

Abstract
Background and Purpose
Voltage‐gated calcium channels are involved in nociception in the CNS and in the periphery. N‐type (Cav2.2) and T‐type (Cav3.1, Cav3.2 and Cav3.3) voltage‐gated calcium channels are particularly important in studying and treating pain and epilepsy.
Experimental Approach
In this study, whole‐cell patch clamp electrophysiology was used to assess the potency and mechanism of action of a novel ortho‐phenoxylanilide derivative, MONIRO‐1, against a panel of voltage‐gated calcium channels including Cav1.2, Cav1.3, Cav2.1, Cav2.2, Cav2.3, Cav3.1, Cav3.2 and Cav3.3.
Key Results
MONIRO‐1 was 5‐ to 20‐fold more potent at inhibiting human T‐type calcium channels, hCav3.1, hCav3.2 and hCav3.3 (IC50: 3.3 ± 0.3, 1.7 ± 0.1 and 7.2 ± 0.3 μM, respectively) than N‐type calcium channel, hCav2.2 (IC50: 34.0 ± 3.6 μM). It interacted with L‐type calcium channels Cav1.2 and Cav1.3 with significantly lower potency (IC50 > 100 μM) and did not inhibit hCav2.1 or hCav2.3 channels at concentrations as high as 100 μM. State‐ and use‐dependent inhibition of hCav2.2 channels was observed, whereas stronger inhibition occurred at high stimulation frequencies for hCav3.1 channels suggesting a different mode of action between these two channels.
Conclusions and Implications
Selectivity, potency, reversibility and multi‐modal effects distinguish MONIRO‐1 from other low MW inhibitors acting on Cav channels involved in pain and/or epilepsy pathways. High‐frequency firing increased the affinity for MONIRO‐1 for both hCav2.2 and hCav3.1 channels. Such Cav channel modulators have potential clinical use in the treatment of epilepsies, neuropathic pain and other nociceptive pathophysiologies.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Abbreviations
- hERG
human ether à‐go‐go related gene
- SSI
steady‐state inactivation
- VGCCs
voltage‐gated calcium channels
Introduction
Sensory nerve conduction relies on the coordinated activation of a suite of ion channels to generate fast and accurate responses to mechanical, thermal and chemical stimuli. In the periphery, harmful signals are perceived by ‘nociceptive’ neurons and relayed centrally via voltage‐activated ion channels (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=81, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=82, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=80) and ligand‐gated ion channels (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=78, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=79, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=77) (Waxman and Zamponi, 2014) as part of the ‘pain‐signalling cascade’ (Gold and Gebhart, 2010). In chronic pain, these pathways are characterized by hyperactivity (Waxman and Zamponi, 2014), and the effects of noxious stimuli are enhanced (hyperalgesia) to the point where non‐noxious stimuli are painful (allodynia) (see Guo and Hu, 2014). Voltage‐gated calcium channels (VGCCs) involved in pain pathways are localized to dendrites of primary sensory neurons and contribute to integrate nociceptive stimuli at the spine by regulating the release of pro‐nociceptive neurotransmitters, including substance P, calcitonin gene‐related peptide and glutamate, at the presynaptic nerve terminus (Simms and Zamponi, 2014). Therefore, modulating specific Cav subtypes could be a promising way to treat chronic pain.
Five distinct voltage‐gated calcium current types have been described in mammalian cells: L‐ (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=528&familyId=80&familyType=IC, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=529&familyId=80&familyType=IC, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=530&familyId=80&familyType=IC and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=531&familyId=80&familyType=IC), P/Q‐ (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=532&familyId=80&familyType=IC), N‐ (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=533&familyId=80&familyType=IC), R‐ (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=534&familyId=80&familyType=IC) and T‐ (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=535&familyId=80&familyType=IC, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=536&familyId=80&familyType=IC, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=537&familyId=80&familyType=IC) types (Catterall et al., 2005). Importantly, each Cav subtype has distinct electrophysiological properties, tissue‐specific localization and pharmacological profiles (Catterall et al., 2005). N‐type calcium channels have been a major focus of research because they are localized at the presynaptic terminals of sensory nerves in the dorsal horn of the spinal cord, where they play a critical role in pain signalling (Saegusa et al., 2001; Lee, 2014). As a result, Cav2.2 channels are strongly implicated in a range of chronic pain conditions including neuropathic pain (pain caused by injury to the nervous system). Specific Cav2.2 channel inhibitors have clinical applications (Altier and Zamponi, 2004; Perret and Luo, 2009; Bourinet et al., 2014), such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2536 (Ziconotide). Ziconotide selectively targets Cav2.2 channels and is used to effectively treat intractable chronic pain conditions (Pope and Deer, 2013). However, due to its chemical nature, Ziconotide must be administered into the cerebrospinal fluid via the intrathecal route and has a relatively narrow therapeutic window (Pope and Deer, 2013).
T‐type calcium channels modulate neuronal excitability in the peripheral and central nervous systems, where they are linked to hyperexcitable states such as epilepsy, pain and tremor disorders like Parkinson's disease (Bourinet et al., 2005; Choi et al., 2007; Kim et al., 2001). Previously characterized T‐type calcium channels inhibitors, such as mibefradil and ethoximide, have analgesic properties, suggesting that T‐type channels may be involved in pain signalling (Matthews and Dickenson, 2001; Todorovic et al., 2002; Flatters and Bennett, 2004; Wen et al., 2010). Unfortunately, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2522 and ethoximide target a wide range of voltage‐gated ion channels, including sodium, potassium and chloride channels (Chouabe et al., 1998; Nilius et al., 1998; Eller et al., 2000).
To overcome this lack of specificity, small molecule T‐type calcium channel inhibitors were designed and have been shown to provide analgesic effects in mouse models of inflammatory and neuropathic pain (Kraus et al., 2010; Reger et al., 2011; Bladen et al., 2015). Of particular note is the analgesic, low MW compound http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7724, which is more potent against inactivated than closed or resting channels (IC50 values of 100 and 10 μM at Cav3.1 respectively), indicating its effects on targets are state‐dependent (Francois et al., 2013). Based on these findings, N‐ and T‐type calcium channels are of particular interest for the study and treatment of chronic pain (see recent reviews by Cheong and Shin, 2013; Lee, 2013; Sekiguchi and Kawabata, 2013; François et al., 2014; Lee, 2014; Simms and Zamponi, 2014).
During the last 5 years, numerous reports have described small molecule Cav2.2 channel inhibitors as alternatives to Ziconotide (see review by Lee, 2014). Regrettably, clinical development of the most promising of these small‐molecules, Z160, a reformulated form of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7765 (Zamponi et al., 2009) (compound 1, Figure 1), was discontinued after it failed to meet end points in phase II clinical trials (Eller et al., 2000). Due to this and the need for effective and safe therapeutics for chronic pain, the development of new classes of small‐molecule Cav channel antagonists is of significant interest.
Figure 1.
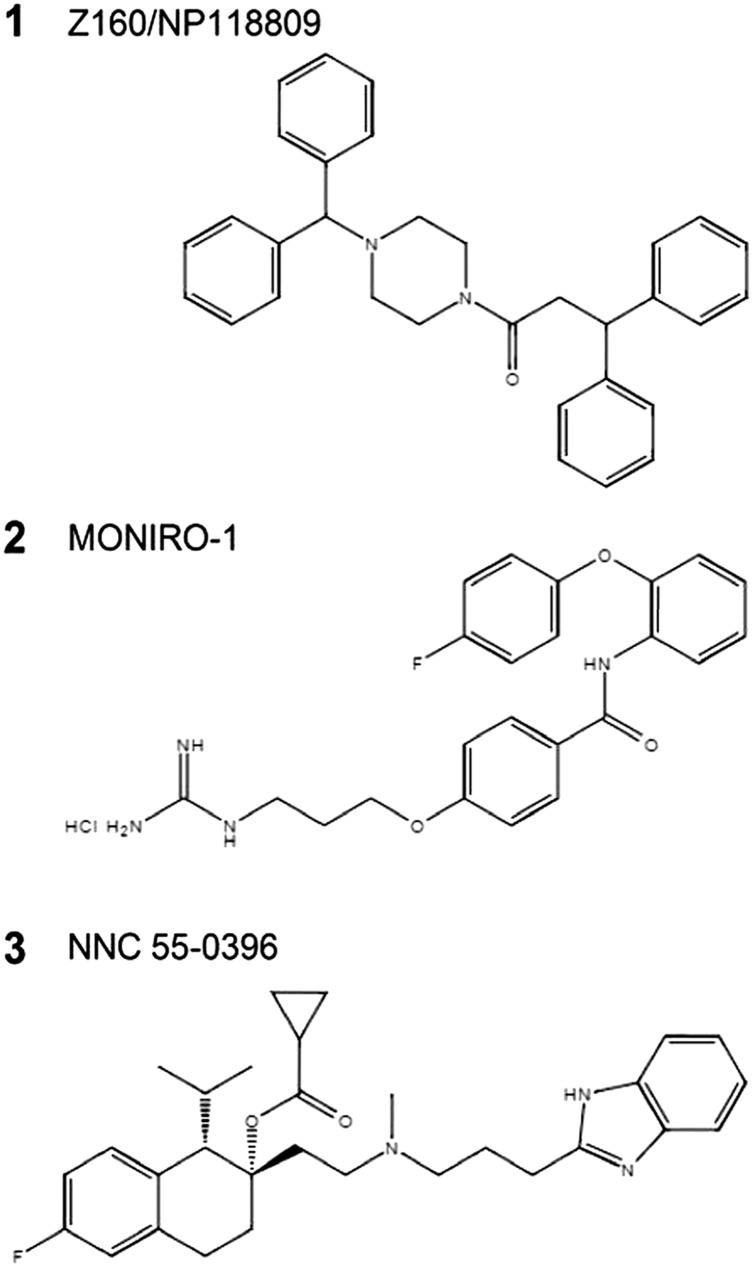
Chemical structures of compounds 1 (Z160/NP118809), 2 (MONIRO‐1) and 3 (NNC 55‐0396) relevant to this study.
The pharmacophore of Cav2.2‐selective http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2535 (McCleskey et al., 1987; Baell et al., 2004; Baell et al., 2006; Andersson et al., 2009; Duggan et al., 2009) has been carefully studied and has led to the identification of a series of compounds that block Cav2.2 channels in a concentration‐dependent manner (Tranberg et al., 2012).
We focused on developing low MW calcium channel inhibitors with enhanced biophysical properties including reversibility, voltage‐ and/or state‐dependence. Compounds acting via these mechanisms can strongly inhibit channels upon membrane depolarization (Hondeghem and Katzung, 1984; Butterworth and Strichartz, 1990) by preferentially targeting the open and/or inactivated states over the resting state. Such compounds would facilitate inhibition of high‐frequency firing or depolarised nerves that can predominate in chronic pain states and could act within a wider therapeutic window.
To develop these calcium channel inhibitors, we screened a small library of related derivatives, using FLIPR Tetra® high‐throughput cellular screening system, for their ability to inhibit Cav2.2‐mediated calcium responses in SH‐SY5Y cells using the fluorescent indicator Calcium 4 dye (Molecular Devices, Sunnyvale, CA, USA) (Gleeson et al., 2015). In this assay, MONIRO‐1 (compound 2, Figure 1) inhibited Ca2+ responses elicited by KCl‐mediated depolarization with an IC50 of 124 μM. Subsequently, we characterized the ortho‐phenoxyanilide derivative MONIRO‐1 electrophysiologically. To directly assess the biophysical properties of MONIRO‐1 inhibition of VGCCs, we used the whole‐cell patch clamp recording technique to study a comprehensive array of Cav channels heterologously or stably expressed in HEK‐293 cells. MONIRO‐1 was 5–100 times more potent against T‐type calcium channels (Cav3.1, Cav3.2 and Cav3.3) than N‐type (Cav2.2) and L‐type (Cav1.2 and Cav1.3) channels. No inhibition of P/Q (Cav2.1) or R‐type currents (Cav2.3) was detected in the presence of up to 100 μM MONIRO‐1. Furthermore, MONIRO‐1 modulated Cav2.2 differently at various stimulation rates and in a state‐dependent fashion, preferentially inhibiting hyperactive channels such as those likely to predominate in pathologically augmented pain states.
Methods
Cell culture and transfection
HEK293 cells containing the SV40 Large T‐antigen (HEK293T) were cultured at 37°C, 5% CO2 in Dulbecco's Modified Eagle's Medium (Invitrogen Life Technologies, Australia) supplemented with 10% fetal bovine serum (Invitrogen), 1% penicillin and streptomycin (Invitrogen). HEK293T cells stably expressing the human Cav2.1 (P/Q‐type) calcium channel (α1A‐2 splice variant; GenBank accession no. AF004883.1), Cav2.2 (N‐type) calcium channel (transcript variant 1, NM_000718) or Cav2.3 (R‐type) calcium channel (neuronal α1E‐3 splice variant, also called α1E‐c, L29385.1) were obtained from Merck and cultured according to procedures described by Dai et al. (2008). These cell lines co‐express recombinant human Cav α2bδ‐1 (M76559.1) and Cav β3a (NM_000725) auxiliary subunits (Dai et al., 2008). Cells are plated onto 12 mm cover glass and incubated at 30°C, 5% CO2 for 24–48 h prior to recording.
In a separate series of experiments, HEK293T cells were transiently co‐transfected with plasmid cDNAs encoding mouse Cav1.2 (provided by Dr D. Lipscombe, Addgene plasmid # 26572; Helton et al., 2005) or human Cav1.3 (provided by Dr G. Zamponi, University of Calgary) with α2δ‐1 and β3, Cav3.1 (provided by Dr G. Zamponi), human Cav3.2 (CACNA1HB; Origene Technologies, Rockville, MD), human Cav3.3 a1Ic‐HE3‐pcDNA3 was a gift from Dr E. Perez‐Reyes (Gomora et al., 2002) (Addgene plasmid # 45810) and Kv11.1 (human ether à‐go‐go related gene [hERG]) (provided by Dr J. Vandenberg, Victor Chang Cardiac Research Institute, Sydney). After transfection, cells were plated on glass coverslips and incubated at 37°C, 5% CO2 overnight. The transfection medium was then replaced with culture medium, and cells were incubated at 30°C, 5% CO2.
Electrophysiology
Electrophysiological experiments were carried out 1–3 days after transfection using the whole‐cell patch clamp recording technique. Cells were superfused with a solution containing 110 mM NaCl, 10 mM CaCl2, 1 mM MgCl2, 5 mM CsCl, 30 mM TEA‐Cl, 10 mM D‐glucose and 10 mM HEPES, pH 7.4 with TEA‐OH (~310 mOsmol kg−1). Fire‐polished borosilicate patch pipettes with tip‐resistance values of 1–3 MΩ were filled with an intracellular solution containing 125 mM K+gluconate, 5 mM NaCl, 2 mM MgCl2, 5 mM EGTA, 4 mM MgATP and 10 mM HEPES, pH 7.2 with CsOH (~300 mOsmol kg−1). Depolarization‐activated calcium currents were recorded at room temperature (21–23°C) using a Multiclamp 700B patch clamp amplifier (Molecular Devices, Sunnyvale, CA, USA) controlled by Digidata 1322A/Clampex 9.2 acquisition systems. MONIRO‐1 stock solution was dissolved in 5% DMSO to a concentration of 5 mM. MONIRO‐1 was further diluted in recording solution to appropriate concentrations. Previous experiments using the maximal concentration of DMSO used, 0.1% at 100 μM, did not modulate VGCC currents in recording solution.
Depolarization‐activated calcium currents (ICa) were evoked from a holding potential of −80 mV, with a 135 ms (50 ms for T‐type) voltage step to a test potential (determined from peak of I‐V curve) applied every 10 s. When evaluating ICa amplitude in the presence and absence of various compounds, test depolarizations of varying durations were applied at a frequency of 0.03–2 Hz, from a holding potential of −60, −80 or −100 mV to a test potential determined from an I–V relationship to elicit peak inward current. To examine the effects of pulse duration on MONIRO‐1 block and recovery from block, pulses of either 45 or 135 ms for Cav2.2 (30 μM) or 15 or 50 ms for Cav3.1 (3 μM) were elicited at a frequency of 0.1 Hz (Vh = −80 mV). Similarly, to study the frequency dependence of MONIRO‐1 inhibition, test pulses (45 ms for Cav2.2 or 15 ms for Cav3.1) were elicited at rates of 0.03, 0.1 and 0.2 Hz for Cav2.2 (10 μM MONIRO‐1) and 0.03, 0.1, 0.2, 1 and 2 Hz for Cav3.1 (3 μM MONIRO‐1). To determine the time constants for activation and inactivation of macroscopic currents, we used a single exponential equation to fit the traces in control and after MONIRO‐1 application.
Activation curves were measured from test pulses from −80 to +80 mV every 5 mV for Cav2.2 and Cav3.1 channels (Vh = −80 mV) before and after MONIRO‐1 application. Steady‐state inactivation (SSI) curves were generated from various pre‐pulse potentials (−120 to 0 mV for Cav2.2; −85 to −20 mV for Cav3.1) of 10 s (Cav2.2) or 1 s (Cav3.1). Recovery from inactivation was examined using a double‐pulse protocol, varying the time between a fully inactivating pre‐pulse and the test pulse repeated every 15 s, using a concentration of MONIRO‐1, which inhibits ~50% of the current. Recovery data were best fit by a double exponential. Currents were filtered at 3 kHz and sampled at 10 kHz. Leak and capacitive currents were subtracted using a −P/4 pulse protocol. Data were stored digitally on a computer for further analysis.
Data analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data reported as mean ± SEM. Data analysis and graphs were built using OriginPro (Origin Lab Corporation, North Hampton, MA, USA). Concentration–response curves were generated by plotting current in the presence of MONIRO‐1 (Icompound) over current prior to application (Icontrol) versus a range of MONIRO‐1 concentrations tested. Typically, four to six concentrations per channel were screened with five to nine cells per concentration. Concentration–response data were plotted using a rectangular hyperbola assuming a Hill coefficient of 1, using the following expression,
where IC50 is the concentration at which half‐maximal inhibition was observed.
Steady‐state activation and inactivation curves were generated using a modified Boltzmann equation,
where Vm is the test potential, V0.5 is the half‐maximal activation potential and κa is the slope factor. SSA and SSI data were each recorded from five to seven individual experiments per channel examined. Recovery from inactivation curves were the results from five to six independent experiments where the complete set of recovery from inactivation time points were acquired. The data were fit using a double exponential of the following equation,
where τ is the fast and slow time constant, t is the recovery time and A is the amplitude of the fast and slow time constants. Statistical significance (P < 0.05) was determined using unpaired Student's t‐tests or one‐way ANOVA with Tukey's post hoc test.
Materials
Compound 2, N‐(2‐(4‐fluorophenoxy)phenyl)‐4‐(3‐guanidinopropoxy)benzamide hydrochloride, MONIRO‐1, was synthesized from 2‐(4‐fluorophenoxy)aniline in four steps, as reported previously (Gleeson et al., 2015). 1H NMR (400 MHz, CD3OD): δ 7.88–7.85 (m, 1H), 7.78–7.76 (m, 2H), 7.21–7.18 (m, 2H), 7.07–6.98 (m, 6H), 6.97–6.94 (m, 1H), 4.13 (t, J = 5.9 Hz, 2H), 3.41 (t, J = 6.8 Hz, 2H), 2.11–2.05 (m, 2H). 13C NMR (100 MHz, CD3OD): δ 168.2, 163.2, 160.2 (d, J = 239 Hz), 158.7, 154.4, 151.0, 130.5, 130.4, 127.9, 127.6, 126.9, 125.0, 120.8 (d, J = 8.3 Hz), 120.1, 117.2 (d, J = 23.4 Hz), 115.4, 66.3, 39.5, 29.5. 19F NMR (376 MHz, CD3OD): δ −120.6, purity >92%, determined by HPLC analysis (λ = 254 nm), all tests were performed using the same batch of compound. The positive control, compound 1 (Z160/NP118809) (Zikolova and Ninov, 1972; Zamponi et al., 2009), was prepared by N‐(3‐dimethylaminopropyl)‐N′‐ethylcarbodiimide‐assisted acylation of N‐benzhydryl piperazine with 3,3‐diphenylpropanoic acid. Control compound NNC 55–0396 dihydrochloride was provided by Alomone Labs (Jerusalem, Israel).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Selective inhibition of human N‐ and T‐type calcium channel subtypes
Whole‐cell calcium currents were elicited by depolarizing pulses at a frequency of 0.1 Hz from a holding potential of −80 mV. MONIRO‐1 reversibly inhibited channel currents mediated by Cav3.2 (IC50 1.7 ± 0.1 μM, n = 5–9 cells per concentration), Cav3.1 (IC50 3.3 ± 0.3 μM, n = 5–6 cells per concentration), Cav3.3 (IC50 7.2 ± 0.3 μM, n = 5 cells per concentration) and Cav2.2 (IC50 34.0 ± 3.6 μM, n = 5–7 cells per concentration) (Figures 2 and 3A). The effects of MONIRO‐1 were further investigated on a panel of VGCCs. At 100 μM, MONIRO‐1 had no apparent effect on Cav2.1 (n = 6) or Cav2.3 (n = 5) (Figure 3B) but inhibited Cav1.2 and Cav1.3‐mediated currents by 46 ± 1% (n = 5) and 27 ± 1% (n = 5) respectively.
Figure 2.
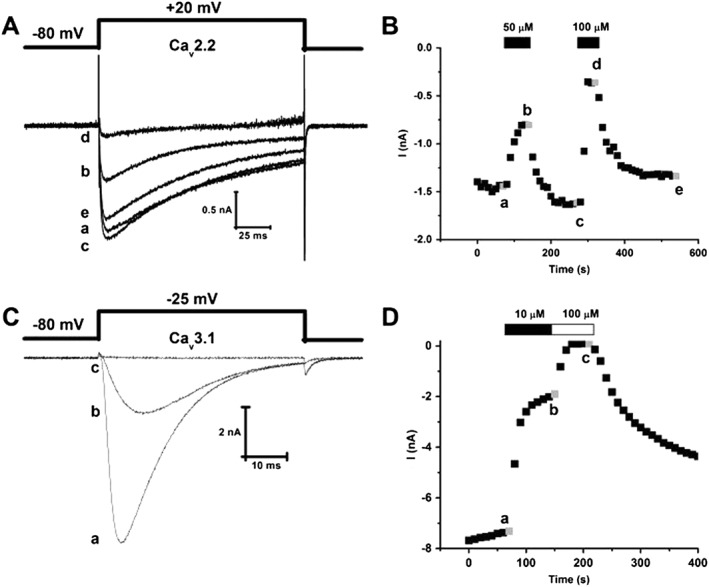
MONIRO‐1 inhibition of human Cav2.2‐ and Cav3.1‐mediated currents in HEK293 cells. (A) Superimposed whole‐cell Ca2+ currents recorded from HEK293 cells stably expressing human N‐type calcium (hCav2.2) channels (α1B + β3 + α2δ1) in response to depolarizing pulses (135 ms duration) to +20 mV from a holding potential of −80 mV. Representative current traces obtained in the absence (a) and presence of 50 (b) and 100 μM (d) MONIRO‐1 and after respective washouts (c, e). (B) Peak inward Ca2+ current amplitude plotted as a function of time. Time course of inhibition and current amplitude recovery in the presence and following washout of 50 and 100 μM MONIRO‐1. Representative currents shown in (A) were obtained at the times indicated by the lower case letters. (C) Superimposed whole‐cell Ca2+ currents recorded from HEK293 cells transiently expressing human T‐type calcium channel, hCav3.1. These Ca2+ currents are in response to depolarizing pulses (50 ms duration) to −25 mV from a holding potential of −80 mV. Representative current traces obtained in the absence (a) and presence of 10 μM (b) and 100 μM (c) MONIRO‐1. (D) Peak inward Ca2+ current amplitude plotted as a function of time. Time course of inhibition and recovery of current amplitude in the absence (a) and presence of 10 (b) and 100 μM (c) MONIRO‐1.
Figure 3.
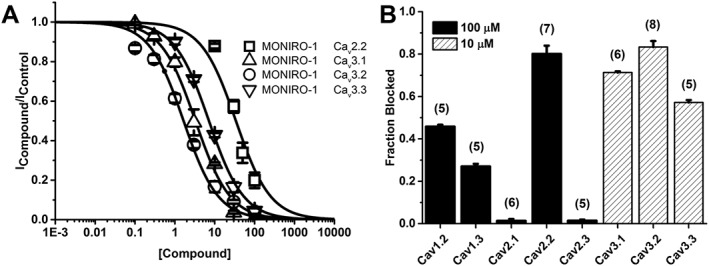
MONIRO‐1 inhibition of voltage‐gated calcium channels. (A) Concentration–response of MONIRO‐1 at hCav2.2, hCav3.1, hCav3.2 and hCav3.3 channels expressed in HEK293 cells. Fitted curves gave IC50 values as shown in the Results. (B) Fraction of current inhibited by 100 (solid) or 10 μM (striped) MONIRO‐1 at Cav1.3, Cav2.1, Cav2.2, Cav2.3, Cav3.1 and Cav3.2 channels. Cells were held at −80 mV and pulsed at 0.1 Hz to the voltage determined to elicit the peak inward current previously determined from an I–V protocol. The number of cells is indicated in parentheses.
In comparison, Z160/NP118809 (Zamponi et al., 2009) inhibited Cav2.2‐mediated currents with an IC50 of 0.28 ± 0.02 μM (n = 5–6 cells per concentration), without affecting Cav3.1 currents (maximal concentration tested 1 μM, Table 1). Application of 10 μM NNC 55‐0396, a T‐type calcium channel inhibitor, inhibited hCav3.2 currents by 96 ± 3% (n = 5). Furthermore, Z160/NP118809 inhibited http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=572 channels with an IC50 of 7.4 μM (Zamponi et al., 2009) whereas 100 μM MONIRO‐1 did not inhibit hERG channel‐mediated K+ currents (Table 1). Table 1 compares the IC50 values obtained for MONIRO‐1 inhibition of L‐, P/Q‐, N‐, R‐ and T‐type calcium channels and the hERG potassium channel, with that of the control compounds, Z160/NP118809 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4269, and other known N‐ and T‐type calcium channel inhibitors where electrophysiology data were obtained across a large portion of the VGCC subtypes.
Table 1.
Comparison of IC50 values obtained for MONIRO‐1 inhibition of L‐, P/Q‐, N‐, R‐ and T‐type calcium channels and the hERG potassium channel, with control compounds Z160/NP118809 and NNC 55‐0396 and other known N‐ and T‐type calcium channel inhibitors
| L‐type (μM) | P/Q‐type (μM) | N‐type (μM) | R‐type (μM) | T‐type (μM) | hERG (μM) | |
|---|---|---|---|---|---|---|
|
Z160/NP118809 (Zamponi et al., 2009) |
12b (Cav1.2) | – | 0.28a | – | – | 7.4b |
| aMONIRO‐1 |
27% @ 100 (Cav1.3) 46% @ 100 (Cav1.2) |
>100 | 34 | >100 |
3.3 (Cav3.1) 1.7 (Cav3.2) 7.2 (Cav3.3) |
>100 |
|
A‐1048400 (Scott et al., 2012) |
28 | 1.3 | 0.8 | – | 0.9 (Cav3.2) | – |
|
Z944 (Tringham et al., 2012) |
32 (Cav1.2) | – | 11 | – |
0.05 (Cav3.1) 0.16 (Cav3.2) |
7.8 |
|
NNC 55–0396 (Chen et al., 2014) |
– | – | – | – |
6.8b (Cav3.1) 96% @10a (Cav3.2) |
– |
|
Ziconotide (Wang et al., 2016) |
– | >10 | 0.18b | – | – | – |
|
TTA‐A2 (Kraus et al., 2010) |
>30b (Cav1.2) | >30b | >30bRecovery from block | >30b |
0.09 (Cav3.1) 0.09 (Cav3.2) |
– |
|
TROX‐1 (Swensen et al., 2012) |
– | 0.29 | 0.19 | 0.28 | – | – |
This work.
Studies were performed with HEK293 cells stably expressing the desired voltage‐gated calcium channel. –, not tested.
The current inhibition properties varied between the different MONIRO‐1‐sensitive Cav subtypes, so we analysed the interaction between MONIRO‐1 and T‐type (Cav3.1) and N‐type (Cav2.2) calcium channels.
Differential modulation of N‐ and T‐type calcium channels: dependence on frequency and holding potential
We examined the inhibitory characteristics of MONIRO‐1 at different test pulse durations and frequencies on Cav2.2 (Figure 4A, B) and Cav3.1 channels (Figure 5A, B), as representatives of the VGCC types targeted by MONIRO‐1. Cav2.2 currents in response to shorter (45 ms) depolarizations were 1.5‐times more sensitive to MONIRO‐1 inhibition than those elicited by a longer (135 ms) pulse. Channels held open by the longer pulse also recovered more completely from block (82 ± 9%, n = 5) than those briefly opened (45 ms; 47 ± 3%, n = 5) (Figure 4A). In agreement with these findings, higher frequency stimulation of Cav2.2 channels rendered a population of channels that was more potently inhibited by MONIRO‐1 (0.03, 0.1 and 0.2 Hz; Figure 4B). At a frequency of 0.2 Hz, MONIRO‐1 inhibited Cav2.2 current by 29 ± 1% (n = 5), whereas at 0.1 and 0.03 Hz, only 21 ± 1% (n = 5) and 16 ± 1% (n = 5) of the current amplitude was inhibited respectively. Interestingly, the effects of MONIRO‐1 on Cav2.2 channels are in marked contrast to its effects on Cav3.1 channels (Figure 5A, B). In the case of Cav3.1 channels, pulse duration did not affect Cav3.1‐mediated current inhibition (% block 15 ms: 43 ± 3%, n = 5; and 50 ms: 51 ± 7%, n = 5), recovery from inhibition (% recovery 15 ms: 95 ± 1.3%, n = 5; and 50 ms: 99 ± 0.8%, n = 5) or frequency dependence of block over the same range as those tested for Cav2.2 channels (% block at 0.2 Hz = 43 ± 1.5%, n = 5; 0.1 Hz = 42 ± 2.3%, n = 5; and 0.03 Hz = 44 ± 1.6%, n = 5). However, at faster rates of 1 and 2 Hz, there was a significant increase in MONIRO‐1 block of Cav3.1 channels (% block at 1 Hz = 61.0 ± 1.5%, n = 6; and 2 Hz = 71.0 ± 2.0%, n = 5). This would increase threefold the affinity for channels firing at faster frequencies of 2 Hz compared with those firing at rates <0.2 Hz.
Figure 4.
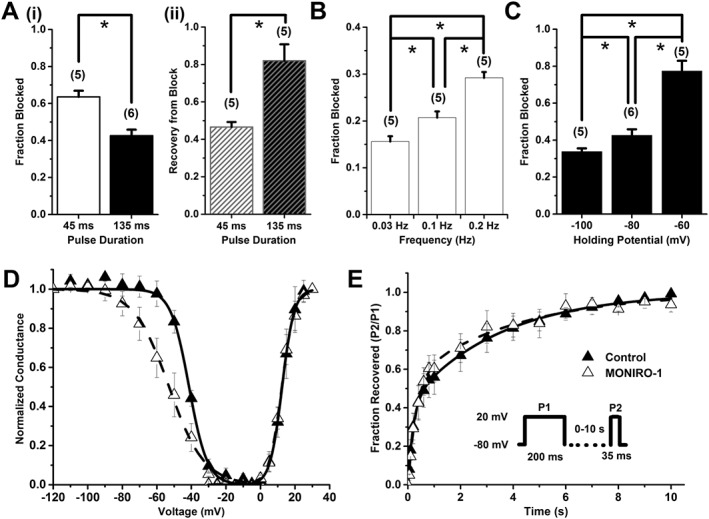
MONIRO‐1 block of human Cav2.2 channels. (A) (i) Fraction blocked and (ii) recovery from block by 30 μM MONIRO‐1, with a depolarizing pulse duration of 45 ms or 135 ms, from a holding potential of −80 mV to a test potential that elicits maximal inward current, repeated every 10 s. The number of cells is indicated in parentheses. (B) Fraction of current inhibited by 10 μM MONIRO‐1 at three different frequencies (0.03, 0.1 and 0.2 Hz; holding potential −80 mV). The number of cells is indicated in parentheses. (C) Effect of holding potential (−100, −80 and −60 mV) on the fraction of current blocked by 30 μM MONIRO‐1 (pulsed at 0.1 Hz). The number of cells is indicated in parentheses. (D) Activation and SSI curves in the absence (control) and presence of 30 μM MONIRO‐1. Activation curves were generated through indicated depolarizing test pulses (135 ms) from a holding potential of −80 mV, repeated every 10 s. For SSI curves, selected pre‐pulse potentials (−120 to 0 mV in 5 mV increments for 10 s) were applied prior to a test pulse (135 ms) determined from the peak current, repeated every 15 s from a holding potential of −80 mV. (E) Recovery from inactivation in the absence (control) and presence of 30 μM MONIRO‐1. A two‐pulse protocol (Inset) was repeated every 15 s, where the time was varied between the first fully inactivating pulse and the test pulse (0–10 s). *P < 0.05, significantly different as indicated; n.s. = not significant; Student's t‐test.
Figure 5.
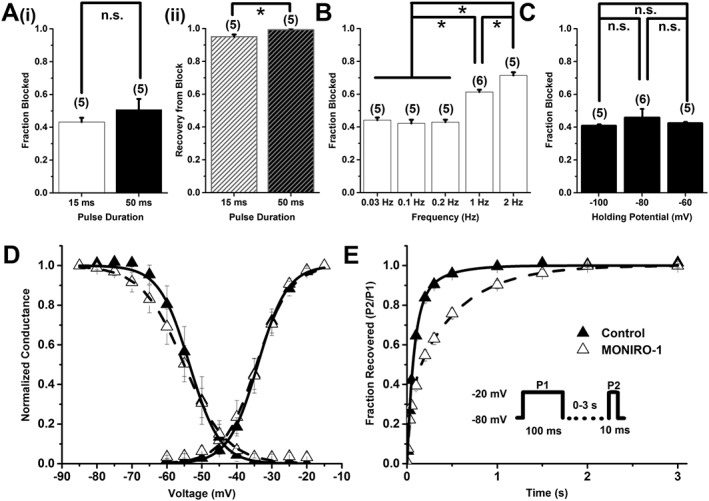
MONIRO‐1 block of human Cav3.1 channels. (A) (i) Bar graph showing fraction blocked and (ii) recovery from block by 3 μM MONIRO‐1, with a depolarizing pulse duration of 15 ms or 50 ms, from a holding potential of −80 mV repeated every 10 s. The number of cells is indicated in parentheses. (B) Bar graph of the fractional current inhibited by 3 μM MONIRO‐1 at five different frequencies (0.03, 0.1, 0.2, 1 and 2 Hz; holding potential −80 mV). The number of cells is indicated in parentheses. (C) Effect of holding potential (−100, −80 and −60 mV) on the fraction of current inhibited by 3 μM MONIRO‐1 (pulsed at 0.1 Hz). The number of cells is indicated in parentheses. (D) Activation and SSI curves in the absence (control) and presence of 3 μM MONIRO‐1. Activation curves were generated through indicated depolarizing test pulses (50 ms) from a holding potential of −80 mV, repeated every 10 s. For SSI curves, selected pre‐pulse potential values (−85 to −20 mV in 5 mV increments for 1 s) were applied prior to a test pulse (50 ms) determined from the peak current, repeated every 10 s from a holding potential of −80 mV. (E) Recovery from inactivation in the absence (control) and presence of 3 μM MONIRO‐1. A two‐pulse protocol (Inset) was repeated every 15 s, where the time was varied between the first fully inactivating pulse and the test pulse. *P < 0.05, significantly different as indicated; n.s. = not significant; Student's t‐test.
To further explore the differential effects of MONIRO‐1 on Cav2.2 and Cav3.1 channels, we investigated the effect of membrane holding potential on block of Cav2.2 and Cav3.1 channels. MONIRO‐1 could more effectively block Cav2.2 channels at more depolarized potentials (block at −100 mV = 34 ± 1.8%; −80 mV = 43 ± 3.2%; and −60 mV = 77 ± 5.5%, n = 5). In contrast, block at all of the holding potentials tested did not affect the degree by which MONIRO‐1 inhibited Cav3.1 channels (block at −100 mV = 41 ± 0.5%; −80 mV = 46 ± 5.1%; and −60 mV = 43 ± 0.6%, n = 5) (Figures 4C and 5C). This result highlights the state‐dependent differences between MONIRO‐1 inhibition of Cav2.2 and Cav3.1 channels.
Effects of MONIRO‐1 on N‐ and T‐type calcium channel activation, inactivation and recovery from inactivation
To identify any effects that MONIRO‐1 may have on calcium current kinetics and to understand the mechanisms behind the enhanced inhibition at depolarised membrane potentials, we examined its effects at half‐maximal concentrations on Cav2.2 and Cav3.1 activation and SSI (Figures 4D and 5D). Exposure to MONIRO‐1 did not affect the voltage‐dependence of activation of Cav2.2 or Cav3.1 channels (Table 2). However, there was a significant ~10 mV hyperpolarizing shift in Cav2.2 SSI. An apparent decrease in the voltage‐dependence of SSI was also seen. However, no significant changes were observed in Cav3.1 SSI in the presence of MONIRO‐1 or voltage‐dependence of SSI. Channel activation and inactivation time constants were examined, with and without MONIRO‐1, for Cav2.2 (30 μM) and Cav3.1 channels (3 μM). MONIRO‐1 had no effect on either the activation or inactivation time constants of Cav2.2 channels (Table 2). However, MONIRO‐1 application increased both activation and inactivation time constants for Cav3.1 channels (Table 2).
Table 2.
Comparison of activation and inactivation parameters between Cav2.2 and Cav3.1 channels before and after application of MONIRO‐1
| Cav2.2 | Cav3.1 | |||
|---|---|---|---|---|
| Control | MONIRO‐1 (30 μM)a | Control | MONIRO‐1 (3 μM)a | |
| τ activation | 1.81 ± 0.15 ms (5) | 1.51 ± 0.17 ms (5) | 1.90 ± 0.08 ms (5) | 3.45 ± 0.51 ms (5)* |
| τ inactivation | 61.75 ± 13.69 ms (5) | 44.45 ± 7.61 ms (5) | 10.78 ± 0.52 ms (5) | 17.91 ± 0.74 ms (5)* |
| V0.5 activation | 12.5 ± 0.5 mV (5) | 12.4 ± 0.4 mV (5) | −34.0 ± 0.4 mV (7) | −34.4 ± 0.6 mV (7) |
| V0.5 inactivation | −41.3 ± 0.7 mV (5) | −52.9 ± 1.3 mV (5)* | −53.5 ± 0.6 mV (6) | −55.1 ± 0.6 mV (6) |
| Slope factor | 5.3 ± 0.6 (5) | 10.2 ± 1.2 (5)* | 4.4 ± 0.6 (6) | 6.1 ± 0.5 (6) |
P < 0.05 compared with control, Students t‐test. Data presented as mean ± SEM and n is given in brackets.
Concentration chosen based on IC50 of individual channel.
Finally, we investigated the effect of MONIRO‐1 on Cav2.2 and Cav3.1 channel recovery from inactivation. MONIRO‐1 had no observed effect on Cav2.2 recovery from inactivation time constants or their relative contribution to either time constants (control τfast = 0.22 ± 0.1 s, Afast = 57 ± 8.5%; τslow = 2.8 ± 0.7 s, Aslow = 45 ± 8.6%; MONIRO‐1 τfast = 0.14± 0.1 s, Afast = 43 ± 7.0%; τslow = 2.4 ± 0.4 s, Aslow = 58 ± 6.2%, n = 5 respectively). However, MONIRO‐1 slowed recovery of Cav3.1 channels from inactivation by decreasing the relative contribution of the fast recovery time constant while increasing the contribution of the slow time constant (control τfast = 0.051 ± 0.008 s, Afast = 80.1 ± 5.1%; τslow = 0.20 ± 0.11 s, Aslow = 21.8 ± 5.4%; MONIRO‐1 τfast = 0.026 ± 0.007 s, Afast = 29.6 ± 4.2%; τslow = 0.33 ± 0.03 s, Aslow = 70 ± 4.1%, n = 6; P < 0.05). Therefore, MONIRO‐1 can inhibit Cav3.1 channels more efficiently at firing frequencies >1 Hz (Figure 5B), as indicated by the slowed recovery from inactivation and frequency dependence, such as those prevailing in hyperexcitable neurons (Cain and Snutch, 2010).
Discussion
In this study, we examined the effects of an ortho‐phenoxyanilide on the inhibition and biophysical properties of VGCCs. This ortho‐phenoxyanilide, MONIRO‐1, is a low MW compound and was selected for its ability to inhibit VGCCs in a high‐throughput calcium influx assay (Gleeson et al., 2015). To ascertain its selectivity and mode of action, we examined its potency of block and biophysical properties on a variety of VGCCs. MONIRO‐1 was shown to be an N‐ and T‐type calcium channel inhibitor with advantageous biophysical properties, including state‐ and use‐dependence. It specifically targets Cav3.2 and Cav2.2 channels, which have been implicated in various pain pathways, and Cav3.1 channels, which are involved in cell signalling and epilepsy (Choi, 2013; Waxman and Zamponi, 2014).
Given that MONIRO‐1 inhibits depolarization‐activated calcium responses from SH‐SY5Y cells (Gleeson et al., 2015), we determined its efficacy and selectivity against several VGCCs expressed in HEK293 cells. After screening representative members of each VGCC family (L‐, N‐, P/Q‐, R‐ and T‐types), we established that MONIRO‐1 preferentially inhibits T‐type channels (Cav3.1, Cav3.2 and Cav3.3 channels), modestly interacts with N‐type Cav2.2 channels (with 5–20 times less affinity compared to T‐type calcium channels) and has poor efficacy for L‐type (Cav1.2 and Cav1.3 channels IC50 > 100 μM). No other VGCCs investigated (P/Q‐type Cav2.1; R‐type Cav2.3) responded to high concentrations of MONIRO‐1. hERG, a typical off‐target channel for low MW compounds, was not modulated by MONIRO‐1 at concentrations up to 100 μM. Subsequently, we characterized the mechanism of MONIRO‐1 inhibition on its primary targets, N‐ and T‐type calcium channels.
In the present study, MONIRO‐1 inhibited Cav3.1 channels with an IC50 of 3 μM, so we examined the mechanistic details of this inhibition. Many T‐type calcium channel inhibitors have been identified in assays looking for inactivation state‐dependent inhibitors. These studies have led to compounds that predominantly bind and alter SSI, including TTA‐A2, A‐1048400 and NNC 55‐0396, unlike MONIRO‐1, which does not affect SSI of Cav3.1 channels. MONIRO‐1 (3 μM) significantly slowed both channel activation and inactivation kinetics, without discernible effects on either V0.5act or V0.5inact. MONIRO‐1 slowed recovery of Cav3.1 from inactivation at rates >1 Hz, which was reflected as an increase in current inhibition at 1 and 2 Hz (Figure 5B, E), suggesting that MONIRO‐1 would preferentially target epileptic firing neurons over normal firing neurons. Taken together, the lack of effect of MONIRO‐1 on other biophysical properties examined, including the lack of effect of holding potential, suggests that it acts by binding to prevent Cav3.1 channels from transitioning from a non‐conducting state to the open state. Therefore, the prolonged recovery from inactivation caused by MONIRO‐1 inhibition of Cav3.1‐mediated currents is likely to be due to the slower dissociation of the compound from the open‐blocked channels or due to its accelerating channel transitions into a slow inactivated channel conformation. A similar phenomenon has been observed for mibefradil inhibition of Cav2.1 channels (Timin et al., 2004).
Cav3.1 channels are key players in the circuitry involved in sleep including the reticular thalamic nucleus, thalamic relay neurons and neocortical pyramidal cells (Chen et al., 2014). Cav3.1 channels are highly expressed in thalamocortical neurons (Kim et al., 2001; Song et al., 2004), where they initiate entry of these neurons into bursting mode – a typical activity mode in absence seizures (Cain and Snutch, 2013). Consequently, Cav3.1 channels have been directly linked to absence of epilepsies (Kim et al., 2001). Disease mutations in Cav3.2 channels have also been linked to both rat models and human idiopathic generalized epilepsy (Khosravani et al., 2005; Powell et al., 2009), suggesting that inhibitors of both Cav3.1 and Cav3.2 channels could decrease neuronal firing induced by hyperactivity of these channels during epilepsy. Compound Z944, which has higher affinity for the inactivated state of T‐type calcium channels over the resting state, reduced seizure activity in terms of time spent in seizure and the total number of seizures per hour (Tringham et al., 2012). During burst firing, VGCCs repetitively open and close at frequencies of 5–10 Hz (Cain and Snutch, 2010). Ideally, an anti‐epileptogenic drug targeting Cav3.1 or Cav3.2 channels in epileptogenic foci, that is, on neurons in bursting mode where the channels are in the open state. Our results suggest that MONIRO‐1 modulates Cav3.1 channels by holding the channel in a non‐conducting state, thus prolonging recovery from inactivation, which makes the compound well suited for the study and/or potentially for the amelioration of Cav3.1 or Cav3.2‐mediated epileptogenesis in neurons entering bursting mode.
Considerable research into pain physiology has focused on voltage‐gated N‐ and T‐type calcium channels. Cav3.2 channels are expressed in peripheral sensory neurons, where they modulate pain processing by controlling dorsal root ganglion neuroexcitability (Carbone and Lux, 1984). Cav2.2 channels are located at presynaptic terminals in the CNS, where they modulate neurotransmitter release and fast synaptic transmission (Wheeler et al., 1994). MONIRO‐1 inhibits Cav2.2 channels under resting state conditions with an IC50 of ~30 μM, which is approximately 10‐fold higher than that observed the T‐type channels. However, the effect of MONIRO‐1 on Cav2.2 channels is enhanced at higher frequencies, where faster pacing of the channel increased inhibition. At frequencies as high as 0.2 Hz (the maximum tested), the apparent affinity increased to ~20 μM, with no observable change to the time course of activation or inactivation of the channel (Figure 4B and Table 2).
Following this observation, we examined the potential state dependence of the activity of MONIRO‐1. Short (45 ms) depolarizing test pulses produced greater inhibition of Cav2.2 channels as well as less recovery after washout compared with a longer (135 ms) test pulse, suggesting a possible decrease in affinity for the open and/or inactivated state (Figure 4A). MONIRO‐1 had a pronounced effect on Cav2.2 channel SSI that was not paralleled in Cav3.1 channels (Figure 4D), along with strong dependence on holding potential. At −60 mV, ~80% of the whole‐cell current was inhibited compared with −100 mV where only ~35% of the current was blocked (Figure 4C). Such a hyperpolarizing shift in SSI and the dependence on holding potential suggests that MONIRO‐1 preferentially binds to the inactivated state of N‐type calcium channels (Bean et al., 1983; Bean, 1984; Sanguinetti and Kass, 1984). Interestingly, unlike its effect on T‐type calcium channels, MONIRO‐1 did not influence N‐type calcium channel recovery from inactivation (Figure 5E), suggesting a distinct mode of action against the two channel families.
Nociceptive stimuli cause depolarization, calcium channel opening, intracellular [Ca2+] increase and neuronal hyper‐excitability, as shown by faster firing rates (Winquist et al., 2005). Nociceptive stimuli reduce the effective open time and increase the frequency of channel opening (Winquist et al., 2005). Sustained neuronal firing in pain syndromes increases cytosolic calcium levels in the dorsal horn and shifts calcium channels into the inactivated state. Therefore, drugs that target Cav2.2 channels are best suited to ‘prefer’ those channels in the inactivated state (predominant in pain syndromes), allowing normal neurotransmitter release signalled by these channels in neurons not in a pain state.
A few compounds have recently reported to exhibit use‐ and/or state‐dependent inhibition of calcium channels involved in pain signalling, including A‐1048400 (Scott et al., 2012) (compound 3, Figure 1), Z160/NP118809 1 (Zamponi et al., 2009) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7766 (Abbadie et al., 2010). To explore MONIRO‐1 inhibitory mechanism of Cav2.2, we examined stimulation conditions comparable with those occurring in the physiological setting. Given that Cav2.2 channels can potentially accumulate in the inactivated state in hyperactive pain state conditions (Winquist et al., 2005), the selective effect of MONIRO‐1 on channels in this state could be a valuable pharmacological property that would enable therapeutic targeting during neuropathic pain treatment. Frequency dependence and preference of MONIRO‐1 for the inactivated Cav2.2 channels suggest that it or related compounds may prove useful in the prevention of high‐frequency firing episodes, an important signalling mode occurring during pain transmission. Therefore, MONIRO‐1 may preferentially inhibit channels in hyperexcitable states in pain‐specific loci, leaving normally firing neurons unaffected (Liao et al., 2011).
In conclusion, we have identified MONIRO‐1 as a low MW antagonist of voltage‐gated calcium channels with 10‐ to 100‐fold higher affinity for Cav3.1, Cav3.2, Cav3.3 and Cav2.2 channels than other classes of VGCCs examined. MONIRO‐1 is a valuable tool for the study of VGCCs because it inhibits N‐ and T‐type calcium channels through distinct mechanisms. Given its state‐ and use‐dependent inhibition of Cav2.2 channels (preferentially inhibits the inactivated state) and Cav3.1 channels (prohibiting channel transitions to the open state), while exhibiting frequency‐dependent block of both channels, MONIRO‐1 is a novel lead compound that could be used to develop Cav channel blockers to treat VGCC pathologies, including neuropathic pain and epilepsy. MONIRO‐1 shows a novel mechanism of block of hCav3.1 channels compared with previously described T‐type calcium channel blockers, in that it slows channel activation and inactivation kinetics and traps channels at high‐frequency firing (>1 Hz) from transitioning to the open state while not altering SSI. Prior to in vivo assessment of their analgesic potential, MONIRO‐1 and analogues need to be characterized at other voltage‐gated ion channels in pain pathways, including Nav1.7 and Nav1.8 channels.
Author contributions
J.R.M., P.J.D., K.L.T. and D.J.A. designed the experiments; J.R.M., L.M., E.C.G. and S.S. performed the experiments; J.R.M. analysed the electrophysiological data; J.R.M., P.J.D., K.L.T. and D.J.A. prepared the figures; J.R.M. wrote the manuscript; J.R.M., R.J.L., P.J.D., K.L.T. and D.J.A. revised the manuscript. All authors reviewed the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank Dr J. E. Graham for helping with chemical synthesis and Dr. R. K. Finol‐Urdaneta for providing valuable comments on drafts of the manuscript. This work was funded by a National Health and Medical Research Council (NHMRC) Program Grant awarded to D.J.A. and R.J.L. (APP1072113). The Monash–CSIRO Collaborative Research Support Scheme and CSIRO's Australian Biotech Growth Partnerships Theme also helped to fund this research.
McArthur, J. R. , Motin, L. , Gleeson, E. C. , Spiller, S. , Lewis, R. J. , Duggan, P. J. , Tuck, K. L. , and Adams, D. J. (2018) Inhibition of human N‐ and T‐type calcium channels by an ortho‐phenoxyanilide derivative, MONIRO‐1. British Journal of Pharmacology, 175: 2284–2295. doi: 10.1111/bph.13910.
References
- Abbadie C, McManus OB, Sun S‐Y, Bugianesi RM, Dai G, Haedo RJ et al. (2010). Analgesic effects of a substituted N‐triazole oxindole (TROX‐1), a state‐dependent, voltage‐gated calcium channel 2 blocker. J Pharmacol Exp Ther 334: 545–555. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altier C, Zamponi GW (2004). Targeting Ca2+ channels to treat pain: T‐type versus N‐type. Trends Pharmacol Sci 25: 465–470. [DOI] [PubMed] [Google Scholar]
- Andersson A, Baell JB, Duggan PJ, Graham JE, Lewis RJ, Lumsden NG et al. (2009). ω‐Conotoxin GVIA mimetics based on an anthranilamide core: effect of variation in ammonium side chain lengths and incorporation of fluorine. Bioorg Med Chem 17: 6659–6670. [DOI] [PubMed] [Google Scholar]
- Baell JB, Duggan PJ, Forsyth SA, Lewis RJ, Lok YP, Schroeder CI (2004). Synthesis and biological evaluation of nonpeptide mimetics of ω‐conotoxin GVIA. Bioorg Med Chem 12: 4025–4037. [DOI] [PubMed] [Google Scholar]
- Baell JB, Duggan PJ, Forsyth SA, Lewis RJ, Lok YP, Schroeder CI et al. (2006). Synthesis and biological evaluation of anthranilamide‐based non‐peptide mimetics of ω‐conotoxin GVIA. Tetrahedron 62: 7284–7292. [Google Scholar]
- Bean BP (1984). Nitrendipine block of cardiac calcium channels: high‐affinity binding to the inactivated state. Proc Natl Acad Sci U S A 81: 6388–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP, Cohen CJ, Tsien RW (1983). Lidocaine block of cardiac sodium channels. J Gen Physiol 81: 613–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladen C, McDaniel SW, Gadotti VM, Petrov RR, Berger ND, Diaz P et al. (2015). Characterization of novel cannabinoid based T‐type calcium channel blockers with analgesic effects. ACS Chem Nerosci 6: 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O et al. (2005). Silencing of the Cav3.2 T‐type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 24: 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, Zamponi GW (2014). Calcium‐permeable ion channels in pain signaling. Physiol Rev 94: 81–140. [DOI] [PubMed] [Google Scholar]
- Butterworth JF, Strichartz GR (1990). Molecular mechanisms of local anesthesia: a review. Anesthesiology 72: 711–734. [DOI] [PubMed] [Google Scholar]
- Cain SM, Snutch TP (2010). Contributions of T‐type calcium channel isoforms to neuronal firing. Channels 4: 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain SM, Snutch TP (2013). T‐type calcium channels in burst‐firing, network synchrony, and epilepsy. Biochimica et Biophysica Acta (BBA) ‐ Biomembranes 1828: 1572–1578. [DOI] [PubMed] [Google Scholar]
- Carbone E, Lux HD (1984). A low voltage‐activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature 310: 501–502. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Perez‐Reyes E, Snutch TP, Striessnig J (2005). International union of pharmacology. XLVIII. Nomenclature and structure‐function relationships of voltage‐gated calcium channels. Pharmacol Rev 57: 411–425. [DOI] [PubMed] [Google Scholar]
- Chen Y, Chen Y, Parker WD, Wang K (2014). The role of T‐type calcium channel genes in absence seizures. Front Neurol 5: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong E, Shin H‐S (2013). T‐type Ca2+ channels in normal and abnormal brain functions. Physiol Rev 93: 961–992. [DOI] [PubMed] [Google Scholar]
- Choi K‐H (2013). The design and discovery of T‐type calcium channel inhibitors for the treatment of central nervous system disorders. Expert Opin Drug Discov 8: 919–931. [DOI] [PubMed] [Google Scholar]
- Choi S, Na HS, Kim J, Lee J, Lee S, Kim D et al. (2007). Attenuated pain responses in mice lacking Cav3.2 T‐type channels. Genes Brain Behav 6: 425–431. [DOI] [PubMed] [Google Scholar]
- Chouabe C, Drici M‐D, Romey G, Barhanin J, Lazdunski M (1998). HERG and KvLQT1/IsK, the cardiac K+ channels involved in long QT syndromes, are targets for calcium channel blockers. Mol Pharmacol 54: 695–703. [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai G, Haedo RJ, Warren VA, Ratliff KS, Bugianesi RM, Rush A et al. (2008). A high‐throughput assay for evaluating state dependence and subtype selectivity of Cav2 calcium channel inhibitors. Assay Drug Dev Technol 6: 195–212. [DOI] [PubMed] [Google Scholar]
- Duggan PJ, Lewis RJ, Phei Lok Y, Lumsden NG, Tuck KL, Yang A (2009). Low molecular weight non‐peptide mimics of ω‐conotoxin GVIA. Bioorg Med Chem Lett 19: 2763–2765. [DOI] [PubMed] [Google Scholar]
- Eller P, Berjukov S, Wanner S, Huber I, Hering S, Knaus HG et al. (2000). High affinity interaction of mibefradil with voltage‐gated calcium and sodium channels. Br J Pharmacol 130: 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatters SJL, Bennett GJ (2004). Ethosuximide reverses paclitaxel‐ and vincristine‐induced painful peripheral neuropathy. Pain 109: 150–161. [DOI] [PubMed] [Google Scholar]
- Francois A, Kerckhove N, Meleine M, Alloui A, Barrere C, Gelot A et al. (2013). State‐dependent properties of a new T‐type calcium channel blocker enhance CaV3.2 selectivity and support analgesic effects. Pain 154: 283–293. [DOI] [PubMed] [Google Scholar]
- François A, Laffray S, Pizzoccaro A, Eschalier A, Bourinet E (2014). T‐type calcium channels in chronic pain: mouse models and specific blockers. Pflugers Arch ‐ Eur J Physiol 466: 707–717. [DOI] [PubMed] [Google Scholar]
- Gleeson EC, Graham JE, Spiller S, Vetter I, Lewis RJ, Duggan PJ et al. (2015). Inhibition of N‐Type calcium channels by fluorophenoxyanilide derivatives. Mar Drugs 13: 2030–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Gebhart GF (2010). Nociceptor sensitization in pain pathogenesis. Nat Med 16: 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomora JC, Murbartian J, Arias JM, Lee JH, Perez‐Reyes E (2002). Cloning and expression of the human T‐type channel CaV3.3: insights into prepulse facilitation. Biophys J 83: 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Hu J (2014). Spinal presynaptic inhibition in pain control. Neuroscience 283: 95–106. [DOI] [PubMed] [Google Scholar]
- Helton TD, Xu W, Lipscombe D (2005). Neuronal L‐type calcium channels open quickly and are inhibited slowly. J Neurosci 25: 10247–10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG (1984). Antiarrhythmic agents: the modulated receptor mechanism of action of sodium and calcium channel‐blocking drugs. Annu Rev Pharmacol Toxicol 24: 387–423. [DOI] [PubMed] [Google Scholar]
- Khosravani H, Bladen C, Parker DB, Snutch TP, McRory JE, Zamponi GW (2005). Effects of CaV3.2 channel mutations linked to idiopathic generalized epilepsy. Ann Neurol 57: 745–749. [DOI] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong M‐J, Kim S‐S et al. (2001). Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking α1G T‐Type Ca2+ channels. Neuron 31: 35–45. [DOI] [PubMed] [Google Scholar]
- Kraus RL, Li Y, Gregan Y, Gotter AL, Uebele VN, Fox SV et al. (2010). In vitro characterization of T‐type calcium channel antagonist TTA‐A2 and in vivo effects on arousal in mice. J Pharmacol Exp Ther 335: 409–417. [DOI] [PubMed] [Google Scholar]
- Lee MS (2014). Chapter Four – recent progress in the discovery and development of N‐type calcium channel modulators for the treatment of pain In: Lawton G, Witty DR. (eds). Prog Med Chem. Elsevier: Oxford, UK, pp. 147–186. [DOI] [PubMed] [Google Scholar]
- Lee S (2013). Pharmacological inhibition of voltage‐gated Ca2+ channels for chronic pain relief. Curr Neuropharm 11: 606–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y‐F, Tsai M‐L, Chen C‐C, Yen C‐T (2011). Involvement of the Cav3.2 T‐type calcium channel in thalamic neuron discharge patterns. Mol Pain 7: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews EA, Dickenson AH (2001). Effects of ethosuximide, a T‐type Ca2+ channel blocker, on dorsal horn neuronal responses in rats. Eur J Pharmacol 415: 141–149. [DOI] [PubMed] [Google Scholar]
- McCleskey EW, Fox AP, Feldman DH, Cruz LJ, Olivera BM, Tsien RW et al. (1987). ω‐Conotoxin: direct and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc Natl Acad Sci U S A 84: 4327–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Voets T, Eggermont J, Droogmans G (1998). Activation of volume‐regulated chloride currents by reduction of intracellular ionic strength in bovine endothelial cells. J Physiol 506: 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perret D, Luo ZD (2009). Targeting voltage‐gated calcium channels for neuropathic pain management. Neurotherapeutics 6: 679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope JE, Deer TR (2013). Ziconotide: a clinical update and pharmacologic review. Expert Opin Pharmacother 14: 957–966. [DOI] [PubMed] [Google Scholar]
- Powell KL, Cain SM, Ng C, Sirdesai S, David LS, Kyi M et al. (2009). A CaV3.2 T‐type calcium channel point mutation has splice‐variant‐specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J Neurosci 29: 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger TS, Yang Z‐Q, Schlegel K‐AS, Shu Y, Mattern C, Cube R et al. (2011). Pyridyl amides as potent inhibitors of T‐type calcium channels. Bioorg Med Chem Lett 21: 1692–1696. [DOI] [PubMed] [Google Scholar]
- Saegusa H, Kurihara T, Zong S, Kazuno A‐A, Matsuda Y, Nonaka T et al. (2001). Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N‐type Ca2+ channel. EMBO J 20: 2349–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Kass RS (1984). Voltage‐dependent block of calcium channel current in the calf cardiac Purkinje fiber by dihydropyridine calcium channel antagonists. Circ Res 55: 336–348. [DOI] [PubMed] [Google Scholar]
- Scott VE, Vortherms TA, Niforatos W, Swensen AM, Neelands T, Milicic I et al. (2012). A‐1048400 is a novel, orally active, state‐dependent neuronal calcium channel blocker that produces dose‐dependent antinociception without altering hemodynamic function in rats. Biochem Pharmacol 83: 406–418. [DOI] [PubMed] [Google Scholar]
- Sekiguchi F, Kawabata A (2013). T‐type calcium channels: functional regulation and implication in pain signaling. J Pharmacol Sci 122: 244–250. [DOI] [PubMed] [Google Scholar]
- Simms BA, Zamponi GW (2014). Neuronal voltage‐gated calcium channels: structure, function, and dysfunction. Neuron 82: 24–45. [DOI] [PubMed] [Google Scholar]
- Song I, Kim D, Choi S, Sun M, Kim Y, Shin H‐S (2004). Role of the α1G T‐type calcium channel in spontaneous absence seizures in mutant mice. J Neurosci 24: 5249–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Herrington J, Bugianesi RM, Dai G, Haedo RJ, Ratliff KS et al. (2012). Characterization of the substituted N‐triazole oxindole TROX‐1, a small‐molecule, state‐dependent inhibitor of CaV2 calcium channels. Mol Pharmacol 81: 488–497. [DOI] [PubMed] [Google Scholar]
- Timin E, Berjukow S, Hering S (2004). Concepts of state‐dependent pharmacology of calcium channels In: McDonough S. (ed). Calcium Channel Pharmacology. Springer: US, pp. 1–19. [Google Scholar]
- Todorovic SM, Meyenburg A, Jevtovic‐Todorovic V (2002). Mechanical and thermal antinociception in rats following systemic administration of mibefradil, a T‐type calcium channel blocker. Brain Res 951: 336–340. [DOI] [PubMed] [Google Scholar]
- Tranberg CE, Yang A, Vetter I, McArthur JR, Baell JB, Lewis RJ et al. (2012). ω‐Conotoxin GVIA mimetics that bind and inhibit neuronal CaV2.2 ion channels. Mar Drugs 10: 2349–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringham E, Powell KL, Cain SM, Kuplast K, Mezeyova J, Weerapura M et al. (2012). T‐type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci Transl Med 4: 121ra19. [DOI] [PubMed] [Google Scholar]
- Wang F, Yan Z, Liu Z, Wang S, Wu Q, Yu S et al. (2016). Molecular basis of toxicity of N‐type calcium channel inhibitor MVIIA. Neuropharmacology 101: 137–145. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Zamponi GW (2014). Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci 17: 153–163. [DOI] [PubMed] [Google Scholar]
- Wen XJ, Xu SY, Chen ZX, Yang CX, Liang H, Li H (2010). The roles of T‐type calcium channel in the development of neuropathic pain following chronic compression of rat dorsal root ganglia. Pharmacology 85: 295–300. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW (1994). Roles of N‐type and Q‐type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264: 107–111. [DOI] [PubMed] [Google Scholar]
- Winquist RJ, Pan JQ, Gribkoff VK (2005). Use‐dependent blockade of CaV2.2 voltage‐gated calcium channels for neuropathic pain. Biochem Pharmacol 70: 489–499. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Feng Z‐P, Zhang L, Pajouhesh H, Ding Y, Belardetti F et al. (2009). Scaffold‐based design and synthesis of potent N‐type calcium channel blockers. Bioorg Med Chem Lett 19: 6467–6472. [DOI] [PubMed] [Google Scholar]
- Zikolova S, Ninov K (1972). Analogs of N1‐benzhydryl‐N4‐cinnamylpiperazine (cinnarizine). II. N1‐Substituted‐N4‐benzhydrylpiperazines. Tr Nauchnoizsled Khim‐Farm Inst 8: 59–67. [Google Scholar]