

Abstract
Osimertinib is a potent, highly selective, irreversible inhibitor of epidermal growth factor receptor (EGFR) and T790M resistance mutation. In vitro metabolism data suggested osimertinib is a substrate of cytochrome P450 (CYP)3A4/5, a weak inducer of CYP3A, and an inhibitor of breast cancer resistance protein (BCRP). A combination of in vitro data, clinical pharmacokinetic data, and drug‐drug interaction (DDI) data of osimertinib in oncology patients were used to develop the physiologically based pharmacokinetic (PBPK) model and verify the DDI data of osimertinib. The model predicted the observed monotherapy concentration profile of osimertinib within 1.1‐fold, and showed good predictability (within 1.7‐fold) to the observed peak plasma concentration (Cmax) and area under the curve (AUC) DDI ratio changes, when co‐administered with rifampicin, itraconazole, and simvastatin, but not with rosuvastatin. Based on observed clinical data and PBPK simulations, the recommended dose of osimertinib when dosed with strong CYP3A inducers is 160 mg once daily. PBPK modeling suggested no dose adjustment with moderate and weak CYP3A inducers.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Based on in vitro studies, osimertinib is a substrate of CYP3A4/5, a weak inducer of CYP3A, and inhibitor of BCRP. Hence, modulators of CYP3A could alter its metabolism and it may cause changes in the exposure of other substrates of CYP3A/BCRP.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addressed the feasibility of building a human PBPK model of osimertinib in Simcyp capable of evaluating the likely impact of administration of CYP3A inhibitors and inducers on the exposure of osimertinib as a victim drug in oncology patients.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study highlights the utility of PBPK modeling and simulation to assess the clinical DDI risk of osimertinib. Our work established that osimertinib is metabolized by multiple CYP isozymes with the relative contribution being the highest for CYP3A. This work provides appropriate dosing recommendations for osimertinib when co‐administered with CYP3A inducers and other dosing recommendations were based on clinical observations.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This PBPK model can be utilized to simulate exposure in scenarios where clinical data may be lacking, such as DDI and special populations.
Osimertinib (Tagrisso) is a highly selective, potent, orally bioavailable, small molecule. It is an irreversible inhibitor of both epidermal growth factor receptor (EGFR), which has been mutated by EGFR tyrosine kinase inhibitors, and EGFR with T790M resistance mutation.1 Osimertinib has been approved by the US Food and Drug Administration (http://www.fda.gov)2 and the European Medicines Agency (http://www.ema.europa.eu)3 for the treatment of patients with metastatic EGFR T790M mutation‐positive non‐small cell lung cancer.
In oncology patients, osimertinib is slowly absorbed with a median time of maximum plasma concentration (Tmax) of 6 hours, an oral clearance of 14.2 L/h, a volume of distribution of around 997 L, and a terminal half‐life of ∼48 hours.4 Following administration of single oral doses of 20 to 240 mg, osimertinib mean peak levels (Cmax) and area under curve profile (AUC) values increased in a dose proportional manner. A threefold accumulation at steady‐state was observed after 15 days of continuous dosing.4
In vitro data indicated that the highest contributor to the metabolism of osimertinib was cytochrome P450 3A (CYP3A4/5; fraction metabolized (fm) CYP3A=54%5) with a smaller contribution by renal clearance. Details of metabolic and disposition pathways of osimertinib are shown in Supplementary Figure S1. Osimertinib may interact with drugs that significantly inhibit or induce CYP3A,5 hence, clinical studies with a strong inhibitor and a strong inducer were conducted. Furthermore, as osimertinib is also a weak inducer of CYP3A in vitro, the possibility that it may cause changes in the exposure of other CYP3A substrates needs to be considered, especially when it is given in multiple doses. In addition, data from in vitro transporter experiments, and a basic static drug‐drug interaction (DDI) analysis (Supplementary Table S1) suggested that the potential for osimertinib to be an inhibitor of breast cancer resistance protein (BCRP) is likely, but unlikely for organic anion‐transporting polypeptide (OATP1B1 and OATP1B3) and organic cation transporter 2 (OCT2). Thus, a clinical study was conducted to understand the effect of osimertinib on exposure of a BCRP substrate, rosuvastatin.
Four clinical DDI studies (Table 1) were conducted to inform the current labeling for osimertinib. Study design of these DDI trials are outlined in Supplementary Figure S2. As a victim drug, minimal CYP3A‐dependent interaction was observed when co‐dosed with itraconazole, whereas significant decrease in exposure was observed when co‐dosed with rifampicin. The results of the DDI study with rifampicin suggest that an osimertinib dose adjustment is recommended when co‐dosed with strong CYP3A inducers. As a perpetrator drug, osimertinib seems to have a minimal to no effect on a CYP3A substrate, simvastatin, and a modest effect on the BCRP substrate, rosuvastatin.
Table 1.
Osimertinib PBPK model input parameters
| Parameters and models | Osimertinib | Source | |
|---|---|---|---|
| Physiochemical properties | Molecular weight | 499.61 | Experimental data |
| Log P | 5.45 | Experimental data | |
| pKa |
Diprotic base pKa 1: 9.50 pKa 2: 4.44 |
Experimental data | |
| B/P ratio | 1 | Experimental data | |
| fu, plasma | 0.0133 | Estimated value | |
| fu, gut | 0.00132 | Predicted by Simcyp | |
| Dosage form | Film coated tablets, immediate release | ||
| Absorption | Absorption model | First order | |
| fa | 0.82 | User defined value, based on Dickinson et al.5 | |
| ka (CV%) | 0.24 | Estimated value from Pop‐PK model Brown et al.7 | |
| Peff,man | 0.187 ×10−4 cm/s | Estimated based on clinical data | |
| Distribution | Distribution model | Full PBPK | |
| Vss (L/kg) | 6.497 | Simcyp Predicted using method 2 | |
| Elimination | Clearance type | Enzyme kinetics | |
| CYP1A2 CLint (µL/min/pmol of isoform) | 0.520 | Retrograde approach | |
| CYP2A6 CLint (µL/min/pmol of isoform) | 1.749 | ||
| CYP2C9 CLint (µL/min/pmol of isoform) | 0.479 | ||
| CYP2E1 CLint (µL/min/pmol of isoform) | 0.111 | ||
| CYP3A4 CLint (µL/min/pmol of isoform) | 0.731 | ||
| CYP3A5 CLint (µL/min/pmol of isoform) | 0.210 | ||
| Renal CL (L/h) | 0.235 | ||
| Interaction | CYP2C8 reversible Inhibition Ki (μM) | 11.4 | Experimental data |
| CYP3A4/5 reversible inhibition Ki (μM) | 2.55 | Experimental data | |
| CYP3A4 time‐dependent inhibition Ki (μM) | 1090 | Experimental data | |
| CYP3A4 time‐dependent inhibition Kinact (hr−1) | 3.70 | Experimental data | |
| CYP3A4/5 induction IC50 (μM) | 0.117 | Experimental data | |
| CYP3A4/5 induction Emax (fold) | 10.78 | Experimental data | |
| Fumic | 0.0261 | Experimental data | |
| Fuinc | 1 | Optimized value based on sensitivity analysis | |
| OATP1B1 inhibition Ki (μM) | 22 | Experimental data | |
| OATP1B1 inhibition Ki (μM) | 52.5 | Experimental data | |
| BCRP inhibition Ki (μM) | 2 | Experimental data | |
| Population | Oncology patients based Cheeti et al.6 | ||
| Clinical data |
D5160C00001 (NCT01802632): Tablet monotherapy data single and multiple dose data D5160C00012 (NCT02157883): DDI study with itraconazole D5160C00013 (NCT02197247): DDI study with rifampicin D5160C00014 (NCT02197234): DDI study with simvastatin D5160C00019 (NCT02317016): DDI study with rosuvastatin |
||
BCRP, breast cancer resistance protein; CL, clearance; CLint, intrinsic clearance; DDI, drug‐drug interaction; Emax, maximum effect; OATP, organic anion‐transporting polypeptide; PBPK, physiologically based pharmacokinetic; Pop‐PK, population pharmacokinetic; Vss, volume of distribution at steady state.
The aim of this study was to build a human physiologically based pharmacokinetic (PBPK) model of osimertinib in Simcyp using in vitro and in vivo pharmacokinetic (PK) parameters. The PBPK model was verified by matching clinical PK plasma profiles generated at different dosage regimens. This verified PBPK model was then used to evaluate the likely impact of administration of CYP3A inhibitors and inducers on the exposure of osimertinib as a victim drug in oncology patients.
The potential interaction between multiple‐oral doses of osimertinib and simvastatin or rosuvastatin were also investigated to assess the potential for osimertinib as a perpetrator to induce or inhibit CYP3A and BCRP inhibition, respectively, and to confirm the predictability of the PBPK model to the clinical data. Finally, the model was used prospectively to predict the likely outcomes of interaction with moderate and weak CYP3A inducers efavirenz and dexamethasone, respectively, and to inform dose adjustment of osimertinib when co‐dosed with strong CYP3A inducers using both clinical DDI data and PBPK model‐based evaluation.
MATERIALS AND METHODS
In vitro studies and input data
A combination of physicochemical parameters, parameters from in vitro absorption, distribution, metabolism, and excretion (ADME) studies and concentration time data from clinical studies with osimertinib tablets (Table 1, 5, 6, 7) were used in order to build a physiologically based model for simulating the exposure of osimertinib in Simcyp version 148 (Simcyp Limited, UK) in virtual oncology populations. Details of the key input parameters used in the PBPK model development are described in Supplementary Methods.
First order absorption model with a predicted human intestinal effective permeability (Peff,human) using Caco2 cell monolayer apparent permeability of 8.64 × 10−6 cm/s value along with observed clearance (CLpo) resulted in misfit to Cmax. Hence, the parameter estimation method using clinical data was used to find appropriate values of the Peff,human and CLpo by fitting the individual subject PK profiles of osimertinib. Optimizing these two values in particular would allow the model to accurately reflect available clinical data with respect to the extent of elimination (CLpo) and the rate of absorption (Peff,human). The estimated Peff,human of 0.19 × 10−4 cm/s and CLpo of 14.75 L/h were used in the model (see Supplemental Section for rationale of using parameter estimation method and results).
At the time of model development, human PK data after intravenous dosing was not available, thus, the volume of distribution at steady state was predicted using full PBPK model Rodgers and Rowland9 method within the Simcyp simulator. Fraction unbound in enterocyte (Fu,gut) value predicted within simulator was 0.00132. The influence of Fu,gut on osimertinib exposure was also studied via sensitivity analysis.
Metabolic clearance of osimertinib and the contribution of CYP enzymes are depicted in Supplementary Figure S1. As human data following oral dosing was available, a decision was made to use the in vivo oral clearance as an estimate of the total clearance (CLpo) in Simcyp. The estimated in vivo oral clearance, 14.75 L/h, observed in the monotherapy study was used to define metabolic clearance of osimertinib with the retrograde method (see Supplementary Section). Table 1, 5, 6, 7 shows the intrinsic metabolic clearance values for each CYP isozyme estimated by retrograde method.
The two active circulating metabolites of osimertinib (AZ5104 and AZ7550) were measured in plasma in all studies. Both metabolites were formed primarily via CYP3A, and seemed to be metabolized via CYP3A as well. Because each of these metabolites contributed to <10% of the total drug‐related exposure, they were not considered in the model for DDI predictions as per the regulatory guidelines.7, 10, 11
The osimertinib PBPK model included the Ki (half‐maximal inhibitory concentration; Ki = IC50/2) for competitive inhibition for CYP2C8 (11.4 µM) and CYP3A4/5 (2.55 µM). As no distinction was made between inhibition of CYP3A4 or CYP3A5, the same values were used for both isoforms. Although there was no anticipated risk of time‐dependent CYP3A inhibition for osimertinib, a Ki (concentration associated with half‐maximal inactivation rate) value 1,090 µM and inactivation rate of the enzyme (Kinact) value of 3.7 h−1 were included in the model for completeness of the available data.
CYP3A induction data parameters Indmax (maximum induction) and IndC50 (induction EC50) from three lots of human hepatocytes could not be estimated due to in vitro cytotoxicity at tested concentrations above 3.3 µM.5 In order to quantify the net effect on a CYP3A substrate due to induction, the Emax was fixed at the maximum concentration at which a value was obtained. This conservative approach provided the worst‐case scenario IndC50 and Indmax estimates for osimertinib. These estimates were calibrated to a positive control (rifampicin), to adjust for both interindividual and interlaboratory variability within the Simcyp simulator (Supplementary Table S2). Calibrated IndC50 and Indmax used in the model were 0.12 uM and 10.8, respectively.
PBPK MODEL DEVELOPMENT, VERIFICATION, AND MODEL APPLICATION
PBPK model development
Simcyp software (Simcyp, version 14, UK) was used to develop and verify a PBPK model of osimertinib.
In the first stage, a “bottom‐up” approach was undertaken, using in vitro and physicochemical properties of osimertinib. Fraction absorbed and first order absorption rate parameters were derived using in vitro permeability data. An in vivo derived estimate of CLpo was used to predict the intrinsic clearance of osimertinib. At this stage, no interaction data was included in the model.
In the second stage, the values of Peff,human and CLpo used in the model were updated using parameter estimation to fit these two parameters (see Supplementary Methods file for methodology of parameter estimation of Peff,human and CLpo) while keeping the remaining parameters fixed.
In the third stage, model estimated clearance was converted to an enzyme kinetics clearance using the retrograde model and the interaction data was included to recover observed clinical data of osimertinib as monotherapy after multiple dosing. This updated model was checked for consistency against the known clinical PK of osimertinib and the model was updated. In particular, the absorption rate constant and fraction unbound in incubation (fuinc) were altered to fit the absorption phase and recover monotherapy steady‐state data well.
The oncology virtual population based on Cheeti et al.,6 was used for simulations. This population is a demography‐based oncology population that included changes to alpha acid glycoprotein (77% higher in patients with cancer), human serum albumin (20% lower in patients with cancer), and hematocrit (10% lower in patients with cancer). The key physiological differences between oncology populations based on Cheeti et al.,6 compared to the healthy volunteer population are shown in Supplementary Table S3. Simulations were performed as per clinical study designs and demographics were matched with trial design of four DDI studies. The effect of food on osimertinib in patients with non‐small cell lung cancer was small and not clinically relevant (D5160C00009). Thus, all simulations were conducted in the fasted state to match the in vivo DDI clinical studies.
PBPK model verification
Predefined acceptance criteria to assess the predictive performance of the PBPK model was set to be twofold, which was based on the clinical relevance. A large therapeutic window is observed with osimertinib, with clinical activity evident at all doses studied (20–240 mg) and no maximum tolerated dose defined. There were no covariates identified that would be expected to increase exposure more than twofold (≥160 mg dose) or decrease below 50% (≤40 mg)7 justifying a twofold window as an appropriate acceptance criteria.
Osimertinib as a victim drug: Simulating interactions with itraconazole and rifampicin
Single doses of osimertinib exposures were simulated in the presence and absence of twice daily dosing of 200 mg of itraconazole. The study design (Supplementary Figure S2) for the simulation replicated the clinical study D5160C00012.12 In the simulation, 200 mg of itraconazole is dosed every 12 hours for 14 days (days 6–18) and 80 mg of osimertinib is dosed alone or 1 hour after the first itraconazole dose on day 5 (i.e., 97 hours after the first itraconazole dose).
Osimertinib exposure was simulated at steady state in the presence and absence of once daily dosing of 600 mg of rifampicin. The study design (Supplementary Figure S2) for the simulation replicated the clinical study D5160C00013.12 In the simulation, 80 mg of osimertinib is dosed every 24 hours for 77 days, and 600 mg of rifampicin is dosed every 24 hours for 21 days starting on day 29 and ending on day 49. On the days when rifampicin is dosed, it is dosed at the same time as osimertinib. In study D5160C00013, the magnitude of the DDI was based on comparing the osimertinib AUC0–t and Cmax on days 28 and 49 (i.e., the last day prior to commencing rifampicin dosing and the last day of rifampicin dosing).
Osimertinib as a perpetrator drug: Simulating interactions with simvastatin and rosuvastatin
Simvastatin and rosuvastatin exposure were simulated in the presence and absence of multiple once daily dosing of 80 mg of osimertinib. The study design for the simulation replicated the clinical studies D5160C00014 and D5160C00019.13 In the simulation, 80 mg of osimertinib was dosed every 24 hours for 30 days and 40 mg of simvastatin or rosuvastatin 20 mg was dosed alone or with the osimertinib dose on day 29 (i.e., 672 hours after the first osimertinib dose).
PBPK MODEL APPLICATION
Predicting the interactions with dexamethasone and efavirenz
The final PBPK model was utilized to simulate changes in the PK profile of osimertinib in the presence of weak to moderate CYP3A inducers that may commonly be co‐administered with osimertinib in the clinic, such as dexamethasone and efavirenz. The dexamethasone PBPK model was developed in‐house and the simulated PK parameters were compared with published clinical data.14 Furthermore, verification using a clinical DDI study15 between dexamethasone and a CYP3A substrate, casopitant,16 showed reasonable predictability (see Supplementary Table S4).
Potential dose adjustment of osimertinib when co‐dosed with strong CYP3A inducers
PBPK simulations for osimertinib as a victim drug were performed to guide the dose adjustment of osimertinib in the presence of a strong CYP3A inducer, such that the osimertinib exposure in the presence of inducer (rifampicin) can be increased to be closer to that without inducer. An osimertinib dose of 160 mg once daily was chosen and the study design was the same as that of the rifampicin DDI study design.
Input parameters of victim and perpetrator drugs and trial design for osimertinib DDI simulations with itraconazole, rifampicin, dexamethasone, efavirenz, simvastatin, and rosuvastatin are detailed in Supplementary Table S5 and Supplementary Table S6.
RESULTS
PBPK model development
Simulating osimertinib exposure alone
To demonstrate the ability of the PBPK model to replicate the plasma concentration‐time profile of osimertinib seen in patients, an example simulation with the same dose regimen as was used in patients dosed at 80 mg in part A of the phase I study D5160C00001 was performed and is shown in Figure 1 and Table 2. The model adequately captures the monotherapy data and seems to be appropriate for the assessments of the magnitude of drug interaction for osimertinib and CYP3A mediated DDI.
Figure 1.
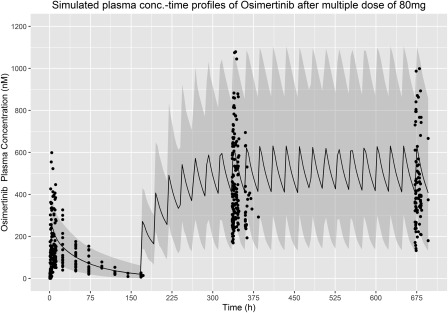
Simulated profile for single osimertinib 80 mg dose (0–168 hours) followed by multiple dosing of osimertinib 80 mg daily for 28 days (168–696 hours). The solid black line is the median prediction using the physiologically based pharmacokinetic model and the shaded area represents the 95% prediction intervals. Closed circles are observed data points from study D5160C00001.
Table 2.
Comparison of the observed clinical exposure to osimertinib monotherapy and summary of PBPK model predictions with known CYP3A4 modulators and the predicted effect of osimertinib on CYP3A4 and BCRP probe substrates
| Osimertinib monotherapy simulations | |||
|---|---|---|---|
| Osimertinib dose | PK parameter | Observed in vivo clinical data | PBPK model predicted (n = 100; 10 subjects × 10 trials) |
| Multiple doses 80 mg once daily |
AUCss (nM*h) Geo.mean (range) [%CV] |
10,360 (4,730 to 22,300) [47] |
11,076 (2,883 to 36,260) [49] |
|
Css,max (nM) Geo.mean (range) [%CV] |
545 (258 to 1,220) [46] |
581 (205 to 1,607) [43] |
|
| DDI simulations | |||
|---|---|---|---|
| Exposure ratio | |||
| Modulators | Interaction mechanism of comedication | AUC ratio with confidence intervals | Cmax ratio with confidence intervals |
| Inhibitor (osimertinib as a victim) | Strong CYP3A4 reversible inhibitor (itraconazole observed)a | 1.24 (1.15 to 1.35) | 0.799 (0.732 to 0.872) |
| Strong CYP3A4 reversible inhibitor (itraconazole predicted)b | 1.79 (1.76 to 1.83) | 1.13 (1.12 to 1.13) | |
| Inducer (osimertinib as a victim) | Strong CYP3A4 inducer (rifampicin observed)a | 0.216 (0.195 to 0.238) | 0.272 (0.244 to 0.303) |
| Strong CYP3A4 inducer (rifampicin predicted)b | 0.357 (0.340 to 0.376) | 0.454 (0.436 to 0.472) | |
| Moderate CYP3A4 inducer (efavirenz predicted)b | 0.580 (0.560 to 0.600) | 0.644 (0.627 to 0.662) | |
| Weak CYP3A4 inducer (dexamethasone predicted)b | 0.999 (0.999 to 0.999) | 0.999 (0.999 to 0.999) | |
| Osimertinib as a perpetrator | CYP3A4 sensitive substrate (simvastatin observed)a | 0.915 (0.772 to 1.084) | 0.771 (0.634 to 0.937) |
| CYP3A4 sensitive substrate (simvastatin predicted)b | 0.678 (0.659 to 0.698) | 0.713 (0.695 to 0.731) | |
| Osimertinib as a perpetrator | BCRP sensitive substrate (rosuvastatin observed)a | 1.35 (1.15 to 1.57) | 1.72 (1.46 to 2.03) |
| BCRP sensitive substrate (rosuvastatin predicted)b | 1.021 (1.020 to 1.023) | 1.060 (1.056 to 1.065) | |
%CV, percentage of coefficient of variation; AUCss, area under the curve at steady‐state; BCRP, breast cancer resistance protein; CI, confidence interval; Cmax, peak plasma concentration; Css, curve at steady‐state; CYP, cytochrome P450; DDI, drug‐drug interaction; PBPK, physiologically based pharmacokinetic.
Observed clinical DDI data with 90% CI.
Predicted DDI data with 95% CI.
The use of experimental fraction unbound in microsomal incubation, (fumic) = 0.0261, as a surrogate of fuinc at induction interaction to obtain free IndC50, showed a significant auto‐induction upon multiple dosing (lower steady‐state AUC and Cmax of osimertinib). Based on the sensitivity analysis results, fuinc = 1 recovered monotherapy multiple dosing data well and, thus, gave the final PBPK model for performing simulations as a victim and perpetrator drug.
PBPK model verification and application
The simulated profiles for the change in AUC from the PBPK simulations investigating the effect of rifampicin and itraconazole on the exposure of osimertinib are shown in Figure 2 and Table 2. The effect of osimertinib on simvastatin and rosuvastatin exposure is depicted in Figure 3 and shown in Table 2. Supplementary Figure S3 shows the predicted PK profiles of victim drugs overlaid with observed interaction data. Simulated PK profiles after oral administration of multiple doses of itraconazole, OH‐itraconazole, and rifampicin were consistent with the observed literature data from similar clinical studies, and would be sufficient to elicit DDI.17, 18, 19 The simulated PK profiles of osimertinib with and without co‐administration of rifampicin and itraconazole were in agreement with the observed data (within 1.5‐fold of observed data from the itraconazole study and within 1.7‐fold of observed data of rifampicin).17, 18, 19 After the verification of the osimertinib PBPK model, it was also applied to predict potential DDI with moderate and weak CYP3A inducers, efavirenz 600 mg or dexamethasone 8 mg once daily. The model predicted that osimertinib AUC would decrease by 42% when the induction effect by efavirenz on both hepatic and gut CYP3A metabolism was considered, but no induction effect was predicted by dexamethasone, as shown in Table 2. The simulated DDI are summarized as a forest plot in Figure 4.
Figure 2.
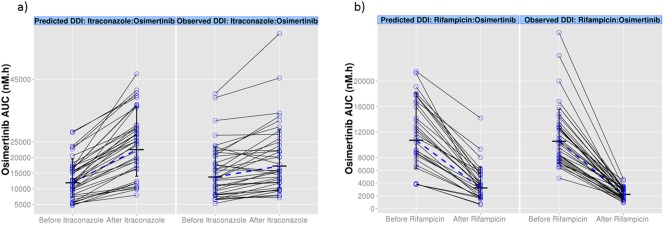
Simulated exposure (area under the curve (AUC)) change for (a) osimertinib (80 mg) in the presence of itraconazole (200 mg once daily) and (b) osimertinib (80 mg) in the presence of rifampicin (600 mg once daily). Values and the dashed line represent the geometric mean of osimertinib AUC for the observed (clinical trial data) and predicted (simulated data). Clinical trial data for the observed osimertinib AUC values with itraconazole and rifampicin were from studies D5160C00012 and D5160C00013, respectively. DDI, drug‐drug interaction.
Figure 3.
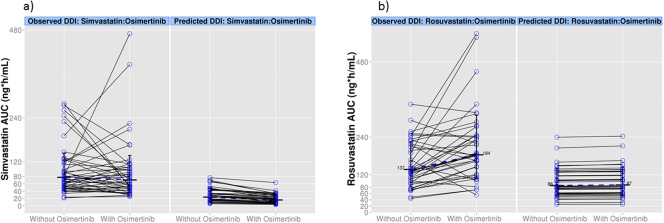
Simulated exposure (area under the curve (AUC)) change for (a) simvastatin (40 mg) or (b) rosuvastatin (20 mg) when co‐administered with osimertinib once daily. Values and the dashed line represent the geometric mean of simvastatin and rosuvastatin AUC for the observed (clinical trial data) and predicted (simulated data). Clinical trial data for the observed simvastatin and rosuvastatin AUC values with osimertinib co‐dosing were from studies D5160C00014 and D5160C00019, respectively. DDI, drug‐drug interaction.
Figure 4.
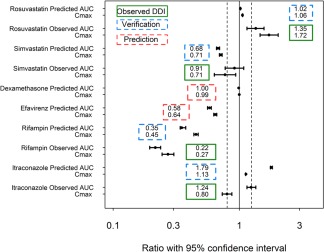
Summary of all drug‐drug interaction (DDI) predictions using osimertinib physiologically based pharmacokinetic model (geometric mean area under the curve (AUC) and peak plasma concentration (Cmax) ratios with 95% confidence interval (CI)). The dashed lines represent the (80% and 125%) interval. Verification and prediction are both model‐based simulations, and also indicate cases in which the actual clinical DDI studies were conducted (verification) or were not (prediction). Observed clinical DDI data with 90% CI; whereas predicted DDI data with 95% CI.
PBPK model application
Prospective simulations suggest that no dose adjustment is required when co‐dosed with moderate and weak CYP3A inducers (Table 2). Table 3 shows the simulation results for potential dose adjustment of osimertinib when co‐dosed with a strong CYP3A inducer and suggest that an osimertinib dose of 160 mg with strong inducer would result in a similar exposure as that of 80 mg osimertinib alone.
Table 3.
PBPK model driven potential osimertinib dose adjustment with known CYP3A inducers
| Osimertinib dose | PK parameter, summary statistic | Observed clinical data |
PBPK model predicted (n = 100) |
Geo.Mean exposure ratio (with inducer vs. 80 mg no inducer) |
|---|---|---|---|---|
| Multiple dose of 80 mg osimertinib without inducer |
AUCss (nM*h) Geo.mean (range) |
10,360 (4,730 to 22,300) | 11,076 (2,883 to 36,260) | 1.07 |
|
Css,max (nM) Geo.mean (range) |
545 (258 to 1,220) | 581 (205 to 1,607) | 1.07 | |
| 160 mg osimertinib dose with rifampicin |
AUCss (nM*h) Geo.mean (range) |
– | 8518 (2,131 to 21,844) | 0.77 |
|
Css,max (nM) Geo.Mean (range) |
– | 557 (190 to 1264) | 0.96 |
AUCss, area under the curve; Css, steady‐state concentration; CYP, cytochrome P450; PBPK, physiologically based pharmacokinetic; PK, pharmacokinetic.
DISCUSSION
A PBPK model was developed based on drug, system‐specific parameters, and study‐specific information, subsequently verified using available DDI clinical data, and used for prospective predictions of osimertinib‐drug combinations for which no clinical data are available. Osimertinib is metabolized by multiple CYP isozymes with the relative contribution being the highest for CYP3A (54%). In addition, a direct conjugation with glutathione, as a minor metabolic pathway, was observed in human hepatocytes. In vitro studies suggested osimertinib to be a weak CYP3A inducer, a weak reversible inhibitor of CYP3A4, and the BCRP transporter. In addition, in vitro studies show osimertinib to be a substrate for the human drug efflux transporter P‐glycoprotein (P‐gp) and BCRP. However, the efflux processes are likely to be saturated at therapeutic doses and, hence, unlikely to be of clinical significance.
Four clinical DDI studies in oncology patients were conducted to understand the clinical implications of osimertinib being both a victim and perpetrator drug (Supplementary Figure S2). Administration of osimertinib orally once daily resulted in approximately threefold accumulation (predictable from its long half‐life) with steady‐state exposures achieved after 15 continuous days of dosing. Hence, the clinical DDI studies and PBPK simulations, where appropriate, were conducted following a multiple dose of osimertinib to understand the maximal impact of osimertinib exposure on co‐dosed compounds, as shown in Supplementary Figure S2.
The osimertinib PBPK model has been robustly defined using a mechanistic approach and the model predicts the in vivo plasma concentration profile for osimertinib tablet formulation dosed alone within 1.1‐fold of the observed monotherapy clinical data (Table 2). Thus, the developed PBPK model seems to be appropriate for the assessments of the magnitude of drug interaction for osimertinib and CYP3A‐mediated DDI. The peak plasma concentrations of osimertinib were achieved with a median (range) Tmax of 6 (3–24) hours.3 This slow absorption leading to a prolonged Tmax was captured well using an estimated Peff,human of 0.19 × 10−4 cm/s value.
Verification of PBPK models with fm,CYP optimization is an important step to justify dose recommendations or substitute DDI studies. Both in vitro phenotyping data and clinical DDI data can be used to assess the robustness of the fm value of the victim drug. In vitro phenotyping data of osimertinib suggested fm,CYP3A of 0.54. Use of in vitro fm,CYP3A (0.54) in the osimertinib PBPK model estimated AUC increase with itraconazole of 1.79‐fold, compared to the observed ratio of 1.24‐fold. The estimated AUC decrease with rifampicin was 0.36‐fold, compared to the observed ratio of 0.22‐fold. The ratio of predicted AUC or Cmax ratio to observed ratio falls within 1.7‐fold, suggesting reasonable concordance between model‐predicted and observed clinical DDI effects. The slight overprediction of DDI with itraconazole and underprediction with rifampicin could be due to a number of factors, which is not limited to:
Simulations utilized itraconazole and rifampicin models provided by Simcyp software platform, which did not include P‐gp inhibition by itraconazole or P‐gp induction potential by rifampicin. In the study of Greiner et al.,20 a 3.5‐fold increase in in vivo P‐gp expression was observed following rifampicin (600 mg daily for 10 days). Due to lack of experimentally determined P‐gp kinetic parameters, the osimertinib PBPK model does not account for the role of P‐gp on osimertinib exposure or P‐gp interaction by rifampicin in the DDI model. Thus, it is possible that underprediction of rifampicin DDI with osimertinib may be related to P‐gp induction by rifampicin, which is not accounted for in the model.21 Similar disagreement among the strong CYP inhibition and induction study results has been reported recently for two anticancer drugs, namely ixazomib22 and tivozanib.23 This apparent discordance between the itraconazole and rifampicin study could be due to rifampicin's pleiotropic mechanism of induction of multiple drug‐metabolizing enzymes and transporters.22 This mechanism could lead to clinically significant reductions in the exposure of osimertinib when co‐dosed with rifampicin, as osimertinib has potential to be metabolized by multiple CYP enzymes, such that the effects of strong CYP3A inhibition are minimal while the effect of induction are significant, as seen in our study.
The oncology population may represent highly variable expressions of CYP3A4 between individuals, which is not adequately captured in the Cheeti et al.,6 population. Recent data24, 25 showed that pooled meta‐analyses could be a more robust way of understanding the drug disease CYP interaction rather than comparing the AUC values from individual study data.
Covalent binding of osimertinib was observed in in vitro systems, such as cryopreserved human hepatocytes, plasma, and serum albumin. Human ADME studies suggested substantial fraction of excretion in feces is of osimertinib‐related material bound to proteins, which could contribute to the clearance of osimertinib. Thus, recombinant CYP studies may have overestimated the contribution of CYP3A metabolism to the clearance of osimertinib in the clinic. Hence, osimertinib may be less susceptible to CYP‐mediated DDI as seen in the clinic with itraconazole DDI study (AUC ratio of 1.24‐fold).
Clinical study D5160C00014 was conducted with a known CYP3A substrate simvastatin. Simvastatin DDI simulations using the PBPK model suggested a net induction of CYP3A (predicted AUC ratio 0.68 vs. observed clinical AUC ratio 0.92), which was within 1.4‐fold of observed data. This slight discrepancy could be due to lack of robust Indmax and IndC50 estimates for osimertinib.
Clinical study D5160C00019 was conducted with a known BCRP substrate rosuvastatin. This study showed an increase in rosuvastatin Cmax ratio by 1.72‐fold, and an AUC change of 1.35‐fold. The PBPK dynamic simulation results did not predict the degree of the interaction that was observed (predicted AUC ratio was 1.02 vs. observed 1.35‐fold) for rosuvastatin. The reason for this underprediction in Simcyp needs to be further evaluated. Nevertheless, the challenge of recovering transporter‐mediated interactions are well noted and it has been observed that the recovery of rosuvastatin interaction is complex with needing a fit‐for‐purpose model either by lowering BCRP Ki values by ∼10‐fold26 or optimizing Fu,gut value. Increasing the enterocyte concentration of osimertinib using higher Fu,gut resulted in good recovery of rosuvastatin Cmax DDI ratio that was observed (Supplementary Figure S4). However, this increase in Fu,gut value had a negative impact on the itraconazole study with AUC ratio overpredicted by fivefold than observed.
A clinical study to investigate the effect of a moderate or weak CYP3A inducer, such as efavirenz or dexamethasone, on exposure to osimertinib has not been conducted. The PBPK model‐based simulations with a moderate inducer (efavirenz) and 80 mg osimertinib, indicated osimertinib geometric mean Cmax ratio decreased to 0.58‐fold; (95% confidence interval (CI) = 0.56–0.60) and geometric mean AUC ratio decreased to 0.64‐fold (95% CI = 0.63–0.66). Based on osimertinib exposure‐response analysis, a less than twofold change in exposure of osimertinib is unlikely to alter its benefit risk ratio.11 In a similar multiple‐dose drug‐interaction simulation with weak inducer (dexamethasone) and 80 mg osimertinib, there was no effect on the exposure of osimertinib. This was confirmed in a retrospective evaluation with clinical data of patients who had received dexamethasone to treat nausea and vomiting when co‐administered with osimertinib in study D5160C00014. In that evaluation, osimertinib observed area under the curve at steady‐state (AUCss) was 11,924 nM.h when co‐administered with a 4 mg q.d. or 8 mg q.d. dose of dexamethasone (n = 3) compared to an AUCss of 11,530 nM.h in other patients (n = 44) who were not dosed with dexamethasone in the same study (data on file). Thus, the clinical data (albeit in a limited number of patients) did not suggest any change in exposure of osimertinib with and without dexamethasone administration consistent with the PBPK predictions. As the primary objective was to recover the rifampicin value to recommend dose adjustments, the PBPK model was more closely fit toward that study while still maintaining the appropriate prediction for other DDI studies. Sensitivity analyses were conducted to depict the worst‐case DDI risks with CYP3A inducers, as shown in Supplementary Figure S5.
The PBPK predictions of simvastatin alone (AUC) were threefold to fourfold lower than the observed values in the study D5160C00014 (Figure 3 a). One of the reasons for this difference could be that the simvastatin compound file supplied within Simcyp version 14 was developed using observed clinical data in healthy subjects and not using oncology patients. Hence, some of the demographic and physiological aspects of cancer are potentially not taken into account in the simvastatin compound file. Moreover, altered CYP‐expression levels in patients with cancer24 could also be contributing to this difference. In our analysis, although the oncology population based on Cheeti et al.6 was used, it does not take into account the altered CYP expression levels. Use of recent virtual oncology populations (Schwenger et al.25), accounting for altered CYP‐expression levels in patients with cancer, resulted in similar DDI ratio (AUC ratio of 0.73) as that of the Cheeti et al.6 population (AUC ratio of 0.68). Although the simvastatin clearance was underpredicted in all oncology populations, the contribution of CYP3A4, the fraction that escaped gut metabolism, and the percentage of active enzyme in the gut and liver in the presence of inhibitor were similar to when healthy volunteer populations were used for DDI simulations. Based on the above, the intrasubject DDI ratio that is simulated using this simvastatin compound file seems to be reasonable. However, this lower simulated exposure compared to observed exposure might warrant further research.
Simulations for osimertinib as a victim drug were performed as a guide to potential dose adjustment of osimertinib in the presence of a strong CYP3A inducer, such that the exposure in the presence of inducer can be increased to be closer to that without inducer. Table 3 shows the predicted exposure and osimertinib dose adjustments in combination with strong CYP3A inducers. When the dose of osimertinib is increased to 160 mg when co‐administered with rifampicin, it is predicted to give a range in exposure that closely matches the observed and PBPK predicted monotherapy data for AUC and Cmax at 80 mg. This falls within twofold of the observed and predicted 80 mg osimertinib monotherapy data.
This PBPK model‐based dose recommendation complemented the clinical DDI study to define an appropriate dose when osimertinib needs to be combined with strong CYP3A inducers (i.e., increase the osimertinib dose to 160 mg when co‐administering with a strong CYP3A4 inducer). No dose adjustments are required when osimertinib is used with moderate and/or weak CYP3A inducers.2 As has been described previously, the AURA phase I study27 demonstrated clinical activity at all doses studied (20–240 mg), with no maximum tolerated dose was reached at the 240 mg dose. As such, based on the exposure response analysis, mean exposure within twofold (lower than that of a 160 mg dose or higher than that of a 40 mg dose) would require no dose adjustments and do not alter the benefit:risk of osimertinib.7
CONCLUSION
The PBPK modeling and simulation approach predicted metabolic interactions appropriately but could not recover the rosuvastatin DDI data. The verified PBPK model was utilized to forecast the DDI risks where no clinical data were available. Simulations suggest that no dose adjustment is required when co‐dosed with moderate and weak CYP3A inducers. With strong CYP3A inducers, in which co‐administration is not avoidable, an increase of the osimertinib dose to 160 mg is likely to provide exposure within the range of the 80 mg dose taken alone.2 Further work should be done to understand the underprediction of rosuvastatin DDI.
Source of Funding
This study was sponsored by AstraZeneca.
Conflict of Interest
Venkatesh Pilla Reddy, Pradeep Sharma, and Karthick Vishwanathan are employees and/or shareholders of AstraZeneca. Michael Walker and Peter Ballard were employees of AstraZeneca at the time of the study and hold AstraZeneca shares. The clinical DDI studies reported in the article were funded by AstraZeneca, Cambridge, UK, the manufacturer of osimertinib. The authors indicate no other conflicts of interest.
Author Contributions
V.P.R., M.W, P.S., P.B., and K.V. wrote the manuscript. V.P.R., P.B., and K.V. designed the research. V.P.R., M.W, P.S., P.B., and K.V. performed the research. V.P.R., M.W., and K.V. analyzed the data.
Supporting information
Supplementary Methods: Details of model input parameters, parameter estimation and retrograde approach to recover metabolic intrinsic clearance
Supplementary Figure S1 Osimertinib elimination pathway in humans and PBPK model assumptions in terms of clearance and relative metabolic (CYP‐mediated) contribution.
Supplementary Figure S2 Clinical DDI study designs of osimertinib with itraconazole (NCT02157883): DDI study with rifampicin (NCT02197247); DDI study with simvastatin (NCT02197234); DDI study with rosuvastatin (NCT02317016)
Supplementary Figure S3 Simulated plasma drug concentration‐time profiles compared to the observed Clinical DDI data (a) with Itraconazole 200mg, (b) Rifampin 600mg, (c) Simvastatin 40mg, (d) Rosuvastatin 20mg. Observed values are dots. Dashed lines are 5th and 95th centile simulated. Solid green line is mean profile with interaction.
Supplementary Figure S4 Sensitivity analyses of osimertinib Fu, gut parameter ranging from 0.00132 to 1. AUC ratio or Cmax ratio (for Rosuvastatin). Results suggests that Fu,gut maybe an important parameter and it requires a more fit for purpose Fu,gut value to recover DDI data from various studies rather than a fixed value
Supplementary Figure S5 Sensitivity analyses with Fu,gut of osimertinib and it's influence on the AUC ratio of osimertinib when dosed with CYP3A inducers. Results suggests as a worst‐case scenario using Fu,gut of 1 for osimertinib resulted in a moderate induction effect by efavirenz and no effect with dexamethasone.
Supplementary Table S1 Interpretation of osimertinib transporter inhibition data using basic static model
Supplementary Table S2 Comparison of the key physiological differences between oncology population based on Cheeti et al.,7 compared to healthy volunteer population of the Simcyp simulator
Supplementary Table S3 Table showing the CYP3A4 Induction EC50 and Emax values from three human hepatocytes and an input parameters used in the simulator after calibration
Supplementary Table S4 Simcyp input parameters for CYP3A/P‐gp modulators and substrates
Supplementary Table S5 Trial design for osimertinib DDI simulations with rifampicin, efavirenz dexamethasone, itraconazole, simvastatin and rosuvastatin
Supplementary Table S6 Dexamethasone PBPK model development and verification with clinical DDI data
Acknowledgments
The authors thank Owen R. Jones, Helen Rollison, Barry Jones, Khanh Bui, Paul Dickinson, and Martin Wild of AstraZeneca, UK, for their scientific feedback in carrying out the study procedures described.
References
- 1. Cross, D.A. et al AZD9291, an irreversible EGFR TKI, overcomes T790M‐mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4, 1046–1061 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.US FDA, AstraZeneca Pharmaceuticals LP TAGRISSOTM (osimertinib) tablet: highlights of prescribing information. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208065s007lbl.pdf> (2017). January 29, 2018.
- 3.TAGRISSO (osimertinib) ‐ EMA ‐ Europa.eu. <http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004124/human_med_001961.jsp&mid=WC0b01ac058001d124>. February 11, 2017.
- 4. Planchard, D. et al Osimertinib Western and Asian clinical pharmacokinetics in patients and healthy volunteers: implications for formulation, dose, and dosing frequency in pivotal clinical studies. Cancer Chemother. Pharmacol. 77, 767–776 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Dickinson, P.A. et al Metabolic disposition of osimertinib in rats, dogs, and humans: insights into a drug designed to bind covalently to a cysteine residue of epidermal growth factor receptor. Drug Metab. Dispos. 44, 1201–1212 (2016). [DOI] [PubMed] [Google Scholar]
- 6. Cheeti, S. , Budha, N.R. , Rajan, S. , Dresser, M.J. & Jin, J.Y. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm. Drug Dispos. 34, 141–154 (2013). [DOI] [PubMed] [Google Scholar]
- 7. Brown, K. et al Population pharmacokinetics and exposure‐response of osimertinib in patients with non‐small cell lung cancer. Br . J. Clin. Pharmacol. 83, 1216–1226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jamei, M. , Marciniak, S. , Feng, K. , Barnett, A. , Tucker, G. & Rostami‐Hodjegan, A. The Simcyp population‐based ADME simulator. Expert Opin. Drug Metab. Toxicol. 5, 211–223 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Rodgers, T. & Rowland, M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm. Res. 24, 918–933 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Guidance for Industry: Drug Interaction Studies ‐ Study Design, Data Analysis, Implications for Dosing, and Labelling Recommendations. <https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf>. February 2012.
- 11.Guideline on the Investigation of Drug Interactions (CPMP/EWP/560/95/Rev. 1 Corr.*) <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf>. 2012.
- 12. Vishwanathan, K. et al Effect of itraconazole or rifampicin on the pharmacokinetics of osimertinib. Br. J. Clin. Pharmacol. (2018). [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harvey, R.D. et al Effect of multiple‐dose osimertinib (AZD9291) on the pharmacokinetics (PK) of simvastatin and rosuvastatin. J. Clin. Oncol. http://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.e14098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marbury, T.C. et al Pharmacokinetics of oral dexamethasone and midazolam when administered with single‐dose intravenous 150 mg fosaprepitant in healthy adult subjects. J. Clin. Pharmacol. 51, 1712–1720 (2011). [DOI] [PubMed] [Google Scholar]
- 15. Johnson, B et al Impact of casopitant, a novel NK‐1 antagonist, on the pharmacokinetics of ondansetron and dexamethasone. Support. Care Cancer 17, 1177–1185 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Motta, P. , Pons, N. , Pagliarusco, S. , Pellegatti, M. & Bonomo, F. Casopitant: in vitro data and SimCyp simulation to predict in vivo metabolic interactions involving cytochrome P450 3A4. Drug Metab. Dispos. 39, 363–372 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Barone, J.A. et al Food interaction and steady‐state pharmacokinetics of itraconazole capsules in healthy male volunteers. Antimicrob. Agents Chemother. 37, 778–784 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahonen, J. , Olkkola, K.T. & Neuvonen, P.J. Effect of itraconazole and terbinafine on the pharmacokinetics and pharmacodynamics of midazolam in healthy volunteers. Br. J. Clin. Pharmacol. 40, 270–272 (1995). [PMC free article] [PubMed] [Google Scholar]
- 19. Prueksaritanont, T. et al Pitavastatin is a more sensitive and selective organic anion‐transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 78, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greiner, B. et al The role of intestinal P‐glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest. 104, 147–153 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wagner, C. , Pan, Y. , Hsu, V. , Sinha, V. & Zhao, P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the US FDA. Clin. Pharmacokinet. 55, 475–483 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Gupta, N. et al Effects of strong CYP3A inhibition and induction on the pharmacokinetics of ixazomib, an oral proteasome inhibitor: results of drug‐drug interaction studies in patients with advanced solid tumors or lymphoma and a physiologically based pharmacokinetic analysis. J. Clin. Pharmacol. 58, 180–192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cotreau, M.M. et al Effects of ketoconazole or rifampin on the pharmacokinetics of tivozanib hydrochloride, a vascular endothelial growth factor receptor tyrosine kinase inhibitor. Clin. Pharmacol. Drug Dev. 4, 137–142 (2015). [DOI] [PubMed] [Google Scholar]
- 24. Coutant, D.E. et al Understanding disease‐drug interactions in cancer patients: implications for dosing within the therapeutic window. Clin. Pharmacol. Ther. 98, 76–86 (2015). [DOI] [PubMed] [Google Scholar]
- 25. Schwenger, E. et al Harnessing meta‐analysis to refine an oncology patient population for physiology‐based pharmacokinetic modeling of drugs. Clin. Pharmacol. Ther. 103, 271–280 (2018). [DOI] [PubMed] [Google Scholar]
- 26. Jamei, M. et al A mechanistic framework for in vitro‐in vivo extrapolation of liver membrane transporters: prediction of drug‐drug interaction between rosuvastatin and cyclosporine. Clin. Pharmacokinet. 53, 73–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jänne, P.A. et al AZD9291 in EGFR inhibitor‐resistant non‐small‐cell lung cancer. N. Engl. J. Med. 30, 1689–1699 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods: Details of model input parameters, parameter estimation and retrograde approach to recover metabolic intrinsic clearance
Supplementary Figure S1 Osimertinib elimination pathway in humans and PBPK model assumptions in terms of clearance and relative metabolic (CYP‐mediated) contribution.
Supplementary Figure S2 Clinical DDI study designs of osimertinib with itraconazole (NCT02157883): DDI study with rifampicin (NCT02197247); DDI study with simvastatin (NCT02197234); DDI study with rosuvastatin (NCT02317016)
Supplementary Figure S3 Simulated plasma drug concentration‐time profiles compared to the observed Clinical DDI data (a) with Itraconazole 200mg, (b) Rifampin 600mg, (c) Simvastatin 40mg, (d) Rosuvastatin 20mg. Observed values are dots. Dashed lines are 5th and 95th centile simulated. Solid green line is mean profile with interaction.
Supplementary Figure S4 Sensitivity analyses of osimertinib Fu, gut parameter ranging from 0.00132 to 1. AUC ratio or Cmax ratio (for Rosuvastatin). Results suggests that Fu,gut maybe an important parameter and it requires a more fit for purpose Fu,gut value to recover DDI data from various studies rather than a fixed value
Supplementary Figure S5 Sensitivity analyses with Fu,gut of osimertinib and it's influence on the AUC ratio of osimertinib when dosed with CYP3A inducers. Results suggests as a worst‐case scenario using Fu,gut of 1 for osimertinib resulted in a moderate induction effect by efavirenz and no effect with dexamethasone.
Supplementary Table S1 Interpretation of osimertinib transporter inhibition data using basic static model
Supplementary Table S2 Comparison of the key physiological differences between oncology population based on Cheeti et al.,7 compared to healthy volunteer population of the Simcyp simulator
Supplementary Table S3 Table showing the CYP3A4 Induction EC50 and Emax values from three human hepatocytes and an input parameters used in the simulator after calibration
Supplementary Table S4 Simcyp input parameters for CYP3A/P‐gp modulators and substrates
Supplementary Table S5 Trial design for osimertinib DDI simulations with rifampicin, efavirenz dexamethasone, itraconazole, simvastatin and rosuvastatin
Supplementary Table S6 Dexamethasone PBPK model development and verification with clinical DDI data