

Abstract
Neuropathic pain remains poorly treated for large numbers of patients, and little progress has been made in developing novel classes of analgesics. To redress this issue, ziconotide (Prialt™) was developed and approved as a first‐in‐class synthetic version of ω‐conotoxin MVIIA, a peptide blocker of Cav2.2 channels. Unfortunately, the impracticalities of intrathecal delivery, low therapeutic index and severe neurological side effects associated with ziconotide have restricted its use to exceptional circumstances. Ziconotide exhibits no state or use‐dependent block of Cav2.2 channels; activation state‐dependent blockers were hypothesized to circumvent the side effects of state‐independent blockers by selectively targeting high‐frequency firing of nociceptive neurones in chronic pain states, thus alleviating aberrant pain but not affecting normal sensory transduction. Unfortunately, numerous drugs, including state‐dependent calcium channel blockers, have displayed efficacy in preclinical models but have subsequently been disappointing in clinical trials. In recent years, it has become more widely acknowledged that trans‐aetiological sensory profiles exist amongst chronic pain patients and may indicate similar underlying mechanisms and drug sensitivities. Heterogeneity amongst patients, a reliance on stimulus‐evoked endpoints in preclinical studies and a failure to utilize translatable endpoints, all are likely to have contributed to negative clinical trial results. We provide an overview of how electrophysiological and operant‐based assays provide insight into sensory and affective aspects of pain in animal models and how these may relate to chronic pain patients in order to improve the bench‐to‐bedside translation of calcium channel modulators.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Abbreviations
- CPM
conditioned pain modulation
- CPP
conditioned place preference
- CRMP2
collapsin response mediator protein 2
- DRG
dorsal root ganglion
- NNT
number needed to treat
- NS
nociceptive specific
- SNL
spinal nerve ligation
- VGCC
voltage‐gated calcium channel
- VPL
ventral posterolateral
- WDR
wide dynamic range
Introduction
Neuropathic syndromes are often characterized by a complex combination of positive and negative sensory phenomena including allodynia (perceiving non‐noxious stimuli as pain), hyperalgesia (increased pain to normally painful stimuli) and also paroxysmal or persistent paraesthesias (abnormal sensations, e.g. numbness and tingling) and dysaesthesias (abnormal and unpleasant sensations, e.g. shocking and paradoxical burning). These symptoms are often confounded by co‐morbidities such as depression, anxiety and sleep disturbances. Meta‐analyses suggest that pain with neuropathic characteristics has a prevalence of 7–10% in the general population (van Hecke et al., 2014) and that large numbers of neuropathic patients fail to achieve adequate relief from currently available treatments (Finnerup et al., 2010; Finnerup et al., 2015). These studies highlight the urgent need for novel classes of therapeutics and better utilization of currently available treatments. In an attempt to address this ongoing challenge, the first‐in‐class drug http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2536 (Prialt™) was developed as a synthetic version of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=533 blocker http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2536, derived from Conus magus, and is licensed for chronic pain. However, due to the narrow therapeutic window and considerable side effects, ziconotide is only administered intrathecally to patients who have failed to respond to other treatments (Sanford, 2013).
As a rare example of bench‐to‐bedside translation, intrathecal ω‐conotoxins were demonstrated to be anti‐nociceptive in neuropathic animals over 20 years ago (Chaplan et al., 1994). Pain has both sensory and affective dimensions but is also a subjective experience; hence by definition, pain in animals must be inferred. As in this study, most animal behavioural experiments rely on evoked reflex withdrawals to assess ‘pain responses’. A binary outcome measure of this nature renders it near impossible to correlate the intensity of a stimulus with the level of response given that a reflex response determines the first point at which a stimulus becomes aversive. As a consequence, the validity of stimulus‐evoked reflexes as predictors of clinical efficacy has been questioned in light of the fact that many pharmacological agents are efficacious in preclinical models, including calcium channel blockers, but fail to translate into the clinic (Percie du Sert and Rice, 2014). The thorny issue of translation is of course far more complex, and there are numerous instances where choice of appropriate endpoints and identifying underlying mechanisms of neuropathy has led to successful forward and back‐translation. Heterogeneity of patient populations is likely to have led to the failure of many clinical trials and sensory profiling and stratification of patients as an alternate approach to aetiological grouping may lead to better indicators of successful treatment (Baron et al., 2012; Freeman et al., 2014; Bouhassira and Attal, 2016). For example, in diabetic neuropathy, low conditioned pain modulation (CPM) predicts efficacy of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=202 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7477 (Yarnitsky et al., 2012; Niesters et al., 2014), and in peripheral neuropathy patients, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7254 was more efficacious in those with the irritable nociceptor versus the non‐irritable phenotype [number needed to treat (NNT) of 4 and 13 respectively] (Demant et al., 2014). As illustrations of successful back‐translation, in the spinal nerve ligation (SNL) model of neuropathy in rats, CPM or diffuse noxious inhibitory controls are absent and can be restored by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4808 and tapentadol, and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5339 inhibits spinal neuronal excitability (Chapman et al., 1998; Bannister et al., 2015). In this respect, the SNL model could represent comparable underlying mechanisms to patients with low CPM or the irritable nociceptor phenotype and thus provides a basis for forward translation. Calcium channel activity could be surmised to be favoured in the SNL model and in patients with the irritable nociceptor phenotype, and so, heterogeneous patient groups based on aetiology only may not provide adequate sensitivity for revealing effects of these drugs.
Most clinical trials continue to rely on ongoing pain scores as a primary clinical endpoint, though more recently in smaller trials, there has been a concerted effort to perform quantitative sensory testing and classify patients according to their sensory profile (Bouhassira and Attal, 2016). This approach attempts to provide a quantitative and qualitative read‐out of the patient's pain state. In terms of a comparable translatable measure, electrophysiological characterizations of neuronal excitability in animals allow reproducible, objective and quantitative measures of sensory neuronal processing to multiple modalities and have the advantage of examining responses to supra‐threshold and brush stimulation which are not particularly amenable to behavioural testing. Ongoing or paroxysmal pain has been difficult to demonstrate in animal models but is a major cause of suffering and poor quality of life in neuropathic patients. As ongoing pain is unpleasant and aversive in its nature, and pain relief is rewarding, a conditioned place preference (CPP) assay in rodents was developed to explore whether injured animals associate with contextual cues affiliated with relief. This method has demonstrated efficacy of clinically used drugs such as intrathecal http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=516 (King et al., 2009).
Calcium channel blockers have been successfully developed for the treatment of absence seizures and are an emerging drug class for the treatment of neurological disorders and chronic pain (reviewed in detail by Zamponi, 2016). In this review, we provide an updated overview of the effects of calcium channel blockers in chronic pain models, with a particular focus on how electrophysiological and CPP studies may provide insight into sensory and affective dimensions of pain. Additionally, we consider the prospects of voltage‐gated calcium channels (VGCCs) as targets for chronic pain and how preclinical data may guide design of clinical trials.
Targeting trafficking of VGCCs
Auxiliary calcium channel subunit α2δ‐1
By far and away, the most frequently prescribed calcium channel modulators for neuropathic pain are the gabapentinoids, gabapentin and pregabalin. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5483 and the newer derivative http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5484 were designed as analogues of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1067. As it transpired, gabapentin was identified as a ligand for the auxiliary voltage‐gated calcium channel subunit α2δ rather than GABA receptors (Gee et al., 1996). Transgenic approaches strongly support that the interaction of gabapentinoids with α2δ‐1 is required for anti‐nociceptive activity in neuropathic conditions given the lack of efficacy in the corresponding null mice (Field et al., 2006; Patel et al., 2013). At the cellular level, it is unclear precisely how gabapentinoids disrupt transmitter release, but a direct block of channel activation seems highly unlikely (Hendrich et al., 2008). Instead, gabapentin has been shown to inhibit axonal trafficking of α2δ‐1, which is elevated in injured primary afferents, and also recycling of calcium channel complexes between intracellular compartments and the synaptic membrane (Bauer et al., 2009; Tran‐Van‐Minh and Dolphin, 2010) (Figure 1). The selective anti‐nociceptive effects in facilitated states imply injury‐induced factors that influence efficacy (reviewed in detail by Patel and Dickenson, 2016). Thus, it would be expected that clinical correlates of these mechanisms would be a determinant of the action of these drugs and so affect their NNT.
Figure 1.

Approaches to calcium channel modulation in chronic pain. To date, targeting trafficking of VGCCs has been the favoured approach to modulating channel activity. The gabapentinoids (GBP) bind to an arginine motif of the α2 subunit of α2δ‐1 and α2δ‐2 and inhibit axonal trafficking of α2δ‐1 and Rab11‐dependent recycling of endosomal channels to the synapse (Bauer et al., 2009; Tran‐Van‐Minh and Dolphin, 2010). Novel approaches to reduce trafficking include utilizing CBD3 peptides to disrupt the interaction between VGCCs and CRMP2. Channel activity can be regulated via inhibitory GPCRs such as α2 adrenoreceptors, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=26 and μ‐opioid receptors. Peptide blockers act by either altering gating by targeting voltage sensitive domains (such as ω‐agatoxin) or by directly blocking the pore (ω‐conotoxins) (Bourinet and Zamponi, 2016).
In behavioural assays, acute, systemically administered, gabapentin exhibits state‐dependent anti‐nociceptive activity when assessed by changes in withdrawal threshold (Field et al., 1997). Both spinal and supra‐spinal mechanisms are likely to be involved as intrathecal and intracerebroventricular pregabalin increase withdrawal thresholds after peripheral nerve injury (Takeuchi et al., 2007). Likewise, when examined against spinal wide dynamic range (WDR) neuronal responses, gabapentin displays state‐dependent inhibitory activity. Increased descending facilitatory drive is key in shaping the inhibitory effects of acute gabapentin treatment after nerve injury, and remarkably, mimicking enhanced descending facilitation in naïve rats recreates the permissive state for gabapentin to act (Suzuki et al., 2005). Increased activity in this pathway underlies the efficacy of pregabalin in a model of opioid‐induced hyperalgesia where no injury exists (Bannister et al., 2011).
In models where central sensitization is evident, even in the absence of pathology or where neuropathy is not the only component, gabapentin and pregabalin reduce spinal neuronal firing to both mechanical‐ and heat‐evoked responses across low and high intensities of stimulation (Donovan‐Rodriguez et al., 2005; Omori et al., 2009; Bannister et al., 2011; Thakur et al., 2012). Patients with osteoarthritis frequently report pain with neuropathic characteristics (Ohtori et al., 2012), and similarly, rodent models of osteoarthritis can be characterized by a neuropathic component leading to cutaneous sensitization outside the injured area which is sensitive to pregabalin inhibition (Rahman et al., 2009; Thakur et al., 2012). In contrast, animal models of osteoarthritis lacking neuropathic characteristics are not sensitive to pregabalin anti‐nociception (Thakur et al., 2012). Correspondingly, there is some evidence that patients reporting pain using neuropathic descriptors respond positively to combination therapy with pregabalin and advocates the benefits of stratification of patients (Ohtori et al., 2013). A complex interplay of inflammatory, neuropathic and disease‐specific components leads to pain chronicity in a rat model of cancer‐induced bone pain, with one consequence being a phenotypic change in the ratio of WDR‐to‐nociceptive specific (NS) neurones in the superficial dorsal horn. Chronic gabapentin not only inhibits enhanced frequencies of firing in this model but normalizes the ratio of neuronal response profiles (Donovan‐Rodriguez et al., 2005). In comparison, spinal nerve‐ligated rats do not display similar increases in neuronal firing frequency in the dorsal horn. Instead, an increase in population coding is proposed to mediate hypersensitivity which subsequently converges onto thalamic relays. In the ventral posterolateral (VPL) thalamus of SNL rats, mechanical‐ and cold‐evoked responses, and to a lesser extent heat responses, of WDR and NS neurones are increased and pregabalin normalizes neuronal coding in a modality selective manner (mechanical and heat) at intensities evoking elevated responses (Patel and Dickenson, 2016). Mechanically evoked responses (brush and punctate stimuli) are inhibited to a greater extent compared with heat stimuli, and this corresponds relatively well with patient psychophysics (Attal et al., 1998), though the contrasting reversal of cold hypersensitivity observed in this latter study may relate to the chronic dosing regimen.
In healthy volunteers, gabapentin is ineffective against temporal summation of pain, and similarly with the analogous process in rats, electrically evoked wind‐up of deep dorsal horn neurones is unaffected (Arendt‐Nielsen et al., 2011). In contrast, in a surrogate model of central sensitization, gabapentin inhibits temporal summation (Arendt‐Nielsen et al., 2007), and there is some evidence that wind‐up is inhibited in animal models where central sensitization is likely to be present such as in cancer‐induced bone pain and after sciatic nerve injury, though surprisingly not after spinal nerve injury (Donovan‐Rodriguez et al., 2005; Curros‐Criado and Herrero, 2007; Bee and Dickenson, 2008; Ding et al., 2014). Wind‐up is a short‐term process reflecting activity‐dependent increases in neuronal excitability and can lead to features shared with central sensitization such as expansion of receptive field size, inducing wind‐up at lower frequencies and overlapping pharmacological dependencies (e.g. reversal by http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=75 antagonists) (Li et al., 1999). Thus, wind‐up could act as a read out of sensitization state, and pharmacological agents that inhibit wind‐up may also reverse central sensitization.
When ongoing pain scores are used as a clinical endpoint, the NNT for gabapentin and pregabalin is approximately 7, although this varies across different aetiologies (Finnerup et al., 2010; Finnerup et al., 2015). Thus, when assuming homogeneity amongst patients, most fail to achieve adequate relief. Post hoc sensory profiling analysis, however, reveals that the presence of pinprick hyperalgesia is a predictor of efficacy in human immunodeficiency virus (HIV)‐induced neuropathy (Simpson et al., 2010) and correlates with surrogate models of sensitization in humans and neuronal characterizations in rodents as gabapentin and pregabalin reduce mechanical hypersensitivity (Werner et al., 2001; Dirks et al., 2002; Segerdahl, 2006; Chizh et al., 2007; Patel and Dickenson, 2016). In CPP assays, neither gabapentin nor pregabalin has rewarding properties in the absence of injury (Andrews et al., 2001), whereas in neuropathic and inflammatory models, gabapentin has been demonstrated to activate reward pathways (Xie et al., 2014; Griggs et al., 2015; Park et al., 2016), though notably this occurs at 10‐fold higher doses than required to inhibit spinal neuronal responses to evoked stimulation (Suzuki et al., 2005). The mechanisms that underpin ongoing pain may be distinct from those that mediate evoked pain. The neural mechanisms by which gabapentin provides relief from ongoing pain are poorly characterized. Despite reports of reductions in spontaneous spinal neuronal activity by gabapentin (Chapman et al., 1998; Suzuki and Dickenson, 2006; Dong et al., 2013; Zhang et al., 2013), pregabalin has no effect on aberrant spontaneous firing in the ventral posterolateral thalamus in SNL rats (Patel and Dickenson, 2016). Spontaneous and evoked activity in the right central nucleus of the amygdala is elevated following a peripheral nerve injury, which can be normalized by pregabalin, suggesting that gabapentinoids may modulate activity within corticolimbic pathways associated with affective dimensions of pain (Goncalves and Dickenson, 2012). Collectively, these data indicate that gabapentinoids poorly relieve ongoing pain in patients but are effective in reducing areas of secondary sensitization and mechanical hyperalgesia. This difference could contribute to high NNTs if these sensory modalities were not subdivided in clinical studies. Importantly, patients have pain scores well above the pain threshold. As gabapentinoids attenuate the firing of spinal and thalamic sensory neurones to noxious intensities at doses that would fully reverse behavioural hypersensitivity to lower intensity stimuli, these may act as a better predictor of their anti‐hyperalgesic effect.
Collapsin response mediator protein 2 (CRMP2)
Even though gabapentinoids can be effective for the management of pain, they are not without adverse effects such as ataxia, nausea, somnolence and dizziness. Thus, new approaches to target channel trafficking have been explored. CaV2.2 channels present in the presynaptic terminals of primary afferent fibres forms part of a large complex of proteins and other molecules including a regulatory protein CRMP2. This protein has been shown to enhance membrane trafficking of Cav2.2 channels, and CRMP2 knockdown reduces calcium currents and transmitter release, suggesting that Cav2.2 channels require the presence of this protein for normal function (Chi et al., 2009). CBD3 is a 15‐amino acid peptide region (ARSRLAELRGVPRGL) of CRMP2 that binds to Cav2.2 channels, and a CBD3 and TAT (HIV‐1 trans‐activator of transcription) conjugated brain penetrant peptide has been shown to block the interaction of CRMP2 and Cav2.2 channels (Francois‐Moutal et al., 2015). Electrophysiological studies in rat spinal cord splices demonstrated that perfusion with TAT‐CBD3 reduces CGRP release (Brittain et al., 2011). Unlike the gabapentinoids, TAT‐CBD3 does not display pathological state‐dependent activity and inhibits acute nociception and inflammation‐induced hypersensitivity but not nerve injury‐induced hypersensitivity (Wilson et al., 2011; Francois‐Moutal et al., 2015). At effective analgesic doses, TAT‐CBD3 does not produce motor impairment, paralysis or cognitive deficits in rats (Wilson et al., 2011). A similar peptide, R9‐CBD3‐A6K, has no aversive or rewarding properties in sham rats but activates reward pathways in a neuropathic state (Xie et al., 2016). Taking into account these results, TAT‐CBD3 could be a potential drug for the treatment of chronic pain, though the poor bioavailability and short half‐life of peptide agents limits their usefulness.
Targeting α1 VGCC subunits
Activation state‐independent channel blockers
http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=80 (L‐type), http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=80 (P/Q‐type), CaV2.2 (N‐type), http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=80 (R‐type) and http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=80 (T‐type) channels are ubiquitously distributed in the central and the peripheral nervous system. In the dorsal horn, CaV2.1 and CaV2.2 channels are predominantly localized in the presynaptic terminals of largely non‐overlapping populations of primary afferent fibres, whereas CaV1 and CaV2.3 channels are more commonly associated with somata and dendrites (Westenbroek et al., 1998) (Figure 2). CaV2.1 and CaV2.2 channels mediate release of neurotransmitters such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1369, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2098 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=695 from primary afferent fibres causing the activation of second‐order neurones (Santicioli et al., 1992; Luebke et al., 1993; Terashima et al., 2013). CaV1 and CaV2.3 channels on the other hand are associated with post‐synaptic excitability; the former appears to underlie the ability of a subset of dorsal horn neurones to mount significant trains of post‐discharge (Morisset and Nagy, 1999), whereas the genetic ablation of the latter demonstrates little role in basal nociception but a reduction of inflammatory hypersensitivity (Saegusa et al., 2000).
Figure 2.
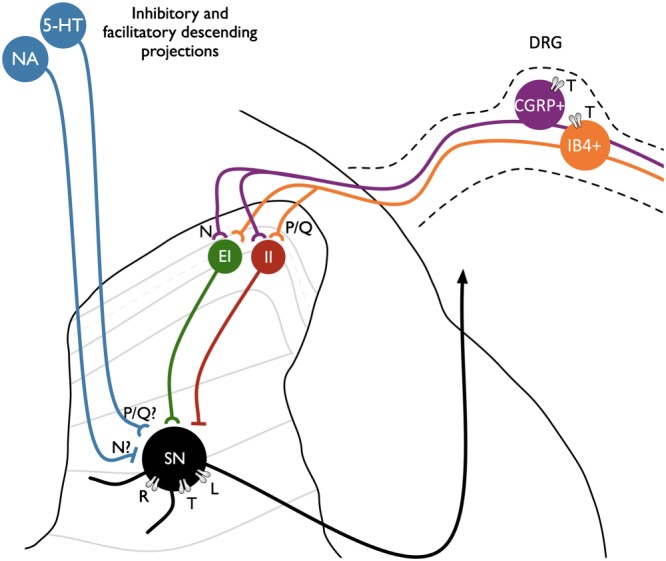
Expression of VGCCs in the dorsal horn. CaV2.1 (P/Q‐type) and CaV2.2 (N‐type) channels are largely expressed in distinct populations of primary afferents with the latter associated with peptidergic transmitters (Westenbroek et al., 1998). CaV3.2 (T‐type) channels are expressed in CGRP+ and IB4+ DRG neurones and also in cutaneous nerve endings (Rose et al., 2013). Both populations synapse with markers of excitatory (EI) and inhibitory (II) interneurons which subsequently converge onto second‐order sensory neurones (SN). The effects of state‐independent blockers on reducing excitatory transmission from primary afferents may be countered by opposing effects in reducing inhibitory transmitter release; for example, CaV2.2 channels have been shown to mediate noradrenaline release from sympathetic neurones, though it is unclear precisely how monoamines are released in the dorsal horn. L‐type (CaV1) and T‐type (CaV3) channels influence post‐synaptic excitability; neuroplasticity in L‐type expression after injury may influence neuronal sensitization (Radwani et al., 2016), whereas spinally expressed T‐type channels, unlike peripherally expressed channels, are not thought to have a pathophysiological role in neuropathy. R‐type (CaV2.3) channels, however, have been implicated in neuronal hyperexcitability in SNL rats (Matthews et al., 2007).
ω‐agatoxin IVA
In trigeminal second‐order neurones, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2529 has a small inhibitory effect on cold‐evoked responses but enhances spontaneous activity in a http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=72 receptor‐sensitive manner (Ebersberger et al., 2004), consistent with a projection to both excitatory and inhibitory interneurones in the dorsal horn (Westenbroek et al., 1998). Spinally application of the CaV2.1 channel blocker, ω‐agatoxin IVA, has a low inhibitory effect on spinal WDR neuronal responses to innocuous and noxious mechanical stimulation in normal rats (Nebe et al., 1997; Matthews and Dickenson, 2001a) and has a similar magnitude of effect in SNL rats (Matthews and Dickenson, 2001a). In contrast, the potency of ω‐agatoxin IVA is enhanced after an acute inflammatory insult (Nebe et al., 1997) and supports a role of CaV2.1 channels in the development of sensitization but argues against a pathophysiological role in a chronic neuropathic state.
CaV3 channel blockers
Likewise, in uninjured conditions, the CaV3 channel blocker http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7182 modestly inhibits WDR responses to mechanical and heat stimulation and exhibits similar potency after peripheral nerve injury (Matthews and Dickenson, 2001b), again arguing against a pathophysiological role of spinal CaV3 channels at least. Unlike CaV2.1 and CaV2.2 channels, CaV3 channels activate at voltages close to resting membrane potential and the gating properties permit regulation of oscillations and burst firing. Intrathalamic delivery of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7718, an activation state‐dependent CaV3.2 blocker, reduces burst firing in the VPL of neuropathic rats, an observation that correlates with the establishment of CPP when Z944 is dosed systemically (LeBlanc et al., 2016). Peripherally expressed CaV3.2 channels may also be important as they modulate excitability of a subset of mechanoreceptors (Shin et al., 2003), and CaV3‐mediated currents are enhanced in injured dorsal root ganglion (DRG) neurones (Jagodic et al., 2008). In SNL rats, the peripherally acting state‐dependent blocker, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7721, reduces spinal WDR neuronal responses to low intensity mechanical stimulation (Jarvis et al., 2014).
ω‐conotoxin GVIA
Studies have shown that, α1B (CaV2.2) deficient mice have a normal life span and normal behaviour, however have impaired basal nociception and development of neuropathy (Hatakeyama et al., 2001; Saegusa et al., 2001). The role this channel plays in the transmission of nociceptive stimuli is further supported by electrophysiological studies demonstrating that http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2535 inhibits mechanical‐ and heat‐evoked responses and wind‐up of spinal WDR neurones consistent with a role in mediating peptide release and consequentially post‐synaptic hyperexcitability. Dural afferents also express CaV2.2 channels, and blockade reduces cold‐evoked and spontaneous activity of trigeminal nucleus neurones (Ebersberger et al., 2004). The increased potency of ω‐conotoxin GVIA after SNL indicates injury‐induced neuroplastic changes in channel expression and/or regulation in the dorsal horn (Matthews and Dickenson, 2001a). In contrast, no change in the effect size of ω‐conotoxin GVIA is observed on spinal WDR neurones innervating the knee following knee inflammation (Neugebauer et al., 1996). Intrathecal ω‐conotoxin MVIIA can induce CPP in SNL rats demonstrating an alleviation of ongoing pain and correlates well with clinical observations (King et al., 2009).
ω‐Conotoxins are small peptides derived from the venom of marine cone snails and block a series of ion channels. These peptides are thought to act by physical occlusion of the α1 ion conducing pore (Bourinet and Zamponi, 2016). In animal models, conotoxins such as GVIA, MVIIA and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2534 have analgesic properties against inflammatory and neuropathic pain; however, they have also been shown to have a series of negative side effects in patients such as dizziness, nausea, nystagmus, gait abnormality, urinary retention, memory impairment and hallucinations. Some of them such as GVIA have irreversible effects once they bind to the pore, which complicates its usage. MVIIA, also known as ziconotide has been the first ω‐conotoxin to enter clinical trials and was approved in 2004 by the FDA, for the management of chronic pain. Initial clinical trials conducted in patients with chronic pain, cancer‐related or AIDS‐related neuropathy refractory to opioid treatment reported improvement of VAS (visual analogue scale) scores (from a mean baseline of >70/100) by 14 to 53%, while the proportion of responders ranged from 16 to 50% (Sanford, 2013). The dosage was often titrated to tolerable levels which may account for some of the variability in effect size. Even though ziconotide does not produce tolerance or addiction like http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627 does, it has adverse neurological side effects that need to be taken into account before prescribing the drug to a patient. In addition, its large amino acid sequence has some limitations. For instance, manufacturing the drug is challenging and expensive. Due to its large size, ziconotide poorly crosses the brain–blood barrier, thus needs to be delivered straight into the spinal cord or in the surrounding cerebrospinal fluid for the treatment of pain.
Due to the limitations of ziconotide, there is a need to develop newer blood–brain barrier penetrable small molecule calcium channel blockers with fewer adverse effects as well as drugs that are orally available. Several new activation state‐dependent blockers have been developed and tested in animal models with some progressing to clinical trials.
Activation state‐dependent channel blockers
ZC88
ZC88 ((N‐[1‐(2,3‐dimethoxyphenyl)‐4‐methyl‐3‐pentene‐1‐yl]‐1‐(5‐bromo‐furfuryl)‐1‐piperidyl‐4‐amine dihydrochloride)) is a non‐peptide, state‐dependent, blocker of CaV2.2 channels and does not affect calcium currents mediated by CaV1, CaV2.1 or CaV2.3 channels in cultured DRG neurones (Zhang et al., 2015). Behavioural studies in a rat chronic constriction injury model of neuropathic pain show a dose‐dependent decrease of mechanical hypersensitivity after oral administration of ZC88 (Meng et al., 2008). However, ZC88 administration has no effect on acute nociception in normal mice in the hot plate test but enhances morphine analgesia. This synergy may arise as opiates can inhibit CaV2.2 via Gi/o protein‐coupled http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 receptors. As chronic treatment with morphine is associated with tolerance and addiction, Meng et al., (2008) tested the effects of ZC88 in animals that had been chronically treated with morphine. In isolation, ZC88 does not produce CPP in uninjured mice. However, in combination with morphine, ZC88 reverses tolerance to chronic morphine in the hot plate test without affecting morphine‐induced CPP. These results are promising, as ZC88 could be a potential drug against neuropathic pain that could be administered together with morphine or used alone.
A‐1264087
A‐1264087 (N 2‐methyl‐N‐{(3aR,4S,6aS)‐2‐[4‐(trifluoromethyl)phenyl]octahydrocyclopenta[c]pyrrol‐4‐yl}‐l‐leucinamide) is a state‐dependent blocker of CaV2.1, CaV2.2 and CaV3 channels. This drug has been shown to block CaV2.2 channels in cultured rat DRG neurones as well as to reduce spontaneous and mechanical‐evoked activity of WDR neurones in SNL animals. Systemic administration reduces mechanically evoked neuronal responses to low‐intensity von Frey filaments as well as Aδ‐fibre and C‐fibre‐evoked responses to electrical stimulation and subsequent wind‐up, selectively in neuropathic rats (Xu et al., 2014). Correspondingly, using withdrawal reflexes as an endpoint, A‐1264087 after oral administration in SNL rats and the iodoacetate model of osteoarthritis attenuates mechanical hypersensitivity (Xu et al., 2014; Zhu et al., 2014). Both of these studies report a decrease in neuronal responses to mechanical stimulation. However, neither of these studies assessed changes in neuronal responses to other modalities or to more noxious intensities of stimulation where C‐fibres are recruited. Thus, it would be interesting to further analyse the effects of A‐1264087 on high intensities of stimulation. Interestingly, in SNL rats, spontaneous spinal neuronal activity is reduced by A‐1264087 when dosed spinally but not when delivered directly to the receptive field (Xu et al., 2014). It would be of value to investigate the effects in operant‐based assays to examine whether A‐1264087 alleviates ongoing as well as evoked pain.
TROX‐1
N‐triazole oxindole (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7766) is an orally bioavailable, blood–brain barrier penetrating, state‐dependent blocker of the CaV2 family. In cultured cell systems, blocking channels under depolarized conditions, TROX‐1 has similar potency across the CaV2 family, thus acting on CaV2.1, CaV2.2 and CaV2.3 channels (Swensen et al., 2012). Behavioural studies demonstrate that in animal models of acute inflammatory pain, oral administration of TROX‐1 reverses behavioural hypersensitivity (Abbadie et al., 2010). However, in CaV2.2 knockout mice, TROX‐1 is ineffective, suggesting that TROX‐1 primarily mediates its effects through CaV2.2 channels in inflammatory conditions, despite evidence that CaV2.3 channels also contribute to spinal neuronal hyperexcitability after injury (Matthews et al., 2007).
TROX‐1 is also anti‐nociceptive in more severe chronic models of pain. Behavioural assessments have also shown that TROX‐1, given by oral, subcutaneous and intrathecal routes, increases paw withdraw thresholds in SNL rats in a dose‐dependent manner with no effect in sham‐operated animals (Abbadie et al., 2010; Patel et al., 2015). In concordance with these behavioural results, in vivo electrophysiological recordings in SNL rats showed a reduction of neuronal‐evoked responses to low intensity and supra‐threshold mechanical stimulation with von Frey filaments after spinal and systemic administration of TROX‐1 (Patel et al., 2015). This effect was modality selective; brush‐, heat‐ and cold‐evoked responses were unaffected by TROX‐1 in the SNL model. Unlike ω‐conotoxin GVIA, TROX‐1 had no effect on neuronal wind‐up. This is despite displaying use dependency in cell lines in vitro using comparable stimulus parameters, which highlights the importance of physiological assays when assessing drug actions.
As described earlier, rodent models of osteoarthritis can share common features with neuropathy resulting from peripheral nerve damage, for example, concurrent time‐dependent increases in descending facilitatory drive and loss of descending noradrenergic inhibition (Rahman et al., 2009; Burnham and Dickenson, 2013). Hence, the effects of TROX‐1 were additionally examined in the rat intra‐articular iodoacetate model of osteoarthritis, where treatment with TROX‐1 reversed behavioural hypersensitivity and weight‐bearing behaviour (Abbadie et al., 2010). In vivo electrophysiological studies have also assessed the efficacy of TROX‐1 in the iodoacetate model of osteoarthritis. Spinal administration of TROX‐1 inhibits evoked WDR neuronal responses to dynamic brush, heat and mechanical stimulation in a dose‐dependent manner (Rahman et al., 2015). Systemic administration of this drug reduces supra‐threshold mechanically evoked responses to von Frey filaments, but does not affect heat and dynamic brush responses, perhaps because a more local administration of the drug (spinal administration) allows for attenuation of these modalities. In sham animals, TROX‐1 does not affect the neuronal responses after evoked stimulation. As observed in SNL rats, TROX‐1 does not reduce wind‐up in arthritic rats; the differential effects of A‐1264087 and TROX‐1 on wind‐up may relate to the selectivity profiles of the drugs. Inactivation of the rostral ventromedial medulla with lidocaine in osteoarthritic rats induces CPP and reveals the presence of ongoing pain (Havelin et al., 2016); as with A‐1264087 and ZC88, it would be of interest to examine whether TROX‐1 modulates affective dimensions of pain. Together, these results suggest that the use of state‐dependent blockers for the treatment of osteoarthritic pain with neuropathic features could be a good alternative to current treatments. The differences between the actions of TROX‐1 in different pain conditions might be due to differences in state‐dependency of the channel such as the depolarizing effect of ionotropic facilitatory http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=68 receptors on pre‐synaptic terminals, altered G‐protein‐mediated regulation by http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=4, GABA‐ and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=1, or changes in the up‐regulation of C‐terminus splice variants of calcium channels which might contribute to changes in biophysical properties of the channel.
When new drugs are developed, the implications of alternative splicing and the effects it has on channel activity are rarely considered. Multiple variants are predicted to exist, but in particular, mutually exclusive exons e37a and e37b of CaV2.2 are highly enriched in DRG neurones. These exons confer different properties to the channel, for instance e37a‐CaV2.2 exhibits larger calcium currents, increased channel open time and slower inactivation than e37b‐CaV2.2 (Castiglioni et al., 2006). VGCCs with these gating properties could be more amenable to block by state‐dependent blockers. However, the role of splice variants in the pathophysiology of neuropathic pain is complex. RNA knockdown has shown that the e37a isoform mediates the transmission of mild mechanical stimulation and heat stimulation and prevents the development of mechanical hypersensitivity in neuropathic and inflammatory models (Altier et al., 2007). Paradoxically, following peripheral nerve injury, there is a decrease in the expression of the e37a isoform (Altier et al., 2007). Neuroplastic shifts in splice variant expression with differences in drug sensitivity and sensitivity to G‐proteins could affect the efficacy of analgesics after injury, including morphine (Jiang et al., 2013), but also, the gabapentinoids as the predominant α2δ‐1 splice variant up‐regulated after nerve injury has reduced affinity for gabapentin (Lana et al., 2014).
Are calcium channels a good target for chronic pain?
Due to their critical role in transmitter release, calcium channels are regarded as an important target in preventing sensory transmission. However, the ubiquitous nature of expression and multitude of processes controlled by calcium channels has complicated the use of blockers for chronic pain (reviewed in detail by Zamponi et al., 2015). Although spinally delivered state‐independent blockers can reduce behavioural and spinal hyperexcitability, micro‐injection of peptide blockers into brainstem regions mediating descending control of pain reveals complex pro‐ and anti‐nociceptive roles of calcium channels and could confound usage of blockers via a systemic route (Figure 3). Targeting nociceptor specific splice variants to improve specificity and reduce side effects seems unlikely to yield positive results given the notorious difficulty in identifying selective low MW blockers of ion channels, let alone selective blockers for splice variants that differ only by small sequences. In theory, state‐dependent blockers would circumvent the confounds of blocking calcium channels by targeting high‐frequency neuronal firing associated with neuropathic pain. In this aspect, compounds such as CNV2197944 (Cav2.2), ABT‐639 (Cav3.2) and Z160 (Cav2.2), which exhibit activation state dependency, were generally well tolerated in phase I clinical trials. Unfortunately, Z160 (NCT1655849) and ABT‐639 (NCT01345045) failed to show analgesic efficacy in phase II trials, whereas results for CNV2197944 have not been reported yet (NCT01848730 and NCT01893125).
Figure 3.

Effects of calcium channel modulators in the pain neuraxis. There are conflicting reports as to whether CaV2.2 channels contribute to ectopic firing in injured primary afferent fibres (Chaplan et al., 1994; Xiao and Bennett, 1995). However, numerous studies demonstrate that state‐dependent and ‐independent blockers can reduce spinal neuronal hyperexcitability when given systemically or spinally. In addition, systemic pregabalin reduces neuronal firing in the amygdala and VPL. Neuropathy can be associated with diminished descending noradrenergic inhibitory control of pain and enhanced serotonergic facilitatory drive. Both CaV2.1 and CaV2.2 channels have complex roles in descending modulation and exert pro‐ and anti‐nociceptive actions when micro‐injected into different regions (Knight et al., 2002; Finn et al., 2003; Urban et al., 2005). However, state‐dependent blockers targeting high‐frequency neuronal firing may avoid the confounding actions of state‐independent blockers (Ce, central nucleus of amygdala; LC, locus coeruleus; PAG, periaqueductal gray; RVM, rostral ventromedial medulla; VP, ventral posterior thalamus).
Z160 is anti‐nociceptive in a range of animal models when examined against withdrawal reflexes (Lee and Snutch, 2013) but failed in phase II trials with ongoing pain as the primary endpoint. It is conceivable that a failure to consider heterogeneity in patients during trial design led to an overall negative outcome. Drawing parallels with anticonvulsant sodium channel blockers which are more efficacious in patients with the irritable nociceptor phenotype, that is, where peripheral afferents are hyperexcitable and patients have evoked hypersensitivity, a state‐dependent calcium channel blocker could also be efficacious in this subgroup (Bouhassira and Attal, 2016; Baron et al., 2017). Likewise, ABT‐639 failed to ameliorate ongoing pain in diabetic polyneuropathy patients (Ziegler et al., 2015). Interestingly, ABT‐639 has no effect on spontaneous firing of WDR neurones in SNL rats but inhibits mechanically evoked firing (Jarvis et al., 2014). Diabetic polyneuropathy is most commonly associated with the ‘sensory loss’ phenotype, whereas a smaller proportion are classified under the ‘mechanical hyperalgesia’ phenotype (Baron et al., 2017); it is possible that ABT‐639 may have had higher efficacy in this subgroup in this trial (Ziegler et al., 2015). The inability of ABT‐639 to cross the blood–brain barrier could also have contributed to an overall negative result.
Conclusion
Despite disappointing clinical trials to date, it would be premature to consign calcium channels as a chronic pain target to the history books. A progressive approach to clinical trial design will be key to the success of future therapeutic approaches. Therapeutic agents displaying pathological state dependency would be preferable to avoid perturbing normal sensory function and to limit side effects. Poly‐pharmacy could be an alternative approach as gabapentin and ω‐conotoxins have demonstrated synergy with morphine (Matthews and Dickenson, 2002; Kolosov et al., 2011). It would be of interest to examine whether state‐dependent calcium channel blockers also exhibit similar synergistic effects. In the short term, patients receiving intrathecal ziconotide may benefit from improved analgesia and fewer side effects with combination therapy.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Conflict of interest
A.H.D has received research funding from Grünenthal GmbH.
Acknowledgements
This work was supported by the Wellcome Trust Pain Consortium (102645 – defining pain circuitry in health and disease) and the BonePain Network (642720 – Marie Sklodowska‐Curie Scholarship).
Patel, R. , Montagut‐Bordas, C. , and Dickenson, A. H. (2018) Calcium channel modulation as a target in chronic pain control. British Journal of Pharmacology, 175: 2173–2184. doi: 10.1111/bph.13789.
References
- Abbadie C, McManus OB, Sun SY, Bugianesi RM, Dai G, Haedo RJ et al. (2010). Analgesic effects of a substituted N‐triazole oxindole (TROX‐1), a state‐dependent, voltage‐gated calcium channel 2 blocker. J Pharmacol Exp Ther 334: 545–555. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altier C, Dale CS, Kisilevsky AE, Chapman K, Castiglioni AJ, Matthews EA et al. (2007). Differential role of N‐type calcium channel splice isoforms in pain. J Neurosci 27: 6363–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews N, Loomis S, Blake R, Ferrigan L, Singh L, McKnight AT (2001). Effect of gabapentin‐like compounds on development and maintenance of morphine‐induced conditioned place preference. Psychopharmacology (Berl) 157: 381–387. [DOI] [PubMed] [Google Scholar]
- Arendt‐Nielsen L, Frokjaer JB, Staahl C, Graven‐Nielsen T, Huggins JP, Smart TS et al. (2007). Effects of gabapentin on experimental somatic pain and temporal summation. Reg Anesth Pain Med 32: 382–388. [DOI] [PubMed] [Google Scholar]
- Arendt‐Nielsen L, Mansikka H, Staahl C, Rees H, Tan K, Smart TS et al. (2011). A translational study of the effects of ketamine and pregabalin on temporal summation of experimental pain. Reg Anesth Pain Med 36: 585–591. [DOI] [PubMed] [Google Scholar]
- Attal N, Brasseur L, Parker F, Chauvin M, Bouhassira D (1998). Effects of gabapentin on the different components of peripheral and central neuropathic pain syndromes: a pilot study. Eur Neurol 40: 191–200. [DOI] [PubMed] [Google Scholar]
- Bannister K, Patel R, Goncalves L, Townson L, Dickenson AH (2015). Diffuse noxious inhibitory controls and nerve injury: restoring an imbalance between descending monoamine inhibitions and facilitations. Pain 156: 1803–1811. [DOI] [PubMed] [Google Scholar]
- Bannister K, Sikandar S, Bauer CS, Dolphin AC, Porreca F, Dickenson AH (2011). Pregabalin suppresses spinal neuronal hyperexcitability and visceral hypersensitivity in the absence of peripheral pathophysiology. Anesthesiology 115: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron R, Forster M, Binder A (2012). Subgrouping of patients with neuropathic pain according to pain‐related sensory abnormalities: a first step to a stratified treatment approach. Lancet Neurol 11: 999–1005. [DOI] [PubMed] [Google Scholar]
- Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G et al. (2017). Peripheral neuropathic pain: a mechanism‐related organizing principle based on sensory profiles. Pain 158: 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CS, Nieto‐Rostro M, Rahman W, Tran‐Van‐Minh A, Ferron L, Douglas L et al. (2009). The increased trafficking of the calcium channel subunit alpha2delta‐1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J Neurosci 29: 4076–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bee LA, Dickenson AH (2008). Descending facilitation from the brainstem determines behavioural and neuronal hypersensitivity following nerve injury and efficacy of pregabalin. Pain 140: 209–223. [DOI] [PubMed] [Google Scholar]
- Bouhassira D, Attal N (2016). Translational neuropathic pain research: a clinical perspective. Neuroscience 338: 27–35. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Zamponi GW (2016). Block of voltage‐gated calcium channels by peptide toxins. Neuropharmacology pii: S0028‐3908(16)30466‐X. https://doi.org/10.1016/j.neuropharm.2016.10.016. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL et al. (2011). Suppression of inflammatory and neuropathic pain by uncoupling CRMP‐2 from the presynaptic Ca(2)(+) channel complex. Nat Med 17: 822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham LJ, Dickenson AH (2013). The antinociceptive effect of milnacipran in the monosodium iodoacetate model of osteoarthritis pain and its relation to changes in descending inhibition. J Pharmacol Exp Ther 344: 696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiglioni AJ, Raingo J, Lipscombe D (2006). Alternative splicing in the C‐terminus of CaV2.2 controls expression and gating of N‐type calcium channels. J Physiol 576: 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Pogrel JW, Yaksh TL (1994). Role of voltage‐dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther 269: 1117–1123. [PubMed] [Google Scholar]
- Chapman V, Suzuki R, Chamarette HL, Rygh LJ, Dickenson AH (1998). Effects of systemic carbamazepine and gabapentin on spinal neuronal responses in spinal nerve ligated rats. Pain 75: 261–272. [DOI] [PubMed] [Google Scholar]
- Chi XX, Schmutzler BS, Brittain JM, Wang Y, Hingtgen CM, Nicol GD et al. (2009). Regulation of N‐type voltage‐gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein‐2 (CRMP‐2) in sensory neurons. J Cell Sci 122: 4351–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizh BA, Gohring M, Troster A, Quartey GK, Schmelz M, Koppert W (2007). Effects of oral pregabalin and aprepitant on pain and central sensitization in the electrical hyperalgesia model in human volunteers. Br J Anaesth 98: 246–254. [DOI] [PubMed] [Google Scholar]
- Curros‐Criado MM, Herrero JF (2007). The antinociceptive effect of systemic gabapentin is related to the type of sensitization‐induced hyperalgesia. J Neuroinflammation 4: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB et al. (2014). The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double‐blind, placebo‐controlled phenotype‐stratified study. Pain 155: 2263–2273. [DOI] [PubMed] [Google Scholar]
- Ding L, Cai J, Guo XY, Meng XL, Xing GG (2014). The antiallodynic action of pregabalin may depend on the suppression of spinal neuronal hyperexcitability in rats with spared nerve injury. Pain Res Manag 19: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks J, Petersen KL, Rowbotham MC, Dahl JB (2002). Gabapentin suppresses cutaneous hyperalgesia following heat‐capsaicin sensitization. Anesthesiology 97: 102–107. [DOI] [PubMed] [Google Scholar]
- Dong L, Crosby ND, Winkelstein BA (2013). Gabapentin alleviates facet‐mediated pain in the rat through reduced neuronal hyperexcitability and astrocytic activation in the spinal cord. J Pain 14: 1564–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan‐Rodriguez T, Dickenson AH, Urch CE (2005). Gabapentin normalizes spinal neuronal responses that correlate with behavior in a rat model of cancer‐induced bone pain. Anesthesiology 102: 132–140. [DOI] [PubMed] [Google Scholar]
- Ebersberger A, Portz S, Meissner W, Schaible HG, Richter F (2004). Effects of N‐, P/Q‐ and L‐type calcium channel blockers on nociceptive neurones of the trigeminal nucleus with input from the dura. Cephalalgia 24: 250–261. [DOI] [PubMed] [Google Scholar]
- Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su T‐Z et al. (2006). Identification of the α2‐δ‐1 subunit of voltage‐dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci 103: 17537–17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MJ, Oles RJ, Lewis AS, McCleary S, Hughes J, Singh L (1997). Gabapentin (neurontin) and S‐(+)‐3‐isobutylgaba represent a novel class of selective antihyperalgesic agents. Br J Pharmacol 121: 1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn DP, Chapman V, Jhaveri MD, Samanta S, Manders T, Bowden J et al. (2003). The role of the central nucleus of the amygdala in nociception and aversion. Neuroreport 14: 981–984. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Attal N, Haroutounian S, Mcnicol E, Baron R, Dworkin RH et al. (2015). Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurol 14: 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerup NB, Sindrup SH, Jensen TS (2010). The evidence for pharmacological treatment of neuropathic pain. Pain 150: 573–581. [DOI] [PubMed] [Google Scholar]
- Francois‐Moutal L, Wang Y, Moutal A, Cottier KE, Melemedjian OK, Yang X et al. (2015). A membrane‐delimited N‐myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain 156: 1247–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman R, Baron R, Bouhassira D, Cabrera J, Emir B (2014). Sensory profiles of patients with neuropathic pain based on the neuropathic pain symptoms and signs. Pain 155: 367–376. [DOI] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN (1996). The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem 271: 5768–5776. [DOI] [PubMed] [Google Scholar]
- Goncalves L, Dickenson AH (2012). Asymmetric time‐dependent activation of right central amygdala neurones in rats with peripheral neuropathy and pregabalin modulation. Eur J Neurosci 36: 3204–3213. [DOI] [PubMed] [Google Scholar]
- Griggs RB, Bardo MT, Taylor BK (2015). Gabapentin alleviates affective pain after traumatic nerve injury. Neuroreport 26: 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama S, Wakamori M, Ino M, Miyamoto N, Takahashi E, Yoshinaga T et al. (2001). Differential nociceptive responses in mice lacking the alpha(1B) subunit of N‐type Ca(2+) channels. Neuroreport 12: 2423–2427. [DOI] [PubMed] [Google Scholar]
- Havelin J, Imbert I, Cormier J, Allen J, Porreca F, King T (2016). Central sensitization and neuropathic features of ongoing pain in a rat model of advanced osteoarthritis. J Pain 17: 374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich J, Van Minh AT, Heblich F, Nieto‐Rostro M, Watschinger K, Striessnig J et al. (2008). Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci U S A 105: 3628–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagodic MM, Pathirathna S, Joksovic PM, Lee W, Nelson MT, Naik AK et al. (2008). Upregulation of the T‐type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J Neurophysiol 99: 3151–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MF, Scott VE, McGaraughty S, Chu KL, Xu J, Niforatos W et al. (2014). A peripherally acting, selective T‐type calcium channel blocker, ABT‐639, effectively reduces nociceptive and neuropathic pain in rats. Biochem Pharmacol 89: 536–544. [DOI] [PubMed] [Google Scholar]
- Jiang YQ, Andrade A, Lipscombe D (2013). Spinal morphine but not ziconotide or gabapentin analgesia is affected by alternative splicing of voltage‐gated calcium channel CaV2.2 pre‐mRNA. Mol Pain 9: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Vera‐Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J et al. (2009). Unmasking the tonic‐aversive state in neuropathic pain. Nat Neurosci 12: 1364–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight YE, Bartsch T, Kaube H, Goadsby PJ (2002). P/Q‐type calcium‐channel blockade in the periaqueductal gray facilitates trigeminal nociception: a functional genetic link for migraine? J Neurosci 22: RC213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolosov A, Aurini L, Williams ED, Cooke I, Goodchild CS (2011). Intravenous injection of leconotide, an omega conotoxin: synergistic antihyperalgesic effects with morphine in a rat model of bone cancer pain. Pain Med 12: 923–941. [DOI] [PubMed] [Google Scholar]
- Lana B, Schlick B, Martin S, Pratt WS, Page KM, Goncalves L et al. (2014). Differential upregulation in DRG neurons of an alpha2delta‐1 splice variant with a lower affinity for gabapentin after peripheral sensory nerve injury. Pain 155: 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc BW, Lii TR, Huang JJ, Chao YC, Bowary PM, Cross BS et al. (2016). T‐type calcium channel blocker Z944 restores cortical synchrony and thalamocortical connectivity in a rat model of neuropathic pain. Pain 157: 255–263. [DOI] [PubMed] [Google Scholar]
- Lee M, Snutch T (2013). Z160: a potent and state‐dependent, small molecule blocker of N‐type calcium channels effective in nonclinical models of neuropathic pain. J Pain 14: S71–S71. [Google Scholar]
- Li J, Simone DA, Larson AA (1999). Windup leads to characteristics of central sensitization. Pain 79: 75–82. [DOI] [PubMed] [Google Scholar]
- Luebke JI, Dunlap K, Turner TJ (1993). Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron 11: 895–902. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Bee LA, Stephens GJ, Dickenson AH (2007). The Cav2.3 calcium channel antagonist SNX‐482 reduces dorsal horn neuronal responses in a rat model of chronic neuropathic pain. Eur J Neurosci 25: 3561–3569. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Dickenson AH (2001a). Effects of spinally delivered N‐ and P‐type voltage‐dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain 92: 235–246. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Dickenson AH (2001b). Effects of ethosuximide, a T‐type Ca(2+) channel blocker, on dorsal horn neuronal responses in rats. Eur J Pharmacol 415: 141–149. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Dickenson AH (2002). A combination of gabapentin and morphine mediates enhanced inhibitory effects on dorsal horn neuronal responses in a rat model of neuropathy. Anesthesiology 96: 633–640. [DOI] [PubMed] [Google Scholar]
- Meng G, Wu N, Zhang C, Su RB, Lu XQ, Liu Y et al. (2008). Analgesic activity of ZC88, a novel N‐type voltage‐dependent calcium channel blocker, and its modulation of morphine analgesia, tolerance and dependence. Eur J Pharmacol 586: 130–138. [DOI] [PubMed] [Google Scholar]
- Morisset V, Nagy F (1999). Ionic basis for plateau potentials in deep dorsal horn neurons of the rat spinal cord. J Neurosci 19: 7309–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebe J, Vanegas H, Neugebauer V, Schaible HG (1997). Omega‐agatoxin IVA, a P‐type calcium channel antagonist, reduces nociceptive processing in spinal cord neurons with input from the inflamed but not from the normal knee joint – an electrophysiological study in the rat in vivo . Eur J Neurosci 9: 2193–2201. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Vanegas H, Nebe J, Rumenapp P, Schaible HG (1996). Effects of N‐ and L‐type calcium channel antagonists on the responses of nociceptive spinal cord neurons to mechanical stimulation of the normal and the inflamed knee joint. J Neurophysiol 76: 3740–3749. [DOI] [PubMed] [Google Scholar]
- Niesters M, Proto PL, Aarts L, Sarton EY, Drewes AM, Dahan A (2014). Tapentadol potentiates descending pain inhibition in chronic pain patients with diabetic polyneuropathy. Br J Anaesth 113: 148–156. [DOI] [PubMed] [Google Scholar]
- Ohtori S, Inoue G, Orita S, Takaso M, Eguchi Y, Ochiai N et al. (2013). Efficacy of combination of meloxicam and pregabalin for pain in knee osteoarthritis. Yonsei Med J 54: 1253–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtori S, Orita S, Yamashita M, Ishikawa T, Ito T, Shigemura T et al. (2012). Existence of a neuropathic pain component in patients with osteoarthritis of the knee. Yonsei Med J 53: 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Kagaya K, Enomoto R, Sasaki A, Andoh T, Nojima H et al. (2009). A mouse model of sural nerve injury‐induced neuropathy: gabapentin inhibits pain‐related behaviors and the hyperactivity of wide‐dynamic range neurons in the dorsal horn. J Pharmacol Sci 109: 532–539. [DOI] [PubMed] [Google Scholar]
- Park HJ, Sandor K, McQueen J, Woller SA, Svensson CI, Corr M et al. (2016). The effect of gabapentin and ketorolac on allodynia and conditioned place preference in antibody‐induced inflammation. Eur J Pain 20: 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Bauer CS, Nieto‐Rostro M, Margas W, Ferron L, Chaggar K et al. (2013). alpha2delta‐1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. J Neurosci 33: 16412–16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Dickenson AH (2016). Neuronal hyperexcitability in the ventral posterior thalamus of neuropathic rats: modality selective effects of pregabalin. J Neurophysiol 116: 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Rutten K, Valdor M, Schiene K, Wigge S, Schunk S et al. (2015). Electrophysiological characterization of activation state‐dependent Ca(v)2 channel antagonist TROX‐1 in spinal nerve injured rats. Neuroscience 297: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percie du Sert N, Rice AS (2014). Improving the translation of analgesic drugs to the clinic: animal models of neuropathic pain. Br J Pharmacol 171: 2951–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwani H, Lopez‐Gonzalez MJ, Cattaert D, Roca‐Lapirot O, Dobremez E, Bouali‐Benazzouz R et al. (2016). Cav1.2 and Cav1.3 L‐type calcium channels independently control short‐ and long‐term sensitization to pain. J Physiol 594: 6607–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman W, Bauer CS, Bannister K, Vonsy JL, Dolphin AC, Dickenson AH (2009). Descending serotonergic facilitation and the antinociceptive effects of pregabalin in a rat model of osteoarthritic pain. Mol Pain 5: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman W, Patel R, Dickenson AH (2015). Electrophysiological evidence for voltage‐gated calcium channel 2 (Cav2) modulation of mechano‐ and thermosensitive spinal neuronal responses in a rat model of osteoarthritis. Neuroscience 305: 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose KE, Lunardi N, Boscolo A, Dong X, Erisir A, Jevtovic‐Todorovic V et al. (2013). Immunohistological demonstration of CaV3.2 T‐type voltage‐gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience 250: 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saegusa H, Kurihara T, Zong S, Kazuno A, Matsuda Y, Nonaka T et al. (2001). Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N‐type Ca2+ channel. EMBO J 20: 2349–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saegusa H, Kurihara T, Zong S, Minowa O, Kazuno A, Han W et al. (2000). Altered pain responses in mice lacking alpha 1E subunit of the voltage‐dependent Ca2+ channel. Proc Natl Acad Sci U S A 97: 6132–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford M (2013). Intrathecal ziconotide: a review of its use in patients with chronic pain refractory to other systemic or intrathecal analgesics. CNS Drugs 27: 989–1002. [DOI] [PubMed] [Google Scholar]
- Santicioli P, Del Bianco E, Tramontana M, Geppetti P, Maggi CA (1992). Release of calcitonin gene‐related peptide like‐immunoreactivity induced by electrical field stimulation from rat spinal afferents is mediated by conotoxin‐sensitive calcium channels. Neurosci Lett 136: 161–164. [DOI] [PubMed] [Google Scholar]
- Segerdahl M (2006). Multiple dose gabapentin attenuates cutaneous pain and central sensitisation but not muscle pain in healthy volunteers. Pain 125: 158–164. [DOI] [PubMed] [Google Scholar]
- Shin JB, Martinez‐Salgado C, Heppenstall PA, Lewin GR (2003). A T‐type calcium channel required for normal function of a mammalian mechanoreceptor. Nat Neurosci 6: 724–730. [DOI] [PubMed] [Google Scholar]
- Simpson DM, Schifitto G, Clifford DB, Murphy TK, Durso‐De Cruz E, Glue P et al. (2010). Pregabalin for painful HIV neuropathy: a randomized, double‐blind, placebo‐controlled trial. Neurology 74: 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Dickenson AH (2006). Differential pharmacological modulation of the spontaneous stimulus‐independent activity in the rat spinal cord following peripheral nerve injury. Exp Neurol 198: 72–80. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Rahman W, Rygh LJ, Webber M, Hunt SP, Dickenson AH (2005). Spinal‐supraspinal serotonergic circuits regulating neuropathic pain and its treatment with gabapentin. Pain 117: 292–303. [DOI] [PubMed] [Google Scholar]
- Swensen AM, Herrington J, Bugianesi RM, Dai G, Haedo RJ, Ratliff KS et al. (2012). Characterization of the substituted N‐triazole oxindole TROX‐1, a small‐molecule, state‐dependent inhibitor of Ca(V)2 calcium channels. Mol Pharmacol 81: 488–497. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Takasu K, Ono H, Tanabe M (2007). Pregabalin, S‐(+)‐3‐isobutylgaba, activates the descending noradrenergic system to alleviate neuropathic pain in the mouse partial sciatic nerve ligation model. Neuropharmacology 53: 842–853. [DOI] [PubMed] [Google Scholar]
- Terashima T, Xu Q, Yamaguchi S, Yaksh TL (2013). Intrathecal P/Q‐ and R‐type calcium channel blockade of spinal substance P release and c‐Fos expression. Neuropharmacology 75: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur M, Rahman W, Hobbs C, Dickenson AH, Bennett DL (2012). Characterisation of a peripheral neuropathic component of the rat monoiodoacetate model of osteoarthritis. PLoS One 7: e33730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran‐Van‐Minh A, Dolphin AC (2010). The alpha2delta ligand gabapentin inhibits the Rab11‐dependent recycling of the calcium channel subunit alpha2delta‐2. J Neurosci 30: 12856–12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban MO, Ren K, Sablad M, Park KT (2005). Medullary N‐type and P/Q‐type calcium channels contribute to neuropathy‐induced allodynia. Neuroreport 16: 563–566. [DOI] [PubMed] [Google Scholar]
- Van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N (2014). Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 155: 654–662. [DOI] [PubMed] [Google Scholar]
- Werner MU, Perkins FM, Holte K, Pedersen JL, Kehlet H (2001). Effects of gabapentin in acute inflammatory pain in humans. Reg Anesth Pain Med 26: 322–328. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hoskins L, Catterall WA (1998). Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci 18: 6319–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SM, Brittain JM, Piekarz AD, Ballard CJ, Ripsch MS, Cummins TR et al. (2011). Further insights into the antinociceptive potential of a peptide disrupting the N‐type calcium channel‐CRMP‐2 signaling complex. Channels (Austin) 5: 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao WH, Bennett GJ (1995). Synthetic omega‐conopeptides applied to the site of nerve injury suppress neuropathic pains in rats. J Pharmacol Exp Ther 274: 666–672. [PubMed] [Google Scholar]
- Xie JY, Chew LA, Yang X, Wang Y, Qu C, Wang Y et al. (2016). Sustained relief of ongoing experimental neuropathic pain by a CRMP2 peptide aptamer with low abuse potential. Pain 157: 2124–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JY, Qu C, Patwardhan A, Ossipov MH, Navratilova E, Becerra L et al. (2014). Activation of mesocorticolimbic reward circuits for assessment of relief of ongoing pain: a potential biomarker of efficacy. Pain 155: 1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chu KL, Zhu CZ, Niforatos W, Swensen A, Searle X et al. (2014). A mixed Ca2+ channel blocker, A‐1264087, utilizes peripheral and spinal mechanisms to inhibit spinal nociceptive transmission in a rat model of neuropathic pain. J Neurophysiol 111: 394–404. [DOI] [PubMed] [Google Scholar]
- Yarnitsky D, Granot M, Nahman‐Averbuch H, Khamaisi M, Granovsky Y (2012). Conditioned pain modulation predicts duloxetine efficacy in painful diabetic neuropathy. Pain 153: 1193–1198. [DOI] [PubMed] [Google Scholar]
- Zamponi GW (2016). Targeting voltage‐gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov 15: 19–34. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A, Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67: 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shao G, Zhang W, Li S, Niu J, Hu D et al. (2013). Gabapentin inhibits central sensitization during migraine. Neural Regen Res 8: 3003–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Yang L, Zhang K, Liu X, Dai W, Zhang C et al. (2015). ZC88, a novel N‐type calcium channel blocker from 4‐amino‐piperidine derivatives state‐dependent inhibits Cav2.2 calcium channels. Brain Res 1605: 12–21. [DOI] [PubMed] [Google Scholar]
- Zhu CZ, Vortherms TA, Zhang M, Xu J, Swensen AM, Niforatos W et al. (2014). Mechanistic insights into the analgesic efficacy of A‐1264087, a novel neuronal Ca(2+) channel blocker that reduces nociception in rat preclinical pain models. J Pain 15: 387 e1–387 14. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Duan WR, An G, Thomas JW, Nothaft W (2015). A randomized double‐blind, placebo‐, and active‐controlled study of T‐type calcium channel blocker ABT‐639 in patients with diabetic peripheral neuropathic pain. Pain 156: 2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]