

Abstract
Hydroxyl radical footprinting (HRF) is a nonspecific protein footprinting method that has been increasingly used in recent years to analyze protein structure. The method oxidatively modifies solvent accessible sites in proteins, which changes upon alterations in the protein, such as ligand binding or a change in conformation. For HRF to provide accurate structural information, the method must probe the native structure of proteins. This requires careful experimental controls since an abundance of oxidative modifications can induce protein unfolding. Fast photochemical oxidation of proteins (FPOP) is a HRF method that generates hydroxyl radicals via photo‐dissociation of hydrogen peroxide using an excimer laser. The addition of a radical scavenger to the FPOP reaction reduces the lifetime of the radical, limiting the levels of protein oxidation. A direct assay is needed to ensure FPOP is probing the native conformation of the protein. Here, we report using enzymatic activity as a direct assay to validate that FPOP is probing the native structure of proteins. By measuring the catalytic activity of lysozyme and invertase after FPOP modification, we demonstrate that FPOP does not induce protein unfolding.
Keywords: fast photochemical oxidation of protein (FPOP), hydroxyl radical footprinting (HRF), oxidative modifications, protein footprinting, native protein structure, mass spectrometry
Introduction
A protein must fold to a native state sampled from an ensemble of states in order to perform its designated function. Misfolding of proteins has been implicated in numerous diseases that lead to dysfunction, hence, it is important to study protein structure to understand the connection between folding and function. High resolution methods such as X‐ray crystallography and NMR have provided a wealth of knowledge on protein structure. These methods are not accessible for the study of all proteins making it necessary to develop other tools for structural analysis. Methods such as cryo‐electron microscopy and fluorescence‐based methods are being increasingly used to interrogate protein structure. Although these methods are not as high resolution as crystallography and NMR, they still provide significant information on the native conformation of proteins.
In recent years, hydroxyl radical footprinting (HRF) coupled with mass spectrometry has emerged as a new tool for analyzing protein structure. In HRF, the comparison of the oxidative modification of solvent accessible residues between two protein states is used to provide information about protein–protein interactions,1, 2 protein–ligand interactions,3, 4 and protein conformational changes.5, 6 Mass spectrometry (MS) is used to identify the modified amino acids and quantitate the extent of oxidation. HRF methods are not limited in the size of proteins they can study, it requires a small amount of material (a few micrograms), and it offers relatively fast analysis in comparison to crystallography and NMR. Frequently, solvent accessibility information from x‐ray crystallography or NMR is combined with the differential HRF experiments to provide more detailed information.3, 7, 8, 9, 10
The efficacy of HRF in providing information on protein structure relies on the method analyzing the native structure of the protein. Since studies have shown that oxidation can induce protein unfolding,11, 12 many HRF methods limit the number of oxidations per protein to minimize over‐oxidation.13 Although there are multiple methods for generating hydroxyl radicals, including synchrotron radiolysis of water,14 laser photolysis of hydrogen peroxide,15, 16 and electrochemistry,17, 18 limiting the total oxidation level to less than 30%, regardless of the method of radical generation, helps maintain structural integrity.19, 20 It has been demonstrated that for synchrotron radiation, exposure times of 50 msec or less are sufficient to oxidatively modify proteins without inducing structural changes to the protein.20 Ion mobility mass spectrometry has also been used as a direct assay to show that synchrotron‐based HRF does not alter the native state of the protein.21 Other experimental assays to validate that HRF methods are probing proteins in their native conformation include measuring the kinetics of oxidation for peptides9, 22, 23 and proteins,13, 24 and analyzing protein structure after modification using circular dichroism13, 16, 22, 25 and NMR.26 These studies tested HRF methods where radicals were generated by synchrotron radiation, γ‐irradiation, a pulsed electron beam, and a nanosecond flash laser.
FPOP is an HRF method that generates hydroxyl radicals via photo‐dissociation of hydrogen peroxide by an excimer laser leading to multiple oxidations per protein. The large number of oxidations for FPOP could possibly lead to over oxidation and protein unfolding. To limit oxidation‐induced unfolding, several experimental controls are utilized. First, a radical scavenger is added to the reaction to reduce the lifetime of the radical to 1 μs. Second, the protein sample is under constant flow, and an exclusion fraction is used to ensure a bolus of sample receives only one pulse of the laser.15 Lastly, samples are collected in a quench solution containing catalase and methionine to quench excess hydrogen peroxide and hydroxyl radicals, respectively. Kinetic calculations15 and dosimetry experiments27 have shown, with the addition of glutamine as a radical scavenger, the lifetime of the hydroxyl radicals is reduced, preventing over‐oxidation of the protein. This ensures the native structure is being probed. However, a recent study by Vahidi and Konermann28 suggest protein oxidation using FPOP could last up to tens of milliseconds due to metastable secondary radicals generated from the glutamine scavenger. Due to this discovery, further validation that FPOP probes the native structure is needed. Gau et al.29 performed the first evaluation of whether FPOP probes the native structure of proteins by confirming that the distribution of the FPOP modifications fit a Poisson, which is indicative of a single conformation modification model. Further validation, using an experimental assay that characterizes the sample after FPOP in a direct manner, would provide evidence that FPOP probes the native conformation of proteins.
Here, we report the use of the catalytic activity of enzymes to monitor whether FPOP modification unfolds proteins. Catalytic activity is an adequate benchmark owing to its sensitivity to protein unfolding which leads to significant activity loss.30, 31 Using two different enzymes, lysozyme and invertase, the catalytic activity was used as a measure to determine retained native structure post FPOP modification. Lysozyme and invertase were used for this study due to the extensive information available for these enzymes and the ease of measuring the enzymatic activity. They contrast each other in structural make‐up and size. Lysozyme is majority alpha‐helix and a relatively small enzyme while invertase has majority beta‐sheets and is much larger. By comparing catalytic activity of seven different conditions, chosen to specifically interrogate the role of the glutamine scavenger in limiting oxidation‐induced conformational change, and localizing the residues with FPOP modifications using bottom‐up proteomics, we determined FPOP does not show a significant structural change and retains the native structure required for efficient catalytic activity.
Results and Discussion
The enzymatic activity of lysozyme is preserved after FPOP
As a first step toward using catalytic activity as a means to elucidate whether FPOP probes the native conformation of proteins, we considered various experimental conditions that would best provide insight into the role of oxidation‐induced protein unfolding. These conditions should take into account the role of the glutamine scavenger in limiting over oxidation as well as how the catalytic activity of the protein would be effected if the protein was unfolded. Seven conditions that provide a comprehensive representation of the effect of oxidation were chosen for this study. These conditions are (i) folded; (ii) unfolded; (iii) typical FPOP sample; (iv) typical FPOP control; (v) over‐oxidized sample; (vi) over‐oxidized control; and (vii) quench condition. The enzyme was in its native conformation and not subjected to FPOP for the folded condition representing a positive control for enzymatic activity. To demonstrate the effect of protein unfolding on catalytic activity, the enzyme was chemically unfolded using 8 M urea and 100 mM DTT for the unfolded condition. For the typical FPOP sample condition, the enzyme was oxidatively modified using standard FPOP conditions (as described in the Materials and Methods) including glutamine as a radical scavenger. In the typical FPOP control, the enzyme was mixed with hydrogen peroxide at concentrations similar to the typical FPOP sample but not exposed to laser irradiation. For the over‐oxidized sample, the enzyme was oxidatively modified in the absence of the glutamine scavenger providing information on the role of the scavenger in limiting oxidation. The typical FPOP and over‐oxidized controls do not get exposed to laser irradiation allowing background oxidation to be subtracted from the laser samples. Finally, the quench condition did not contain hydrogen peroxide and was not exposed to laser irradiation but did include glutamine and the quench reagents, methionine and dimethylthiourea (DMTU). The quench reagents were added to all of the FPOP samples to stop the FPOP reaction. The presence of these reagents and glutamine in the typical FPOP samples might have an effect on the enzymatic assay which this condition takes into account.
The catalytic activity of lysozyme was tested immediately after FPOP oxidation. The rate of enzymatic activity, measured as a change in absorbance over time in the presence of substrate, in the seven different conditions are shown in Figure 1(A). First, protein concentration was used to normalize the enzymatic activity, and then activity was further normalized to the folded sample which was set at 100% retained activity [Fig. 1(B)]. In parallel to the enzymatic assay, far‐UV circular dichroism25 spectra were collected for the folded and unfolded samples to assess protein structure (Supporting Information Fig. S1). Using the structure predictive software K2D2,32 the folded samples consisted of 84% alpha‐helical and 0.6% beta strand secondary structure compared to 15% alpha‐helical and 7.7% beta strand secondary structure in the unfolded samples. Unfolded samples retained <1% of its catalytic activity due to loss of secondary structure [Fig. 1(B), Supporting Information Table S1]. The typical FPOP sample condition retained 47%±0.2 activity which was eightfold higher than the over‐oxidized sample condition which only retained 6% ± 0.02 activity. The typical FPOP control, over‐oxidized control, and quench condition samples retained 106 ± 1.2, 49 ± 1.7, and 87 ± 2.5% activity, respectively.
Figure 1.
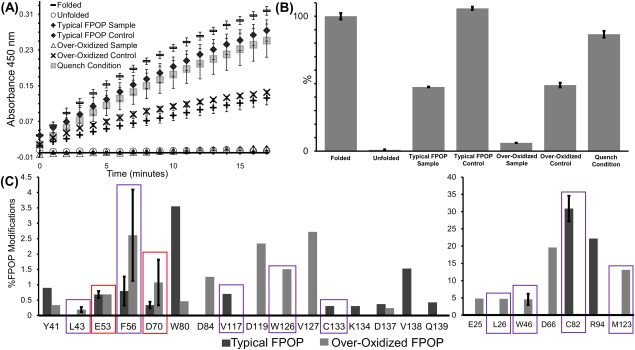
(A) Absorbance at 450 nm was tested every minute for 15 minutes to determine the activity of lysozyme. The highest activity was in the folded lysozyme sample followed by typcial FPOP control, quench condition, over‐oxidized control, typical FPOP sample, over‐oxidized sample, and unfolded sample, respectively. (B) For each condition, the activity was normallized using their protein concentration and compared to the folded lysozyme, which was set to 100% activity. (C) Extent of FPOP modifications for lysozyme on the residue‐level for the typical FPOP and over‐oxidization condition. Red boxes highlight the residues involved with catalytic activity and purple boxes highlight the buried residues.
To further elucidate the effect of the glutamine scavenger on protein modification, we performed ESI‐MS analysis on the intact protein after FPOP. In both the absence and presence of glutamine, the unmodified protein was the most abundant followed by several oxidations that are multiples of +16 Da (Fig. 2). In the absence of the glutamine scavenger, lysozyme displays an abundance of the singly modified protein (∼95% of the unmodified protein) and a multitude of multiply oxidized proteins [Fig. 2(A)]. In comparison, when glutamine is present in the FPOP reaction, the singly modified protein comprises only 30% of the unmodified protein [Fig. 2(B)]. This is comparable to other HRF studies where appreciable protein structural damage was not observed when the singly modified protein was between 30 and 50% of the unmodified protein.19 In addition, in the typical FPOP sample, only eight multiply oxidized proteins are present, which is less than the eleven modified peaks observed in the condition lacking glutamine. The abundance of these eleven peaks are significantly higher than any of the modified species when glutamine is present. The excess of modifications in the absence of glutamine denotes an increased radical lifetime leading to over‐oxidation. This, in conjunction with the almost complete loss of catalytic activity after FPOP without glutamine present, indicates the high levels of oxidation leads to disruption of the native structure of the protein. The retention of some activity in the typical FPOP sample suggests lysozyme remains folded after FPOP.
Figure 2.
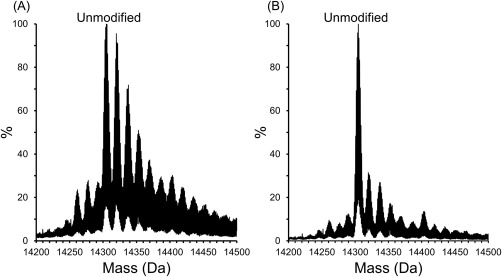
Intact MS analysis show multiple oxidation states of lysozyme after FPOP in the (A) over‐oxidized sample and (B) typical FPOP sample.
To ensure the retained activity in each condition is not solely due to the presence of unmodified protein in the sample, the peak areas of the intact protein spectra were calculated. For the typical FPOP sample, typical FPOP control, over‐oxidized sample, and over‐oxidized control, the unmodified protein makes up 40%, 60%, 20%, and 59% of the total area, respectively (Supporting Information Fig. S2). These areas do not correlate with the retained activity of any of the conditions indicating the modified protein is also contributing to the enzymatic activity.
Tandem MS reveals localized sites of oxidation
To investigate the effects of over oxidation on protein structure, FPOP modifications were localized using tandem MS after trypsin digestion, and the extent of FPOP modification at the residue level was calculated [Fig. 1(C)]. The residues modified in the typical FPOP and over‐oxidized conditions were mapped onto the crystal structures of lysozyme [Fig. 3(A,B)]. There were seven residues, Y41, E53, F56, D70, W80, C82, and D137 that were modified in both conditions. Six residues, C94, V117, C133, K134, V138, and Q139 were uniquely modified in the typical FPOP condition. An additional ten residues, E25, L26, L43, W46, D66, D84, D119, M123, W126, and V127 were uniquely modified in the over‐oxidized condition. The increased number of modifications in the over‐oxidized condition correlates well with the intact MS data and underscores the increase in modifications observed in the absence of a radical scavenger. Furthermore, based on the solvent accessible surface area (SASA) of each residue calculated from the x‐ray crystal structure (pdb ID: 4WMG), more buried residues (SASA ≤0.11) were modified in the over‐oxidized condition [Fig. 3(A,B)]. These buried residues include L26, L43, W46, W123, and M126 that were modified only in the over‐oxidized condition, V117 and C113 were modified in the typical FPOP condition, and F56 and C82 modified in both conditions. In the typical FPOP samples buried residue C82 is highly modified (>30%), however the other three buried residues F56, V117, and C133 all have less than 1% modifications [Fig.1(C)]. In contrast, three of the buried residues modified in the over‐oxidized condition, L26, W46, and M123, have greater than ∼5% oxidation. Residues L43, F56, C82, and W126 have oxidations of 0.2%, 2.6%, 0.4%, and 1.5%, respectively. The increased number of modified buried residues with a higher overall modification indicates the longer radical lifetime leads to conformational changes that expose these buried residues.
Figure 3.
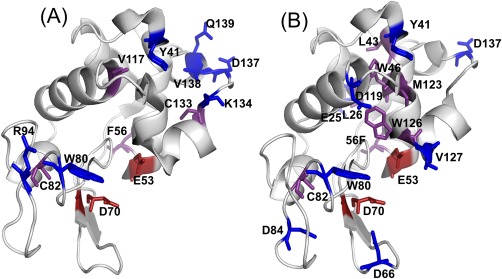
FPOP modifications from lysozyme mapped on a crystal structure (pdb ID: 4WMG) for the (A) typical FPOP condition and (B) over‐oxidized condition. The modified active site residues are highlighted in red, and the modified residues with a SASA value ≤0.11 are highlighted in purple.
The limited oxidation of buried residues in the typical FPOP condition compared to the over‐oxidized condition suggests the protein structure remains intact after FPOP in the presence of glutamine. However, the complete retention of activity in the typical FPOP control indicates that FPOP is contributing to the observed twofold loss in activity in the typical FPOP sample. The catalytic residues of lysozyme, E53 and D70, and a residue involved in substrate binding, W80, was oxidatively modified in the typical FPOP sample and over‐oxidized control. The oxidation of these residues could be contributing to the decrease in activity in the typical FPOP samples.
In a previous study by Lin et al.,33 carboiimide labeling was used to modify the carboxyl groups in lysozyme. Performing the labeling procedure in the presence of the substrate led to all carboxyl residues being modified with the exception of catalytic residues Glu53 and Asp70, which were protected from labeling by the substrate interaction, and a retention of 56.5% catalytic activity. In a sequential step with the same sample, the substrate was removed, and the labeling reaction was repeated. Both Glu53 and Asp70 were modified, and a complete loss of enzymatic activity was observed demonstrating modification at these residues leads to a loss of catalytic activity. In our study, a complete loss of activity was not observed when the catalytic residues were oxidatively modified. Under typical FPOP conditions, Glu53 and Asp70 were only modified at 0.7 and 0.3% [Fig. 1(C)] which may explain having only a twofold loss of catalytic activity. In comparison, Lin et al. observed 30 and 70% labeling for Glu35 and Asp70, respectively, leading to a complete loss of activity.33 The no laser control for the over‐oxidized condition also has a twofold loss of catalytic activity [Fig. 2(B)]. This can be attributed to the background oxidation of Asp70 (Supporting Information Table S2) further correlating the loss of catalytic activity with the oxidation of active site residues. For the typical FPOP control condition, which shows no loss of catalytic activity [Fig. 2(B)], neither of the active site residues were modified. Another consideration for the loss of enzyme activity is the improper substrate binding with lysozyme.
For proper substrate binding, a major shift in the main chain and side chains of residues 88 to 94 takes place owing to changes in the hydrogen bonding network.34, 35 In the absence of substrate, residues W80 and W81 form hydrogen bonds within this region. For substrate binding, W80 shifts to form a new hydrogen bond with the substrate. This shift along with other dielectric constant changes leads to the shifting of residues 88–94 toward the active‐site cleft.34 The typical FPOP sample showed 3.6% oxidation of W80 [Fig. 1(C)]. This oxidation would affect the hydrogen bonding network between this residue and the substrate, leading to lysozyme forming a weaker bond with the substrate. This further explains the loss of enzymatic activity observed in the typical FPOP sample. It is important to note that W80 is not modified in the typical FPOP control samples where minimal enzymatic activity was lost. Furthermore, a second residue in this important 88–94 region, C94, is modified in both the typical FPOP sample [Fig. 1(C)] and over‐oxidized control (Supporting Information Table S2) which may lead to other changes in the active‐site cleft. The retention of 47.5% catalytic activity after typical FPOP sample in comparison to an almost complete loss of activity for the chemically unfolded protein and over‐oxidized FPOP sample demonstrates that FPOP does not induce protein unfolding.
Invertase retains enzymatic activity after FPOP
To further verify FPOP does not alter the native conformation of proteins, a second enzyme, invertase, was tested. The same seven conditions as lysozyme were applied to invertase and studied by using an enzymatic assay. Far‐UV CD spectra collected for folded and unfolded invertase were collected (Supporting Information Fig. S3). The folded invertase samples consisted of 36% beta strands and 14% alpha‐helices compared to the unfolded samples with 29% beta stands and 8.2% alpha‐helices. After 24 hr incubation with 100 mM DTT and 8 M urea, the protein still retained a significant amount of secondary structure. This is evident in the enzymatic assay where the unfolded sample retained 45% ± 1.1 of its catalytic activity [Fig. 4(A), Supporting Information Table S3]. This is similar to the typical FPOP sample which retained 34% ± 2.8 activity. The over‐oxidized sample had a significant loss in activity with only 9% ± 1.3 retention. Both typical FPOP and over‐oxidized controls displayed a decrease in activity with 61 ± 4.2 and 38% ± 10.2 retention, respectively [Fig. 4(A), Supporting Information Table S3].
Figure 4.
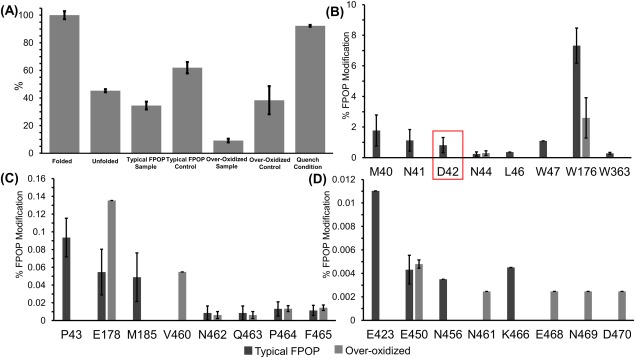
(A) Normalized catalytic activity of invertase with folded protein set to 100% activity. (B‐D) Extent of FPOP modifications for invertase on the residues level for typical FPOP condition and over‐oxidized condition.
The specific residues that were modified in the typical FPOP and over‐oxidized conditions were mapped onto the crystal structure of invertase [Fig. 5(A,B)]. The typical FPOP condition displayed 19 modified residues compared to 13 modified residues in the over‐oxidized condition [Fig. 4(B–D)], which contradicts the fact that the loss of catalytic activity in the over‐oxidized condition is caused by the increase in oxidations. The increase in oxidations in the absence of a radical scavenger may have led to protein unfolding and increasing exposure to the backbone leading to backbone cleavages, which would limit the number of oxidized residues identified. Previously, Aye et al. observed that oxidatively modifying unfolded ubiquitin led to backbone cleavages owing to the increased solvent accessibility of the backbone in the unfolded state.16
Figure 5.
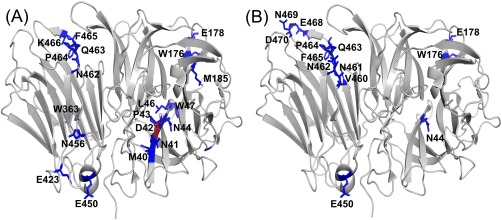
FPOP modifications from invertase mapped on a crystal structure (pdb ID: 4EQV) for the (A) typical FPOP condition and (B) over‐oxidized condition. The modified residue in the active site is highlighted in red.
An important residue in the catalytic activity of invertase, Asp42, is modified in the typical FPOP sample which would account for the loss of activity in that sample. A previous study by Reddy and Maley demonstrated the importance of Asp42 in the catalytic process by changing it to asparagine using site‐directed mutagenesis.36 The mutated protein had a 20‐fold decrease in activity when the neutral residue was present, and the residual activity observed was contamination from the wild type enzyme from endogenous to the host yeast cells.36 Under typical FPOP conditions, Asp42 undergoes a loss of CO2, as evidenced by a −44 Da mass shift in the b5 and y10 ion in the MS/MS spectrum, resulting in a neutral residue in the active site (Fig. 6). This modification was not very abundant at only 0.82% leading to only a twofold loss in activity (Fig. 4). The role of Asp42 in the decrease in catalytic activity is further demonstrated in the typical FPOP and over‐oxidized controls where Asp42 is oxidatively modified in both samples (Supporting Information Table S4), which have a decrease in activity of 1.6‐ and 2.6‐fold, respectively [Fig. 4(A)]. For the typical FPOP control, Asp42 had 0.10% background oxidation and the over‐oxidized control had 0.65% background oxidation (Supporting Information Table S4). Asp42 was also modified in the over‐oxidized sample at similar levels, 0.8%, to the typical laser sample, but an almost complete loss of catalytic activity is observed indicating that an additional component, protein unfolding, is contributing to the loss of catalytic activity.
Figure 6.
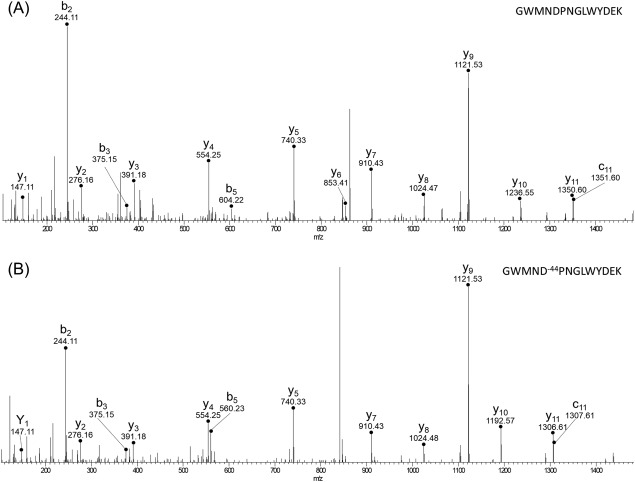
The MS/MS spectra for the (A) unmodified peptide and the (B) modified peptide. A 44 Dalton lose for both the b5 and y10 ion on the modified peptide corresponds to a CO2 loss on Asp42.
Further consideration of modified residues adjoined to the active site effecting enzymatic activity is needed. In each condition, residues M40, N41, and N44 were modified, while P43 was modified in the typical FPOP sample, typical FPOP control, and over‐oxidized control (Supporting Information Table S4). Due to the proximity of these residues (Supporting Information Fig. S4), various hydrophobic interactions or Van der Waals forces could be formed or disrupted altering the dielectric response between the active residue and substrate and possibly the flexibility of the binding pocket. To fully conclude the effects of oxidation near the active site and loss of activity, additional investigation is needed, but the correlation between the oxidation of these four residues and the decrease in activity in the typical FPOP sample, typical FPOP control, and over‐oxidized control points to a possible connection.
We have demonstrated, using enzymatic assays, that FPOP performed under strict experimental conditions does not significantly alter the native structure of proteins. The inclusion of the radical scavenger to reduce the radical lifetime limits oxidation‐induced unfolding of the protein as evidence by retained catalytic activity after FPOP in the presence of glutamine. This study strengthens the assertion of Gau et al.29 that the method is analyzing proteins in their native conformation.
Materials and Methods
In vitro FPOP
The lyophilized lysozyme (Sigma‐Aldrich) and invertase (Sigma‐Aldrich) was resuspended in 66 mM potassium phosphate (Sigma‐Aldrich) and water, respectively. For each protein, two different FPOP conditions were used. In the first condition, the typical FPOP condition, FPOP was performed as previously described.8 Briefly, immediately prior to infusion, 7.5 mM hydrogen peroxide (Thermo Fisher Scientific) was added to a sample of 0.18 mg/mL of protein with 10 mM glutamine (Thermo Fisher Scientific). Samples were infused through a flow tube and exposed to laser irradiation at 248 nm. Samples were collected in a vial containing 100mM dimethylthiolurea (DMTU) (Sigma‐Aldrich) and 10 mM methionine (Thermo Fisher Scientific) to quench the reaction. For this study, DMTU was used in place of catalase to quench hydrogen peroxide to reduce interference in the enzymatic assay. Three technical replicates were performed for this condition as well as three technical replicates without laser irradiation (typical FPOP control). In the second condition, the over‐oxidized condition, sample specifications were the same as above except that glutamine was omitted from the reaction thus extending the lifetime of the hydroxyl radicals. Samples were infused and exposed to laser irradiation similar to the typical FPOP condition. Three technical replicates were performed for this condition as well as three technical replicates without laser irradiation (over‐oxidized control). For laser irradiation, samples were passed through a 3 mm irradiation window at 39.76 µL/min with a laser frequency of 10 Hz allowing a 20% exclusion factor. A 248‐nm KrF excimer laser (GAM Laser Inc.) was used to irradiate the samples at 100 mJ/pulse.
Enzymatic activity assays
After FPOP, the enzymatic activity was measured for each protein. Three separate controls were used to monitor the effects FPOP on the activity of the protein. Control one was the protein in buffer, for lysozyme, and water, for invertase (folded condition). Control two was the unfolded protein (unfolded condition). To unfold the protein, samples were incubated in 8 M urea and 100 mM DTT for 48 and 24 hours for lysozyme and invertase, respectively, at room temperature. Control three had the normal FPOP components but omitted hydrogen peroxide to consider the effects of scavenger and quench (quench condition). The catalytic activity of lysozyme and invertase was determined by using a lysozyme activity kit (Sigma Aldrich) and an invertase activity kit (Sigma Aldrich), respectively, according to the manufacturer's instructions. For each sample, a blank, where protein was omitted, was analyzed to remove matrix effects. Approximately, 0.22 µg of lysozyme was added to 0.05% of Micrococcus lysodeikticus cells. The absorbance at 450 nm was recorded every minute for 15 minutes. For invertase, 0.07 µg/mL of protein was incubated with a sucrose solution and the Master Reaction Mix. The absorbance at 570 nm was measured after 20 minutes. Owing to observed protein loss after infusion through silica tubing during FPOP, the activity of each sample was normalized by concentration. For both lysozyme and invertase activity, the control sample with folded protein was set to 100% enzymatic activity, and all samples were compared to it.
MS analysis
Intact MS analysis was completed using a nanoAcquity UPLC (Waters) and a Q Exactive (Thermo Fisher Scientific) mass spectrometer. Protein was loaded onto a MassPREP Micro Desalting column (Waters) and washed with 0.1% formic acid in water for 5 minutes and eluted protein at 60% acetonitrile and 0.1% formic acid for 5 minutes with a rate of 5 µL/min. Using MagTran, the area of the top 8 Gaussians were calculated to determine the amount of modified and unmodified protein.
For bottom‐up analysis, protein was digested as previously described.37 FPOP samples and controls were dried using a vacuum centrifuge and resuspended in 8 M Urea and 100 mM Tris‐HCl pH 8.5. The samples were reduced with tris(2‐carboxyethyl)phosphine (TCEP) (Sigma‐Aldrich), alkylated with iodoacetamide (IAA) (Sigma‐Aldrich), and the reaction quenched with dithiothreitol (DTT) (Sigma‐Aldrich). Samples were then subjected to a tryptic digest (Thermo Fisher Scientific) overnight at 37°C. The digestion was quenched with formic acid (Thermo Fisher Scientific) with a total concentration of 5%. Samples were desalted using NuTip C‐18 media packed zip tips (Glygen Corporation). After clean‐up, the samples were dried and resuspended in 20 µL of 10% acetonitrile with 0.1% formic acid (Thermo Fisher Scientific). MS/MS analysis was completed using a nanoAcquity UPLC (Waters) and Q Exactive mass spectrometer (Thermo Fisher Scientific). Samples were loaded on an Acquity UPLC C18 Trap Column (Waters) and washed for 10 minutes at 5 µL/min with 1% acetonitrile 0.1% formic acid. Samples were separated on an in house packed 75 µM diameter column with 20 cm bed of Magic 5 µm C18 particles (Michrom Bioresources Inc.) with a 70‐minute gradient reaching 45% acetonitrile 0.1% formic acid. The total run time was 106 minutes including loading, washing, and equilibrating. For MS1 the AGC target was set to 3e6 and for data‐dependent MS2 it was set to 1e5.
Data analysis
For lysozyme, the tandem MS data was searched using Byonic (Protein Metrics). The data was searched in two stages. In the first stage, a single no laser dataset was searched against a Swiss Prot human database which contains 20,165 proteins with the carbomidomethylation of cysteines as the only modification with a parent ion tolerance of 5 ppm and fragment tolerance of 0.2 Da. A target decoy database search was also performed and the false discovery rate (FDR) was set to 1%. From this search, a focused database was generated that included decoy sequences. In the second stage, all data files were searched against the focused database will all known hydroxyl radical side‐chain reactions products38 as variable modifications. The FDR was set to 1%. The software Byologic (Protein Metrics) was used to quantitate the extent of FPOP modification using Eq. 1:
The EIC area modified is the area of the peptide with a modified residue and EIC area is the total area of the same peptide with and without the modified residue.
For invertase, MS/MS data analysis was completed like previously described.8, 37 Samples were searched using Proteome Discoverer (Thermo Fisher Scientific) with Sequest HT (Matrix Sciences Ltd.) against a yeast FASTA database which contains 6,566 proteins. Carbomidomethylation of cysteine was searched as a static modification while the FPOP modifications searched as dynamic modifications with a parent ion mass tolerance of 10 ppm and the fragment mass tolerance of 0.02 Da. The fractional oxidation per residue was determined using Eq. 1.
Conflict of Interests
There is no competing interest.
Supporting information
Supporting Information
Acknowledgments
Special thanks to the University of Maryland School of Pharmacy mass spectrometry center (SOP1841‐IQB2014). The authors would like to thank Upneet Kaur, Jessica Espino, and Danté Johnson for help in editing.
Statement: Hydroxyl radical footprinting is increasingly being used to study protein interactions. Fast photochemical oxidation of proteins (FPOP) utilizes an excimer laser to photo‐dissociate hydrogen peroxide to form hydroxyl radicals. With the addition of a glutamine scavenger, the hydroxyl radicals are quenched in a microsecond, faster than proteins unfold. But further validation is needed to conclude FPOP labels the native protein structure. Testing the catalytic activity of lysozyme and invertase after FPOP showed the oxidative modifications do not induce protein unfolding.
References
- 1. Gau B, Garai K, Frieden C, Gross ML (2011) Mass spectrometry‐based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4. Biochemistry 50:8117–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jones LM, Sperry JB, Carroll JA, Gross ML (2011) Fast photochemical oxidation of proteins for epitope mapping. Anal Chem 83:7657–7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang H, Gau BC, Jones LM, Vidavsky I, Gross ML (2011) Fast photochemical oxidation of proteins for comparing structures of protein− ligand complexes: the calmodulin− peptide model system. Anal Chem 83:311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Z, Moniz H, Wang S, Ramiah A, Zhang F, Moremen KW, Linhardt RJ, Sharp JS (2015) High structural resolution hydroxyl radical protein footprinting reveals an extended Robo1‐heparin binding interface. J Biol Chem 290:10729–10740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kiselar JG, Janmey PA, Almo SC, Chance MR (2003) Structural analysis of gelsolin using synchrotron protein footprinting. Mol Cell Proteomics 2:1120–1132. [DOI] [PubMed] [Google Scholar]
- 6. Vahidi S, Stocks BB, Liaghati‐Mobarhan Y, Konermann L (2012) Mapping pH‐induced protein structural changes under equilibrium conditions by pulsed oxidative labeling and mass spectrometry. Anal Chem 84:9124–9130. [DOI] [PubMed] [Google Scholar]
- 7. Jones LM, Zhang H, Cui W, Kumar S, Sperry JB, Carroll JA, Gross ML (2013) Complementary MS methods assist conformational characterization of antibodies with altered S–S bonding networks. J Am Soc Mass Spectrom 24:835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rinas A, Jones LM (2015) Fast photochemical oxidation of proteins coupled to multidimensional protein identification technology (MudPIT): expanding footprinting strategies to complex systems. J Am Soc Mass Spectrom 26:540–546. [DOI] [PubMed] [Google Scholar]
- 9. Kiselar JG, Mahaffy R, Pollard TD, Almo SC, Chance MR (2007) Visualizing Arp2/3 complex activation mediated by binding of ATP and WASp using structural mass spectrometry. Proc Natl Acad Sci USA 104:1552–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li X, Grant OC, Ito K, Wallace A, Wang S, Zhao P, Wells L, Lu S, Woods RJ, Sharp JS (2017) Structural analysis of the glycosylated intact HIV‐1 gp120–b12 antibody complex using hydroxyl radical protein footprinting. Biochemistry 56:957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao J, Yin DH, Yao Y, Sun H, Qin Z, Schöneich C, Williams TD, Squier TC (1998) Loss of conformational stability in calmodulin upon methionine oxidation. Biophys J 74:1115–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dalle‐Donne I, Rossi R, Giustarini D, Gagliano N, Di Simplicio P, Colombo R, Milzani A (2002) Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radical Biol Med 32:927–937. [DOI] [PubMed] [Google Scholar]
- 13. Venkatesh S, Tomer KB, Sharp JS (2007) Rapid identification of oxidation‐induced conformational changes by kinetic analysis. Rapid Commun Mass Spectrom 21:3927–3936. [DOI] [PubMed] [Google Scholar]
- 14. Chance MR, Sclavi B, Woodson SA, Brenowitz M (1997) Examining the conformational dynamics of macromolecules with time‐resolved synchrotron X‐ray ‘footprinting’. Structure 5:865–869. [DOI] [PubMed] [Google Scholar]
- 15. Hambly DM, Gross ML (2005) Laser flash photolysis of hydrogen peroxide to oxidize protein solvent‐accessible residues on the microsecond timescale. J Am Soc Mass Spectrom 16:2057–2063. [DOI] [PubMed] [Google Scholar]
- 16. Aye TT, Low TY, Sze SK (2005) Nanosecond laser‐induced photochemical oxidation method for protein surface mapping with mass spectrometry. Anal Chem 77:5814–5822. [DOI] [PubMed] [Google Scholar]
- 17. McClintock C, Kertesz V, Hettich RL (2008) Development of an electrochemical oxidation method for probing higher order protein structure with mass spectrometry. Anal Chem 80:3304–3317. [DOI] [PubMed] [Google Scholar]
- 18. Monroe EB, Heien ML (2013) Electrochemical generation of hydroxyl radicals for examining protein structure. Anal Chem 85:6185–6189. [DOI] [PubMed] [Google Scholar]
- 19. Maleknia SD, Downard KM (2014) Advances in radical probe mass spectrometry for protein footprinting in chemical biology applications. Chem Soc Rev 43:3244–3258. [DOI] [PubMed] [Google Scholar]
- 20. Maleknia SD, Downard K (2001) Radical approaches to probe protein structure, folding, and interactions by mass spectrometry. Mass Spectrom Rev 20:388–401. [DOI] [PubMed] [Google Scholar]
- 21. Downard KM, Maleknia SD, Akashi S (2012) Impact of limited oxidation on protein ion mobility and structure of importance to footprinting by radical probe mass spectrometry. Rapid Commun Mass Spectrom 26:226–230. [DOI] [PubMed] [Google Scholar]
- 22. Sharp JS, Sullivan DM, Cavanagh J, Tomer KB (2006) Measurement of multisite oxidation kinetics reveals an active site conformational change in Spo0F as a result of protein oxidation. Biochemistry 45:6260–6266. [DOI] [PubMed] [Google Scholar]
- 23. Kiselar JG, Maleknia SD, Sullivan M, Downard KM, Chance MR (2002) Hydroxyl radical probe of protein surfaces using synchrotron X‐ray radiolysis and mass spectrometry. Int J Radiat Biol 78:101–114. [DOI] [PubMed] [Google Scholar]
- 24. Sharp JS, Tomer KB (2007) Analysis of the oxidative damage‐induced conformational changes of apo‐ and holocalmodulin by dose‐dependent protein oxidative surface mapping. Biophys J 92:1682–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDaniel EW, Martin DW, Barnes WS (1962) Drift tube‐mass spectrometer for studies of low‐energy ion‐molecule reactions. Rev Sci Instrum 33:2–7. [Google Scholar]
- 26. Watson C, Janik I, Zhuang T, Charvatova O, Woods RJ, Sharp JS (2009) Pulsed electron beam water radiolysis for submicrosecond hydroxyl radical protein footprinting. Anal Chem 81:2496–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Niu B, Zhang H, Giblin D, Rempel DL, Gross ML (2015) Dosimetry determines the initial OH radical concentration in fast photochemical oxidation of proteins (FPOP). J Am Soc Mass Spectrom 26:843–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vahidi S, Konermann L (2016) Probing the time scale of FPOP (fast photochemical oxidation of proteins): radical reactions extend over tens of milliseconds. J Am Soc Mass Spectrom 27:1156–1164. [DOI] [PubMed] [Google Scholar]
- 29. Gau BC, Sharp JS, Rempel DL, Gross ML (2009) Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal Chem 81:6563–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsou C‐L (1993) Conformational flexibility of enzyme active sites. Science 262:380–381. [DOI] [PubMed] [Google Scholar]
- 31. Ahern TJ, Klibanov AM (1985) The mechanism of irreversible enzyme inactivation at 100C. Science 228:1280–1284. [DOI] [PubMed] [Google Scholar]
- 32. Perez‐Iratxeta C, Andrade‐Navarro MA (2008) K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol 8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin T‐Y, Koshland D (1969) Carboxyl group modification and the activity of lysozyme. J Biol Chem 244:505–508. [PubMed] [Google Scholar]
- 34. Perkins SJ, Johnson LN, Machin PA, Phillips DC (1978) Crystal structures of egg‐white lysozyme of hen in acetate‐free medium and of lysozyme complexes with N‐acetylglucosamine and beta‐methyl N‐acetylglucosaminide. Biochem J 173:607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kelly JA, Sielecki AR, Sykes BD, James MNG, Phillips DC (1979) X‐ray crystallography of the binding of the bacterial cell wall trisaccharide NAM‐NAG‐NAM to lysozyme. Nature 282:875. [DOI] [PubMed] [Google Scholar]
- 36. Reddy VA, Maley F (1990) Identification of an active‐site residue in yeast invertase by affinity labeling and site‐directed mutagenesis. J Biol Chem 265:10817–10820. [PubMed] [Google Scholar]
- 37. Rinas A, Espino JA, Jones LM (2016) An efficient quantitation strategy for hydroxyl radical‐mediated protein footprinting using Proteome Discoverer. Anal Bioanal Chem 408:3021–3031. [DOI] [PubMed] [Google Scholar]
- 38. Xu G, Chance MR (2007) Hydroxyl radical‐mediated modification of proteins as probes for structural proteomics. Chem Rev 107:3514–3543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information