

Abstract
Background and Purpose
Oxycodone, a prescription opioid, is a major drug of abuse, especially in the USA, and contributes significantly to opioid overdose deaths each year. Overdose deaths result primarily from respiratory depression. We have studied respiratory depression by oxycodone and have characterized how tolerance develops on prolonged exposure to the drug. We have investigated the role of PKC in maintaining tolerance and have examined whether ethanol or pregabalin reverses oxycodone‐induced tolerance.
Experimental Approach
Respiration was measured in male CD‐1 mice by whole‐body plethysmography. Mice were preinjected with oxycodone then implanted with mini‐pumps (s.c.) delivering 20, 45 or 120 mg·kg−1·day−1 oxycodone for 6 days and subsequently challenged with oxycodone (3 mg·kg−1, i.p.) or morphine (10 mg·kg−1, i.p.) to assess the level of tolerance.
Key Results
Oxycodone‐treated mice developed tolerance to oxycodone and cross tolerance to morphine‐induced respiratory depression. Tolerance was less with 20 mg·kg−1·day−1 than with 45 or 120 mg·kg−1·day−1 oxycodone treatment. At doses that do not depress respiration, ethanol (0.3 g·kg−1), pregabalin (20 mg·kg−1) and calphostin C (45 μg·kg−1) all reversed oxycodone‐induced tolerance resulting in significant respiratory depression. Reversal of tolerance was less in mice treated with oxycodone (120 mg·kg−1·day−1). In mice receiving ethanol and calphostin C or ethanol and pregabalin, there was no greater reversal of tolerance than seen with either drug alone.
Conclusion and Implications
These data suggest that oxycodone‐induced tolerance is mediated by PKC and that reversal of tolerance by ethanol or pregabalin may be a contributory factor in oxycodone overdose deaths.
Abbreviation
- NorBNI
norbinaltorphimine
Introduction
Opioid overdose deaths are a prominent public health issue in many countries around the world. In the USA, deaths involving prescription opioids and illicit http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9082 increased fivefold from 1999 to 2015 (Hedegaard et al., 2017) despite considerable efforts aimed at combating the problem (Okie, 2010). In 2015, the number of overdose deaths involving prescription opioids in the USA (~17 000) was greater than that from illicit heroin (~13 000). Death due to opioid overdose results primarily from respiratory depression (White and Irvine, 1999). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7093 (Oxycontin) is one of the most commonly prescribed opioid analgesics in the USA and forms a significant percentage of the hospital admissions for opioid overdose (Pfister et al., 2016) and prescription opioid overdose deaths (Inciardi et al., 2007; Kenan et al., 2012; Sgarlato and deRoux, 2015). In most instances of prescription opioid overdose, other drugs such as alcohol and benzodiazepines were also found to be present (Sgarlato and deRoux, 2015). We and others have suggested that polydrug use (i.e. using other drugs such as alcohol, benzodiazepines or http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5484 in addition to opioids) may increase the likelihood of opioid overdose (Darke and Hall, 1995, 2003; Hickman et al., 2007; Brecht et al., 2008; Hill et al., 2016; Lyndon et al., 2017).
We have recently reported that in mice, tolerance develops to the respiratory depressant effects of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627, an active metabolite of heroin (Hill et al., 2016). The tolerance induced by morphine was reversed by a single low dose of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2299 that itself did not induce respiratory depression, thus resulting in significant respiratory depression to a dose of morphine in otherwise morphine‐tolerant animals. In contrast, the tolerance induced by prolonged treatment with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5458 was not reversed by ethanol. This may be explained by morphine and methadone inducing http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 (μ receptor) desensitization and tolerance by different cellular mechanisms (for review, see Williams et al., 2013). There is substantial evidence that μ receptor desensitization and tolerance to morphine is mediated primarily by a http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=286&familyType=ENZYME‐dependent mechanism (Bailey et al., 2006; 2009a,b) whereas desensitization and tolerance to methadone is likely mediated by a http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=283 (GRK)‐ and arrestin‐dependent mechanism (McPherson et al., 2010). We have recently gone on to show that tolerance to the respiratory depressant effects of morphine can also be reversed by acute injection of the gabapentoid drug pregabalin (Lyndon et al., 2017).
Oxycodone depresses respiration in man (Leino et al., 1999; Chang et al., 2010) and in rodents (Kuo et al., 2015; Whiteside et al., 2016). In the present experiments, we have characterized the respiratory depressant effects of oxycodone in mice. We have examined whether prolonged oxycodone administration results in the development of tolerance to respiratory depression, the role of PKC in this tolerance, and whether acute administration of a low dose of ethanol or pregabalin, drugs that may be taken by people also using oxycodone, could reverse oxycodone‐induced tolerance.
Methods
Animals
Male CD‐1 mice (Harlan Laboratories, Bicester, UK) weighing approximately 30 g were group housed, four to six per cage and maintained at 22°C on a reversed 12 h dark–light cycle with food and water available ad libitum. Experiments were performed in the dark (active) phase. A total of 291 mice were used in the study. All procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986, the European Communities Council Directive (2010/63/EU) and the University of Bristol ethical review document. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Measurement of respiration
Respiration was measured in freely moving animals using plethysmography chambers (EMKA Technologies, France) supplied with a 5% CO2 in air mixture (BOC Gas Supplies, UK). For a detailed description of measuring respiratory function by whole‐body plethysmography see Lim et al. (2014). Animals were randomly ascribed to treatment groups with the experimenter blinded to drug treatment until after subsequent data analyses had been performed. Rate and depth of respiration were recorded and averaged over 5 min periods (except immediately after drug injection when the time period was 3 min) and converted to minute volume (rate × tidal volume). Data are presented both as minute volume and as percentage change from the pre‐drug minute volume baseline, calculated for each mouse individually before mean data were plotted. Presenting data as percentage change from the pre‐drug levels has been performed to control for variation between treatment groups that may have different baseline levels of respiration. In our experience, variation in baseline respiration levels do not influence the extent of opioid depression of respiration (Hill and Henderson, unpublished data).
Data from previous experiments where respiratory depression was measured either following acute opioid administration in naïve mice or following pump implantation were subjected to post hoc power analyses using G*Power (version 3.1.9). Our calculations indicated that n = 6 (acute experiments) or n = 7 (pump experiments) for each individual group would produce a significant result if an actual effect occurred.
The acute effects of opioid agonists (oxycodone, morphine and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1655) on respiration (minute volume) were monitored for 30 min following drug administration. The opioid antagonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1638, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1641 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1642 (NorBNI) were administered 30 min prior to the opioid agonists.
Induction of opioid tolerance
We have previously reported that in the mouse tolerance to the respiratory, depressant effects of morphine did not develop with repeated twice daily doses of morphine (Hill et al., 2016) but did develop with continuous administration of morphine from either a morphine pellet or a s.c. implanted osmotic mini‐pump (Withey et al., 2017). Oxycodone is metabolized more rapidly in rodents than in man (Raehal and Bohn, 2011). In the present experiments, oxycodone tolerance was induced by priming mice with three i.p. injections of oxycodone at 12 h intervals followed by s.c. implantation on the dorsal flank of an osmotic mini‐pump (ALZET®) containing oxycodone for 6 days. This follows the protocol first described for induction of opioid tolerance in mice by Quillinan et al. (2011). Implantation of osmotic mini‐pumps was done under http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2505 general anaesthesia. Three schedules of oxycodone treatment were used:
Low‐dose treatment – 3 × 30 mg·kg−1 i.p injections at 12 h intervals followed by 6 days of 20 mg·kg−1·day−1 s.c. The oxycodone dosing is based on the relative potency of oxycodone and morphine to depress respiration in mice (see Figure 1) and the protocol described by Withey et al. (2017) for induction of morphine tolerance.
Moderate‐dose treatment – 3 × 100 mg·kg−1 i.p injections at 12 h intervals followed by 6 days of 45 mg·kg−1·day−1 s.c. In this treatment, the doses of oxycodone are the same as the doses of morphine used by Withey et al. (2017).
High‐dose treatment – 3 × 100 mg·kg−1 i.p injections at 12 h intervals followed by 6 days of 120 mg·kg−1·day−1 s.c. In this treatment, the daily dose of oxycodone released from the pump has been increased sixfold from the low‐dose treatment.
Figure 1.
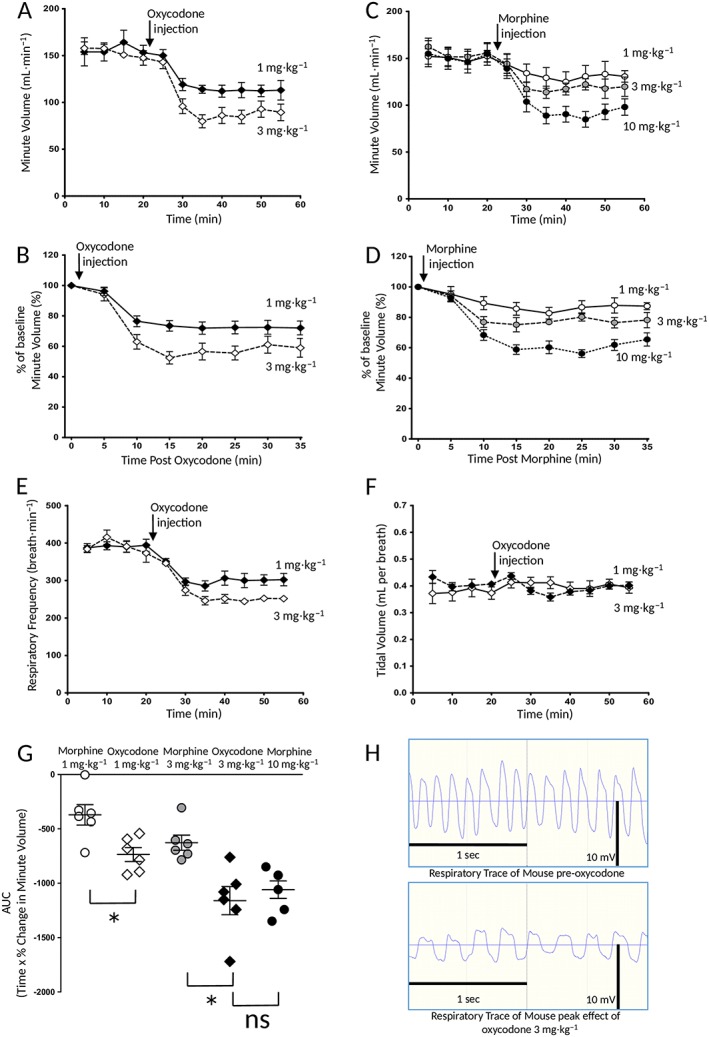
Depression of respiration by oxycodone and morphine. Oxycodone (1 and 3 mg·kg−1, i.p.) and morphine (1–10 mg·kg−1, i.p.) dose‐dependently depressed mouse respiration. Saline did not depress respiration. In (A) and (C), data are presented as minute volume whereas in (B) and (D), the level of respiratory depression seen following drug injection is expressed as a percentage change in the pre‐drug minute volume baseline, calculated for each mouse individually before mean data were plotted. In (E) and (F), the effect of oxycodone on respiratory rate and tidal volume is shown. Data are from the same groups of mice as in (A) and (B). In (G), the data in (B) and (D) have been recalculated and plotted as the AUC. The AUC for the percentage change in minute volume has been calculated for each individual animal before the mean AUC has been calculated. The data were analysed using one‐way ANOVA with Bonferroni's comparison (P < 0.05, n = 6 for all groups). In (A–G), data are expressed as mean ± SEM. In (H), raw respiration traces recorded from a single mouse before and after administration of oxycodone (3 mg·kg−1, i.p.) are shown. The thin horizontal line indicates the point of pressure inflexion. On the respiration traces, inspiration is downwards. *P < 0.05; ns, non significant; n = 6 for all groups.
Assessment of opioid tolerance
To assess the level of tolerance induced by the oxycodone treatments, mice were injected on day 6 following oxycodone pump implantation with a challenge dose of morphine (10 mg·kg−1, i.p.) or oxycodone (3 mg·kg−1, i.p.) and respiration monitored for 30 min. The degree of respiratory depression observed in opioid‐treated animals was compared to that observed in control animals that had received saline priming injections and been implanted with saline‐filled osmotic mini‐pumps rather than opioid drug and which had been challenged with morphine (10 mg·kg−1, i.p.). We used the same morphine challenge to assess oxycodone‐induced tolerance as we have used in previous studies of tolerance induced by prolonged morphine and methadone treatments (Hill et al., 2016; Lyndon et al., 2017; Withey et al., 2017), in order that the levels of tolerance induced by different opioids can be directly compared. In addition, we have also used an oxycodone challenge to determine the degree of tolerance that prolonged oxycodone treatment induces to oxycodone itself.
Reversal of tolerance
The ability of ethanol (0.3 g·kg−1, i.p.), pregabalin (20 mg·kg−1, i.p.) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5156 (45 μg·kg−1, i.p.) to reverse tolerance to respiratory depression induced by prolonged oxycodone treatment was investigated. Ethanol and pregabalin were administered simultaneously with the acute morphine challenge whereas calphostin C was administered as a pretreatment 30 min before the acute morphine challenge. In control experiments, the appropriate vehicle was injected with the morphine challenge.
Measurement of brain and plasma oxycodone levels
Mice were killed using escalating CO2 and blood samples collected from the descending abdominal aorta. Blood samples were centrifuged at 3000× g for 10 min at 4°C and the aliquoted plasma supernatant stored at −20°C. Immediately after blood sampling, mice were decapitated and the head placed on ice. After removal from the skull, the brains were flash frozen in liquid nitrogen before storage at −80°C. Subsequently, plasma and brain levels of oxycodone were measured by GC‐MS analysis at the Virginia Commonwealth University (Richmond, VA, USA) by the method described in Jacob et al. (2017).
Data analysis
AUC was determined using a 100% baseline as described previously (Hill et al., 2016). Overall changes from a single factor were analysed using one‐way ANOVA with Bonferroni's post test. Interaction between prolonged drug treatment and challenge drug was analysed using a two‐way ANOVA in a two‐by‐two factorial. Changes in groups over time with repeat measurements were analysed using a two‐way repeated measures ANOVA with Bonferroni's post test to analyse drug effect over time. GraphPad Prism 5 was used for all statistical analyses. All data are displayed as mean ± SEM. P values <0.05 were considered statistically significant. The data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Drugs and chemicals
Morphine hydrochloride (Macfarlan Smith), naloxone hydrochloride, naltrindole hydrochloride, NorBNI dihydrochloride, oxycodone hydrochloride and (+)‐(5α,7α,8β)‐N‐methyl‐N‐[7‐(1‐pyrrolidinyl)‐1‐oxaspiro[4.5]dec‐8‐yl]‐benzeneacetamide (U69593) (all from Sigma Aldrich, UK) as well as S‐pregabalin (extracted and purified from Lyrica tablets by Dr Erica Burnell, University of Bristol) were dissolved in sterile saline. Ethanol (Sigma Aldrich, UK) was diluted in sterile saline. Calphostin C (Tocris, UK) was dissolved in 100% DMSO and diluted in distilled water such that the final concentration of DMSO was 0.5%.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Results
Comparison of the respiratory depressant effects of oxycodone and morphine
We have previously demonstrated the respiratory depressant effect of morphine (3 and 10 mg·kg−1, i.p.) in mice breathing 5% CO2 in air (Hill et al., 2016). Acute administration of oxycodone (1 or 3 mg·kg−1, i.p.) produced dose‐dependent depression of respiration (Figure 1A, B, E, G). Oxycodone was slightly more potent in depressing respiration than morphine, in that 3 mg·kg−1 oxycodone produced a similar degree of respiratory depression as that to 10 mg·kg−1 morphine (Figure 1G). No mice were insensitive to the respiratory depressant effects of oxycodone at the doses tested (Figure 1G). The depression of respiration by oxycodone resulted from a decrease in the rate of respiration (control rate 550 ± 33 breaths per min vs. 291 ± 22 breaths·min−1 at the peak of inhibition by oxycodone 3 mg·kg−1) (Student's unpaired t‐test, P < 0.05), rather than a decrease in tidal volume (control tidal volume 0.29 ± 0.02 vs. 0.28 ± 0.01 mL·min−1 at the peak of inhibition by oxycodone mg·kg−1) (Student's unpaired t‐test, P < 0.05). As previously reported for morphine (Hill et al., 2016), although tidal volume was maintained in the presence of oxycodone, the duration of inspiration was prolonged whilst the depth of respiration was reduced (Figure 1E, F, H). Tidal volume was maintained because the duration of inspiration was prolonged by an apneustic compensation (see also Hill et al., 2016). Mice did not appear to exhibit ribcage muscle stiffness during oxycodone treatment.
Prior administration of the opioid receptor antagonist naloxone (1 mg·kg−1, i.p., 30 min prior to opioid agonist) inhibited the respiratory depressant effects of both oxycodone (3 mg·kg−1) and morphine (10 mg·kg−1) whereas the selective http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=317&familyId=50&familyType=GPCR (δ receptor; also known as DOPr) antagonist naltrindole (20 mg·kg−1, i.p., 30 min prior to opioid agonist) had no effect on the respiratory depressant effect of either opioid (Figure 2A, B). Treatment with the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=318 (κ receptor; also known as KOPr) antagonist NorBNI (10 mg·kg−1, i.p.) 24 h prior to the opioid administration did not affect the respiratory depressant effect of oxycodone (3 mg·kg−1) or morphine (10 mg·kg−1) (Figure 2A, B) but did, however, prevent the antinociceptive response to the κ receptor agonist U69593 (20 mg·kg−1, i.p.) in the tail flick latency assay (latencies to withdrawal – control 3.6 ± 0.9 s; U69593 6.1 ± 0.5 s; NorBNI + U69593 3.3 ± 0.5 s) (n = 6 in all groups) (U69593 vs. control or NorBNI + U69593 P < 0.05, one‐way ANOVA with Bonferroni's post test) proving that the antagonist was active in these in vivo experiments.
Figure 2.
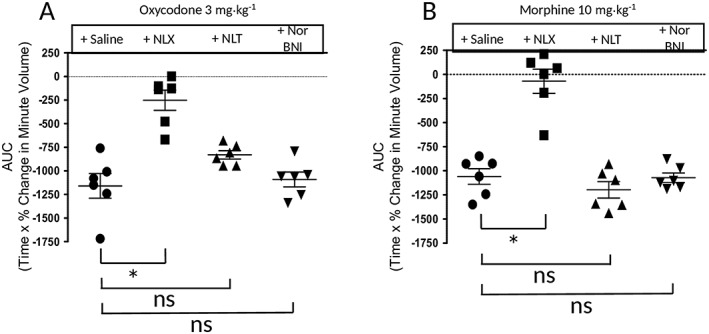
Effect of opioid receptor antagonists on the depression of respiration by oxycodone and morphine. Naloxone (NLX; 1 mg·kg−1, i.p.), administered as a 30 min pretreatment, markedly reduced the respiratory depression induced by oxycodone (3 mg·kg−1, i.p.) and abolished that induced by morphine (10 mg·kg−1, i.p.). Naltrindole (NLT; 20 mg·kg−1, i.p.), administered as a 30 min prtreatment, had no effect on the respiratory depression induced by oxycodone or morphine. Treatment with NorBNI (10 mg·kg−1, i.p.) 24 h prior to opioid administration did not affect respiratory depression induced by oxycodone or morphine. Data are expressed as mean ± SEM and were compared using one‐way ANOVA with Bonferroni's comparison. *P < 0.05; ns, non significant; n = 6 for all groups.
Development of tolerance to oxycodone and cross‐tolerance to morphine following prolonged oxycodone administration
To induce tolerance with oxycodone, we used a similar schedule of priming doses followed by pump implantation to that we have used previously for morphine and methadone (Withey et al., 2017). We examined three oxycodone treatment schedules – low dose, moderate dose and high dose (for details of the dosage regimes, see Methods). The plasma and brain levels of oxycodone measured on day 6 of treatment are given in Table 1. By day 6, the respiratory rate of mice in each oxycodone treatment group was not different from that of saline‐treated control mice [minute volume values (mL·min−1) 184 ± 12 saline‐treated; 174 ± 17 low‐dose oxycodone; 175 ± 10 medium‐dose oxycodone; 186 ± 13 high‐dose oxycodone] (one‐way ANOVA with Bonferroni's post test, P > 0.05). Without removing the oxycodone‐containing pump, mice were then challenged with oxycodone (3 mg·kg−1, i.p.) or morphine (10 mg·kg−1, i.p.) to assess the degree of tolerance that had been induced. Prolonged medium‐dose oxycodone treatment reduced the response to acute oxycodone challenge by 68% (Figure 3E, F). Prolonged low‐dose oxycodone treatment reduced the response to acute morphine challenge by 42% whereas prolonged medium‐ and high‐dose oxycodone treatment decreased the response to acute morphine challenge by over 88 and 80% respectively (Figure 3A–D). The reductions in the morphine challenge responses induced by the prolonged medium‐ and high‐dose oxycodone treatments were not statistically different from each other.
Table 1.
Plasma and brain levels of oxycodone following prolonged (6 days) treatments
| Oxycodone treatment | Low | Medium | High |
|---|---|---|---|
| Plasma concentration (ng·mL−1) | 81 ± 8 | 284 ± 147 | 1670 ± 635 |
| Brain level (ng·g−1) | 248 ± 126 | 426 ± 87 | 1703 ± 665 |
In all cases n = 6, except the plasma concentration following medium oxycodone treatment where n = 5 (one sample lost)
Figure 3.
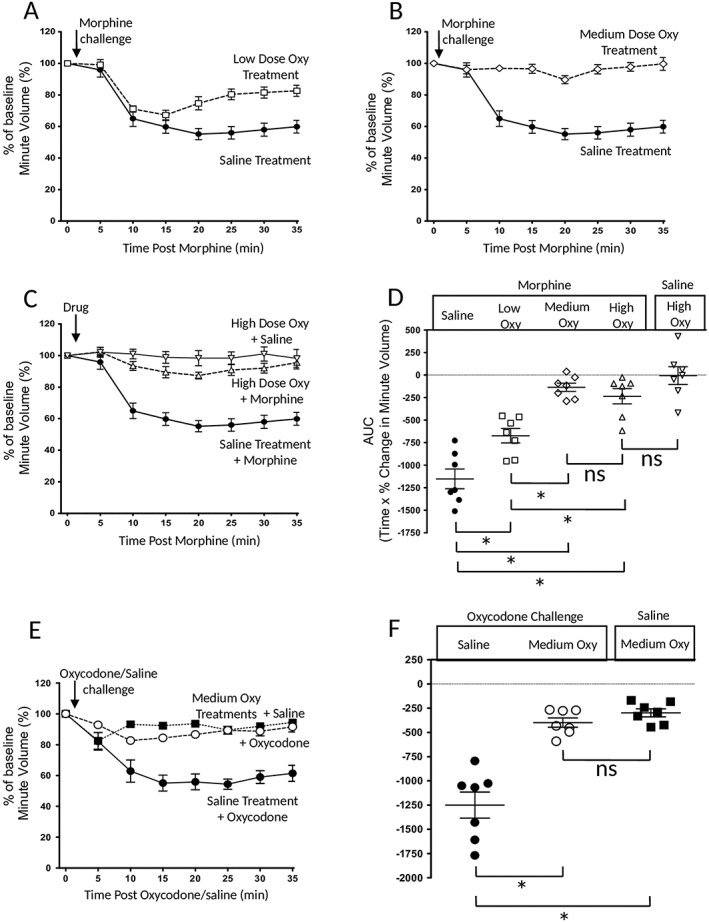
Tolerance to respiratory depression induced by prolonged oxycodone treatment. (A–C) The depression of respiration induced by a challenge dose of morphine (10 mg·kg−1, i.p.) in mice that had received prolonged low‐, medium‐ or high‐dose oxycodone treatment. In (D), the AUC for the percentage change in minute volume in (A–C) has been determined for each individual animal before the mean AUC has been calculated and compared to that observed in animals that had not received prolonged oxycodone treatment. The depression of respiration induced by the challenge dose of morphine (10 mg·kg−1, i.p.) was attenuated by oxycodone pretreatment. (E) The depression of respiration induced by a challenge dose of oxycodone (3 mg·kg−1, i.p.) in mice that had received prolonged medium oxycodone treatment. In (F), the AUC for the percentage change in minute volume in (E) has been determined for each individual animal before the mean AUC has been calculated and compared to that observed in animals that had not received prolonged oxycodone treatment. The depression of respiration induced by the challenge dose of oxycodone (3 mg·kg−1, i.p.) was attenuated by oxycodone pretreatment. Data were analysed using one‐way ANOVA with Bonferroni's comparison. *P < 0.05; ns, non significant; n = 7 for all groups. In all graphs, data are expressed as mean ± SEM.
Ethanol and pregabalin reversal of tolerance induced by oxycodone
We have previously reported that a single, low dose of ethanol reverses morphine‐induced tolerance to respiratory depression in mice, but did not reverse either the tolerance induced by prolonged methadone treatment or the blockade of morphine‐induced respiratory depression produced by prolonged treatment with buprenorphine (Hill et al., 2016). Similarly, we have shown that a single dose of S‐pregabalin reversed morphine‐induced tolerance to respiratory depression in mice (Lyndon et al., 2017). We therefore examined the ability of ethanol and pregabalin to reverse tolerance induced by prolonged oxycodone treatment. To do this, we used doses of ethanol (0.3 g·kg−1, i.p.) and pregabalin (20 mg·kg−1, i.p.) that do not depress respiration (Hill et al., 2016; Lyndon et al., 2017).
After prolonged treatment with medium‐ or high‐dose oxycodone, mice that received ethanol (0.3 g·kg−1, i.p.) simultaneously with the challenge dose of morphine (10 mg·kg−1) showed significantly greater respiratory depression compared to mice that received only morphine challenge following prolonged oxycodone treatment (Figure 4A), indicating that ethanol had reversed the oxycodone‐induced tolerance. This low dose of ethanol alone did not cause significant respiratory depression in medium‐or high‐dose oxycodone pump‐implanted mice (Figure 4). Tolerance induced by the medium‐dose oxycodone treatment was reversed by ethanol to 84% of control. Tolerance induced by high‐dose oxycodone treatment was reversed to 55% of control.
Figure 4.
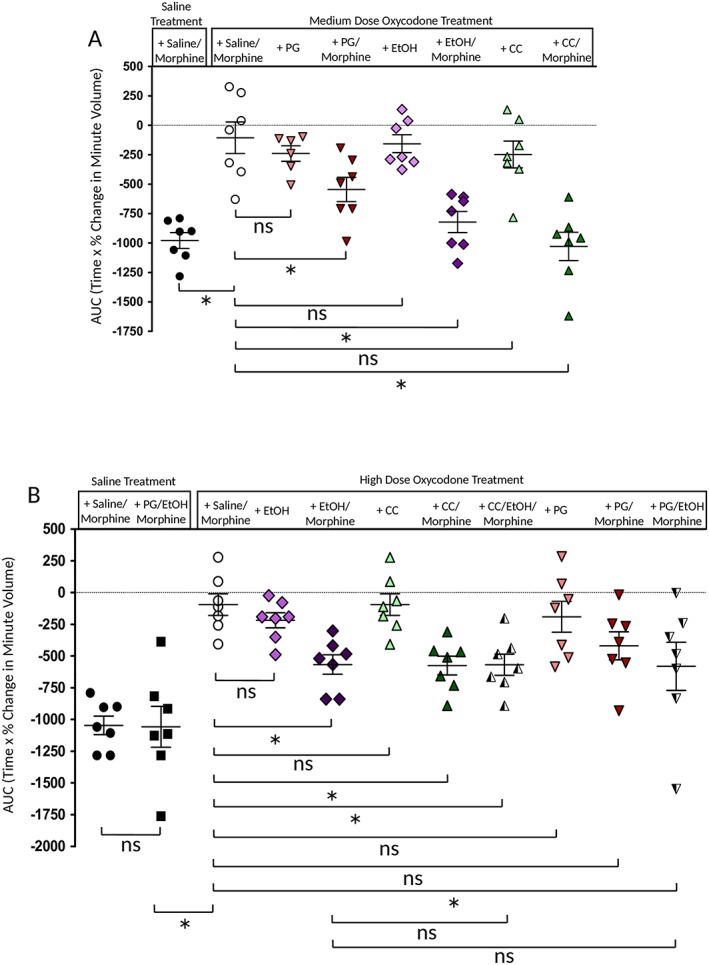
Reversal by ethanol, pregabalin and calphostin C of tolerance to respiratory depression induced by prolonged treatment with oxycodone. (A). In mice that received prolonged treatment with medium‐dose oxycodone, administration of ethanol (EtOH: 0.3 g·kg−1), pregabalin (PG: 20 mg·kg−1) or calphostin C (CC: 45 μg·kg−1) with the acute morphine challenge resulted in greater depression of respiration than in mice challenged with morphine alone. Data are expressed as mean ± SEM and were compared using a two‐way ANOVA with Bonferroni's comparison. * Indicates P < 0.05 compared to medium oxycodone treatment plus saline/morphine challenge; n = 7 for all groups. (B) Effect of ethanol (0.3 g·kg−1), pregabalin (20 mg·kg−1) or calphostin C (45 μg·kg−1) in mice that had undergone the high‐dose oxycodone treatment. In mice receiving both ethanol and calphostin C or ethanol and pregabalin, there was no greater effect than that seen with either drug alone, that is, additivity was not observed. In each graph, the AUC for the percentage change in minute volume has been determined for each individual animal before the mean AUC has been calculated. Data are expressed as mean ± SEM and were compared using a two‐way ANOVA with Bonferroni's comparison. Statistical comparisons indicated by a bar in (B) are as follows: *P < 0.05, and ns, P > 0.05; n = 7 for all groups.
In mice that had received the medium‐dose oxycodone treatment, acute administration of pregabalin (20 mg·kg−1, i.p.) partially restored the respiratory depressant effect of the challenge dose of morphine (to 55% of control) (Figure 4A). Pregabalin alone did not cause significant respiratory depression in medium‐ or high‐dose oxycodone pump‐implanted mice (Figure 4). In mice that had received the high‐dose oxycodone treatment, the restoration of the respiratory depressant effect of morphine, when pregabalin was co‐administered, did not reach statistical significance (Figure 4B).
We next examined whether ethanol and pregabalin would act additively to reverse tolerance induced by high‐dose oxycodone treatment. Oxycodone‐treated mice received an acute injection of pregabalin (20 mg·kg−1 i.p) and ethanol (0.3 g·kg−1, i.p.) along with morphine (10 mg·kg−1, i.p.). With the combination of ethanol and pregabalin, there was no greater reversal of oxycodone‐induced tolerance than by ethanol alone (Figure 4B).
Reversal of oxycodone‐induced tolerance by calphostin C and lack of additivity with ethanol
There is substantial evidence for the role of PKC in desensitization of μ receptors by morphine and subsequent development of cellular tolerance on prolonged exposure to morphine (Bailey et al 2006; 2009a,b; Levitt and Williams, 2012; Williams et al., 2013). Similarly, PKC has been shown to be involved in the development of tolerance to the antinociceptive and respiratory depressant effects of morphine (Smith et al., 2007; Withey et al., 2017). We therefore sought to investigate the role of PKC in tolerance induced by oxycodone using calphostin C, a brain‐penetrant drug that inhibits both conventional and novel isoforms of PKC (Kobayashi et al., 1989).
Calphostin C (45 μg·kg−1, i.p.) by itself had no direct effect on respiration in näive mice nor did it alter the acute respiratory depressant effect of morphine (10 mg·kg−1) (Withey et al., 2017). However, in mice that had received the chronic medium‐dose oxycodone treatment, an acute injection of calphostin C (45 μg·kg−1, i.p.) 30 min prior to an acute morphine challenge completely restored the respiratory depressant effect of the morphine challenge (Figure 4A). This effect was not observed in oxycodone‐treated mice injected with the vehicle for calphostin (0.5% DMSO in saline) prior to the morphine challenge (data not shown). As with ethanol and pregabalin, calphostin C alone did not cause significant respiratory depression in medium‐dose oxycodone pump‐implanted mice (Figure 4). These data are consistent with calphostin C reversing oxycodone‐induced tolerance to respiratory depression. In mice that had received the high‐dose oxycodone treatment, the acute injection of calphostin C (45 μg·kg−1, i.p.) resulted in partial reversal of oxycodone‐induced tolerance (to 55% of control) (Figure 4B).
We next examined whether ethanol and calphostin C would act additively to reverse tolerance induced by high‐dose oxycodone treatment. Oxycodone‐treated mice received an acute injection of calphostin C (45 μg·kg−1 i.p) 30 min prior to administration of ethanol (0.3 g·kg−1, i.p.) and morphine (10 mg·kg−1, i.p.). With the combination of ethanol and calphostin C, there was no greater reversal of oxycodone‐induced tolerance than by either drug alone (Figure 4B).
Discussion
The results of this study demonstrate that oxycodone, a prescription opioid and major drug of abuse, induces marked respiratory depression; that with prolonged oxycodone administration tolerance develops to this effect; and most interestingly that this tolerance is reversed by inhibition of PKC, by low‐dose ethanol, and by pregabalin. Thus, this study highlights the potential dangers of polydrug abuse when taking oxycodone either as a prescribed drug or as a drug of abuse.
We observed that in the mouse, oxycodone was more potent than morphine in depressing respiration. The depression of respiration by oxycodone was reversed by naloxone but not significantly altered by the δ receptor antagonist naltrindole nor by the κ receptor antagonist NorBNI indicating that the effect was mediated through activation of μ receptor. Yang et al. (2016) had reported that in μ receptor knockout mice, high doses of oxycodone (>40 mg·kg−1 s.c.) induce naltrindole‐reversible antinociception.
We have previously observed that tolerance to the respiratory depressant effects of morphine does develop, but does so more slowly than tolerance to antinociception (Hill et al., 2016). The present study demonstrates that for respiratory depression, prolonged (6 day) exposure to oxycodone also results in tolerance to oxycodone as well as cross tolerance to morphine. The degree of cross tolerance to morphine observed increased as the oxycodone treatment was increased. Oxycodone is eliminated in the rodent more rapidly than morphine (Raehal and Bohn, 2011) which may explain why we needed to use higher doses of oxycodone than morphine to induce equivalent levels of tolerance (compare the levels of tolerance induced by oxycodone in this paper with those observed for morphine in Hill et al., 2016).
The tolerance to respiratory depression induced by oxycodone was reversed by calphostin C, a brain‐penetrant inhibitor of PKC (Kobayashi et al., 1989). The fact that calphostin C reversed oxycodone‐induced tolerance after 6 days of oxycodone treatment indicates that ongoing PKC activity is required to maintain oxycodone tolerance. We have previously reported that calphostin C reversed morphine‐ but not methadone‐induced tolerance (Withey et al., 2017). Morphine and oxycodone have similar lower intrinsic agonist efficacy at MOPr for G protein activation whereas methadone has higher intrinsic efficacy (McPherson et al., 2010). The agonist efficacy of oxycodone and morphine is still sufficient, however, for them to induce both analgesia and respiratory depression in man. It appears likely therefore that PKC is involved in tolerance to lower efficacy μ receptor agonists but not to higher efficacy agonists. Multiple isoforms of PKC (PKC‐α, ‐γ and ‐ε) have been identified as mediators of tolerance to the antinociceptive actions of morphine (Smith et al., 2007). Of related interest, expression of constitutively active PKCα or PKCε isoforms in the pre‐Bötzinger complex, a group of neurons involved in the generation of respiratory rhythm that are inhibited by μ receptor agonists, increased the development of tolerance to respiratory depression induced by morphine, an effect that afforded increased protection from death by overdose (Lin et al., 2012).
The potential for other non‐opioid drugs to contribute to overdose deaths involving opioids has long been recognized (see Hickman et al., 2007). In the present study, we focused on two drugs, ethanol and pregabalin. Ethanol is frequently detected at post mortem along with oxycodone and other prescription opioids (Cone et al., 2004; Häkkinen et al., 2012). Pregabalin is also used in the treatment of severe pain states, sometimes in combination with opioids. It has recently become a drug of abuse popular with heroin users and increasingly associated with opioid overdose deaths (Häkkinen et al., 2014; Lyndon et al., 2017). In England and Wales, there has been a dramatic increase in acute drug deaths involving pregabalin and its congener, gabapentin, in recent years (Lyndon et al., 2017). Data for 2016 reveal that in over 90% of deaths in which pregabalin or http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5483 was present at post mortem, an opioid was also present (Office for National Statistics, 2017 2016).
Ethanol is one of the most commonly co‐abused drugs by opioid users, despite long‐standing warnings that ethanol and opioids pose a significant health risk when taken together. Low doses of ethanol have previously been shown to reverse morphine‐induced cellular tolerance (Llorente et al., 2013), antinociception tolerance (Hull et al., 2013) and tolerance to respiratory depression (Hill et al., 2016). Ethanol also reverses oxycodone‐induced antinociception tolerance (Jacob et al., 2017) as well as tolerance to respiratory depression as shown in this paper.
Acute ethanol administration in mice at doses that reversed tolerance did not alter brain concentrations of either oxycodone (Jacob et al., 2017) or morphine (Hill et al., 2016), suggesting that ethanol reversal of tolerance to these opioids is not due to altered drug distribution or other pharmacokinetic changes. Reversal of oxycodone‐induced tolerance by ethanol did not summate with the reversal induced by calphostin C. This may indicate that these two drugs act to reduce PKC activity. Attempts to demonstrate ethanol inhibition of PKC have produced mixed results. Reneau et al. (2011) demonstrated that ethanol (1–100 mM) inhibited PKCα activity in vitro by up to 40%, although we and others found less inhibition of PKCα activity by ethanol (Slater et al., 1997; Rex et al., 2008; Llorente et al., 2013). However, such in vitro studies using purified PKC are likely to be hindered by the absence of cofactors that are required for ethanol inhibition of the enzyme and also do not examine the initial translocation of PKC from the cytoplasm to the plasma membrane.
There have been several recent reports that gabapentin and pregabalin are being misused with subsequent development of dependence (Papazisis and Tzachanis, 2014; Mersfelder and Nichols, 2016; Schjerning et al., 2016; Smith et al., 2016). Additionally, pregabalin misuse is increasingly observed to be associated with opioid use, in particular, heroin users report that pregabalin reinforces or enhances the rewarding effects of heroin (Grosshans et al., 2013; McNamara et al., 2015; Smith et al., 2015; Bastiaens et al., 2016; Evoy et al., 2017; Lyndon et al., 2017). We have previously demonstrated that in mice, a dose of pregabalin that did not in itself depress respiration reversed tolerance to respiratory depression induced by prolonged morphine treatment, thus increasing opioid‐induced respiratory depression (Lyndon et al., 2017). At higher doses, pregabalin alone depressed respiration, an effect that summated with that of morphine. In the present study, we have shown that pregabalin partially reversed tolerance to respiratory depression induced by oxycodone. We are not aware of any evidence to date that pregabalin or gabapentin directly inhibit PKC, but gabapentin has been shown to reduce PKC translocation to the plasma membrane in rat spinal cord (Zhang et al., 2015). However, the mechanism by which pregabalin reverses oxycodone tolerance remains to be elucidated. Our observation that pregabalin reverses oxycodone tolerance highlights the potential risk of concomitant use of pregabalin and oxycodone either in patients taking these drugs to treat pain states or in people misusing these drugs.
Conclusions
Our data provide compelling evidence for an interaction between ethanol or pregabalin and oxycodone to reverse tolerance to the respiratory depressant effects of oxycodone. We conclude from this study that ethanol or pregabalin reversal of tolerance to the respiratory depression induced by oxycodone may be an important contributing factor in deaths involving this drug.
Author contributions
R.H., W.L.D., E.K. and G.H. participated in research design. R.H. performed the experiments. R.H. and G.H. analysed the data. R.H., W.L.D., E.K. and G.H. participated in writing the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank Dr J. Jacob and Mr J Poklis (Virginia Commonwealth University) for performing the measurements of oxycodone plasma and brain levels. The work described in this paper was supported by a grant from NIH (RO1DA036975) to W.L.D. and G.H. The funder of the study had no role in the study design, data collection, data analysis, data interpretation or writing of the report.
Hill, R. , Dewey, W. L. , Kelly, E. , and Henderson, G. (2018) Oxycodone‐induced tolerance to respiratory depression: reversal by ethanol, pregabalin and protein kinase C inhibition. British Journal of Pharmacology, 175: 2492–2503. doi: 10.1111/bph.14219.
References
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to Pharmacology 2017/18: G‐protein coupled receptors. Br J Pharmacol 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to Pharmacology 2017/18: Enzymes. Br J Pharmacol 174 (Suppl 1): S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Smith FL, Kelly E, Dewey WL, Henderson G (2006). How important is protein kinase C in μ‐opioid receptor desensitization and morphine tolerance? Trends Pharmacol Sci 27: 558–565. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Llorente J, Gabra BH, Smith FL, Dewey WL, Kelly E et al (2009a). Role of protein kinase C and μ‐opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci 29: 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Oldfield S, Llorente J, Caunt CJ, Teschemacher AG, Roberts L et al (2009b). Involvement of PKC alpha and G‐protein‐coupled receptor kinase 2 in agonist‐selective desensitization of μ‐opioid receptors in mature brain neurons. Br J Pharmacol 158: 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaens L, Galus J, Mazur C (2016). Abuse of gabapentin is associated with opioid addiction. Psychiatry Q 87: 763–767. [DOI] [PubMed] [Google Scholar]
- Brecht M, Huang D, Evans E, Hser YI (2008). Polydrug use and implications for longitudinal research: ten‐year trajectories for heroin, cocaine, and methamphetamine users. Drug Alcohol Depend 96: 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Maney KM, Phillips JP, Langford RM, Mehta V (2010). A comparison of the respiratory effects of oxycodone versus morphine: a randomised, double‐blind, placebo‐controlled investigation. Anaesthesia 65: 1007–1012. [DOI] [PubMed] [Google Scholar]
- Cone EJ, Fant RV, Rohay JM, Caplan YH, Ballina M, Reder RF et al (2004). Oxycodone involvement in drug abuse deaths. II. Evidence for toxic multiple drug‐drug interactions. J Anal Toxicol 28: 616–624. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darke S, Hall W (1995). Levels and correlates of polydrug use among heroin users and regular amphetamine users. Drug Alcohol Depend 39: 231–235. [DOI] [PubMed] [Google Scholar]
- Darke S, Hall W (2003). Heroin overdose: research and evidence‐based intervention. J Urban Health 80: 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evoy KE, Morrison MD, Saklad SR (2017). Abuse and misuse of pregabalin and gabapentin. Drugs 77: 403–426. [DOI] [PubMed] [Google Scholar]
- Grosshans M, Lemenager T, Vollmert C, Kaemmerer N, Schreiner R, Mutschler J et al (2013). Pregabalin abuse among opiate addicted patients. Eur J Clin Pharmacol 69: 2021–2025. [DOI] [PubMed] [Google Scholar]
- Häkkinen M, Launiainen T, Vuori E, Ojanperä I (2012). Comparison of fatal poisonings by prescription opioids. Forensic Sci Int 222: 327–331. [DOI] [PubMed] [Google Scholar]
- Häkkinen M, Vuori E, Kalso E, Gergov M, Ojanperä I (2014). Profiles of pregabalin and gabapentin abuse by postmortem toxicology. Forensic Sci Int 241: 1–6. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard H, Warner M, Miniño AM (2017). Drug overdose deaths in the United States, 1999–2015. NCHS data brief, no 273. Hyattsville, MD: National Center for Health Statistics. [Google Scholar]
- Hickman M, Carrivick S, Paterson S, Hunt N, Zador D, Cusick L et al (2007). London audit of drug‐related overdose deaths: characteristics and typology, and implications for prevention and monitoring. Addiction 102: 317–323. [DOI] [PubMed] [Google Scholar]
- Hill R, Lyndon A, Withey S, Roberts J, Kershaw Y, Maclachlan J et al (2016). Ethanol reversal of tolerance to the respiratory depressant effects of morphine. Neuropsychopharmacology 41: 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull LC, Gabra BH, Bailey CP, Henderson G, Dewey WL (2013). Reversal of morphine analgesic tolerance by ethanol in the mouse. J Pharmacol Exp Ther 345: 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inciardi JA, Surratt HL, Lugo Y, Cicero TJ (2007). The diversion of prescription opioid analgesics. Law Enforc Exec Forum 7: 127–141. [PMC free article] [PubMed] [Google Scholar]
- Jacob JC, Poklis JL, Akbarali HI, Henderson G, Dewey WL (2017). Ethanol reversal of tolerance to the antinociceptive effects of oxycodone and hydrocodone. J Pharmacol Exp Ther 362: 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo A, Wyse BD, Meutermans W, Smith MT (2015). In vivo profiling of seven common opioids for antinociception, constipation and respiratory depression: no two opioids have the same profile. Br J Pharmacol 172: 532–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenan K, Mack K, Paulozzi L (2012). Trends in prescriptions for oxycodone and other commonly used opioids in the United States, 2000‐2010. Open Med 6: e41–e47. [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi E, Ando K, Nakano H, Lida T, Ohno H, Morimoto M et al (1989). Calphostins (UCN‐1028), novel and specific inhibitors of protein kinase C. I. Fermentation, isolation, physico‐chemical properties and biological activities. J Antibiot 42: 1470–1474. [DOI] [PubMed] [Google Scholar]
- Leino K, Mildh L, Lertola K, Seppälä T, Kirvelä O (1999). Time course of changes in breathing pattern in morphine‐ and oxycodone‐induced respiratory depression. Anaesthesia 54: 835–840. [DOI] [PubMed] [Google Scholar]
- Levitt ES, Williams JT (2012). Morphine desensitization and cellular tolerance are distinguished in rat locus ceruleus neurons. Mol Pharmacol 82: 983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R, Zavou MJ, Milton PL, Chan ST, Tan JL, Dickinson H et al (2014). Measuring respiratory function in mice using unrestrained whole‐body plethysmography. J Vis Exp 2014 (90): e51755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HY, Law PY, Loh HH (2012). Activation of protein kinase C (PKC)α or PKCε as an approach to increase morphine tolerance in respiratory depression and lethal overdose. J Pharmacol Exp Ther 341: 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente J, Withey S, Rivero G, Cunningham M, Cooke A, Saxena K et al (2013). Ethanol reversal of cellular tolerance to morphine in rat locus coeruleus neurons. Mol Pharmacol 84: 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyndon A, Audrey S, Wells C, Burnell ES, Ingle S, Hill R et al (2017). Risk to heroin users of polydrug use of pregabalin or gabapentin. Addiction 112: 1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara S, Stokes S, Kilduff R, Shine A (2015). Pregabalin abuse amongst opioid substitution treatment patients. Ir Med J 108: 309–310. [PubMed] [Google Scholar]
- McPherson J, Rivero G, Baptist M, Llorente J, Al‐sabah S, Krasel C et al (2010). μ‐Opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol 78: 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersfelder TL, Nichols WH (2016). Gabapentin: abuse, dependence, and withdrawal. Ann Pharmacother 50: 229–233. [DOI] [PubMed] [Google Scholar]
- Office for National Statistics (2017). Number of drug‐related deaths involving gabapentin or pregabalin with or without an opioid drug, England and Wales, 2016. Available at: https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/adhocs/007306numberofdrugrelateddeathsinvolvinggabapentinorpregabalinwithorwithoutanopioiddrugenglandandwales2016 (accessed 7th September 2017) (Archived at http://www.webcitation.org/6tIOoqWUg on 7th September 2017).
- Okie S (2010). A flood of opioids, a rising tide of deaths. N Engl J Med 363: 1981–1985. [DOI] [PubMed] [Google Scholar]
- Papazisis G, Tzachanis D (2014). Pregabalin's abuse potential: a mini review focusing on the pharmacological profile. Int J Clin Pharmacol Ther 52: 709–716. [DOI] [PubMed] [Google Scholar]
- Pfister GJ, Burkes RM, Guinn B, Steele J, Kelley RR, Wiemken TL et al (2016). Opioid overdose leading to intensive care unit admission: epidemiology and outcomes. J Crit Care 35: 29–32. [DOI] [PubMed] [Google Scholar]
- Quillinan N, Lau EK, Virk M, von Zastrow M, Williams JT (2011). Recovery from μ‐opioid receptor desensitization after chronic treatment with morphine and methadone. J Neurosci 31: 4434–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM (2011). The role of β‐arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 60: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reneau J, Reyland ME, Popp RL (2011). Acute ethanol exposure prevents PMA‐mediated augmentation of N‐methyl‐D‐aspartate receptor function in primary cultured cerebellar granule cells. Alcohol 45: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex EB, Rankin ML, Ariano MA, Sibley DR (2008). Ethanol regulation of D1 dopamine receptor signaling is mediated by protein kinase C in an isozyme‐specific manner. Neuropsychopharmacology 33: 2900–2911. [DOI] [PubMed] [Google Scholar]
- Schjerning O, Rosenweig M, Pottegard A, Damkier P, Nielsen J (2016). Abuse potential of pregabalin: a systematic review. CNS Drugs 30: 9–25. [DOI] [PubMed] [Google Scholar]
- Sgarlato A, deRoux SJ (2015). Prescription opioid related deaths in New York City: a 2 year retrospective analysis prior to the introduction of the New York State I‐STOP law. Forensic Sci Med Pathol 11: 388–394. [DOI] [PubMed] [Google Scholar]
- Slater SJ, Kelly MB, Larkin JD, Ho C, Mazurek A, Taddeo FJ et al (1997). Interaction of alcohols and anaesthetics with protein kinase Cα. J Biol Chem 272: 6167–6173. [DOI] [PubMed] [Google Scholar]
- Smith RV, Havens JR, Walsh SL (2016). Gabapentin misuse, abuse and diversion: a systematic review. Addiction 111: 1160–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RV, Lofwall MR, Havens JR (2015). Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry 172: 487–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FL, Gabra BH, Smith PA, Redwood MC, Dewey WL (2007). Determination of the role of conventional, novel and atypical PKC isoforms in the expression of morphine tolerance in mice. Pain 127: 129–139. [DOI] [PubMed] [Google Scholar]
- White JM, Irvine RJ (1999). Mechanisms of fatal opioid overdose. Addiction 94: 961–972. [PubMed] [Google Scholar]
- Whiteside GT, Hummel M, Boulet J, Beyenhof JD, Strenkowski B, John JD et al (2016). Robustness of arterial blood gas analysis for assessment of respiratory safety pharmacology in rats. J Pharmacol Toxicol Methods 78: 32–41. [DOI] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, Von Zastrow M, Schulz S et al (2013). Regulation of μ‐opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65: 223–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withey SL, Hill R, Lyndon A, Dewey WL, Kelly E, Henderson G (2017). Effect of tamoxifen and brain‐penetrant protein kinase C and c‐Jun N‐terminal kinase inhibitors on tolerance to opioid‐induced respiratory depression in mice. J Pharmacol Exp Ther 361: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PP, Yeh GC, Yeh TK, Xi J, Loh HH, Law PY et al (2016). Activation of δ‐opioid receptor contributes to the antinociceptive effect of oxycodone in mice. Pharmacol Res 111: 867–876. [DOI] [PubMed] [Google Scholar]
- Zhang YB, Guo ZD, Li MY, Fong P, Zhang JG, Zhang CW et al (2015). Gabapentin effects on PKC‐ERK1/2 signaling in the spinal cord of rats with formalin‐induced visceral inflammatory pain. PLoS One 10: e0141142. [DOI] [PMC free article] [PubMed] [Google Scholar]