs. *P < 0.05, significantly different from control; Wilcoxon signed‐rank test; n = 13. (C) This was also evident using cumulative histogram analysis. *P < 0.05, significantly different from control; Kolmogorov–Smirnov test. (D) Decrease of sEPSC amplitude was not significant using cumulative amplitude analysis. (E) Recording of dorsal root stimulation‐evoked EPSC from one neuron before and during 20:4‐NAPE (20 μM) application. (F) Acute application of 20:4‐NAPE (20 μM) significantly decreased the mean amplitude of eEPSCs. *P < 0.05, significantly different from control; Wilcoxon signed‐rank test; n = 15.
As observed with the sEPSCs, 20:4‐NAPE (20 μM, 4 min) also significantly decreased the amplitude of eEPSCs (Figure 2E,F). The reduction of eEPSCs amplitude (>15%) was present in 8 of 15 recorded neurons. Together, these findings indicate that application of 20:4‐NAPE had a robust inhibitory effect on the excitation of superficial spinal dorsal horn neurons in naive conditions.
Inhibition by 20:4‐NAPE of spontaneous activity was prevented by blocking the CB1 receptors but not TRPV1 channels
In the next experiments, we investigated whether the inhibitory effects of 20:4‐NAPE were mediated through either of anandamide's main targets, the CB1 receptors or the TRPV1 channels.
Application of the highly selective CB1 receptor antagonist PF514273 (0.2 μM, 6 min) by itself caused a small and non significant increase in sEPSCs frequency (Figure 3A,C). However, in the presence of PF514273, 20:4‐NAPE (20 μM, 4 min) no longer changed the frequency of sEPSCs, compared with the control value or with the antagonist alone (Figure 3A,C,F). The amplitude of sEPSCs in control conditions (20.0 ± 1.7 pA, n = 11) was not affected either by PF514273 alone (18.8 ± 1.8 pA) or by 20:4‐NAPE in the presence of PF514273 (18.6 ± 2.1 pA). The failure of 20:4‐NAPE to reduce either the frequency or the amplitude of sEPSCs in the presence of PF514273, contrasted with the effects of 20:4‐NAPE alone, as shown in Figure 2B.
Figure 3.
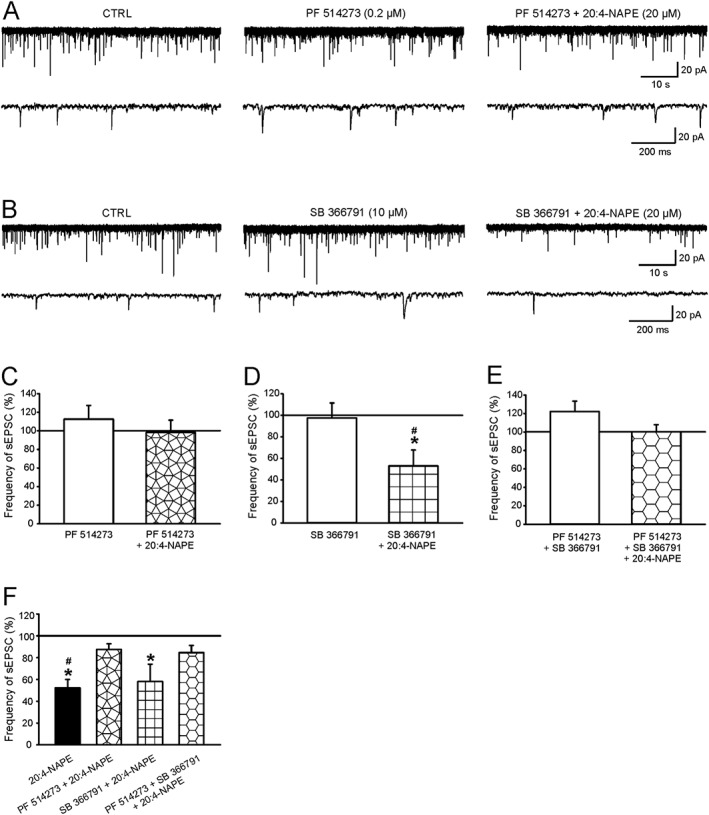
The effect of CB1 receptor and TRPV1 channel antagonists on the 20:4‐NAPE‐induced inhibition of sEPSC frequency in naive slices. (A, C) The application of PF514273 (0.2 μM) alone did not change the frequency of sEPSCs (n = 11). The presence of PF514273 (0.2 μM) prevented 20:4‐NAPE (20 μM) from inhibiting the sEPSCs frequency (B, D) SB366791 (10 μM, n = 10) alone, did not change sEPSCs frequency. The presence of SB366791 (10 μM) did not prevent 20:4‐NAPE (20 μM) from decreasing sEPSCs frequency. *P < 0.05, significantly different from control, # P < 0.05, significantly different from SB366791 alone repeated measures ANOVA on ranks followed by Student–Newman–Keuls test. (E) The application of both antagonists, PF514273 (0.2 μM) with SB366791 (10 μM), did not change the frequency of sEPSCs (n = 8). Subsequent co‐application of PF514273 (0.2 μM), SB366791 (10 μM) with 20:4‐NAPE (20 μM) prevented the 20:4‐NAPE induced inhibition. (F) The same data are expressed as a percentage of previous recording conditions. For 20:4‐NAPE alone; *P < 0.05, significantly different from basal frequency of sEPSCs; Wilcoxon signed‐rank test; n = 13. For SB366791 + 20:4‐NAPE ;, *P < 0.05, significantly different from SB366791 pretreatment; Wilcoxon signed‐rank test; n = 10. # P < 0.05, significant difference between 20:4‐NAPE alone and PF514273 + 20:4‐NAPE; one‐way ANOVA followed by Student–Newman–Keuls test; n = 11.
Pretreatment of slices with the selective antagonist of TRPV1 channels, SB366791 (10 μM, 6 min) alone, did not change the frequency of sEPSCs (Figure 3B,D) and 20:4‐NAPE (20 μM, 4 min) in the presence of SB366791 still reduced the frequency of sEPSCs in 8 out of 10 recorded neurons (Figure 3B, D), compared with the control values. This reduction was not different from that observed with 20:4‐NAPE alone (Figure 2B). The amplitude of sEPSCs was not affected by SB366791 alone (control: 29.0 ± 41 pA, SB366791: 24.6 ± 4.0 pA) or by 20:4‐NAPE in the presence of SB366791 (25.2 ± 4.6 pA, n = 10).
We then tested the combination of the CB1 receptor and the TRPV1 channel antagonists. This combination, given alone, caused a small and non‐significant increase of the sEPSCs frequency (Figure 3E). Application of 20:4‐NAPE in the presence of the combination of both antagonists did not change the frequency of sEPSCs, compared with control values (Figure 3E). The average amplitude of sEPSCs was not changed by any of the conditions (control: 20.8 ± 2.0 pA, PF514273 + SB366791: 19.2 ± 1.6 pA, PF514273 + SB366791 + 20:4 NAPE: 17.4 ± 1.1 pA, n = 8) in this set of experiments.
To compare all the experimental situations and to correct for any effects of the antagonists alone, we have also expressed these data as a percentage of the values obtained with antagonists, singly or combined, given alone set to 100% (Figure 3F). This analysis showed that the inhibition by 20:4‐NAPE of the frequency of sEPSCs was mediated by activation of CB1 receptors, but not by TRPV1 channels.
20:4‐NAPE‐induced decrease of eEPSCs amplitude was prevented by antagonism of CB1 receptors but not that of TRPV1 channels, in slices from naive animals.
Here we have studied the effects of PF514273 and SB366791 on the EPSCs evoked by stimulation of the dorsal root (eEPSCs). Application of PF514273 (0.2 μM, 6 min) alone did not change the amplitude of eEPSCs (Figure 4A,C) but this antagonist did block the effect of 20:4‐NAPE, compared to both the control (Figure 4C) and PF514273 pretreatment (Figure 4E) values. Out of these neurons, seven exhibited a lack of reduction. Four of these seven recorded neurons were not affected by 20:4‐NAPE application, and in remaining three neurons, the amplitude increased >15%. Overall, these results showed that the inhibition by 20:4‐NAPE of eEPSC amplitude was mediated by CB1 receptors, in the group of superficial spinal dorsal horn neurons.
Figure 4.
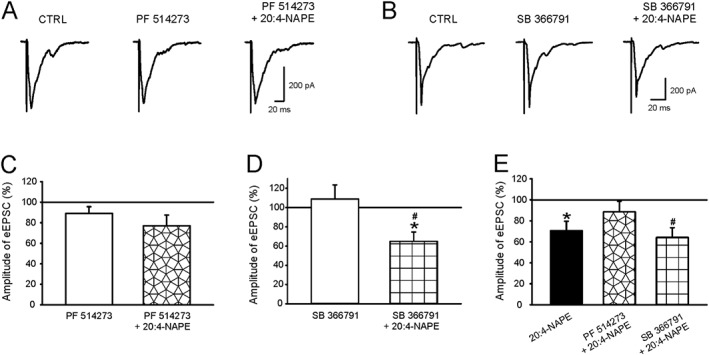
Effect of CB1 receptor and TRPV1 channel antagonists on 20:4‐NAPE‐induced inhibition of eEPSC amplitude in naive slices. (A, C) Pretreatment with PF514273 (0.2 μM, n = 13) did not change the amplitude of the recorded eEPSC in spinal cord slices prepared from naive animals. In the presence of PF514273 (0.2 μM), 20:4‐NAPE (20 μM) also did not change the amplitude of eEPSC (n = 13). (B, D) Pretreatment with SB366791 (10 μM) did not change the control eEPSC amplitude. In the presence of SB366791 (10 μM), 20:4‐NAPE (20 μM) decreased the eEPSC amplitude. *P < 0.05, significantly different from control, # P < 0.05, significantly different from SB366791 pretreatment; repeated measures ANOVA on ranks followed by Student–Newman–Keuls test; n = 10. (E) Data are shown as a percentage of previous condition to eliminate the effect of antagonist activity. *P < 0.05, significantly different from eEPSC basal amplitude; Wilcoxon signed‐rank test; n = 15. # P < 0.05, significantly different from SB366791 pretreatment; Wilcoxon signed‐rank test; n = 10.
Inhibition of TRPV1 channels with SB366791 (10 μM, 6 min) alone did not change the eEPSC amplitude (Figure 4B,D) and 20:4‐NAPE was still able to decrease the amplitude of eEPSCs, in the presence of SB366791, compared with control (Figure 4D) and SB366791 pretreatment (Figure 4E) values. The amplitude reduction was evident in 8 of 10 neurons, and it did not change in the two remaining neurons. The degree of 20:4‐NAPE‐induced reduction in the presence of SB366791 was not significantly different from that produced by 20:4‐NAPE alone (Figure 4E). These findings indicate that TRPV1 channels were not involved in mediating the inhibitory effect of 20:4‐NAPE on eEPSC amplitude.
Application of 20:4‐NAPE reduced the frequency of sEPSCs in spinal dorsal horn neurons under inflammatory conditions
Peripheral inflammation was induced by subcutaneous injection of carrageenan in the hindpaws, 24 h before behavioural testing. Signs of inflammation (redness, hypersensitivity and swelling) were present in the hind paws of all animals. The paw withdrawal latency to thermal stimuli was significantly decreased from 11.8 ± 0.6 s to 8.3 ± 0.5 s (n = 12, P < 0.05, paired t‐test). The control sEPSC frequency (1.28 ± 0.24 Hz; n = 56) recorded in neurons 1 day after the inflammation induction was higher but not statistically different, when compared with the control sEPSC frequency recorded in naive animals.
Application of 20:4‐NAPE (20 μM, 4 min) to slices prepared from the spinal cord of these animals strongly inhibited the sEPSC frequency in seven of the nine recorded neurons (Figure 5). This effect of 20:4‐NAPE on the frequency of sEPSC under inflammatory conditions was not significantly different from that observed in naive rats, i.e.,without inflammation (Figure 2B). Application of 20:4‐NAPE also reduced the amplitude of sEPSCs from 21.4 ± 2.3 to 18.4 ± 1.6 pA (n = 9, P = 0.05, paired t‐test). However, a decrease of more than 15% was present only in 3 from 9 recorded neurons.
Figure 5.
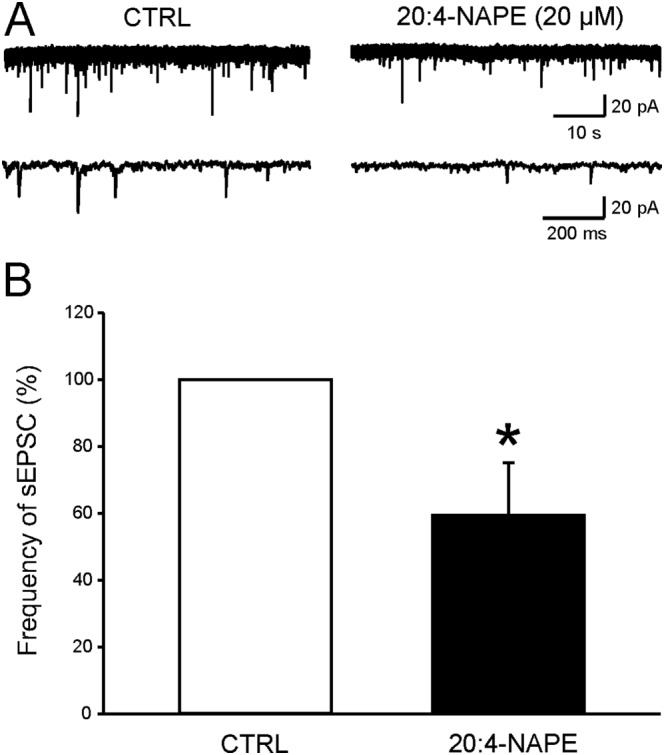
Application of 20:4‐NAPE decreased the frequency of sEPSCs under inflammatory conditions. (A) Native recording from one superficial dorsal horn neuron before and during application of 20:4‐NAPE (20 μM) to spinal cord slice dissected 24 h after the induction of peripheral inflammation. (B) Application of 20:4‐NAPE (20 μM) decreased the frequency of sEPSCs. *P < 0.05, significantly different from control; Wilcoxon signed‐rank test; n = 9.
Inhibition of sEPSCs by 20:4‐NAPE, under inflammatory conditions, was mediated through CB1 receptors with contributions from TRPV1 channels.
We tested the effects of PF514273 and SB366791 on 20:4‐NAPE‐induced inhibition in slices from rats with peripheral inflammation. Pretreatment with PF514273 (0.2 μM, 6 min) alone did not change the sEPSC frequency (Figure 6A,C) but did block the inhibition produced by 20:4‐NAPE, compared with the control value (Figure 6A,C). In 9 of these 16 recorded neurons, the frequency of sEPSC increased; in four neurons, it did not change; and it decreased in three neurons. The amplitude of sEPSCs was not significantly affected by PF514273 application alone (control: 27.4 ± 2.4 pA, PF514273: 24.6 ± 2.5 pA) or PF514273 with 20:4‐NAPE co‐application (24.1 ± 2.2 pA, n = 16).
Figure 6.
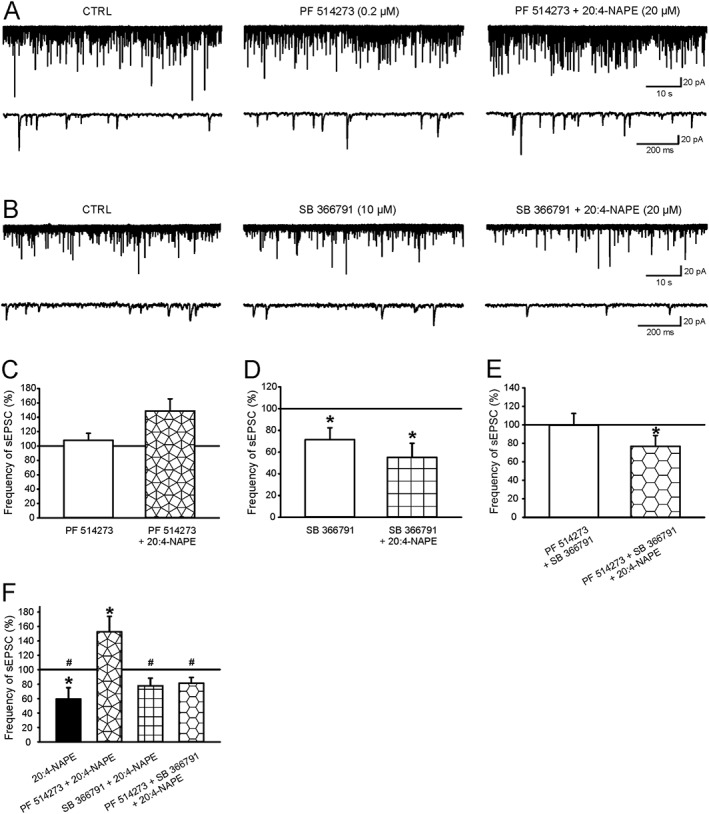
The effect of CB1 and TRPV1 antagonists on 20:4‐NAPE‐induced inhibition of sEPSC frequency under inflammatory conditions: (A, C) The application of PF514273 (0.2 μM, n = 16) did not change the frequency of sEPSCs. Subsequent co‐application of PF514273 (0.2 μM) and 20:4‐NAPE (20 μM) did not significantly change the frequency of sEPSCs, compared with control. (B, D) The frequency of sEPSCs significantly decreased during application of SB366791 (10 μM). *P < 0.05, significantly different from control; repeated measures (RM) ANOVA on ranks followed by Student–Newman–Keuls test; n = 16, In the presence of SB366791 (10 μM), 20:4‐NAPE (20 μM) induced a stronger decrease of sEPSC frequency. *P < 0.05, significantly different from control; RM ANOVA on ranks followed by Student–Newman–Keuls test. (E) The combined application of PF514273 (0.2 μM) and SB366791 (10 μM) did not change the frequency of sEPSCs. In the presence of both antagonists, 20:4‐NAPE (20 μM) significantly decreased the frequency of sEPSCs, compared with control. *P < 0.05, significantly different from control; RM ANOVA on ranks followed by Student–Newman–Keuls test; n = 15. (F) The same data are shown as a percentage of previous recording conditions: 20:4‐NAPE (n = 9) versus sEPSC basal frequency, PF514237 + 20:4‐NAPE versus PF514273 (n = 16) pretreatment, SB366791 + 20:4‐NAPE versus SB366791 (n = 16) pretreatment and PF514237 + SB366791 + 20:4‐NAPE versus both antagonists pretreatment (n = 15). *P < 0.05, significantly different from pretreatment, Wilcoxon signed‐rank test. # P < 0.05, significantly different from PF514237 + 20:4‐NAPE co‐application, one‐way ANOVA followed by Student–Newman–Keuls test.
In the inflamed slices, application of SB366791 (10 μM, 6 min) alone significantly decreased the frequency of sEPSCs (Figure 6B,D) and 20:4‐NAPE induced a further decrease of the sEPSC frequency compared with control values (Figure 6B,D). When responses of the individual cells were assessed, 11 of the 16 neurons exhibited decreased sEPSC frequency and it did not change in the rest of the neurons tested. The amplitude of sEPSCs was not significantly changed during the entire experiment (control: 24.2 ± 1.7 pA, SB366791: 24.2 ± 1.6 pA, SB366791 + 20:4‐NAPE: 21.8 ± 1.2 pA, n = 16).
The effect of combining both antagonists was evaluated in further experiments. Pretreatment with the combination did not change sEPSC frequency, when all the neurons were pooled together (Figure 6E). Although in 9 of these 15 recorded neurons,the combination decreased the sEPSC frequency (64.9 ± 4.8%, P < 0.05, repeated measures ANOVA on ranks with Student–Newman–Keuls test), in five neurons, it increased the sEPSC frequency (160.6 ± 11.2%, P > 0.05). In the presence of the combination, 20:4‐NAPE decreased the sEPSC frequency (Figure 6E) when compared with control values. Antagonist co‐treatment prevented the inhibitory effect of 20:4‐NAPE application in 7 of the 15 cells. The amplitude of sEPSCs was not significantly changed during different recording conditions (control: 24.9 ± 3.7 pA, SB366791 + PF514273: 23.9 ± 3.2 pA, SB366791 + PF514273 + 20:4‐NAPE: 20.0 ± 2.8 pA, n = 15).
These data have again been presented as percentages of control or antagonist(s) alone (Figure 6F). From this analysis, it emerged that 20:4‐NAPE alone induced inhibition of sEPSC frequency, compared with the control, which was comparable to that from naive (non‐inflamed) slices. However, PF514237 completely reversed the effects of 20:4‐NAPE and significantly increased sEPSC frequency. SB366791 did not block the inhibitory actions of 20:4‐NAPE. However, when SB366791 was combined with PF514237, 20:4‐NAPE was again an inhibitor of sEPSC frequency.
These results suggest that, under these inflammatory conditions, the inhibitory effect induced by 20:4‐NAPE application on sEPSC frequency is primarily mediated by activation of CB1 receptors (Figure 6F). Moreover, when the CB1 receptors were blocked, application of 20:4‐NAPE increased sEPSC frequency, which was prevented by TRPV1 channel blockade.
The reduction of the eEPSC amplitude induced by application of 20:4‐NAPE was prevented by blocking either the CB1 or TRPV1 receptors under the inflammatory conditions
We recorded eEPSCs in dorsal horn neurons after dorsal root stimulation in spinal cord slices prepared 24 h after induction of peripheral inflammation. In these slices, application of 20:4‐NAPE (20 μM) decreased eEPSCs amplitude (Figure 7). This decrease (>15%) was present in 9 of the 14 recorded neurons.
Figure 7.
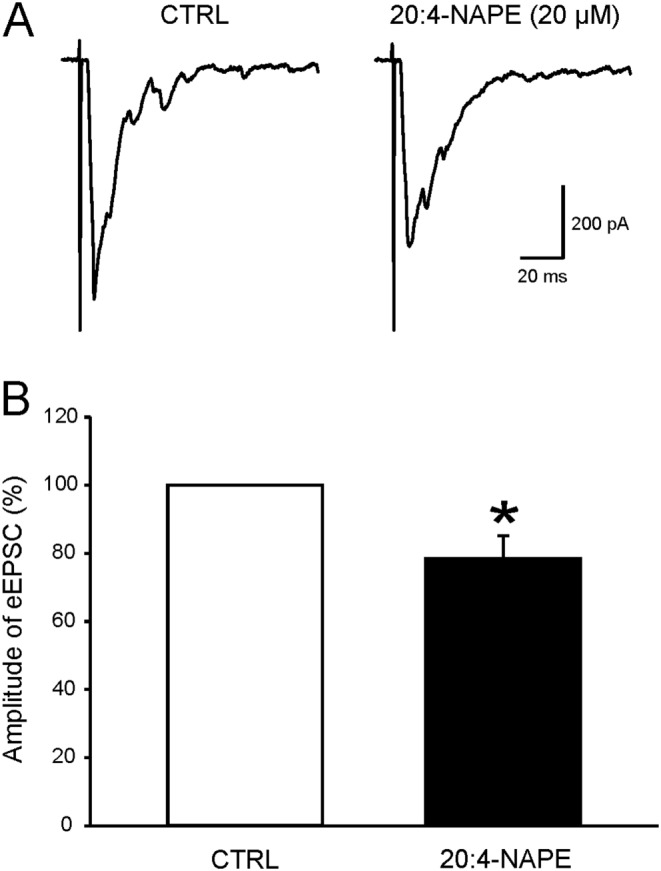
Application of 20:4‐NAPE decreased the amplitude of evoked EPSCs in superficial dorsal horn neurons under inflammatory conditions. (A) An example of native recording from one nociceptive neuron before and during application of 20:4‐NAPE (20 μM) to acute spinal cord slice prepared 24 h after intraplantar injection of carrageenan. (B) Application of 20:4‐NAPE (20 μM) decreased the amplitude of eEPSCs. *P < 0.05, significantly different from control; Wilcoxon signed‐rank test; n = 14.
Treatment of the slices with the CB1 receptor antagonist PF514273 (0.2 μM, 6 min) did not change the amplitude of eEPSCs (Figure 8A,C). Subsequent co‐application of PF514273 (0.2 μM, 4 min) and 20:4‐NAPE (20 μM) increased the amplitude of eEPSCs without reaching statistical significance (Figure 8A,C), compared with control values. This CB1 receptor antagonist thus prevented the inhibitory effect of 20:4‐NAPE in 11 of 16 neurons.
Figure 8.
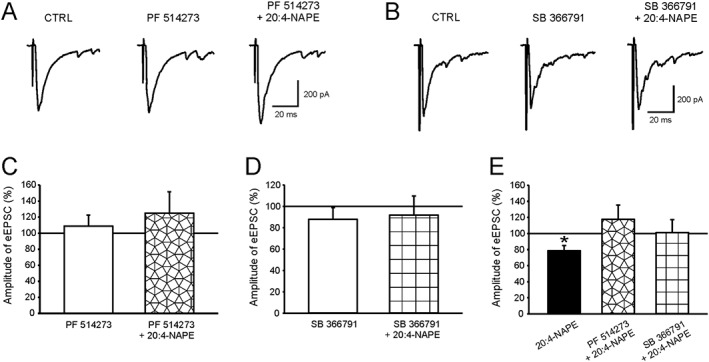
Antagonists of CB1 receptors and TRPV1 channels blocked the 20:4‐NAPE‐induced decrease of eEPSC amplitude under inflammatory conditions. (A, C) PF514273 alone (0.2 μM, n = 16) did not change the amplitude of eEPSCs. In the presence of PF514273, 20:4‐NAPE (20 μM) did not significantly affect the amplitude of eEPSCs. (B, D) Pretreatment with SB366791 (10 μM, n = 11) did not change the amplitude of eEPSC. In the presence of SB366791 (10 μM), 20:4‐NAPE (20 μM) also did not change the eEPSC amplitude. (E) The same data are shown as a percentage of previous recording conditions: *P < 0.05, significantly different from eEPSC basal amplitude; Wilcoxon signed‐rank test; n = 14.
While application of the TRPV1 channel antagonist SB366791 (10 μM, 6 min) alone, in another group of neurons it, did not change the eEPSC amplitude either (Figure 8B,D), did block the inhibitory effect of 20:4‐NAPE (Figure 8B,D), compared with control values. As obsereved with PF514237, the TRPV1 channel antagonist prevented 20:4‐NAPE‐induced inhibition in the majority (8 of 11) of the recorded superficial dorsal horn neurons.
Expressing these data as percentages of control or antagonist alone (Figure 8E), it is clear that, under these inflammatory conditions, the inhibition by 20:4‐NAPE of eEPSC amplitude was mediated by both CB1 receptors or TRPV1 channels
Discussion
Here, we report that 20:4‐NAPE inhibited the excitatory nociceptive synaptic transmission, as demonstrated by decrease of sEPSC frequency and reduction of dorsal root stimulation‐evoked EPSC amplitude in the superficial spinal dorsal horn. This inhibition was observed both in naive conditions and following the development of hindpaw inflammation. The differential effects of CB1 receptor and TRPV1 channel antagonists indicated that the mechanisms underlying 20:4‐NAPE‐induced inhibition were different in those two conditions.
20:4‐NAPE and anandamide synthesis
20:4‐NAPE is a substrate for anandamide synthesis in enzyme preparations and cultured primary sensory neurons (Wang et al., 2006; Varga et al., 2014). Here, we found that spinal cord slices also produce anandamide after 20:4‐NAPE application. Although direct effects of 20:4‐NAPE or indirect effects through a metabolite other than anandamide on some other receptors cannot be categorically excluded, we propose that at least the great majority of the effects induced by 20:4‐NAPE as described here, was mediated through the synthesis of anandamide acting on CB1 receptors and TRPV1 channels.
Anandamide activation of CB1 receptors and TRPV1 channels
CB1 receptors and TRPV1 channels constitute the main targets for anandamide (Devane et al., 1992; Zygmunt et al., 1999) and our data show that these two targets mediate, at least, the majority of the effects of 20:4‐NAPE. However, as anandamide is a highly promiscuous molecule, the involvement of other molecules including PPARsα and γ, Na+ and T‐type Ca2+ channels (Chemin et al., 2001; Kim et al., 2005; O'Sullivan, 2007; Okura et al., 2014) cannot be ruled out. Nevertheless, because of the marked effects of the CB1 receptor and TRPV1 channel antagonists, here we addressed the contribution of only these two targets.
The inhibition by 20:4‐NAPE of sEPSC frequency and eEPSC amplitude was mediated preferentially by activation of CB1 receptors under normal, naive conditions. Although expression of post‐synaptic CB1 receptors in the spinal cord has been reported (Farquhar‐Smith et al., 2000), most studies suggest exclusive pre‐synaptic location either on DRG neuron terminals or terminals of GABAergic inhibitory interneurons (Nyilas et al., 2009; Pernia‐Andrade et al., 2009; Hegyi et al., 2012; Veress et al., 2013). Activation of CB1 receptors at both locations leads to reduced transmitter release (Morisset et al., 2001; Nyilas et al., 2009; Pernia‐Andrade et al., 2009). In our preparations, the inhibitory synaptic transmission was pharmacologically blocked. Therefore, it seems reasonable to suggest that the CB1 receptors mediated the inhibitory effects of 20:4‐NAPE application, through anandamide‐mediated CB1 receptor activation and subsequent reduction of transmitter release from spinal terminals of DRG neurons.
Under naive conditions, there was a tendency for the antagonist of CB1 receptors, per se, to non‐significantly increase the frequency of sEPSC, whereas the TRPV1 channel antagonist did not affect the superficial dorsal horn neurons sEPSC frequency, as previously reported (Spicarova et al., 2014a). Nevertheless, moderate TRPV1 channel‐mediated sEPSC tonic activity was reported in lamina II neurons in mice (Park et al., 2011).
The robust decrease of sEPSC frequency mediated by AMPA receptors (Spicarova and Palecek, 2010) induced by 20:4‐NAPE, was also accompanied by moderate reduction of sEPSC amplitude in some neurons. In our preparations, the recorded neurons have contacts with numerous synapses, which spontaneously release glutamate and induce sEPSCs. The robust decrease of sEPSC frequency could elicit strong attenuation of glutamate release from specific, 20:4‐NAPE‐responsive, afferents, leading to an average decrease of sEPSC amplitude, without affecting the postsynaptic mechanisms.
The effect of peripheral inflammation
In slices taken after the induction of peripheral inflammation, 20:4‐NAPE induced a significant inhibition of sEPSC frequency, similar to that in the naive preparations. However, SB366791 reduced sEPSC frequency, suggesting tonic activation of presynaptic TRPV1 channels. The effect of SB366971 per se is consistent with inflammation‐induced tonic activity (Lappin et al., 2006) and increased sensitivity to endogenous agonists (Spicarova and Palecek, 2009) of presynaptic TRPV1 channels in the spinal cord dorsal horn. These channels are expressed in the overwhelming majority of spinal C‐fibre terminals in the superficial dorsal horn (Caterina et al., 1997; Guo et al., 1999). Consistent with this high level of expression of TRPV1 channels, regulation (activation, desensitization and inhibition) of TRPV1 channels has a marked effect on glutamate release from these afferents (Spicarova et al., 2014b). It has been suggested that modulation of TRPV1 channels in the dorsal horn could underlie several pathological pain states (Kanai et al., 2005; Spicarova et al., 2011; Spicarova et al., 2014a).
Tonic activation of presynaptic CB1 receptors was not detected under the inflammatory conditions. However, the CB1 receptor antagonist prevented inhibition by 20:4‐NAPE of sEPSC frequency. Moreover, 20:4‐NAPE significantly increased the frequency of sEPSCs, when CB1 receptors were blocked, and this potentiating effect was prevented by blockade of TRPV1 channels (Figure 6F). This indicates that, under inflammatory conditions, 20:4‐NAPE‐induced inhibition of the sEPSC frequency was mediated by CB1 receptors while the potentiating effect mediated by TRPV1 channels was unmasked only when the CB1 receptors were blocked.
The CB1 receptor‐mediated block of the inhibition by 20:4‐NAPE of eEPSC amplitude, was maintained after the development of inflammation. However, this effect of 20:4‐NAPE was prevented by blocking either CB1 receptors or TRPV1 channels, indicating involvement of both pathways. We did not observe a significant reduction of eEPSC amplitude after antagonism of TRPV1 channels, as with the sEPSC. While it is possible that activation of TRPV1 channels under these conditions did not play such an important role, it needs also to be taken into account that the electrical stimulation of dorsal roots could activate also myelinated primary afferents that do not express TRPV1 channels (Caterina et al., 1997; Guo et al., 1999). The effects of the TRPV1 channel antagonist thus could be ‘diluted’.
In contrast to potentiation of the spontaneous transmitter release by TRPV1 channel agonists, the release induced by action potentials evoked by dorsal root electrical stimulation may be blocked by activation of TRPV1 channels (Yang et al., 1999; Baccei et al., 2003). Thus, it is possible that activation of these channels on presynaptic terminals of DRG neurons by 20:4‐NAPE, reduced the glutamate release from primary afferents and thus contributed to the decrease of evoked EPSC amplitude in the recorded postsynaptic neuron. In addition, rapid internalization of voltage‐activated Ca2+ channels by activation of TRPV1 channels (Wu et al., 2005) could underlie the reduction of synchronous transmitter release. Although the vast majority of spinal TRPV1 channels are localized on terminals of primary sensory neurons, postsynaptic expression of these channels was also described in some GABAergic neurons, in which TRPV1 channel activation induces long‐term depression through the reduction of AMPA channels in the plasma membrane (Caterina et al., 1997; Guo et al., 1999; Kim et al., 2012). We cannot exclude the possibility that our neurons recorded in laminae I and II(outer) could include GABAergic cells in which the postsynaptic TRPV1 channel‐mediated modulation under the inflammatory conditions could occur, though it would change only the EPSC amplitude.
The role of 20:4‐NAPE and anandamide in nociceptive modulation
In summary, our data indicate that application of exogenous 20:4‐NAPE induced mainly CB1 receptor‐mediated inhibitory effects on excitatory transmission in naive animals while TRPV1 channel‐mediated mechanisms were also involved after peripheral inflammation. We propose, that if the effects of 20:4‐NAPE are indeed mediated through anandamide synthesis, balanced signalling by anandamide and its targets are involved in preventing the spread of nociceptive signals into supraspinal structures and this balance may be compromised during inflammation.
Anandamide, due to its lipophilic nature, would, most likely, be produced in close proximity to its target. The TRPV1 channel‐expressing primary sensory neurons indeed express Ca2+‐sensitive and Ca2+‐insensitive anandamide‐synthesizing pathways (Varga et al., 2014; Sousa‐Valente et al., 2014b; Sousa‐Valente et al., 2017). Further, transcripts of several anandamide‐synthesizing enzymes are expressed in the spinal dorsal horn (Malek et al., 2014). Enhanced activity after inflammation and during the electrical stimulation of primary afferent fibres resulted in increased concentration of Ca2+ in presynaptic terminals and could induce or increase the enzymic activity of NAPE‐PLD. Furthermore, Ca2+ influx through postsynaptic AMPA receptors could be also involved through promoting anandamide synthesis from 20:4‐NAPE, as NAPE‐PLD is expressed in post‐synaptic dendrites in the spinal dorsal horn (Hegyi et al., 2012). Ca2+‐insensitive pathways and NAPE‐PLD activity as a part of a retrograde inhibitory mechanism (Katona and Freund, 2008) could be also involved in this anandamide synthesis. NAPE‐PLD and other anandamide‐synthesizing enzymes may be particularly important for regulating nociceptive spinal processing under inflammatory conditions.
Application of 20:4‐NAPE, in our experiments, also provided a distinctive opportunity to study the role of the spinal endocannabinoid system, by application of substrate for anandamide synthesis instead of anandamide directly. By this approach, physiological mechanisms of anandamide synthesis played an important role, including the level of their activity and local distribution, thus changing localised anandamide concentrations. By flooding the preparation, by applying anandamide directly, it is more likely that other receptors and biological pathways would have been activated. This method of local ‘on demand’ anandamide production from its precursor may prove to be of advantage also in the clinical settings for pain treatment. Especially now as clinical trials focused to increase anandamide levels, by reducing its hydrolysis with inhibitors of fatty acid amide hydrolase did not show clinical efficacy (Mallet et al., 2016).
Author contributions
J.P. conceived and designed the study. V.N., P.M. and P.A. conducted experiments, V.N., P.M. and D.S. analysed the data. V.N., I.N., D.S. and J.P. participated in writing the manuscript. All authors read and approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by GACR 15‐11138S, MSMT LH15279, CZ.1.05/1.1.00/02.0109, RVO67985823, GAUK138215. Authors would like to thank Professor Ivan Miksik for the anandamide mass spectrometry analysis.
Nerandzic, V. , Mrozkova, P. , Adamek, P. , Spicarova, D. , Nagy, I. , and Palecek, J. (2018) Peripheral inflammation affects modulation of nociceptive synaptic transmission in the spinal cord induced by N‐arachidonoylphosphatidylethanolamine. British Journal of Pharmacology, 175: 2322–2336. doi: 10.1111/bph.13849.
References
- Ahluwalia J, Urban L, Bevan S, Nagy I (2003). Anandamide regulates neuropeptide release from capsaicin‐sensitive primary sensory neurons by activating both the cannabinoid 1 receptor and the vanilloid receptor 1 in vitro. Eur J Neurosci 17: 2611–2618. [DOI] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Capogna M, Bevan S, Nagy I (2000). Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience 100: 685–688. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkaitis MS, Solorzano C, Landry RP, Piomelli D, DeLeo JA, Romero‐Sandoval EA (2010). Evidence for a role of endocannabinoids, astrocytes and p38 phosphorylation in the resolution of postoperative pain. PLoS One 5: e10891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Oh‐hashi K, Naruse Y, Iijima N, Ueda M, Shimosato G et al. (2003). Local inflammation increases vanilloid receptor 1 expression within distinct subgroups of DRG neurons. Brain Res 963: 190–196. [DOI] [PubMed] [Google Scholar]
- Amaya F, Shimosato G, Kawasaki Y, Hashimoto S, Tanaka Y, Ji RR et al. (2006). Induction of CB1 cannabinoid receptor by inflammation in primary afferent neurons facilitates antihyperalgesic effect of peripheral CB1 agonist. Pain 124: 175–183. [DOI] [PubMed] [Google Scholar]
- Baccei ML, Bardoni R, Fitzgerald M (2003). Development of nociceptive synaptic inputs to the neonatal rat dorsal horn: glutamate release by capsaicin and menthol. J Physiol 549 (Pt 1): 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczynski MW, Svensson CI, Dumlao DS, Fitzsimmons BL, Shim JH, Scherbart TJ et al. (2010). Inflammatory hyperalgesia induces essential bioactive lipid production in the spinal cord. J Neurochem 114: 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipatikom K et al. (2004). Cultured rat microglial cells synthesize the endocannabinoid 2‐arachidonylglycerol, which increases proliferation via a CB2 receptor‐dependent mechanism. Mol Pharmacol 65: 999–1007. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997). The capsaicin receptor: a heat‐activated ion channel in the pain pathway. Nature 389: 816–824. [DOI] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Perez‐Reyes E, Nargeot J, Lory P (2001). Direct inhibition of T‐type calcium channels by the endogenous cannabinoid anandamide. EMBO J 20: 7033–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa B, Bettoni I, Petrosino S, Comelli F, Giagnoni G, Di Marzo V (2010). The dual fatty acid amide hydrolase/TRPV1 blocker, N‐arachidonoyl‐serotonin, relieves carrageenan‐induced inflammation and hyperalgesia in mice. Pharmacological Res 61: 537–546. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G et al. (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258: 1946–1949. [DOI] [PubMed] [Google Scholar]
- Dow RL, Carpino PA, Hadcock JR, Black SC, Iredale PA, DaSilva‐Jardine P et al. (2009). Discovery of 2‐(2‐chlorophenyl)‐3‐(4‐chlorophenyl)‐7‐(2,2‐difluoropropyl)‐6,7‐dihydro‐2H‐pyraz olo[3,4‐f][1,4]oxazepin‐8(5H)‐one (PF‐514273), a novel, bicyclic lactam‐based cannabinoid‐1 receptor antagonist for the treatment of obesity. J Med Chem 52: 2652–2655. [DOI] [PubMed] [Google Scholar]
- Farquhar‐Smith WP, Egertova M, Bradbury EJ, McMahon SB, Rice AS, Elphick MR (2000). Cannabinoid CB(1) receptor expression in rat spinal cord. Mol Cell Neurosci 15: 510–521. [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM et al. (2004). Identification and characterisation of SB‐366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology 46: 133–149. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R (1999). Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci 11: 946–958. [DOI] [PubMed] [Google Scholar]
- Hegyi Z, Hollo K, Kis G, Mackie K, Antal M (2012). Differential distribution of diacylglycerol lipase‐alpha and N‐acylphosphatidylethanolamine‐specific phospholipase d immunoreactivity in the superficial spinal dorsal horn of rats. Glia 60: 1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Nakazato E, Fujiuchi A, Hara T, Imai A (2005). Involvement of an increased spinal TRPV1 sensitization through its up‐regulation in mechanical allodynia of CCI rats. Neuropharmacology 49: 977–984. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF (2008). Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med 14: 923–930. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HI, Kim TH, Shin YK, Lee CS, Park M, Song JH (2005). Anandamide suppression of Na+ currents in rat dorsal root ganglion neurons. Brain Res 1062: 39–47. [DOI] [PubMed] [Google Scholar]
- Kim YH, Back SK, Davies AJ, Jeong H, Jo HJ, Chung G et al. (2012). TRPV1 in GABAergic interneurons mediates neuropathic mechanical allodynia and disinhibition of the nociceptive circuitry in the spinal cord. Neuron 74: 640–647. [DOI] [PubMed] [Google Scholar]
- Kwon SG, Roh DH, Yoon SY, Moon JY, Choi SR, Choi HS et al. (2014). Blockade of peripheral P2Y1 receptors prevents the induction of thermal hyperalgesia via modulation of TRPV1 expression in carrageenan‐induced inflammatory pain rats: involvement of p38 MAPK phosphorylation in DRGs. Neuropharmacology 79: 368–379. [DOI] [PubMed] [Google Scholar]
- Lappin SC, Randall AD, Gunthorpe MJ, Morisset V (2006). TRPV1 antagonist, SB‐366791, inhibits glutamatergic synaptic transmission in rat spinal dorsal horn following peripheral inflammation. Eur J Pharmacol 540: 73–81. [DOI] [PubMed] [Google Scholar]
- Lever IJ, Robinson M, Cibelli M, Paule C, Santha P, Yee L et al. (2009). Localization of the endocannabinoid‐degrading enzyme fatty acid amide hydrolase in rat dorsal root ganglion cells and its regulation after peripheral nerve injury. J Neurosci 29: 3766–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Cheng J, Han JS, Wan Y (2004). Change of vanilloid receptor 1 expression in dorsal root ganglion and spinal dorsal horn during inflammatory nociception induced by complete Freund's adjuvant in rats. Neuroreport 15: 655–658. [DOI] [PubMed] [Google Scholar]
- Malek N, Kucharczyk M, Starowicz K (2014). Alterations in the anandamide metabolism in the development of neuropathic pain. Biomed Res Int 2014: 686908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet C, Dubray C, Duale C (2016). FAAH inhibitors in the limelight, but regrettably. Int J Clin Pharmacol Ther 54: 498–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisset V, Ahluwalia J, Nagy I, Urban L (2001). Possible mechanisms of cannabinoid‐induced antinociception in the spinal cord. Eur J Pharmacol 429: 93–100. [DOI] [PubMed] [Google Scholar]
- Morisset V, Urban L (2001). Cannabinoid‐induced presynaptic inhibition of glutamatergic EPSCs in substantia gelatinosa neurons of the rat spinal cord. J Neurophysiol 86: 40–48. [DOI] [PubMed] [Google Scholar]
- Nagy I, Friston D, Valente JS, Torres Perez JV, Andreou AP (2014). Pharmacology of the capsaicin receptor, transient receptor potential vanilloid type‐1 ion channel. Prog Drug Res 68: 39–76. [DOI] [PubMed] [Google Scholar]
- Nyilas R, Gregg LC, Mackie K, Watanabe M, Zimmer A, Hohmann AG et al. (2009). Molecular architecture of endocannabinoid signaling at nociceptive synapses mediating analgesia. Eur J Neurosci 29: 1964–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okura D, Horishita T, Ueno S, Yanagihara N, Sudo Y, Uezono Y et al. (2014). The endocannabinoid anandamide inhibits voltage‐gated sodium channels Nav1.2, Nav1.6, Nav1.7, and Nav1.8 in Xenopus oocytes. Anesth Analg 118: 554–562. [DOI] [PubMed] [Google Scholar]
- O'Sullivan SE (2007). Cannabinoids go nuclear: evidence for activation of peroxisome proliferator‐activated receptors. Br J Pharmacol 152: 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CK, Lu N, Xu ZZ, Liu T, Serhan CN, Ji RR (2011). Resolving TRPV1‐ and TNF‐alpha‐mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J Neurosci 31: 15072–15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernia‐Andrade AJ, Kato A, Witschi R, Nyilas R, Katona I, Freund TF et al. (2009). Spinal endocannabinoids and CB1 receptors mediate C‐fiber‐induced heterosynaptic pain sensitization. Science 325: 760–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Dubner R (1999). Inflammatory Models of Pain and Hyperalgesia. ILAR J 40: 111–118. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Kilo S, Hargreaves KM (1998). Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain 75: 111–119. [DOI] [PubMed] [Google Scholar]
- Snider NT, Walker VJ, Hollenberg PF (2010). Oxidation of the endogenous cannabinoid arachidonoyl ethanolamide by the cytochrome P450 monooxygenases: physiological and pharmacological implications. Pharmacol Rev 62: 136–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa‐Valente J, Andreou AP, Urban L, Nagy I (2014a). Transient receptor potential ion channels in primary sensory neurons as targets for novel analgesics. Br J Pharmacol 171: 2508–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa‐Valente J, Varga A, Ananthan K, Khajuria A, Nagy I (2014b). Anandamide in primary sensory neurons: too much of a good thing? Eur J Neurosci 39: 409–418. [DOI] [PubMed] [Google Scholar]
- Sousa‐Valente J, Varga V, Torres Perez JVT, Jenes A, Wahba J, Mackie K et al. (2017). Inflammation of peripheral tissues and injury to peripheral nerves induce diferring effects in the expression of the calcium‐sensitive anandamide‐synthesising enzyme and related molecules in rat primary sensory neurons. J Comp Neurol 525: 1778–1796. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicarova D, Adamek P, Kalynovska N, Mrozkova P, Palecek J (2014a). TRPV1 receptor inhibition decreases CCL2‐induced hyperalgesia. Neuropharmacology 81: 75–84. [DOI] [PubMed] [Google Scholar]
- Spicarova D, Nerandzic V, Palecek J (2011). Modulation of spinal cord synaptic activity by tumor necrosis factor alpha in a model of peripheral neuropathy. J Neuroinflammation 8: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicarova D, Nerandzic V, Palecek J (2014b). Update on the role of spinal cord TRPV1 receptors in pain modulation. Physiol Res 63 (Suppl 1): S225–S236. [DOI] [PubMed] [Google Scholar]
- Spicarova D, Palecek J (2009). The role of the TRPV1 endogenous agonist N‐Oleoyldopamine in modulation of nociceptive signaling at the spinal cord level. J Neurophysiol 102: 234–243. [DOI] [PubMed] [Google Scholar]
- Spicarova D, Palecek J (2010). Modulation of AMPA excitatory postsynaptic currents in the spinal cord dorsal horn neurons by insulin. Neuroscience 166: 305–311. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Trevisani M, Vellani V, De Petrocellis L, Schiano Moriello A, Campi B et al. (2005). Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J 24: 3026–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognetto M, Amadesi S, Harrison S, Creminon C, Trevisani M, Carreras M et al. (2001). Anandamide excites central terminals of dorsal root ganglion neurons via vanilloid receptor‐1 activation. J Neurosci 21: 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda N, Tsuboi K, Uyama T (2013). Metabolism of endocannabinoids and related N‐acylethanolamines: canonical and alternative pathways. FEBS J 280: 1874–1894. [DOI] [PubMed] [Google Scholar]
- Varga A, Jenes A, Marczylo TH, Sousa‐Valente J, Chen J, Austin J et al. (2014). Anandamide produced by Ca(2+)‐insensitive enzymes induces excitation in primary sensory neurons. Pflugers Archiv 466: 1421–1435. [DOI] [PubMed] [Google Scholar]
- Vellani V, Petrosino S, De Petrocellis L, Valenti M, Prandini M, Magherini PC et al. (2008). Functional lipidomics. Calcium‐independent activation of endocannabinoid/endovanilloid lipid signalling in sensory neurons by protein kinases C and A and thrombin. Neuropharmacology 55: 1274–1279. [DOI] [PubMed] [Google Scholar]
- Veress G, Meszar Z, Muszil D, Avelino A, Matesz K, Mackie K et al. (2013). Characterisation of cannabinoid 1 receptor expression in the perikarya, and peripheral and spinal processes of primary sensory neurons. Brain Struct Funct 218: 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Okamoto Y, Morishita J, Tsuboi K, Miyatake A, Ueda N (2006). Functional analysis of the purified anandamide‐generating phospholipase D as a member of the metallo‐beta‐lactamase family. J Biol Chem 281: 12325–12335. [DOI] [PubMed] [Google Scholar]
- Wang J, Ueda N (2009). Biology of endocannabinoid synthesis system. Prostaglandins Other Lipid Mediat 89: 112–119. [DOI] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL (2005). Transient receptor potential vanilloid type 1 activation down‐regulates voltage‐gated calcium channels through calcium‐dependent calcineurin in sensory neurons. J Biol Chem 280: 18142–18151. [DOI] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H, Li YQ, Yoshimura M (1999). Action of capsaicin on dorsal root‐evoked synaptic transmission to substantia gelatinosa neurons in adult rat spinal cord slices. Brain Res 830: 268–273. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V et al. (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400: 452–457. [DOI] [PubMed] [Google Scholar]