

Abstract
Background and Purpose
The photo‐isomerizable local anaesthetic, quaternary ammonium–azobenzene–quaternary ammonium (QAQ), provides rapid, optical control over pain signalling without involving genetic modification. In darkness or in green light, trans‐QAQ blocks voltage‐gated K+ and Na+ channels and silences action potentials in pain‐sensing neurons. Upon photo‐isomerization to cis with near UV light, QAQ blockade is rapidly relieved, restoring neuronal activity. However, the molecular mechanism of cis and trans QAQ blockade is not known. Moreover, the absorption spectrum of QAQ requires UV light for photo‐control, precluding use deep inside neural tissue.
Experimental Approach
Electrophysiology and molecular modelling were used to characterize the binding of cis and trans QAQ to voltage‐gated K+ channels and to develop quaternary ammonium–ethylamine–azobenzene–quaternary ammonium (QENAQ), a red‐shifted QAQ derivative controlled with visible light.
Key Results
trans QAQ was sixfold more potent than cis QAQ, in blocking current through Shaker K+ channels. Both isomers were use‐dependent, open channel blockers, binding from the cytoplasmic side, but only trans QAQ block was slightly voltage dependent. QENAQ also blocked native K+ and Na+ channels preferentially in the trans state. QENAQ was photo‐isomerized to cis with blue light and spontaneously reverted to trans within seconds in darkness, enabling rapid photo‐control of action potentials in sensory neurons.
Conclusions and Implications
Light‐switchable local anaesthetics provide a means to non‐invasively photo‐control pain signalling with high selectivity and fast kinetics. Understanding the mode of action of QAQ and related compounds will help to design of drugs with improved photo‐pharmacological properties.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Abbreviations
- AAQ
acrylamide–azobenzene–quaternary ammonium
- ACSF
artificial CSF solution
- BzAQ
benzoyl‐azobenzene‐quaternary ammonium
- CAP
compound action potential
- DRG
dorsal root ganglion
- Ip
peak current
- Iss
steady‐state current
- MEA
multi‐electrode array
- ORI
oocyte Ringer's solution
- PI
photo‐switching index
- QAQ
quaternary ammonium–azobenzene–quaternary ammonium
- QENAQ
quaternary ammonium–ethylamine–azobenzene–quaternary ammonium
- Shaker‐IR
Shaker‐inactivation removed
- TEA
tetraethyl ammonium
- TG
trigeminal ganglion
Introduction
Local anaesthetics decrease pain sensation by silencing the firing of nociceptor neurons. However, local anaesthetics can also silence other sensory and motor neurons, leading to serious side effects, especially when the drugs are applied for an extended period of time. We have been developing light‐sensitive local anaesthetics that can be controlled with spatio‐temporal precision and which act specifically on nociceptors, avoiding side effects.
Opto‐pharmacology (or photo‐pharmacology) refers to the use of photo‐sensitive drugs that change structure upon irradiation with light (Kramer et al., 2013; Velema et al., 2014; Lerch et al., 2016). The aim is to establish precise, optical control over the activity, the kinetics and the site of action of a bioactive compound. Opto‐pharmacological agents are rationally designed to incorporate in their structure a chemical photo‐switch, which confers to the drug its photo‐sensitivity. Azobenzene is a commonly used photo‐switch, owing to the considerable geometrical difference between its bent, cis and straight, trans configuration (Fehrentz et al., 2011; Szymański et al., 2013). Upon photo‐isomerization, azobenzene‐containing drugs change shape, affecting their binding to target receptors and hence their biological activity. Azobenzene can be rapidly switched back and forth between their two isomers to enable control over receptors with high temporal precision (ms). Light can be delivered with high spatial precision to restrict the action of azobenzene‐containing drugs to a tightly localized region in biological tissue. Finally, the wavelength and/or intensity of incident light can be adjusted to change the ratio between trans and cis isomers, altering the effective concentration of the active form of the compound interacting with the receptor.
We have developed a series of photo‐switchable blockers for http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=81, called XAQs (Banghart et al., 2009; Mourot et al., 2011, 2012, 2013a,b). XAQs possess a quaternary ammonium group (Q), which binds to the cytoplasmic tetraethyl ammonium (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2343) binding site on K+ channels, a central azobenzene photo‐switch (A) and a terminal aliphatic chain of variable structure (X). XAQs are permanently charged, yet most are sufficiently hydrophobic to passively cross the membrane bilayer and reach the cytoplasmic lumen on K+ channels (Banghart et al., 2009). They have been used to optically control action potential firing in dissociated neurons (Banghart et al., 2009), brain slices (Mourot et al., 2011) and in the retina for restoring visual responses in blind mice (Polosukhina et al., 2012; Tochitsky et al., 2014, 2016). One of these photo‐switchable blockers, quaternary ammonium–azobenzene–quaternary ammonium (QAQ) (Figure 1A) has two important features that distinguish it from other XAQs: (i) it contains two permanently charged quaternary ammonium groups and therefore cannot passively cross the membrane bilayer, and (ii) it is not selective for voltage‐gated K+ channels but also photo‐sensitizes http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=82 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=80 (Mourot et al., 2012). QAQ can be loaded into neurons by two means: either artificially using the patch pipette or ‘naturally’ by passing through open ion channels with large pores that are endogenous to some neurons, such as the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=484 and the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=507. Once inside neurons, trans QAQ blocks many voltage‐gated ion channels and silences neuronal activity in the dark, while illumination with violet light isomerises the molecule to cis, relieving blockade and restoring excitability.
Figure 1.
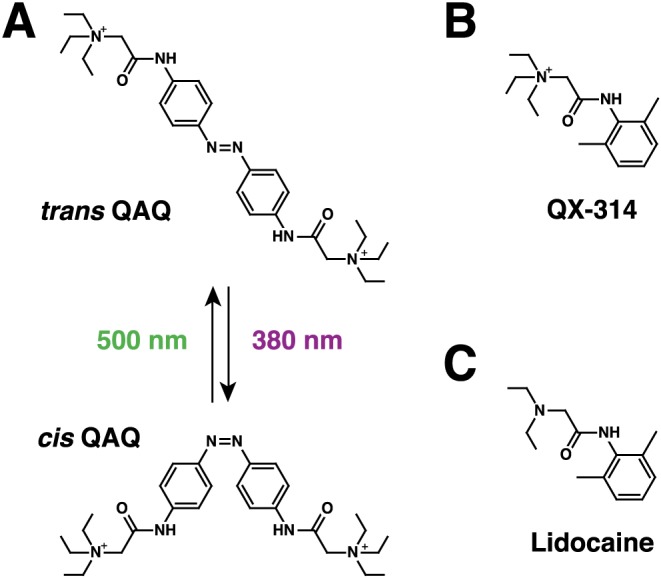
QAQ, a photo‐switchable local anaesthetics. (A) Chemical structure of trans and cis QAQ. QAQ photo‐isomerizes to cis upon illumination with near‐UV light (380 nm). Isomerization back to trans occurs rapidly upon illumination with green light (500 nm), or slowly (min) in the dark. (B) Chemical structure of QX‐314. (C) Chemical structure of the local anaesthetic lidocaine.
QAQ is structurally and functionally similar to the membrane‐impermeant, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2623 derivative http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2405 (Figure 1B; Strichartz, 1973), with the additional benefit of being photo‐controllable. Lidocaine (Figure 1C) is a potent local anaesthetic that prevents signal propagation to the brain, by blocking voltage‐gated Na+ and other ion channels (Fozzard et al., 2011). Because lidocaine is membrane permeant when deprotonated, however, it silences not only pain‐sensing neurons but also other sensory and motor neurons. In contrast, both QX‐314 and QAQ fail to block Na+ channels when applied outside cells, but are potent blockers once introduced inside cells. In a clever experimental design, Binshtok et al. (2007) achieved nociceptive‐selective analgesia using QX‐314. They found that this molecule could enter nociceptors selectively by passing through the pore of noxious‐heat sensitive TRPV1 channels, which are highly expressed in nociceptors but scarcely present in other neuronal cell types (Binshtok et al., 2007). Similarly, we found that QAQ could selectively silence pain‐sensing neurons, while leaving other sensory modalities unaffected. But unlike QX‐314 that produces long‐lasting analgesia (Binshtok et al., 2009), the effect of QAQ can be reversed within seconds using flashes of near‐UV light (Mourot et al., 2012). QAQ can therefore function as a targeted, light‐tunable local anaesthetic, both ex vivo and in vivo.
However, how the two isomers of QAQ block ion channels is unclear. In addition, the short wavelength of light necessary to effectively switch QAQ to cis can be cell damaging and poorly penetrates intact tissue. Therefore, we sought a mechanistic understanding of QAQ action on ion channels, to inform chemical modifications that could improve photo‐control in neural tissue. We probed cis and trans QAQ binding using electrophysiology and molecular modelling. We chose the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=540, a prototypical voltage‐gated ion channel with well‐understood biophysical properties, as a model system to study QAQ‐ion channel interactions. Having established how QAQ interacts with Shaker K+ channels, we have designed a red‐shifted version of QAQ (QENAQ) that photo‐isomerizes to cis using blue light and reverts back to trans within seconds in darkness. We further show that QENAQ can be used to optically control action potential firing in intact dorsal root ganglia (DRGs). Insights from QAQ and QENAQ will inform the design of related compounds with tuned properties, expanding this group of optical tools for neuronal control, both for gaining scientific insights and as potential therapeutic agents.
Methods
Synthesis of QAQ and QENAQ
QAQ was synthesized as described in (Mourot et al., 2012). QENAQ was synthesized as described in Scheme 1. Reactions were carried out under nitrogen atmosphere and magnetically stirred in oven‐dried glassware.
Scheme 1.
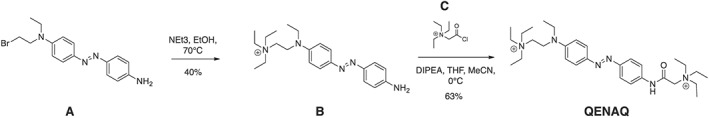
Synthesis of QENAQ.
(E)‐2‐((4‐((4‐aminophenyl)diazenyl)phenyl)(ethyl)amino)‐N,N,N‐triethylethan‐1‐aminium (B): 4‐((4‐aminophenyl)diazenyl)‐N‐(2‐bromoethyl)‐N‐ethylaniline (A, 63 mg, 0.182 mmol) (Kienzler et al., 2013) was transferred to a heavy‐walled pressure vessel and dissolved in ethanol (4 mL), dichloromethane (0.5 mL) and triethylamine (0.2 mL). The mixture was sparged with nitrogen gas, and the flask was sealed and then heated to 80°C for 5 days. The reaction mixture was concentrated in vacuo, and the residue purified by reverse phase (C18 silica gel) flash column chromatography (0 to 40% methanol in 0.1% aqueous formic acid) to yield 26.5 mg (40%) of B as a red solid. 1H NMR (500 MHz, methanol‐d 4) δ 7.77 (d, J = 8.6 Hz, 2H), 7.63 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 6.74 (d, J = 8.6 Hz, 2H), 3.85 (t, J = 7.7 Hz, 2H), 3.54 (q, J = 6.9 Hz, 2H), 3.44 (m, 8H), 1.35 (t, J = 7.2 Hz, 9H), 1.23 (t, J = 6.9 Hz, 3H); HRMS (ESI+) m/z calcd for C22H34N5 [M]+: 368.2808; found 368.2809.
QENAQ: Freshly prepared 2‐chloro‐N,N,N‐triethyl‐2‐oxoethan‐1‐aminium [C, 150 mg, 0.842 mmol (Banghart et al., 2009)] dissolved in acetonitrile (5 mL) was slowly added to a stirred flask containing (E)‐2‐((4‐((4‐aminophenyl)diazenyl)phenyl)(ethyl)amino)‐N,N,N‐triethylethan‐1‐aminium (B, 18.5 mg, 0.050 mmol) dissolved in acetonitrile (5 mL) and diisopropylethylamine (0.2 mL), all of which was cooled to 0°C in an ice bath. The reaction was stirred and allowed to warm to room temperature overnight. The reaction mixture was concentrated in vacuo, and the residue purified by reverse phase (C18 silica gel) flash column chromatography (0 to 20% methanol in 0.1% aqueous formic acid) to yield 14 mg (55%) of QENAQ as a red solid. 1H NMR (600 MHz, methanol‐d 4) δ 7.88 (d, J = 9.1 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 9.2 Hz, 2H), 4.22 (s, 2H), 3.90 (s, 2H), 3.69 (q, J = 7.3 Hz, 6H), 3.58 (q, J = 7.1 Hz, 2H), 3.50–3.42 (m, 8H), 1.40 (t, J = 7.2 Hz, 9H), 1.37 (t, J = 7.2 Hz, 9H), 1.26 (t, J = 7.0 Hz, 3H); 13C NMR (151 MHz, Methanol‐d 4) δ 163.14, 151.22, 150.86, 145.75, 140.33, 126.20, 124.07, 121.58, 113.65, 55.88, 55.85, 54.47, 49.57, 46.83, 44.03, 12.45, 7.99, 7.90; (ESI+) m/z calcd for C30H50O1N6 [M]2+: 255.2021; found 255.2018.
UV/Vis spectra were measured at room temperature using a SmartSpec Plus photometer (Bio‐Rad, Hercules, CA, USA).
Animals
All animal care and experimental procedures were approved by the University of California (UC) Berkeley Institutional Animal Care and Use Committee and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Male wild‐type mice (C57BL/6J strain, Jackson Laboratory) age 0–6 months were used in all experiments.
Expression of ion channel proteins in Xenopus laevis oocytes
Oocytes of maturation stages I to IV from Xenopus leavis frogs were surgically removed from female adults by laparotomy. Frogs were housed in a facility approved by Office of Laboratory Animal Care (OLAC) of UC Berkeley. The animals were anaesthetized by tricaine methane‐sulfonate (MS‐222, Syndel, Ferndale, WA, USA) at neutral pH, and individual frogs were used up to six times to harvest eggs. Removed oocytes were predigested with 1–2 mg·mL−1 collagenase (Sigma, St Louis, MO, USA) to support defolliculation, and connective membranes were manually removed. Oocytes with intact vitelline membranes were kept at 16°C in oocyte Ringer's solution (ORI; composition in mM: 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, adjusted to pH 7.4 with NaOH). Expression of the channel protein, Shaker‐inactivation removed (Shaker‐IR), was accomplished by injection of mRNA constructs which were synthesized from linearized DNA plasmids (mMessage mMachine T7 kit, Applied Biosystems, Foster City, CA, USA) containing Shaker‐IR and a T7 site as previously described (Smart and Krishek, 1995). RNA injection took place 12 to 48 h after oocyte harvest with a Nanoinject II microinjector (Drummond Scientific, Droomall, PA, USA), and injected oocytes were stored in ORI for 2–3 days at 16°C before recording; ORI buffer was replaced daily. Injection pipettes were pulled from borosillicate capillaries (Microcaps: inner diameter 0.70 mm, outer diameter 0.97 mm, Drummond Scientific) with a micropipette puller (P‐97, Sutter Instruments, Novato, CA, USA). Tips were broken over a tissue cloth to have an approximate tip diameter of 15 μm, backfilled with coloured mineral oil and loaded with RNA solution. RNA was diluted in ORI to a final concentration of 0.05–1 ng·nL−1, and 25–50 nL was injected totaling 2.5–5 ng of RNA. For optimal expression and oocyte health, RNA was injected at the midline, the equatorial band and between animal and vegetal pole (Smart and Krishek, 1995) of stage IV oocytes.
Voltage‐clamp inside‐out recordings from Xenopus oocytes
Before recording, the outer vitelline membrane was removed by placing the oocyte in hypertonic stripping solution (in mM: 200 K+ asparate, 20 KCl, 1 MgCl, 10 EGTA, 10 HEPES, pH 7.4) for a few minutes (Brown et al., 2008) and carefully peeling off the outer membrane with fine forceps avoiding cell damage. Oocytes were then transferred into a recording chamber filled with a bath solution matching the intracellular ion composition (in mM: 160 KCl, 0.5 MgCl2, 1 EGTA and 10 HEPES, adjusted pH 7.4 with KOH). Filamented, heat polished borosillicate glass pipettes (Warner Instruments; outer diameter 1.5 mm and inner diameter 1.17 mm) with 1–2 MΩ resistance were filled with pipette solution mimicking extracellular ion composition (in mM: 150 NaCl, 10 KCl, 10 HEPES, 1 MgCl2 and 3 CaCl2, adjusted to pH 7.4 with NaOH, filtered 0.2 μm). All electrophysiology experiments were conducted at room temperature. A high seal resistance was established before the pipette was retracted rapidly ripping out a piece of membrane covering the opening of the pipette forming an inside‐out patch. Electrophysiological signals were amplified by a Patch‐Clamp PC‐501A amplifier (Warner Instruments), low‐pass filtered at 1 kHz, digitized at 5 kHz by a Digidata 1322A converter (Molecular Devices, Sunnyvale, CA, USA) and acquired with Clampex 10 software (Molecular Devices). A custom‐made Matlab program (MathWorks, Natick, MA, USA) was used to convert, concatenate and analyse Clampex files. A seal test (−60 to −80 mV for 50 ms) verified the stability of the recording before each voltage step. A self‐made line connector was used to quickly apply different solutions to the membrane patch that was guided into a polyethylene tube for fast solution exchange. Dose–response curves were created utilizing the following model equations:
| (1) |
where I c is current at concentration c, A 1 is bottom and A 2 is top asymptote; the Matlab regression model was programmed to let A 1 → 0 for c → infinity and A 2 → 100 for c → 0. A non‐parametric Ranksum (Mann–Wilcoxon) test was used to test the difference between the IC50s.
For modelling voltage dependence of QAQ block, the following equation was applied:
| (2) |
where K(0) is the zero‐voltage dissociation constant, [B] is the blocker concentration (here 100 μM), z the molecular charge (here 1), δ the fraction of the total potential drop at the binding site, F Faraday constant, R gas constant absolute temperature and T absolute temperature (Woodhull, 1973). For the molecular charge, we used z = 1, because it is very unlikely that the two positive charges of the molecule sense the electric field of the membrane, especially in the trans configuration where the two ammonium groups are located about 19 Å away from each other.
Voltage‐clamp whole‐cell recordings from dissociated trigeminal neurons
Trigeminal ganglion (TG) neurons from neonatal mice were prepared as described in (McKemy et al., 2002). Briefly, TGs were dissected, and neurons were dissociated with collagenase and trypsin and plated on poly(l‐lysine)‐coated coverslips. TG neurons were kept in minimum essential medium containing 5% horse serum, MEM vitamins (Invitrogen, Waltham, Massachusetts, USA), glutamine and penicillin–streptomycin for one night before measurements. Patch clamp recordings were performed at room temperature. Bath solution contained (in mM): 138 NaCl, 1.5 KCl, 1.2 MgCl2, 2.5 CaCl2, 5 HEPES and 10 glucose. Pipette solution contained (in mM): 10 NaCl, 135 K+ gluconate, 10 HEPES, 2 MgCl2, 2 MgATP and 1 EGTA. All solutions were adjusted to pH 7.4 and filtered (0.2 μm). Patch pipettes resistances were 2–4 MΩ. Electrophysiological measurements were performed with a Patch‐Clamp PC505B amplifier (Warner Instruments, Hamden, CT, USA), digitized with a Digidata 1200 interface (Molecular Devices) and low‐pass filtered at 2 kHz. Na+ channel currents were corrected by P/N leak subtraction. For intracytoplasmic application through the patch pipette, QENAQ was dissolved in intracellular solution (200 μM), and measurements were started after 5–10 min of equilibration time.
Illumination for voltage‐clamp experiments
Illumination of membrane patches or cells was performed by a Lambda‐LS xenon lamp (Sutter Instruments) with 379 ± 17 nm and 500 ± 8 nm band pass filters and a 125 W light source. The light beam was led through a 20× objective [Nikon Fluor, Tokyo, Japan, Numerical Aperture (NA) 0.75]. Light intensities, measured at the objective using a hand‐held power meter (Newport Corporation, Irvine, CA, USA), were 3.85 mW·cm−2 for 380 nm light, 4 mW·cm−2 for 480 nm light and 500 μW·cm−2 for 500 nm light.
Preparation of intact dorsal root ganglia (DRGs) tissue from adult mouse
Adult mice (wild‐type C57BL/6J, Jackson Laboratory, on regular diet, housed in a mouse facility approved by OLAC of UC Berkeley) were deeply anaesthetized with isoflurane and quickly killed by cervical dislocation. The skin covering the dorsal side of the mouse was removed, and the peripheral sciatic nerves were isolated from the surrounding muscle. Fibres were transected about 5 mm distal to the sciatic plexus preserving about 2.5–3 cm of total nerve fibre. Following transection, the spinal column from the upper cervical vertebrae to the lower lumbar region was carefully detached from the mouse and placed in cold (4°C) artificial CSF solution (ACSF, buffered at pH 7.4, in mM: 124 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 26 NaHCO3, 20 d‐glucose, 2 Na pyruvate and 0.4 ascorbic acid), constantly equilibrated with 95% O2 and 5% CO2. A laminectomy from the thorax to the sacrum revealed the spinal column that was subsequently removed exposing the DRGs. Finally, bone structures of the spinal column were gently opened to take out the lumbar L3–L6 DRGs. Only the most outer layer of the epineurium was removed, leaving the tissue intact otherwise. After dissection, the DRG preparations were placed on a floating nitrocellulose membrane (Sartorius Stedim Biotech, Concord, CA, USA) in an oxygenation chamber keeping the tissue at the air‐solution interface of equilibrated ACSF at room temperature (18–22°C) to recover for 30–60 min before the start of recordings.
Multi‐electrode array recordings from intact dorsal root ganglia
Intact DRG tissue was mounted onto a multi‐electrode array (MEA) chip (MEA60‐200 three‐dimensional (3D) GND, Qwane Bioscience, Lausanne, Switzerland) consisting of sixty 3D tip‐shaped platinum electrodes penetrating 60 μm into tissue spaced at 200 μm (Heuschkel et al., 2002). The tissue is secured in place to ensure optimal contact using a ‘harp’ made from dialysis membrane stretched over thick platinum wire and bonded with super glue; the wire was U‐shaped to allow the nerve to exit without being crushed. The MEA chip was mounted on an MEA1060‐UP‐BC amplifier (Multi Channel Systems, Germany) and placed on the stage of an upright microscope (Labophot‐2, Nikon, Japan). Peripheral nerves were electrically stimulated using a suction electrode driven by an external, battery driven DS2 stimulus isolator (Digitimer, Welwyn Garden City, UK). The nerve bundle was gently led into a manipulator‐mounted glass suction electrode of appropriate size. Stimulation mode (constant current vs. constant voltage), blanking time of the amplifier (200–2000 μs), stimulus pulse length (20–600 μs) and stimulation strength were optimized to yield extracellular responses with maximal response amplitude while minimizing electrical artefacts at the beginning of recordings. DRGs were checked for response to stimulation at 1 Hz prior to recording. Evoked responses were recorded at 20 kHz sampling rate with MC_Rack v4.5.12 software (Multi Channel Systems, Reutlingen, Germany). Raw voltage traces were filtered with a second‐order Butterworth high‐pass filter at 300 Hz and a low‐pass filter of 10 kHz. Illumination was provided by a Spectra‐LCR‐3X‐A2 LED light source (Lumencor, OR, USA) routed through a 4× objective (NA 0.13, Nikon), yielding an intensity of 13.2 mW·cm−2 for 475 nm light. Stimulator, recording software and light source were centrally controlled by a custom made Matlab (MathWorks) program triggering TTL pulses through a BNC‐2110 I/O card (National Instruments, Austin, TX, USA). The MEA chamber was continuously perfused with oxygenated ACSF at about 3 mL·min−1, and recordings were performed at constant 34°C. QENAQ (300 μM in ACSF) was incubated with DRGs for 5 min. DRGs were then washed with ACSF for 10 min after incubation, before experiments. Recordings were done at a stimulation rate of 10 Hz while illuminating the DRG with 480 nm light or in darkness. The start of recordings was synchronized to the onset of the stimulus pulse, and recordings lasted 50 to 90 ms for each stimulation sweep. Each experiment consisted of 5 cycles of 30 s under 480 nm light followed by 30 s in the dark. The DRG was stimulated for the last 5 s under illumination or in the dark, allowing 25 s to recover from adaptation in between stimulation episodes.
Analysis of multi‐electrode data
Multi‐electrode data were imported into Matlab using the import functionality of the FIND program bundle (University of Freiburg, Germany; Meier et al., 2008). Subsequent analysis was performed by a custom made Matlab program package. Stimulation artefacts were excluded from the analysis, and evoked signals were detected by a threshold manually set beyond the noise level. Overall responses were divided in negative and positive deflections, and each deflection was analysed separately. A Matlab algorithm conservatively excluded small and/or irregular signals. We chose the area under the voltage trace as parameter to calculate photo‐sensitivity. This value was averaged over the five recording cycles in each wavelength. As QAQ and QENAQ function as open‐channel blockers (Mourot et al., 2012), photo‐sensitivity increases over the duration of a stimulation episode. Therefore, the last 1.5 s of the stimulation protocol was used to calculate a measure of normalized photo‐sensitization, the photo‐switching index (PI), which is defined as (area in 480 nm − area in dark)/(area in 480 nm + area in dark). The distribution of PI values followed a non‐normal distribution, and we therefore applied the non‐parametric Mann–Whitney U‐test (for two comparisons) or the Kruskal–Wallis test with Dunn's post hoc test (for more than two comparisons). The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Molecular docking in silico
The three‐dimensional structure of the open channel Kv1.2–2.1 chimera (PDB ID 2R9R (Long et al., 2007) was used to dock cis and trans QAQ molecules into the lumen of the channel. Molecular docking was carried out in Glide 5.7 (Halgren et al., 2004) implemented in Maestro 9.2 (Schrodinger Inc., Portland, OR, USA). The X‐ray structure was used to create a grid after the addition of hydrogen atoms and the removal of non‐protein moieties. A QAQ 3D structure (cis or trans configuration) was docked into this grid using the standard precision algorithm.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Concentration dependence of QAQ blockade
Intracellular QAQ blocks voltage‐gated K+, Na+ and Ca2+ channels in darkness (Mourot et al., 2012). To investigate QAQ binding to the internal TEA binding site, we have used the Shaker‐IR K+ channel, a mutant that does not inactivate during brief depolarization (Hoshi et al., 1990; Demo and Yellen, 1991). This channel lacks the N‐terminal ‘ball and chain’ motif, which by binding to the internal TEA binding site (Zhou et al., 2001) might interfere with QAQ blockade. Because QAQ does not cross the lipid bilayer (Mourot et al., 2012), we used the inside‐out configuration from excised patches to directly expose the photo‐switchable blocker to the cytosolic side of Shaker‐IR and characterize cis and trans QAQ blockade (Figure 2A). Shaker‐IR opening was triggered with depolarizing voltage steps (−60 to +40 mV) for 200 ms at 1 Hz. QAQ was applied until blockade of current reached steady state (Figure 2B). Then, QAQ was photo‐isomerized to cis using 380 nm light, which resulted in a rapid increase in K+ current. Illumination with 500 nm light reverted QAQ to trans and restored blockade. Using 100 μM QAQ, 61.0 ± 5.5% of the Shaker‐IR steady‐state current (I ss) was blocked under green light, and blockade was reduced to 16.4 ± 3.8% under 380 nm light (Figure 2C, D). At this illumination condition, >96% of the QAQ molecules are converted to cis (Mourot et al., 2013b). Hence, while both isomers of QAQ can block Shaker‐IR current, the effect is more profound in trans than in cis. A full dose–response curve of steady‐state current blockade showed that trans QAQ has a sixfold lower IC50 value (65 ± 17 μM) compared with cis QAQ (380 nm light: >96% cis, IC50 = 390 ± 111 μM, P = 0.008 Mann‐Wilcoxon test, Figure 2D).
Figure 2.
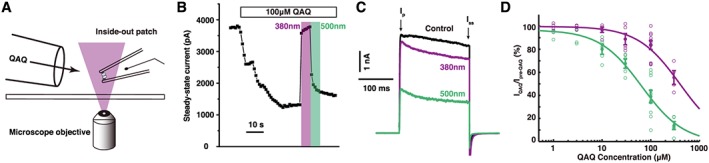
Concentration dependence of cis and trans QAQ blockade on Shaker‐IR K+ channels. (A) Schematic representation of an inside‐out patch recording with local perfusion tube and illumination through the microscope objective. QAQ has direct access to the internal lumen on Shaker‐IR. (B) Representative Shaker‐IR current recorded at 1 Hz frequency, upon application of QAQ 100 μM and under illumination with 380 and 500 nm light. Current was elicited by a 200 ms depolarization from −60 to +40 mV. Steady‐state current (I ss), that is, current at the end of the 200 ms pulse, is plotted. (C) Representative K+ current elicited by the depolarization to +40 mV, before QAQ application and during QAQ application under green and violet light illumination. I p, current at the peak; I ss, steady state current. (D) Concentration dependence of QAQ block (1 Hz) under 380 nm (violet) and 500 nm light (green). IC50trans = 65 ± 17 μM, IC50cis = 390 ± 111 μM (mean ± 95% confidence interval); open circles represent individual experiments, and filled circles mean ± SEM, n 1 μM = 4, n 3 μM = 4, n 10 μM = 5, n 30 μM = 5, n 100 μM = 10 and n 300 μM = 5 patches; IC50 curve derived from data range [10, 300].
Voltage dependence of QAQ blockade
Because QAQ is a charged molecule, its interaction with K+ channels may be sensitive to the electric field across the membrane. We investigated the effect of membrane potential on cis and trans QAQ block with incremental voltage steps from −80 to +40 mV (Figure 3A). QAQ blockade was more potent under 500 nm than 380 nm light, for all membrane potentials tested (Figure 3A, B). To quantify the voltage dependence of blockade, we measured the reduction in current by both trans and cis QAQ (100 μM) at voltages where channel activation is complete, that is, greater than −20 mV (see black trace in Figure 3C). Blockade with trans QAQ was slightly voltage dependent and favoured by more positive potentials (Figure 3C). Voltage dependence of block can be described by the ‘effective valence’ value, and the fraction δ of the membrane potential QAQ traverses to reach its binding site (Woodhull, 1973). A fit of Equation (2) (see the section on Methods) yields δ trans = 0.18, which agrees closely with the δ value (0.15) reported for TEA block on K+ channels (French and Shoukimas, 1981; Yellen et al., 1991). In contrast, blockade with cis QAQ was not voltage dependent and was fitted with a linear regression (mcis = 0.0004 mV−1, n.s. to m = 0 with P = 0.32, Student's t‐test). This suggests that cis QAQ blocks K+ current, but without sensing the electrical field of the membrane.
Figure 3.
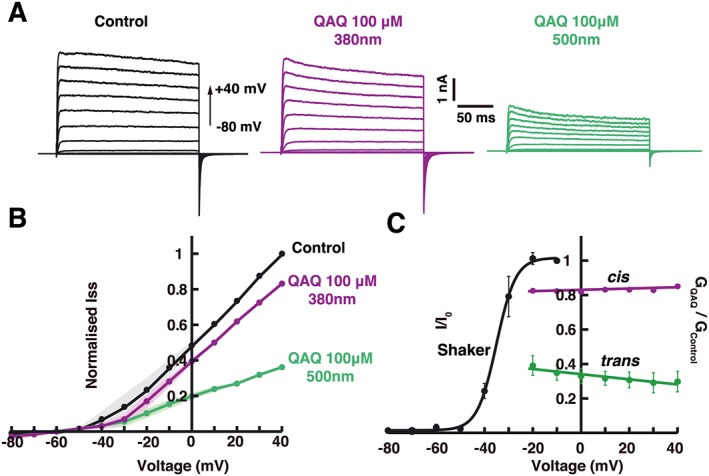
Voltage dependence of cis and trans QAQ blockade on Shaker‐IR K+ channels. (A) Representative current traces at different holding voltages (from −80 to +40 mV) before QAQ application and in the presence of QAQ (100 μM) under 380 or 500 nm light. B) Current–voltage relationships of Shaker‐IR before QAQ application, or during QAQ application (100 μM) under 380 nm or 500 nm light. Currents were normalized to pool multiple patch recordings (n = 5); solid line represents mean values, and SEMs are represented by shaded areas. (C) Voltage dependence of cis and trans QAQ blockade, compared with the voltage dependence of Shaker‐IR gating. The amplitudes of Shaker current I are normalized (I/I 0) so that I ss at −10 mV is unity. The ratio of Shaker I ss current in the presence of 100 μM QAQ in either wavelength was plotted over a voltage range where Shaker‐IR is fully open, that is, for voltages greater than −20 mV. The cis QAQ shows no significant voltage dependence of block (P > 0.05, Student's t‐test), and solid lines represents linear regression fit (m cis = 0.0002 ± 0.00008 mV−1). In contrast, trans QAQ blockade exhibited a slight voltage dependence (zδ trans = 0.18 and K(0)trans = 58 μM). Error bars represent SEM, n = 5.
Use and time dependence of QAQ blockade
A common property of quaternary ammonium pore blockers is that they bind to the pore only after the channel has opened (Choi et al., 1993). To test whether this is true for cis and trans QAQ, we have perfused 300 μM QAQ under 380 or 500 nm light for 1 min, while keeping the membrane potential at −60 mV to ensure a very low opening probability. This perfusion time is normally sufficient to achieve potent blockade (Figure 2B). Shaker‐IR currents were then monitored at 1 Hz (+40 mV depolarization pulses), under both wavelengths of light, and compared with currents recorded before QAQ application. The first depolarizing test pulse elicited a nearly‐full sized I ss current, but block rapidly accumulated over time with subsequent pulses under both wavelengths of light similar to QAQ application while Shaker is constantly opened (Figure 4A). This indicates that neither cis nor trans QAQ has access to the pore of Shaker‐IR while the channel is closed. Like other quaternary ammonium pore blockers, QAQ produced a use‐dependent accumulative blockade in response to a train of depolarizing pulses, under both wavelengths of light (Figure 4B). Higher frequencies produced more blockade for both cis and trans QAQ, while blockade with cis QAQ was negligible for frequencies <1 Hz. Hence, increased opening of K+ channels allows increased access of QAQ to the pore. The blockade of both cis and trans QAQ was also time dependent, that is, blockade developed non‐instantaneously during a voltage pulse (Figure 4C), which corresponds to the association of QAQ with the open channel. The current block with trans QAQ followed an exponential decline and increased with QAQ concentration (Figure 4D), similar to other open‐channel blockers, including long alkyl‐chain quaternary ammonium (Choi et al., 1993) and the N‐terminal ball and chain motif of K+ channels (Demo and Yellen, 1991). In contrast, the blockade of cis QAQ could not be fitted with an exponential function.
Figure 4.
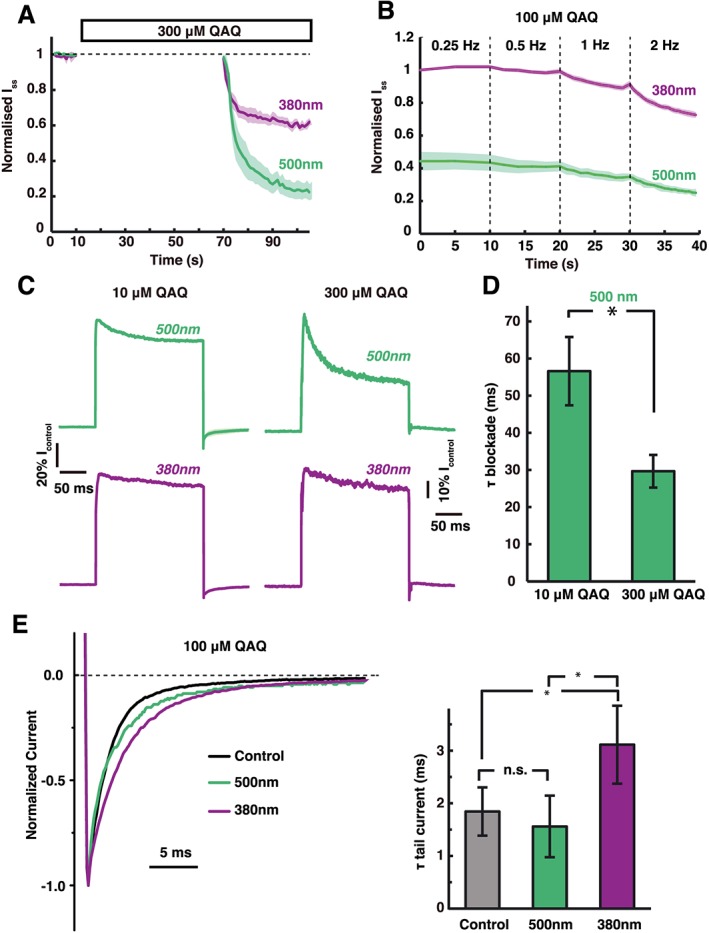
State‐dependent interaction of QAQ with Shaker‐IR. (A) QAQ does not associate with closed Shaker‐IR channels, neither in the cis nor in the trans form. Shaker‐IR channels were kept close at negative potential (−60 mV), while QAQ was perfused either under green or under violet light. QAQ block accumulated only after channels were open using depolarization to +40 mV. Shaded areas represent SEM, n = 4. (B) The cis and trans QAQ are use‐dependent blockers of Shaker‐IR K+ channels. Blockade accumulated with increased opening frequency of the channels (0.25 to 2 Hz). Average of three examples is shown. (C) Representative traces for association rates of trans and cis QAQ (10 and 300 μM) to open Shaker‐IR channels. Current kinetics within a 200 ms pulse (+40 mV) were normalized to control (pre‐QAQ) and compared for 10 and 300 μM QAQ. (D) Blockade kinetics increased with trans QAQ concentration, fitting an exponential function. Data shown are means± SEM; * P< 0.05, significantly different as indicated; Student's t‐test, n = 6. (E) Closing kinetics of Shaker‐IR in the absence and presence of cis or trans QAQ. Representative tail currents are plotted after normalization. Inset: trans QAQ (500nm) did not affect channel closing rate. n.s, P > 0.05, Student's t‐test, n = 6. In contrast, cis QAQ (380nm) significantly slowed channels closing compared with trans QAQ and also with control values. *P < 0.05, significantly different as indicated, Student's t‐test, n = 6.
Interaction of QAQ with the gating mechanism of Shaker‐IR
We next tested whether QAQ interacts with the gating mechanism of the Shaker‐IR channel. Most quaternary ammonium blockers do not affect the activation kinetics of the channel, but slow down deactivation because they must dissociate before the channel can close (Armstrong, 1971; Choi et al., 1993; Holmgren et al., 1997). Similarly, we found that the activation kinetics of Shaker‐IR were not affected by the presence of QAQ, in either wavelength (see Figure 2C for instance). To look at deactivation kinetics, we measured the exponential decline in tail current after repolarization to −60 mV and found that channel closing was indeed slowed when cis QAQ was bound to the pore (Figure 4E), consistent with a ‘foot in the door’ mechanism (Armstrong, 1971). Surprisingly, trans QAQ had no significant effect on channel deactivation kinetics, which may indicate a faster dissociation kinetics of the elongated trans isomer, compared with the bent cis QAQ.
Binding modes of cis and trans QAQ
The two QAQ isomers block Shaker‐IR K+ channels with different potency, voltage dependence and time dependence, and only the cis isomer affects channel gating, suggesting that cis and trans QAQ bind the internal lumen with different orientations. The crystal structure of the K+ channel might provide additional insights into light‐dependent QAQ blockade. We used a docking simulation to explore the predicted fit of cis or trans QAQ to a chimeric K+ channel, whose pore domain is derived from Kv1.2, a mammalian homologue of Shaker (Long et al., 2007). This channel was crystallized in the open conformation, which should allow QAQ access and binding. Figure 5 shows one of the best docking poses for each isomer. In the trans configuration, a quaternary ammonium of QAQ is directly beneath the selectivity filter, on the central fourfold axis of the channel (Figure 5A, B). The second quaternary ammonium is directed away towards the exit, while the azobenzene interacts with hydrophobic residues from the S5 and S6 helices. This pose is similar to quaternary ammonium blockers complexed with the KcsA K+ channel (Zhou et al., 2001; Lenaeus et al., 2005; Faraldo‐Gómez et al., 2007) and is consistent with electrophysiology experiments indicating two functionally distinct subsites for blockers with a TEA headgroup and a hydrophobic tail (Baukrowitz and Yellen, 1996). Binding of cis QAQ was drastically different: due to its bent configuration, this isomer failed to position a charged ammonium in a central position below the selectivity filter, in none of the best poses (Figure 5 A, B). These in silico results are in agreement with our electrophysiology providing a plausible explanation for why blockade of cis QAQ is less potent (Figure 2D) and not voltage sensitive (Figure 3C).
Figure 5.
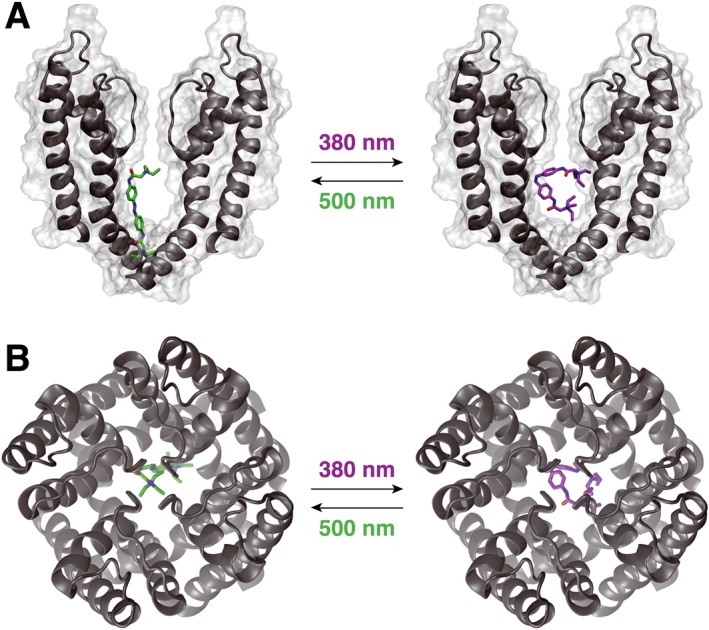
Molecular docking of cis and trans QAQ to open K+ channel. Crystal structure of the open KV1.2–2.1 chimera with docked QAQ isomers. The voltage‐sensing domains of the K+ channel are not represented, for clarity. (A) Side view, perpendicular to the membrane. Note the position of the quaternary ammonium ion just below the selectivity filter for trans, but not for cis QAQ. Only two of the four subunits are shown, for clarity purposes. (B) Top view, from the extracellular side.
Tuning the photo‐physical properties of QAQ
QAQ has been used in vivo as an optically regulated local anaesthetic, but only in the cornea that is fully transparent, thereby facilitating light access. Optical control of nociception in the skin or deeper into organ tissues requires a QAQ derivative that photo‐switches at longer wavelengths of light. The photo‐sensitive core of QAQ (Figure 1A) is made of a ‘classical’ azobenzene group flanked with two mildly electron‐withdrawing acylamino moieties. In the dark, trans QAQ predominates, with a strong π–π* band at 362 nm in water (Figure 6B). Irradiation with near UV light (380 nm) isomerizes the azobenzene group to the higher‐energy cis conformer. The cis QAQ is metastable and relaxes to the lower‐energy trans state in the dark, with a half‐life of 7–8 min (Mourot et al., 2012). Isomerization back to trans can be accelerated upon irradiation with a longer wavelength of light (500 nm). The limitation of using UV light for in vivo and therapeutic applications prompted us to design a red‐shifted version of QAQ. The incorporation of electron‐donating groups in the ortho position of the azobenzene ring shifts the wavelength for maximal photo‐switching to a longer wavelength and increases the rate of spontaneous cis to trans relaxation in the dark (Beharry et al., 2011; Fehrentz et al., 2012; Samanta et al., 2013; see Dong et al., 2015). Yet efficient red shifting is achieved with the introduction of bulky electron‐donating substituents, which can impair the binding of the ligand moiety to the protein (Fehrentz et al., 2012). Azobenzenes with an electron‐donating group in the para position also absorb at longer wavelengths of light (blue range) and display fast kinetics of thermal relaxation in the dark (ms‐s) (Sadovski et al., 2009; Mourot et al., 2011; Kienzler et al., 2013). We reasoned that the steric hindrance induced by a para substitution might be better tolerated than that induced by an ortho substitution and that ion channel blockade might be less affected. We designed a red‐shifted version of QAQ (QENAQ) with minimal variation to the structure, by replacing the electron‐withdrawing acylamino with an electron‐donating ethylamino group in the para position (Figure 6A). QENAQ was synthetized as shown in Scheme 1. As expected, QENAQ displayed a red shift in its absorption spectrum by about 80 nm (λ max = 445 nm) in aqueous solution (Figure 6B), similar to that observed for related red‐shifted K+ channel blockers (Mourot et al., 2011).
Figure 6.
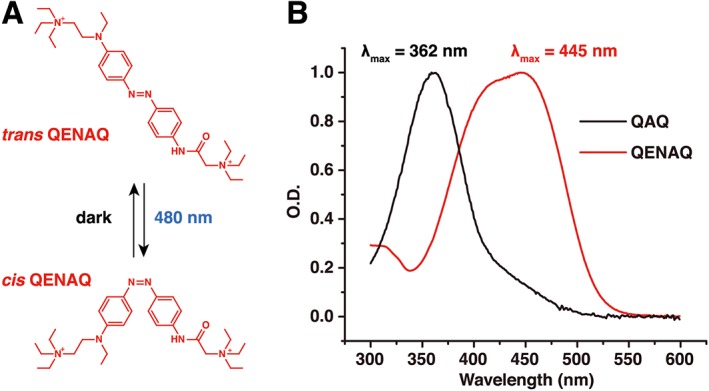
QENAQ, a red‐shifted QAQ. (A) Chemical structure of trans and cis QENAQ. QENAQ photo‐isomerizes to cis under illumination with blue light (480 nm) and reverts back to trans rapidly (seconds) in the dark. (B) Absorption spectra of QAQ and QENAQ in the dark (trans isomer) in phosphate buffer saline, pH 7.4. λ max = 362 nm for QAQ and 445 nm for QENAQ.
QENAQ photo‐sensitizes native voltage‐gated K+ and Na+ channels
QAQ blocks not only K+ channels but also Na+ and Ca2+ channels (Mourot et al., 2012). The initial red‐shifted compounds we had designed were potent K+ channel blockers but failed to photo‐sensitize Na+ channels and therefore could not silence neurons (Mourot et al., 2011). To test whether QENAQ photo‐sensitizes voltage‐gated Na+ and K+ channels, we included it in the patch pipette, to allow direct access of the drug to the cytoplasmic binding site. Voltage‐gated Na+ current from primary sensory neurons were recorded under 480 nm light and in darkness. We found that Na+ currents were larger under blue light, when QENAQ (200 μM) was partially in cis, than in darkness, when QENAQ was fully in trans (Figure 7A). Illumination with blue light rapidly increased current, while blocking was restored within seconds in darkness (Figure 7B). We initially tested the concentration used for QAQ (100 μM) but did not observe reliable photo‐switching (not shown). QENAQ (200 μM) also photo‐sensitized native voltage‐gated K+ channels from primary sensory neurons. K+ currents were larger under blue light than in darkness (Figure 7C) and blockade occurred within seconds after light was shut off (Figure 7D). Hence, like QAQ, QENAQ blocks voltage‐gated K+ and Na+ channels in the trans configuration and unblocks them in cis. But unlike QAQ, conversion to cis occurred upon illumination with visible light, instead of near‐UV light needed for QAQ. In addition, cis QENAQ is not thermally stable in the dark, and conversion back to trans occurs spontaneously, within seconds as judged by the recurrence of Na+ and K+ channel blockade. Hence, a single wavelength of light, in the blue range, is required to rapidly toggle QENAQ in and out of its binding site on ion channels.
Figure 7.
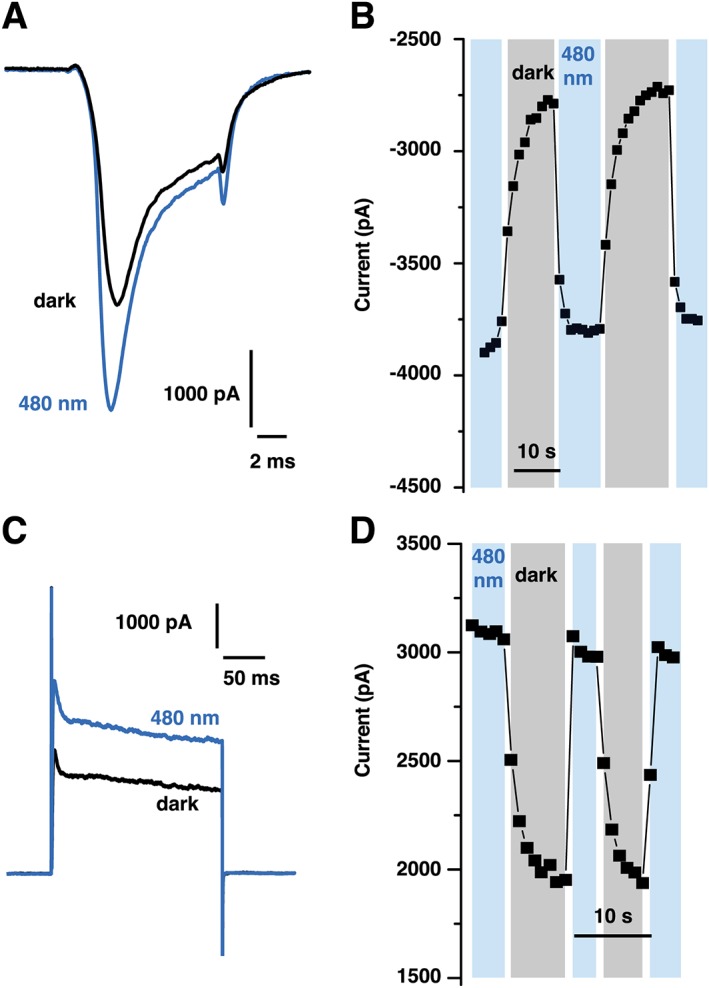
Optical control of native Na+ and K+ channels in dissociated trigeminal neurons with QENAQ. (A) Representative voltage‐gated Na+ current from a TG neuron filled with 200 μM QENAQ, under 480 nm light and in the dark. Opening of the channels was triggered at 1 Hz using a 10 ms depolarization from −60 to −10 mV. (B) Reversibility of Na+ peak current photo‐switching. The cis QENAQ was unstable in the dark and reversed to trans within seconds. (C) Representative voltage‐gated K+ current from a TG neuron filled with 200 μM QENAQ, under 480 nm light and in the dark. Opening of the channels was triggered at 1 Hz using a 200 ms depolarization from −60 to +40 mV. (D) Reversibility of K+ steady‐state current photo‐switching.
QENAQ photo‐sensitizes intact DRG neurons
QENAQ, like QAQ, photo‐sensitizes voltage‐gated Na+ and K+ channels in primary sensory neurons. To check whether QENAQ could be used to optically control the electrical activity of these neurons, we turned to a system we have developed for QAQ (Mourot et al., 2012). We recorded the activity of tens of sensory neurons at once within an intact DRG, while simultaneously controlling the isomeric state of the photo‐switch using the optics of a microscope (Figure 8A). Whole intact DRGs were placed on a 3D MEA, consisting of 60 spine‐shaped electrodes that protrude into the ganglion (Figure 8B). Neuronal activity was evoked by electrically stimulating action potentials in axons of the peripheral nerve. Each electrode recorded the integrated extracellular activity of several neurons, appearing as a compound action potential (CAP; Figure 8C). Most signals recorded originate from C‐fibre neurons, including nociceptors, which can be identified by their slow‐conduction velocity (Mourot et al., 2012). After treatment with QENAQ, the amplitude of evoked CAPs was larger in 480 nm light than in darkness (Figure 8C, D). Moreover, the CAP amplitude diminished more rapidly in darkness than in 480 nm light during a stimulus train (Figure 8D, E), consistent with cumulative activity‐dependent blockade of voltage‐gated channels. Photo‐sensitivity was quantified by a measure called PI of firing (Figure 8E inset and methods), which reflects the amount of photo‐switch compound loaded into neurons. QAQ is a membrane‐impermeant molecule, but it can enter neurons by passing through open TRPV1 channels or other conduit channels that open transiently in dissected DRGs (Mourot et al., 2012). Similarly, QENAQ alone photo‐sensitized neurons, while application of the TRPV1 agonist capsaicin (1 μM) promoted QENAQ loading into neurons, as shown by an increased PI (Figure 8F). Hence, QENAQ could accumulate in neurons through activated nociceptive ion channels and enable photo‐control of action potential firing with visible light.
Figure 8.
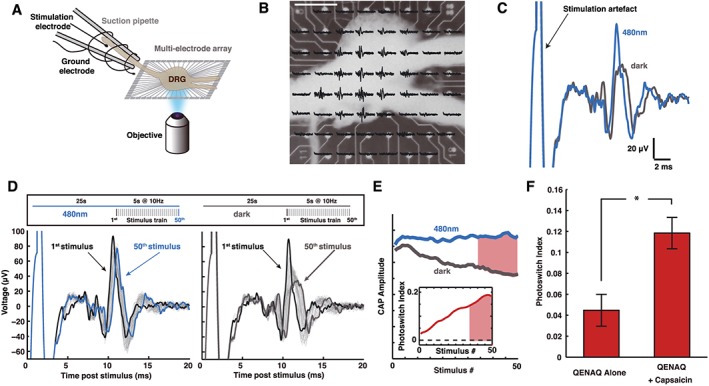
Optical control of intact DRGs using visible light. (A) Schematic representation of the experimental arrangement for measuring DRG photo‐sensitization. An intact, acutely dissected mouse DRG is positioned on an MEA with 60 electrodes. The electrodes have a 3D tip shape in order to penetrate the DRG and detect signals from neurons. The peripheral nerve bundle was led into a suction electrode and electrically stimulated using a battery‐driven isolator. A 4× objective was used to focus light (480 nm) onto the DRG. (B) Image of an intact DRG mounted onto a MEA chip with superimposed nerve stimulation responses (CAPs). Scale bar 400 μm. (C) CAP signals recorded from one electrode, in darkness and under 480 nm light. The DRG was pre‐incubated with 300 μM QENAQ. The amplitude of evoked CAPs was smaller in darkness (trans QENAQ) than under illumination with blue light (cis QENAQ). (D) Adaptation of CAP amplitude of a QENAQ‐treated DRG under blue light and in the dark over the course of a stimulus train (5 s at 10 Hz, with 25 s pauses in between trains, upper panel). (E) Change in CAP amplitude during a stimulus train. Because of the use‐dependence of block by QENAQ in darkness, the decrease in CAP amplitude accumulates over time during a stimulus train. We measured the mean CAP amplitude for each wavelength over the last 1.5 s of the stimulus train (time period shown in red) to calculate the PI that increases during a stimulus train (inset). (F) The TRPV1 agonist capsaicin promotes QENAQ loading into sensory neurons. Photo‐sensitization, shown as PI, with QENAQ is greater when QENAQ is co‐applied with capsaicin (1 μM) (n = 83 signals, n = 6 DRGs) than QENAQ alone (n = 180 signals, n = 6 DRGs).* P < 0.05, significantly different as indicated; non‐parametric ranksum test.
Discussion
QAQ and QENAQ are potent photo‐switchable blockers of voltage‐gated K+ and Na+ channels. They provide non‐invasive and rapid silencing of firing by pain‐sensing neurons and therefore offer promise both for understanding and for treating pain. As a consequence, characterizing the biophysics of QAQ binding to voltage‐gated ion channels may increase the utility of QAQ as a fundamental research tool, while rationally tuning the photochemical properties of QAQ should improve the translational value of such compounds.
Mechanism of QAQ blockade
We found that both isomers of QAQ are use‐dependent open‐channel blockers for voltage‐gated K+ channels. The mechanism of trans QAQ block resembles that of hydrophobic quaternary ammonium blockers, with a slight voltage dependence, possibly due to the positioning of the TEA group just beneath the selectivity filter. The mechanism of cis QAQ blockade is quite different: it shows no voltage dependence, in agreement with the TEA groups positioned away from the central path for K+ ions. The cis QAQ is less potent than trans QAQ at blocking ion conduction. This can be explained either by a decreased affinity, or by a decreased efficacy of block of the cis isomer, or both. The fact that cis QAQ drastically affects the closing kinetics of the channel at a concentration (100 μM) that yields only 16% block favours the hypothesis of cis QAQ occupying the lumen, but blocking K+ conduction inefficiently. This hypothesis is also in agreement with our docking simulations and an unusual binding of the quaternary ammonium groups. However, as we technically were unable to perfuse cis QAQ at a saturating concentration (Figure 2D), we could not verify whether blockade with cis QAQ is complete or not, and therefore, we cannot completely rule out the possibility that the affinity of cis QAQ is lower than that of trans QAQ. QAQ is sixfold more potent in blocking K+ channels in the trans than in the cis configuration. This is significantly less than light‐dependent block by two other azobenzene photo‐switches AAQ or BzAQ (30‐ and 60‐fold trans/cis blocking ratio respectively) (Banghart et al., 2009). A large difference in IC50 between the two isomers is important to avoid residual blockade with the cis isomer, for a large range of concentrations. Nevertheless, the appropriate concentration (100 μM), the appropriate stimulation frequency (<1 Hz) and sufficiently bright light can ensure no blockade of K+ channels in 380 nm light and nearly complete block in 500 nm light (see Figure 4B). Similarly, 100 μM QAQ ensures no block of Na+ channels in the dark and nearly full unblock in 380 nm light at low frequency stimulation (Mourot et al., 2012). Together, these actions on channels enable reversible control of action potential firing.
Silencing primary sensory neurons with light
Optical silencing of nociceptor firing can also been achieved through exogenous expression of optogenetic tools (Montgomery et al., 2016). Genetic strategies can be used to restrict opsin expression to a subset of neurons, for example, to nociceptors expressing http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 (Daou et al., 2013) or TRPV1 receptors (Li et al., 2015). Intrasciatic viral injection of AAV6 viruses carrying inhibitory opsins has been shown to selectively transduce nociceptors in wild‐type animals, resulting in effective optical inhibition of acute pain, even though only a small fraction of nociceptors could be transduced (Iyer et al., 2014). Our method for silencing neurons is different: QAQ and QENAQ block the intrinsic neuronal ion channels that are responsible for the initiation and propagation of action potentials. We found that the mechanism of QAQ blockade is indeed very similar to that of classical local anaesthetics (Hille, 2001; Fozzard et al., 2011), which are highly potent analgesics. QAQ and QENAQ block both K+ and Na+ channels, reducing action potential amplitude and increasing its half width (Mourot et al., 2012). Broadening action potentials increases the open time of Na+ channels, potentiating channel blockade and resulting in a net inhibition of neuronal activity (Drachman and Strichartz, 1991). QAQ, which potently photo‐sensitizes ion channels at a concentration of 100 μM, is slightly more potent than QENAQ. Both molecules can be applied to study neuronal systems, as with QX‐314, while adding the ability to precisely photo‐control ion channel blockade. QAQ and QENAQ photo‐sensitize native tissue within minutes, as opposed to days or weeks for viral expression of opsins. Moreover, because they are highly soluble small molecules, QAQ and QENAQ diffuse readily through tissue, reaching all cells that are susceptible to photo‐sensitization.
Red‐shifting QAQ for safer, potent optical control deep into tissue
Light penetration through biological tissue critically depends on the wavelength, with short wavelengths being scattered and absorbed more strongly (Yizhar et al., 2011). QENAQ has a red‐shifted absorbance spectrum and therefore photo‐switches with visible light, which should facilitate control with transdermal illumination. In addition, because QENAQ relaxes back to trans rapidly in the dark (seconds), only a single wavelength is required for rapidly switching the molecule between its active and inactive forms. As a downside, continuous high‐intensity light is required to reduce the concentration of the trans isomer sufficient to relieve blockade. However, a derivative of QENAQ with a higher thermal stability, as has been achieved with an ortho‐substituted QAQ derivatives (half‐life of the cis isomer from hours to days) (Mourot et al., 2013b) would be an ideal candidate for long‐lasting optical control of pain signalling. A major issue remains light delivery in the periphery, where classical optical fibres are not suitable. Multiple technologies are emerging, including wirelessly powered miniature LEDs (Kim et al., 2013; Montgomery et al., 2015; Park et al., 2015) and wireless optofluidic systems for localized drug delivery and photo‐stimulation (Jeong et al., 2015). This latter system could prove extremely useful for opto‐pharmacological applications using QENAQ or other photo‐sensitive drugs.
Potential clinical implications for chronic pain
The emergence of opto‐pharmacology offers interesting therapeutic promise in pain research, by achieving high temporal and spatial precision of drug action (Lerch et al., 2016). Several photo‐pharmacological agents have been introduced targeting specific transduction channels and neurotransmitter receptors in the pain pathway, including TRPV1 agonists and antagonists (Stein et al., 2013; Frank et al., 2015), a http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 agonist (Schönberger and Trauner, 2014), a http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5464‐derived anaesthetic (Stein et al., 2012) and an allosteric modulator of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=292 (Zussy et al., 2016). QAQ and QENAQ represent a different strategy for achieving optical control over nociception. While these compounds act on voltage‐gated ion channels found in all neurons, they only enter and accumulate in nociceptors, which have large‐pore ion channels such as TRPV1. This gives QAQ and QENAQ potentially interesting clinical value for controlling chronic pain while simultaneously minimizing unwanted side effects in other neurons. Indeed, it has been shown that, in animal models of neuropathic pain, TRPV1 channels are hyperactive in central terminals of the dorsal horn (Kim et al., 2014). Hence, by exploiting this cell‐entry mechanism, QAQ and QENAQ could be self‐targeted to hyperactive neurons, that is, the ones that need to be silenced the most, and afford pain‐selective analgesia. In addition, because ion channel blockade can be finely tuned with light intensity and/or wavelength, analgesia could potentially be photo‐titrated at will. QAQ and QENAQ may not fulfil all the prerequisites for clinical use. In particular, it will be important to develop compounds with greater IC50 difference between the two isomers and that can be photo‐isomerized in both directions with wavelengths of light in therapeutic window (600–1200 nm). Nevertheless, QAQ and QENAQ represent a useful starting point for the development of light‐switchable local anaesthetics.
Author contributions
A.M. and C.H. performed electrophysiology and molecular modelling and analysed data. A.M. and M.A.K. designed and M.A.K. synthesized QENAQ. A.M., C.H. and R.H.K. designed the experiments and wrote the manuscript.
Conflict of interest
R.H.K. is a SAB member and consultant of Photoswitch Biosciences, Inc., which is developing commercial uses for chemical photo‐switches. The other authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by US National Institutes of Health grants R01EY02433, R01NS100911 and U01NS090527 to R.H.K. C.H. was supported by a predoctoral fellowship of the Howard Hughes Medical Institute. We thank Sarah Mondoloni (UPMC Paris) for additional experiments, not included.
Mourot, A. , Herold, C. , Kienzler, M. A. , and Kramer, R. H. (2018) Understanding and improving photo‐control of ion channels in nociceptors with azobenzene photo‐switches. British Journal of Pharmacology, 175: 2296–2311. doi: 10.1111/bph.13923.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM (1971). Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J Gen Physiol 58: 413–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banghart MR, Mourot A, Fortin DL, Yao JZ, Kramer RH, Trauner D (2009). Photochromic blockers of voltage‐gated potassium channels. Angew Chem Int Ed Engl 48: 9097–9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz TT, Yellen G (1996). Two functionally distinct subsites for the binding of internal blockers to the pore of voltage‐activated K+ channels. Proc Natl Acad Sci 93: 13357–13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beharry AA, Sadovski O, Woolley GA (2011). Azobenzene photoswitching without ultraviolet light. J Am Chem Soc 133: 19684–19687. [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Bean BP, Woolf CJ (2007). Inhibition of nociceptors by TRPV1‐mediated entry of impermeant sodium channel blockers. Nature 449: 607–610. [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Gerner P, Oh SB, Puopolo M, Suzuki S, Roberson DP et al. (2009). Coapplication of lidocaine and the permanently charged sodium channel blocker QX‐314 produces a long‐lasting nociceptive blockade in rodents. Anesthesiology 111: 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Johnson BE, Goodman MB (2008). Patch clamp recording of ion channels expressed in Xenopus oocytes. JoVE. pii: 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KL, Mossman CC, Aubé JJ, Yellen G (1993). The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron 10: 533–541. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daou I, Tuttle AH, Longo G, Wieskopf JS, Bonin RP, Ase AR et al. (2013). Remote optogenetic activation and sensitization of pain pathways in freely moving mice. J Neurosci 33: 18631–18640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo SDS, Yellen G (1991). The inactivation gate of the Shaker K+ channel behaves like an open‐channel blocker. Neuron 7: 743–753. [DOI] [PubMed] [Google Scholar]
- Dong M, Babalhavaeji A, Samanta S, Beharry AA, Woolley GA (2015). Red‐shifting azobenzene photoswitches for in vivo use. Acc Chem Res 48: 2662–2670. [DOI] [PubMed] [Google Scholar]
- Drachman DD, Strichartz GG (1991). Potassium channel blockers potentiate impulse inhibition by local anesthetics. Anesthesiology 75: 1051–1061. [DOI] [PubMed] [Google Scholar]
- Faraldo‐Gómez JD, Kutluay E, Jogini V, Zhao Y, Heginbotham L, Roux B (2007). Mechanism of intracellular block of the KcsA K+ channel by tetrabutylammonium: insights from X‐ray crystallography, electrophysiology and replica‐exchange molecular dynamics simulations. J Mol Biol 365: 649–662. [DOI] [PubMed] [Google Scholar]
- Fehrentz T, Kuttruff CA, Huber FME, Kienzler MA, Mayer P, Trauner D (2012). Exploring the pharmacology and action spectra of photochromic open‐channel blockers. Chembiochem 13: 1746–1749. [DOI] [PubMed] [Google Scholar]
- Fehrentz T, Schönberger M, Trauner D (2011). Optochemical genetics. Angew Chem Int Ed Engl 50: 12156–12182. [DOI] [PubMed] [Google Scholar]
- Fozzard HA, Sheets MF, Hanck DA (2011). The sodium channel as a target for local anesthetic drugs. Front Pharmacol 2: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank JA, Moroni M, Moshourab R, Sumser M, Lewin GR, Trauner D (2015). Photoswitchable fatty acids enable optical control of TRPV1. Nat Commun 6: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French RJ, Shoukimas JJ (1981). Blockage of squid axon potassium conductance by internal tetra‐N‐alkylammonium ions of various sizes. Biophys J 34: 271–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47: 1750–1759. [DOI] [PubMed] [Google Scholar]
- Heuschkel MO, Fejtl M, Raggenbass M, Bertrand D, Renaud P (2002). A three‐dimensional multi‐electrode array for multi‐site stimulation and recording in acute brain slices. J Neurosci Methods 114: 135–148. [DOI] [PubMed] [Google Scholar]
- Hille BB (2001). Ion Channels of Excitable Membranes, 3rd edn. Sinauer Associates Inc: Sunderland, MA, USA. [Google Scholar]
- Holmgren MM, Smith PL, Yellen G (1997). Trapping of organic blockers by closing of voltage‐dependent K+ channels: evidence for a trap door mechanism of activation gating. J Gen Physiol 109: 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW (1990). Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250: 533–538. [DOI] [PubMed] [Google Scholar]
- Iyer SM, Montgomery KL, Towne C, Lee SY, Ramakrishnan C, Deisseroth K et al. (2014). Virally mediated optogenetic excitation and inhibition of pain in freely moving nontransgenic mice. Nat Biotechnol 32: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J‐W, McCall JG, Shin G, Zhang Y, Al‐Hasani R, Kim M et al. (2015). Wireless optofluidic systems for programmable in vivo pharmacology and optogenetics. Cell : 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienzler MA, Reiner A, Trautman E, Yoo S, Trauner D, Isacoff EY (2013). A red‐shifted, fast‐relaxing azobenzene photoswitch for visible light control of an ionotropic glutamate receptor. J Am Chem Soc 135: 17683–17686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TI, McCall JG, Jung YH, Huang X, Siuda ER, Li Y et al. (2013). Injectable, cellular‐scale optoelectronics with applications for wireless optogenetics. Science 340: 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Chu Y, Han L, Li M, Li Z, LaVinka PC et al. (2014). Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain. Neuron 81: 873–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer RH, Mourot A, Adesnik H (2013). Optogenetic pharmacology for control of native neuronal signaling proteins. Nat Neurosci 16: 816–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaeus MJ, Vamvouka M, Focia PJ, Gross A (2005). Structural basis of TEA blockade in a model potassium channel. Nat Struct Mol Biol 12: 454–459. [DOI] [PubMed] [Google Scholar]
- Lerch MM, Hansen MJ, van Dam GM, Szymański W, Feringa BL (2016). Emerging targets in photopharmacology. Angew Chem Int Ed Engl 55: 10978–10999. [DOI] [PubMed] [Google Scholar]
- Li B, Yang X‐Y, Qian F‐P, Tang M, Ma C, Chiang L‐Y (2015). A novel analgesic approach to optogenetically and specifically inhibit pain transmission using TRPV1 promoter. Brain Res 1609: 12–20. [DOI] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB, MacKinnon R (2007). Atomic structure of a voltage‐dependent K+ channel in a lipid membrane‐like environment. Nature 450: 376–382. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKemy DDD, Neuhausser WMW, Julius D (2002). Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416: 52–58. [DOI] [PubMed] [Google Scholar]
- Meier R, Egert U, Aertsen A, Nawrot MP (2008). FIND – a unified framework for neural data analysis. Neural Netw 21: 1085–1093. [DOI] [PubMed] [Google Scholar]
- Montgomery KL, Iyer SM, Christensen AJ, Deisseroth K, Delp SL (2016). Beyond the brain: optogenetic control in the spinal cord and peripheral nervous system. Sci Transl Med 8: 337rv5. [DOI] [PubMed] [Google Scholar]
- Montgomery KL, Yeh AJ, Ho JS, Tsao V, Iyer SM, Grosenick L et al. (2015). Wirelessly powered, fully internal optogenetics for brain, spinal and peripheral circuits in mice. Nat Methods : 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourot A, Fehrentz T, Kramer RH (2013a). Photochromic potassium channel blockers: design and electrophysiological characterization In: Methods in Molecular Biology. Humana Press: Totowa, NJ, pp. 89–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourot A, Fehrentz T, Le Feuvre Y, Smith CM, Herold C, Dalkara D et al. (2012). Rapid optical control of nociception with an ion‐channel photoswitch. Nat Methods 9: 396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourot A, Kienzler MA, Banghart MR, Fehrentz T, Huber FME, Stein M et al. (2011). Tuning photochromic ion channel blockers. ACS Chem Nerosci 2: 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourot A, Tochitsky I, Kramer RH (2013b). Light at the end of the channel: optical manipulation of intrinsic neuronal excitability with chemical photoswitches. Front Mol Neurosci 6: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SI, Brenner DS, Shin G, Morgan CD, Copits BA, Chung HU et al. (2015). Soft, stretchable, fully implantable miniaturized optoelectronic systems for wireless optogenetics. Nat Biotechnol 33: 1280–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polosukhina A, Litt J, Tochitsky I, Nemargut J, Sychev Y, De Kouchkovsky I et al. (2012). Photochemical restoration of visual responses in blind mice. Neuron 75: 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadovski O, Beharry AA, Zhang F, Woolley GA (2009). Spectral tuning of azobenzene photoswitches for biological applications. Angew Chem Int Ed Engl 48: 1484–1486. [DOI] [PubMed] [Google Scholar]
- Samanta S, Beharry AA, Sadovski O, McCormick TM, Babalhavaeji A, Tropepe V et al. (2013). Photoswitching azo compounds in vivo with red light. J Am Chem Soc 130610143333001. [DOI] [PubMed] [Google Scholar]
- Schönberger M, Trauner D (2014). A photochromic agonist for μ‐opioid receptors. Angew Chem Int Ed Engl 53: 3264–3267. [DOI] [PubMed] [Google Scholar]
- Smart TG, Krishek BJ (1995). Xenopus oocyte microinjection and ion‐channel expression In: Boulton A, Baker G, Walz W. (eds). Neuromethods: Patch‐Clamp Applications and Protocols. Humana Press: New Jersey, pp. 259–305. [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Breit A, Fehrentz T, Gudermann T, Trauner D (2013). Optical control of TRPV1 channels. Angew Chem Int Ed Engl 52: 9845–9848. [DOI] [PubMed] [Google Scholar]
- Stein M, Middendorp SJ, Carta V, Pejo E, Raines DE, Forman SA et al. (2012). Azo‐propofols: photochromic potentiators of GABAA receptors. Angew Chem Int Ed Engl 51: 10500–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strichartz GR (1973). The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J Gen Physiol 62: 37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymański W, Beierle JM, Kistemaker HAV, Velema WA, Feringa BL (2013). Reversible photocontrol of biological systems by the incorporation of molecular photoswitches. Chem Rev 113: 6114–6178. [DOI] [PubMed] [Google Scholar]
- Tochitsky I, Helft Z, Meseguer V, Fletcher RB, Vessey KA, Telias M et al. (2016). How azobenzene photoswitches restore visual responses to the blind retina. Neuron 92: 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochitsky I, Polosukhina A, Degtyar VE, Gallerani N, Smith CM, Friedman A et al. (2014). Restoring visual function to blind mice witha photoswitch that exploits electrophysiological remodeling of retinal ganglion cells. Neuron 81: 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velema WA, Szymański W, Feringa BL (2014). Photopharmacology: beyond proof of principle. J Am Chem Soc 136: 2178–2191. [DOI] [PubMed] [Google Scholar]
- Woodhull AM (1973). Ionic blockage of sodium channels in nerve. J Gen Physiol 61: 687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G, Jurman ME, Abramson T, MacKinnon R (1991). Mutations affecting internal TEA blockade identify the probable pore‐forming region of a K+ channel. Science 251: 939–942. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K (2011). Optogenetics in neural systems. Neuron 71: 9–34. [DOI] [PubMed] [Google Scholar]
- Zhou MM, Morais‐Cabral JH, Mann SS, MacKinnon R (2001). Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 411: 657–661. [DOI] [PubMed] [Google Scholar]
- Zussy C, Gómez‐Santacana X, Rovira X, De Bundel D, Ferrazzo S, Bosch D et al. (2016). Dynamic modulation of inflammatory pain‐related affective and sensory symptoms by optical control of amygdala metabotropic glutamate receptor 4. Mol Psychiatry. https://doi.org/10.1038/mp.2016.223. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]