

Abstract
Background and Purpose
Conditions such as hypertension and renal allograft rejection are accompanied by chronic, agonist‐independent, signalling by angiotensin II AT1 receptors. The current treatment paradigm for these diseases entails the preferred use of inverse agonist AT1 receptor blockers (ARBs). However, variability in the inverse agonist activities of common biphenyl‐tetrazole ARBs for the active state of AT1 receptors often leads to treatment failure. Therefore, characterization of robust inverse agonist ARBs for the active state of AT1 receptors is necessary.
Experimental Approach
To identify the robust inverse agonist for active state of AT1 receptors and its molecular mechanism, we performed site‐directed mutagenesis, competition binding assay, inositol phosphate production assay and molecular modelling for both ground‐state wild‐type AT1 receptors and active‐state N111G mutant AT1 receptors.
Key Results
Although candesartan and telmisartan exhibited weaker inverse agonist activity for N111G‐ compared with WT‐AT1 receptors, only eprosartan exhibited robust inverse agonist activity for both N111G‐ and WT‐ AT1 receptors. Specific ligand–receptor contacts for candesartan and telmisartan are altered in the active‐state N111G‐ AT1 receptors compared with the ground‐state WT‐AT1 receptors, suggesting an explanation of their attenuated inverse agonist activity for the active state of AT1 receptors. In contrast, interactions between eprosartan and N111G‐AT1 receptors were not significantly altered, and the inverse agonist activity of eprosartan was robust.
Conclusions and Implications
Eprosartan may be a better therapeutic option than other ARBs. Comparative studies investigating eprosartan and other ARBs for the treatment of diseases caused by chronic, agonist‐independent, AT1 receptor activation are warranted.
Abbreviations
- Ang II
angiotensin II
- ARB
AT1 receptor blocker
- ECL2
extracellular loop 2
- IP
inositol phosphate
- TM
transmembrane
- WT
wild‐type
Introduction
GPCRs comprise one of the largest superfamilies of integral membrane proteins in the human genome and are commonly characterized by their seven‐transmembrane (TM) α‐helix structure (Fredriksson et al., 2003). GPCRs respond to a wide variety of ligands, such as photons, tastants, ions, monoamines, purines, lipids, peptides and proteins, promoting intracellular signalling cascades in numerous physiological and pathological processes. It has been reported that ~26% of commercially available drugs are known to target GPCRs (Garland, 2013).
The http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=34&familyId=6&familyType=GPCR belongs to the rhodopsin family of GPCRs and is involved in the regulation of various physiological and pathological processes. For example, physiological activation of AT1 receptors regulates vascular tone, water–electrolyte balance and cardiac function and maintains cardiovascular homeostasis. However, excessive AT1 receptor activation causes a wide variety of human diseases, such as hypertension, cardiac hypertrophy, coronary artery disease, stroke and diabetic nephropathy (Khan, 2011; Vijayaraghavan and Deedwania, 2011; Lee et al., 2012; Vejakama et al., 2012). Although the AT1 receptor is traditionally activated by its agonist Ang II, recent studies revealed that mechanical stress and AT1 receptor‐directed autoantibodies can activate this receptor without the need for Ang II stimulation, in diseases such as hypertension, cardiac hypertrophy, pre‐eclampsia, graft rejection for renal transplantation, primary aldosteronism and systemic sclerosis (Mederos y Schnitzler et al., 2011; Riemekasten et al., 2011; Storch et al., 2012; Unal et al., 2012; Rossitto et al., 2013; Gunther et al., 2014; Wallukat and Schimke, 2014; Li et al., 2015). Therefore, robust blockade of AT1 receptors in the clinical setting requires not only antagonist activity against Ang II binding but also robust inverse agonist activity for the Ang II‐independent active state of AT1 receptors, to yield enhanced therapeutic effects against the various disease states.
As described in our recent study, the most commonly prescribed AT1 receptor blockers (ARBs), namely, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=590, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=586, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3937 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=589, exhibit robust inverse agonist activity for the ground state of AT1 receptors. However, the inverse agonist activity of these four ARBs strongly decreases upon transition of AT1 receptors to the active state (Takezako et al., 2015). A robust inverse agonist towards the active state of AT1 receptors has not yet been discovered.
Most ARBs exhibit a common chemical structure, namely, a biphenyl‐tetrazole moiety, and are therefore referred to as biphenyl‐tetrazole ARBs (Figure 1A). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=587, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=591, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6901, losartan, EXP3174, valsartan and irbesartan belong to this biphenyl‐tetrazole class of ARBs. Among these, candesartan exhibits two unique structural features, namely, a benzimidazole ring and an ethoxy group substituent on the imidazole core (Figure 1A), and is known to cause insurmountable antagonism of AT1 receptors (Fierens et al., 1999; Takezako et al., 2004). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=592 is a non‐biphenyl‐tetrazole ARB and is also known to cause insurmountable antagonism of AT1 receptors (Le et al., 2007). Telmisartan contains a carboxyl group instead of a tetrazole moiety at the 2′‐position of the biphenyl moiety, in addition to bulky bis‐benzimidazole rings (Figure 1A). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=588 is an also non‐biphenyl‐tetrazole ARB and is known to cause surmountable antagonism of AT1 receptors (Timmermans, 1999). Eprosartan exhibits a unique structure composed of carboxyphenyl and thiophenepropanoic acid moieties (Figure 1A). In this study, we have examined whether these structural differences affect the inverse agonist activities of candesartan, telmisartan and eprosartan for the ground and active states of AT1 receptors . Specifically, we determined how the transition of AT1 receptors to the active‐state influences the inverse agonist activities of these ARBs and found that the non‐biphenyl‐tetrazole ARB eprosartan is a unique and robust inverse agonist for the active state of AT1 receptors.
Figure 1.
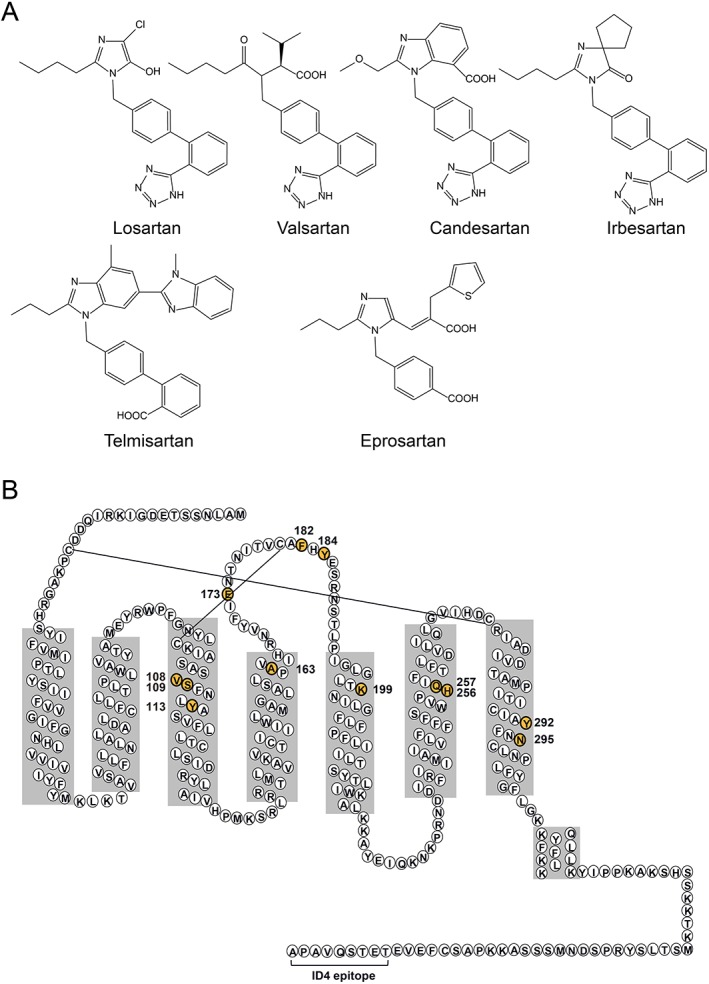
Structures of ARBs and AT1 receptors. (A) The chemical structures of losartan, valsartan, candesartan, irbesartan, telmisartan and eprosartan. Four of the six ARBs exhibit a common structure, namely, a biphenyl‐tetrazole moiety, while telmisartan and eprosartan are non‐biphenyl‐tetrazole ARBs. (B) Secondary structure model of rat AT1 receptors, revised on the basis of the crystal structure of human AT1 receptors. Residues that are numbered and highlighted in yellow indicate residues mutated in this study. The epitope tag attached to the C‐terminus in underlined and allowed for detection by the ID4 monoclonal antibody.
Methods
Mutagenesis, expression and membrane preparation
The synthetic rat AT1 receptor gene, cloned in the shuttle expression vector pMT‐3, was used for expression and mutagenesis, as previously described (Noda et al., 1996). Residues previously identified as ARB‐binding site residues were mutated. For each mutation, we substituted amino‐acid residues with residues containing a side chain of nearly the same size and/or chemical characteristics, as previously described (Takezako et al., 2015). For expression of AT1 receptors, 10 μg of purified plasmid DNA was transfected into same passage of COS‐1 cells for each experiment. Since experiment was performed in same cells, our samples do not need to be randomized. Membranes were prepared by transfecting COS‐1 cells cultured in DMEM supplemented with 10% FBS using the FuGENE6 transfection reagent. The transfected cells were cultured for 48 h and subsequently harvested. Cell membranes were prepared using the nitrogen Parr bomb disruption method in the presence of protease inhibitors. Receptor expression was assessed by immunoblot analysis and 125I‐[Sar1, Ile8]Ang II saturation binding analysis.
Competition binding assay
Binding experiments using 125I labelled [Sar1, Ile8]Ang II were carried out under equilibrium conditions, as previously described (Takezako et al., 2004).
Inositol phosphate production assay
The inositol phosphate (IP) production assay was carried out as previously described (Takezako et al., 2015). Briefly, semiconfluent AT1 receptor‐transfected COS‐1 cells were seeded in six‐well plates and subsequently labelled with myo‐[2‐3H(N)]‐inositol (1.5 μCi·mL−1; specific activity, 22 μCi·mol−1) for 24 h at 37°C in DMEM supplemented with 10% FBS. Labelled cells were washed twice with DMEM and subsequently incubated with DMEM containing 10 mM LiCl and vehicle or one of the various ARBs for 120 min at 37°C. Following incubation, the medium was removed, and perchloric acid was used to extract the total soluble IP from the cells, as previously described (Noda et al., 1996). EC50 and IC50 values were determined by non‐linear regression analysis using GraphPad Prism. The inverse agonist activities of the ARBs were calculated for each AT1 receptor mutant as a percent of receptor activity of vehicle‐treated cells expressing each AT1 receptor mutant (constitutive activity of each mutant). We defined the constitutive activity of each mutant receptor in vehicle‐treated cells as 0%. Therefore, an inverse agonist activity of −10% reflects a constitutive activity of 90%, while an inverse agonist activity of −100% reflects a constitutive activity of 0%. In other words, an inverse agonist activity of −100% reflects complete suppression of constitutive activity for the examined wild‐type (WT) or mutant AT1 receptor.
Models of AT1 receptor ARB‐binding pockets
Models of the ligand‐binding pockets for candesartan, telmisartan and eprosartan were constructed as described in Zhang et al. (2015). The AT1 receptor crystal structure was used to dock the ARBs via an energy‐based docking protocol using the ICM molecular modelling software suite from Molsoft (San Diego, CA, USA). The initial model for each ARB was first optimized by adding side‐chain hydrogen atoms, followed by optimization of the resultant conformations and subsequent generation of soft potential maps in a 30 × 30 × 30 Å3 box, which covered the extracellular half of the AT1 receptor. Two‐dimensional representations of the compounds were used to generate the molecular models, and their three‐dimensional geometry was optimized using the MMFF‐94 force field (Halgren, 1995). Biased probability Monte Carlo optimization of the internal coordinates of the ligand in the grid potentials of the receptor was employed for molecular docking (Abagyan and Totrov, 1997). Five independent docking runs were carried out for each ligand starting from a random conformation. Monte Carlo sampling and optimization were performed with the high thoroughness parameter set to 30. The Lys1995.42 side chain was treated as a flexible group in the receptor, allowing this side chain's rotamers to freely sample the space. Up to 30 alternative complex conformations of the ligand–receptor complex were generated. The conformations were rescored using the ICM binding score function, which accounts for van der Waals, electrostatic, H‐bonding, non‐polar and polar atom solvation energy differences between bound and unbound states, ligand internal strain, conformational entropy and ligand‐independent and receptor‐independent constants. The results of individual docking runs for each ligand were considered consistent if at least three of the five docking runs produced similar conformations (RMSD <2.0 Å) and binding scores of <−20.0 kJ·mol−1. No distance restraints or any other a priori derived information for the ligand–receptor interactions were used in the unbiased docking procedure.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as mean ± SEM. Multiple comparisons were made using a one‐way ANOVA followed by Bonferroni's or Dunnett's post hoc tests using StatView Software (SAS Institute Inc., Cary, NC, USA) as statistical program. P values of <0.05 were considered to be statistically significant. Although operator and data analyst were not blinded, analysed data were confirmed by other co‐authors.
Materials
Ang II and [Sar1, Ile8]Ang II were purchased from Bachem (Bubendorf, Switzerland). 125I‐[Sar1, Ile8]Ang II (specific activity, 2200 Ci·mmol−1) was purchased from Dr Robert Speth (The University of Mississippi Peptide Radioiodination Service Center, MS). Candesartan, telmisartan and eprosartan were gifts from Takeda Pharma (Tokyo, Japan), Boehringer Ingelheim Pharmaceuticals (Biberach an der Riss, Germany) and Solvay Pharmaceuticals (Hannover, Germany) respectively. Myo‐[2‐3H(N)]inositol was purchased from GE Healthcare Life Sciences (Little Chalfont, UK). COS‐1 cells were purchased from the European Collection of Cell Culture (Salisbury, UK). The FuGENE 6 transfection reagent was purchased from Roche Diagnostics (Indianapolis, IN, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Eprosartan is a unique and robust inverse agonist for the active state of AT1 receptors
We investigated the binding affinities of ARBs for both WT‐ AT1 receptors, which are representative of the ground‐state receptor, and the constitutively active N111G mutant of the AT1 receptor (N111G‐ AT1 receptor), which mimics the active state of AT1 receptors (Boucard et al., 2003; Martin et al., 2004; Martin et al., 2007; Unal et al., 2013). The pharmacological properties of losartan, valsartan, irbesartan, candesartan, telmisartan and eprosartan were compared for both WT‐ and N111G‐ AT1 receptors. Data for losartan, valsartan and irbesartan were taken from our recent study (Takezako et al., 2015). The binding affinities of all six ARBs were higher for WT‐AT1 receptors than for N111G‐AT1 receptors. The order of binding affinity for WT‐AT1 receptors was determined to be candesartan > irbesartan > telmisartan > valsartan = eprosartan > losartan (Table 1) (Takezako et al., 2015). In contrast, the order of binding affinity for N111G‐AT1 receptors was candesartan > irbesartan > telmisartan > eprosartan > valsartan > losartan (Table 2) (Takezako et al., 2015). The inverse agonist activity of all six ARBs increased in a concentration‐dependent manner for both WT‐ and N111G‐AT1 receptors (Figure 2). The order of potency of the six ARBs for WT‐AT1 receptors was candesartan > valsartan = irbesartan = telmisartan > eprosartan > losartan (Figure 2A). In contrast, the order of potency of the six ARBs for N111G‐ AT1 receptors was candesartan > irbesartan > telmisartan > valsartan > losartan > eprosartan (Figure 2A). Although five of the six ARBs exhibited weaker maximal inverse agonist activity for N111G‐ AT1 receptors compared with the WT receptors, only eprosartan exhibited robust inverse agonist activity for both N111G‐ and WT‐ AT1 receptors (Figure 2B). Thus, we identified eprosartan as a unique and robust inverse agonist of the active state of AT1 receptors.
Table 1.
Binding properties of ARBs for the WT‐AT1 receptors and various mutants
| Candesartan | Telmisartan | Eprosartan | ||||
|---|---|---|---|---|---|---|
| Mutation | Ki (nM) | ΔKi | Ki (nM) | ΔKi | Ki (nM) | ΔKi |
| WT‐AT1 receptors | 0.08 ± 0.02 | 1 | 0.5 ± 0.02 | 1 | 3.1 ± 0.5 | 1 |
| V108I | 6.9 ± 1.1 | 86.3 | 63.3 ± 4.4 | 127 | 13.8 ± 0.6 | 4.5 |
| S109T | 70.7 ± 14.4 | 884 | 15.7 ± 0.5 | 31 | 3.9 ± 0.3 | 1.3 |
| Y113A | 45.7 ± 11.7 | 571 | 56.8 ± 7.8 | 114 | 16.4 ± 7.0 | 5.3 |
| A163T | 0.5 ± 0.1 | 6.3 | 0.05 ± 0.01 | 0.1 | 2.9 ± 0.8 | 0.9 |
| E173A | 0.2 ± 0.01 | 2.5 | 0.6 ± 0.1 | 1.2 | 3.4 ± 0.2 | 1.1 |
| F182A | 0.3 ± 0.06 | 3.8 | 0.2 ± 0.02 | 0.4 | 4.4 ± 2.1 | 1.4 |
| Y184A | 0.1 ± 0.03 | 1.3 | 0.1 ± 0.03 | 0.2 | 4.1 ± 1.2 | 1.3 |
| K199A | 2.8 ± 0.4 | 35 | 0.4 ± 0.2 | 0.8 | 18.4 ± 0.2 | 5.9 |
| K199Q | 2.3 ± 0.4 | 29 | 1.0 ± 0.3 | 2 | 4.6 ± 0.3 | 1.5 |
| H256A | 0.4 ± 0.1 | 5.0 | 0.4 ± 0.08 | 0.8 | 28.5 ± 1.0 | 9.2 |
| Q257A | 5.6 ± 0.9 | 70 | 16.2 ± 4.2 | 32 | 66.6 ± 12.9 | 21.5 |
| Q257E | 1.2 ± 0.3 | 15 | 2.6 ± 0.3 | 5.2 | 39.9 ± 9.0 | 12.9 |
| Y292A | 0.6 ± 0.2 | 7.5 | 1.3 ± 0.4 | 2.6 | 18.4 ± 0.2 | 5.9 |
| N295A | 5.2 ± 1.0 | 65 | 22.0 ± 4.3 | 44 | 180 ± 10.4 | 58.1 |
| S109T/A163T | 15.0 ± 1.2 | 188 | 0.5 ± 0.09 | 1 | 2.9 ± 0.5 | 0.9 |
| S109T/H256A | 54.8 ± 6.9 | 685 | 0.6 ± 0. 2 | 1.2 | 96.9 ± 7.8 | 31.3 |
| S109T/N295A | 3183 ± 365 | 39 787 | 50.9 ± 4.1 | 102 | 392 ± 110 | 127 |
| A163T/H256A | 0.3 ± 0.02 | 3.8 | 0.03 ± 0.01 | 0.06 | 44.8 ± 3.1 | 14.5 |
| A163T/N295A | 16.0 ± 4.5 | 200 | 6.3 ± 1.3 | 12.6 | 168 ± 45.4 | 54.2 |
| K199Q/H256A | 2.1 ± 0.4 | 26 | 2.3 ± 1.2 | 4.6 | 1335 ± 213 | 431 |
| H256A/N295A | 0.3 ± 0.2 | 3.8 | 8.4 ± 1.6 | 16.8 | 582 ± 6.7 | 188 |
Ligand binding properties for WT‐AT1 receptors and various mutants. Values are presented as mean ± SEM of at least three independent experiments performed in duplicate. The effect of the mutations on the binding affinity is expressed as ΔKi = Ki (mutant)∕Ki (WT‐AT1 receptor).
Table 2.
Binding properties of ARBs for various mutants of N111G‐AT1 receptors
| Candesartan | Telmisartan | Eprosartan | ||||
|---|---|---|---|---|---|---|
| Mutation | Ki (nM) | ΔKi | Ki (nM) | ΔKi | Ki (nM) | ΔKi |
| N111G | 1.4 ± 0.5 | 1 | 38.8 ± 4.9 | 1 | 44.1 ± 6.1 | 1 |
| N111G/V108I | 119 ± 19.0 | 85 | 627 ± 114 | 16.2 | 353 ± 36.4 | 8 |
| N111G/S109T | 281 ± 57.5 | 201 | 268 ± 27.6 | 6.9 | 124 ± 17.1 | 2.8 |
| N111G/A163T | 15.6 ± 4.9 | 11.1 | 4.0 ± 0.1 | 0.1 | 102 ± 10.5 | 2.3 |
| N111G/E173A | 1.3 ± 0.2 | 0.9 | 51.4 ± 3.6 | 1.3 | 46.9 ± 8.0 | 1.1 |
| N111G/F182A | 1.7 ± 0.6 | 1.2 | 11.1 ± 2.1 | 0.3 | 59.1 ± 10.1 | 1.3 |
| N111G/Y184A | 1.2 ± 0.08 | 0.9 | 17.6 ± 2.8 | 0.5 | 87.4 ± 14.9 | 2.0 |
| N111G/K199A | 148 ± 3.4 | 106 | 202 ± 11.6 | 5.2 | 334 ± 45.9 | 7.6 |
| N111G/K199Q | 82.1 ± 11.3 | 58.6 | 205 ± 46.4 | 5.3 | 446 ± 91.1 | 10.1 |
| N111G/H256A | 0.8 ± 0.2 | 0.6 | 47.0 ± 14.7 | 1.2 | 1262 ± 87 | 28.6 |
| N111G/Q257A | 4.8 ± 0.7 | 3.4 | 66.0 ± 13.5 | 1.7 | 8061 ± 648 | 183 |
| N111G/Q257E | 10.9 ± 5.3 | 7.8 | 258 ± 44.1 | 6.6 | 975 ± 284 | 22.1 |
| N111G/Y292A | 0.005 ± 0.001 | 0.004 | 0.7 ± 0.2 | 0.02 | 44.7 ± 11.6 | 1 |
| N111G/N295A | 6.7 ± 0.08 | 4.8 | 193 ± 30.9 | 5 | 360 ± 20.7 | 8.2 |
| N111G/S109T/A163T | 310.0 ± 21.4 | 221 | 150 ± 15.5 | 3.9 | 153 ± 8.8 | 3.5 |
| N111G/S109T/H256A | 168 ± 5.8 | 120 | 166 ± 24.7 | 4.3 | 1737 ± 258 | 39.4 |
| N111G/S109T/N295A | 1274 ± 139 | 1231 | 449 ± 41.2 | 11.6 | 494 ± 95 | 11.2 |
| N111G/A163T/H256A | 45.6 ± 17.5 | 32.6 | 19.0 ± 6.1 | 0.5 | 1410 ± 288 | 32 |
| N111G/A163T/N295A | 20.2 ± 4.4 | 14.4 | 6.9 ± 1.8 | 0.2 | 541 ± 105 | 12.3 |
| N111G/K199Q/H256A | 240 ± 5.5 | 171 | 209 ± 4.8 | 5.4 | 31 631 ± 728 | 717 |
| N111G/H256A/N295A | 0.06 ± 0.02 | 0.04 | 19.5 ± 0.9 | 0.5 | 612 ± 49.2 | 13.9 |
Ligand‐binding properties of various mutants of N111G‐AT1 receptors. Values are presented as mean ± SEM of at least three independent experiments performed in duplicate. The effect of the mutations on the binding affinity is expressed as ΔKi = Ki (mutant)∕Ki (N111G‐AT1 receptor).
Figure 2.
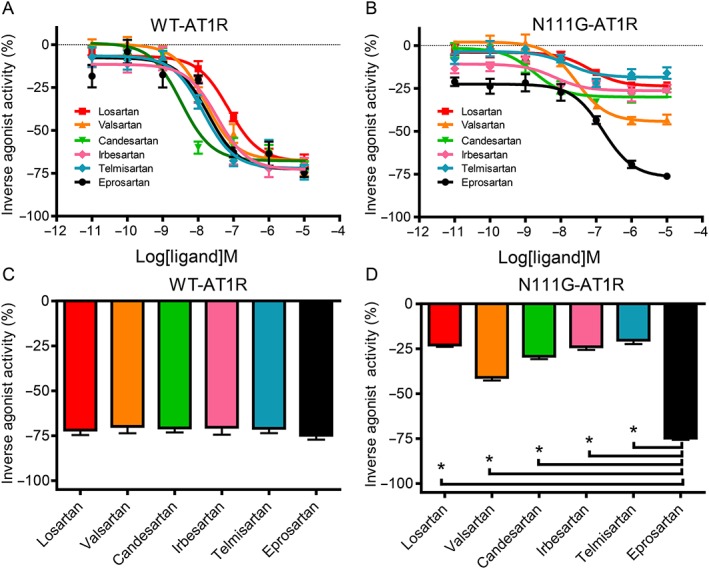
Differences in the inverse agonist activities of the six ARBs for WT‐ AT1 receptors and the N111G‐ AT1 receptor mutant, as measured by the IP assay. The concentration‐dependent inverse agonist activities of losartan, valsartan, candesartan, irbesartan, telmisartan and eprosartan for (A) WT‐ AT1 receptors and (B) N111G‐ AT1 receptors transfected into COS‐1 cells. Maximal inverse agonist activities of losartan, valsartan, candesartan, irbesartan, telmisartan and eprosartan for (C) WT‐ AT1 receptors and (D) N111G‐ AT1 receptors, measured at a concentration of 10 μM for each ARB. Data for losartan, valsartan and irbesartan are taken from our recent study and used with permission from Molecular Pharmacology (Takezako et al., 2015). The inverse agonist activities of the six ARBs are expressed as a percentage of the constitutive activity of vehicle‐treated WT‐ AT1 receptor‐transfected and N111G‐AT1 receptor‐transfected COS‐1 cells. The constitutive activities of vehicle‐treated cells expressing WT‐ AT1 receptors and N111G‐ AT1 receptors were defined as 0%. Data represent mean ± SEM of independent experiments (n = 6 for each groups). * P < 0.05, significant differences between the groups; one‐way ANOVA followed by Bonferroni's or Dunnett's post hoc tests.
Identification of ARB‐binding residues of WT‐ AT1 receptors
To identify the amino‐acid residues involved in ligand binding to WT‐ AT1 receptors, we examined the effects of various mutations introduced in the WT‐receptor on the binding affinities of candesartan, telmisartan and eprosartan (Table 1). All mutants introduced in the WT‐ AT1 receptors are known to alter the binding affinity and/or inverse agonist activity of the biphenyl‐tetrazole ARBs (Takezako et al., 2015), because these mutants might be predicted to alter the binding affinities of not only candesartan but also telmisartan and eprosartan. As mutation of some non‐interacting residues could result in small reductions in ligand binding affinity, we used the effect of a known change to set a threefold change as the cut‐off for reduction in binding affinity, as previously described (Takezako et al., 2015). The mutations V108I, S109T, Y113A, A163T, F182A, K199A, K199Q, H256A Q257A, Q257E, Y292A and N295A reduced the binding affinity of candesartan. The mutations V108I, S109T, Y113A, Q257A, Q257E and N295A reduced the binding affinity of telmisartan, whereas A163T and Y184A mutations increased the binding affinity of telmisartan. The mutations V108I, Y113A, K199A, H256A, Q257A, Q257E, Y292A and N295A reduced the binding affinity of eprosartan. Taken together, these results suggest that the residues Val108TM3, Ser109TM3, Tyr113TM3, Ala163TM4, Phe182ECL2, Lys199TM5, His256TM6, Gln257TM6, Tyr292TM7 and Asn295TM7 in WT‐ AT1 receptors are involved in candesartan binding, while the residues Val108TM3, Ser109TM3, Tyr113TM3, Ala163TM4, Tyr184ECL2, Gln257TM6 and Asn295TM7 are involved in telmisartan binding, and the residues Val108TM3, Tyr113TM3, Lys199TM5, His256TM6, Gln257TM6, Tyr292TM7 and Asn295TM7 are involved in eprosartan binding.
To elucidate combinational interactions between the AT1 receptor residues involved in binding of candesartan, telmisartan and eprosartan, the effects of seven double mutations on the binding affinities of the ARBs were examined (Table 1). As the ARB‐binding site residues Ser109TM3, Ala163TM4, Lys199TM5, His256TM6 and Asn295TM7 in AT1 receptors are located on different TM helices, we selected combinations of S109T, A163T, K199Q, H256A and N295A mutations to evaluate the combined effects of different TM helices on both binding affinity and inverse agonism, as previously described (Takezako et al., 2015). The S109T/N295A mutation synergistically reduced the binding affinities of candesartan and eprosartan and additively reduced the binding affinity of telmisartan. The A163T/N295A mutation synergistically reduced the binding affinity of candesartan. The S109T/H256A, K199Q/H256A and H256A/N295A mutations synergistically reduced the binding affinity of eprosartan. These results indicate that interactions between Ser109TM3 and Asn295TM7 are important for the binding of all three ARBs, while interactions between Ala163TM4 and Asn295TM7 are important for candesartan binding, and those between Ser109TM3 and His256TM6, between Lys199TM5 and His256TM6 and between His256TM6 and Asn295TM7 are important for eprosartan binding.
Identification of ARB‐binding residues of N111G‐ AT1 receptors
To identify the amino‐acid residues in N111G‐ AT1 receptors involved in ligand binding, we examined the effects of various mutations introduced in these receptors on the binding affinities of candesartan, telmisartan and eprosartan (Table 2). All mutants introduced in the N111G‐ AT1 receptors alter the binding affinity and/or inverse agonist activity of the biphenyl‐tetrazole ARBs (Takezako et al., 2015), because these mutants might be predicted to alter the binding affinities of not only candesartan but also telmisartan and eprosartan. As previously described, the N111G/Y113A mutant did not show any detectable binding activity, so that the effects of this mutant could not be examined (Takezako et al., 2015).
The effects of most of the examined N111G‐ AT1 receptor mutants on the binding affinities of the three ARBs were quite different from those observed for WT‐ AT1 receptor mutants. The mutations N111G/V108I, N111G/S109T, N111G/A163T, N111G/K199A, N111G/K199Q, N111G/Q257A, N111G/Q257E and N111G/N295A reduced the binding affinity of candesartan. However, the effects of other N111G‐ AT1 receptor mutants on the binding affinity of candesartan were different from those observed for WT‐ AT1 receptor mutants. Contrary to the effects observed for the F182A, H256A and Y292A mutations in WT‐ AT1 receptor, the N111G/F182A and N111G/H256A mutations did not reduce the binding affinity of candesartan, while the N111G/Y292A mutation increased the binding affinity of candesartan. The mutations N111G/V108I, N111G/S109T, N111G/Q257E and N111G/N295A reduced the binding affinity of telmisartan. However, the effects of other N111G‐ AT1 receptor mutants on the binding affinity of telmisartan were different from those observed for WT‐ AT1 receptor mutants. Two additional mutations, namely, N111G/K199A and N111G/K199Q, reduced the binding affinity of telmisartan. Contrary to the effects observed for the A163T, Y184A and Q257A mutations in WT‐AT1 receptors, the N111G/A163T mutation increased the binding affinity of telmisartan, while the N111G/Y184A and N111G/Q257A mutations did not alter the binding affinity of telmisartan. Contrary to the effects observed for the Y292A mutation in WT‐AT1 receptors, the N111G/Y292A mutation increased the binding affinity of telmisartan. The mutations N111G/V108I, N111G/K199A, N111G/K199Q, N111G/H256A, N111G/Q257A, N111G/Q257E and N111G/N295A reduced the binding affinity of eprosartan. However, the effects of other N111G‐ AT1 receptor mutants on the binding affinity of eprosartan were different from those observed for WT‐ receptor mutants. Contrary to the effects observed for the Y292A mutation in WT‐ AT1 receptor, the N111G/Y292A mutation did not alter the binding affinity of eprosartan. Taken together, these results suggest that the residues Val108TM3, Ser109TM3, Ala163TM4, Lys199TM5, Gln257TM6 and Asn295TM7 in N111G‐ AT1 receptors are involved in candesartan binding, while Val108TM3, Ser109TM3, Ala163TM4, Phe182ECL2, Lys199TM5, Gln257TM6, Tyr292TM7 and Asn295TM7 are involved in telmisartan binding, and Val108TM3, Lys199TM5, His256TM6, Gln257TM6 and Asn295TM7 are involved in eprosartan binding.
To resolve the putative interactions between the residues in N111G‐AT1 receptors involved in the binding of candesartan, telmisartan and eprosartan, we examined the effects of seven triple mutations on the binding affinities of all three ARBs (Table 2). The N111G/S109T/N295A mutation synergistically reduced the binding affinity of candesartan and additively reduced the binding affinities of telmisartan and eprosartan. The N111G/K199Q/H256A mutation synergistically reduced the binding affinities of candesartan and eprosartan. The N111G/S109T/H256A mutation additively reduced the binding affinities of eprosartan. Contrary to the effects observed for the H256A/N295A mutation, the N111G/H256A/N295A mutation did not show combined effects on the binding affinity of eprosartan. No other triple mutations exhibited combined effects on the binding affinities of any of the three ARBs. These results indicate that interactions between Ser109 and Asn295 in N111G‐AT1 receptors are important for binding of all three ARBs and that interactions between Ser109 and His256 are important for eprosartan binding, while interactions between Lys199 and His256 are important for both candesartan and eprosartan binding.
Critical residues in WT‐ AT1 receptors responsible for the inverse agonist activities of candesartan, telmisartan and eprosartan
To identify the critical amino‐acid residues in WT‐ AT1 receptors responsible for the inverse agonist activities of candesartan, telmisartan and eprosartan, we examined the effects of various mutations introduced in these WT‐receptors on the inverse agonist activities of the ARBs. As previously shown, the V108I, S109T, A163T, E173A, F182A, Q257A, Y292A and N295A mutations demonstrated sufficient constitutive activity (Takezako et al., 2015), and thus, the effects of these mutations on inverse agonism were examined (Figure 3). As the Y113A, K199A, K199Q and H256A mutations displayed only subtle constitutive activity, the effects of these mutations on inverse agonism could not be examined. The S109T, F182A, Q257A, Y292A and N295A mutations significantly decreased the inverse agonist activity of candesartan. The F182A, Q257A, Y292A and N295A mutations significantly decreased the inverse agonist activity of telmisartan. The S109T, E173A, F182A, Q257A, Y292A and N295A mutations significantly decreased the inverse agonist activity of eprosartan. Other mutations did not alter the inverse agonist activity of any of the three ARBs. These results suggest that Ser109TM3, Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 in WT‐AT1 receptors are critical residues responsible for the inverse agonist activities of candesartan, while Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 are critical residues responsible for the inverse agonist activity of telmisartan, and Ser109TM3, Glu173ECL2, Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 are critical residues responsible for the inverse agonist activity of eprosartan.
Figure 3.
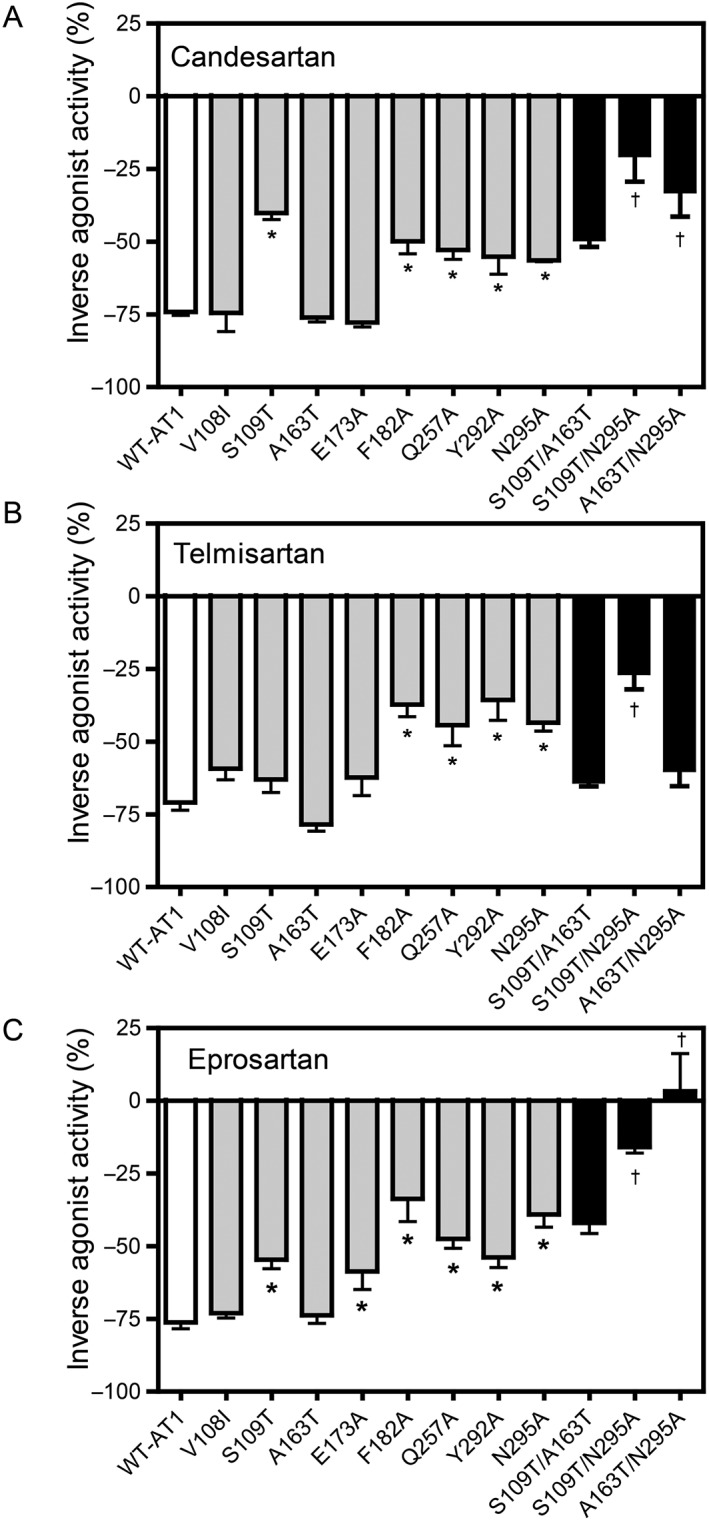
Effects of the AT1 receptor mutants on the inverse agonist activities of candesartan, telmisartan and eprosartan in cells expressing various mutants of WT‐AT1 receptors, as measured by the IP assay. The inverse agonist activities of (A) candesartan, (B) telmisartan and (C) eprosartan at a concentration of 10 μM for each ARB in COS‐1 cells transfected with WT‐AT1 receptors, single mutants and double mutants are shown. The double mutants were constructed using two independent mutants that significantly attenuated the inverse agonist activity. The inverse agonist activities are expressed as a percentage of the constitutive activity of either WT‐AT1 receptors or each mutant. The constitutive activities of the vehicle‐treated cells expressing WT‐AT1 receptors and each mutant were defined as 0% for each. Data represent mean ± SEM of independent experiments [candesartan: n = 8 (WT‐AT1 receptor) and n = 6 (all mutants); telmisartan: n = 12 (Y292A), n = 10 (WT‐ AT1 receptor, E173A and Q257A), n = 8 (N295A) and n = 6 (all other mutants); and eprosartan: n = 12 (WT‐ AT1 receptor), n = 10 (E173A, Q257A, Y292A and N295A) and n = 6 (all other mutants)]. Group sizes are not equal in Figure 3. The reason for this is that data of some groups showed large SEM when n = 6 per group, and we had to examine additional experiments to confirm exact value for these groups. * P < 0.05, significant difference from WT‐ AT1 receptors †, additive effect; one‐way ANOVA followed by Bonferroni's or Dunnett's post hoc tests.
To elucidate the combinational interactions between the residues in WT‐ AT1 receptors responsible for the inverse agonist activities of candesartan, telmisartan and eprosartan, the effects of double mutations on the inverse agonist activities were examined. The S109T/A163T, S109T/N295A and A163T/N295A mutations demonstrated sufficient constitutive activity, as previously described (Takezako et al., 2015), and thus, the effects of these mutations on the inverse agonist activities were examined (Figure 3). The S109T/N295A mutation additively decreased the inverse agonist activities of all three ARBs. The A163T/N295A mutation additively decreased the inverse agonist activity of candesartan and synergistically decreased the inverse agonist activity of eprosartan. The S109T/A163T mutation did not demonstrate any combined effects on the inverse agonist activities of any of the three ARBs. These results suggest that combinational interactions between Ser109TM3 and Asn295TM7 in WT‐ AT1 receptors are important for the inverse agonist activities of all three ARBs, while those between Ala163TM4 and Asn295TM7 are important for the inverse agonist activities of candesartan and eprosartan.
Critical residues in N111G‐ AT1 receptors responsible for the inverse agonist activities of candesartan, telmisartan and eprosartan
To identify the critical residues in N111G‐ AT1 receptors responsible for the inverse agonist activities of the various ARBs, the effects of various mutations introduced in the N111G‐receptors on inverse agonist activities were examined. All mutations showed significantly higher constitutive activity than WT‐ AT1 receptors, as previously described (Takezako et al., 2015), and thus, the effects of these mutations on the inverse agonist activities were examined (Figure 4). The effects of different mutations in N111G‐ AT1 receptors on the inverse agonist activities of the three ARBs were quite different from those observed for different mutations in the WT‐ receptors.
Figure 4.
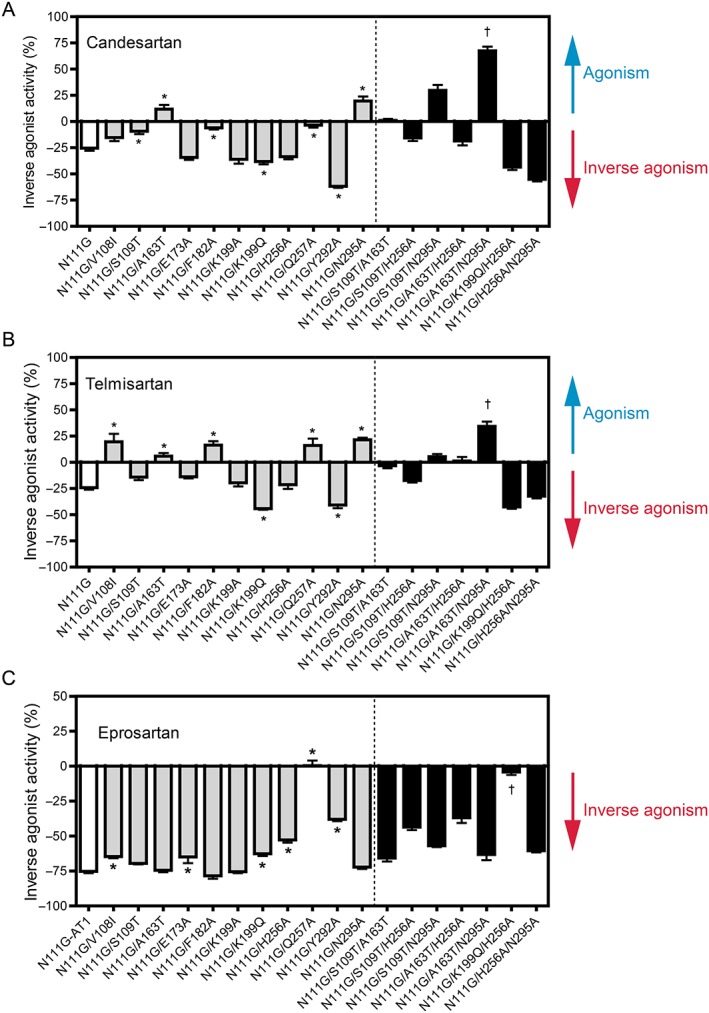
Effects of N111G‐ AT1 receptor mutants on the activities of candesartan, telmisartan and eprosartan. The activities (inverse agonism or activity switch from inverse agonism towards agonism) of (A) candesartan, (B) telmisartan and (C) eprosartan at a concentration of 10 μM for each ARB in COS‐1 cells transfected with N111G‐ AT1 receptors, single mutants in N111G‐ AT1 receptors and double mutants in N111G‐ AT1 receptors are shown. Double mutants were constructed by making two additional independent mutations in N111G‐AT1 receptors that significantly attenuated the inverse agonist activity or switched activity from inverse agonism towards agonism. The agonist and inverse agonist activities are expressed as a percentage of the constitutive activity of vehicle‐treated cells expressing N111G‐ AT1 receptors and each N111G‐ AT1 receptor mutant. The constitutive activities of vehicle‐treated cells expressing N111G‐AT1 receptors and each N111G‐AT1 receptor mutant were defined as 0%. Data represent mean ± SEM of independent experiments [candesartan: n = 16 (N111G/K199Q, N111G/H256A and N111G/Q257A), n = 12 (N111G), n = 8 (N111G/V108I, N111G/A163T, N111G/E173A and N111G/K199Q/H256A) and n = 6 (all other mutants); telmisartan: n = 12 (N111G), n = 8 (N111G/K199Q, N111G/H256A, N111G/Q257A, N111G/S109T/A163T and N111G/A163T/N295A) and n = 6 (all other mutants); and eprosartan: n = 10 (N111G), n = 8 (N111G/K199Q, N111G/H256A, N111G/Q257A, N111G/S109T/A163T and N111G/A163T/N295A) and n = 6 (all other mutants)]. Group sizes are not equal in Figure 4. The reason for this is that data of some groups showed large SEM when n is 6 per group, and we had to examine additional experiments to confirm exact value for these groups. * P < 0.05, significantly different from N111G‐ AT1 receptors; †, additive effect; one‐way ANOVA followed by Bonferroni's or Dunnett's post hoc tests.
The N111G/S109T, N111G/F182A and N111G/Q257A mutations significantly decreased the inverse agonist activity of candesartan. However, the effects of other mutations in N111G‐ AT1 receptors on the inverse agonist activity of candesartan were different from those observed for similar mutations in WT‐ AT1 receptors. Contrary to the effects observed for the A163T, Y292A and N295A mutations, the N111G/A163T and N111G/N295A mutations switched activity towards agonism for candesartan, while the N111G/Y292A mutation significantly increased the inverse agonist activity of candesartan. The inverse agonist activity of candesartan was significantly increased by an additional mutation, namely, N111G/K199Q. The effects of all mutations in N111G‐ AT1 receptors on the inverse agonist activity of telmisartan were completely different from those observed for similar mutations in WT‐ AT1 receptors. Contrary to the effects observed for the V108I, A163T, F182A, Q257A, Y292A and N295A mutations in WT‐ AT1 receptors, the N111G/V108I, N111G/A163T, N111G/F182A, N111G/Q257A and N111G//N295A mutations switched activity towards agonism for telmisartan, while the N111G/Y292A mutation significantly increased the inverse agonist activity of telmisartan. The inverse agonist activity of telmisartan was significantly increased by an additional mutation, namely, N111G/K199Q. The N111G/V108I, N111G/E173A, N111G/K199Q, N111G/H256A, N111G/Q257A and N111G/Y292A mutations significantly decreased the inverse agonist activity of eprosartan. However, the effects of other mutations in N111G‐AT1 receptors on the inverse agonist activity of eprosartan were different from those observed for mutations in WT‐ AT1 receptors. Contrary to the effects observed for the V108I, S109T and N295A mutations in the WT‐ receptors, the N111G/V108I mutation significantly decreased the inverse agonist activity of eprosartan, while the N111G/S109T and N111G/N295A mutations did not alter the inverse agonist activity of eprosartan. Other mutations did not alter the inverse agonist activities of any of the three ARBs. These results suggest that Ser109TM3, Phe182ECL2 and Gln257TM6 in N111G‐ AT1 receptors are critical residues responsible for the inverse agonist activity of candesartan, while Lys199TM5 and Tyr292TM7 influence the inverse agonist activity of candesartan, and Ala163TM4 and Asn295TM7 modulate the activity switch from inverse agonism towards agonism for candesartan. On the other hand, Lys199TM5 and Tyr292TM7 in N111G‐ AT1 receptors influence the inverse agonist activity of telmisartan, while Val108TM3, Ala163TM4, Phe182ECL2, Gln257TM6 and Asn295TM7 modulate the activity switch from inverse agonism towards agonism for telmisartan. Finally, Val108TM3, Glu173ECL2, Lys199TM5, His256TM6, Gln257TM6 and Tyr292TM7 in N111G‐ AT1 receptors are critical residues responsible for the inverse agonist activity of eprosartan.
To determine the combinational interactions between the residues responsible for inverse agonism of N111G‐ AT1 receptors, the effects of triple mutations on the inverse agonist activities of the various ARBs were examined. The mutations N111G/S109T/A163T, N111G/S109T/H256A, N111G/S109T/N295A, N111G/A163T/H256A, N111G/A163T/N295A, N111G/K199Q/H256A and N111G/H256A/N295A demonstrated sufficient constitutive activity, as previously described (Takezako et al., 2015), and thus, the effects of these mutations on the inverse agonist activities of the ARBs were examined (Figure 4). The N111G/K199Q/H256A mutation additively decreased the inverse agonist activity of eprosartan. The N111G/A163T/N295A mutation additively increased the activity switch from inverse agonism towards agonism for both candesartan and telmisartan. No other triple mutations exhibited combined effects on the inverse agonist activities of any of the three ARBs. These results suggest that interactions between Lys199TM5 and His256TM6 in N111G‐AT1 receptors are important for the inverse agonist activity of eprosartan, while interactions between Ala163TM4 and Asn295TM7 are important for the activity switch from inverse agonism towards agonism for candesartan and telmisartan.
Molecular model of docking of ARBs to WT‐AT1 receptors
To examine whether the residues targeted in our study actually interact with candesartan, telmisartan and eprosartan, molecular models of AT1 receptors were employed. The molecular models were developed based on the crystal structure of human AT1 receptors bound to the biphenyl‐tetrazole ARB ZD7155, as described in the Methods section (Zhang et al., 2015). We used the human AT1 receptors structure, as the overall sequence homology of rat and human AT1 receptors is 95%, and all of the residues examined in this study are the same as those of human AT1 receptors. The docking models of the three ARBs are shown in Figure 5. The binding poses for the three ARBs in AT1 receptors were predicted by energy‐based docking simulation studies. The nature of the interaction with AT1 receptors is different for each of the ARBs owing to their distinct chemical structures. However, all three ARBs bind in similar orientations and engage in interactions with critical residues, including Arg167ECL2 (Takezako et al., 2015; Zhang et al., 2015). As we previously demonstrated that Arg167ECL2 is critical for binding ARBs and Ang II (Noda et al., 1995; Takezako et al., 2004; Zhang et al., 2015; Takezako et al., 2017), the effects of Arg167ECL2 mutations on the binding affinities and inverse agonist activities of the ARBs were not investigated in the present study, to avoid duplication of negative binding results. Of the 12 residues examined in this study, Val108TM3, Ser109TM3, Tyr113TM3, Ala163TM4, Phe182ECL2, Tyr184ECL2, Lys199TM5, His256TM6, Gln257TM6, Tyr292TM7 and Asn295TM7 were found to be present in the common ARB‐binding pocket. One residue, namely, Glu173ECL2, lacks reliable X‐ray diffraction density in the AT1 receptor structure; therefore, this residue is not indicated in Figure 5A–D.
Figure 5.
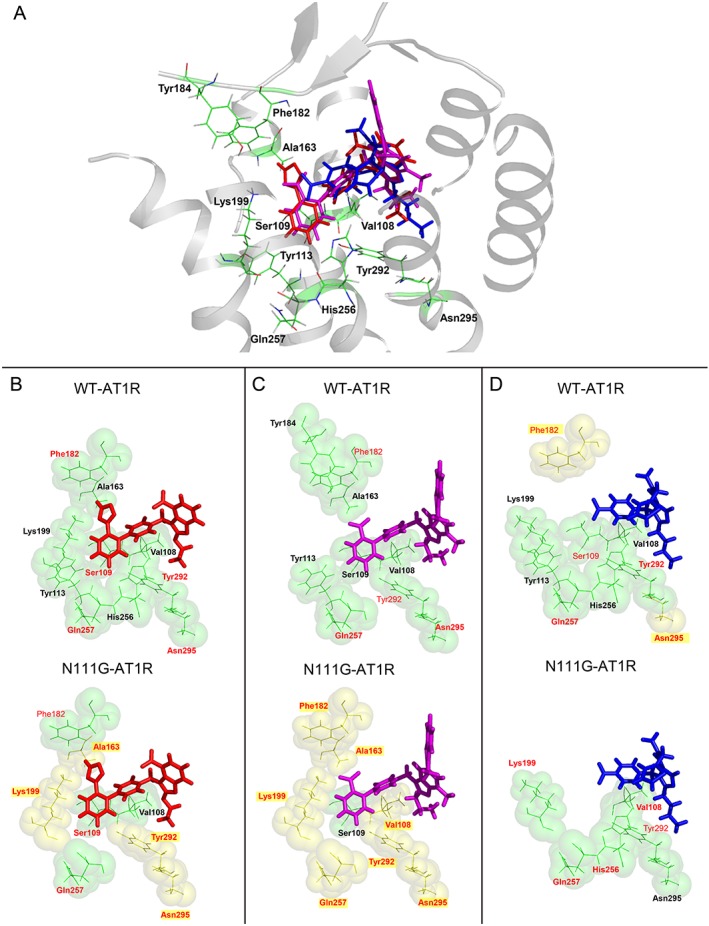
(A) Molecular model of candesartan (red), telmisartan (magenta) and eprosartan (blue) docking to AT1 receptors. Comparison of the AT1 receptor binding pocket interactions with (B) candesartan, (C) telmisartan and (D) eprosartan in the ground state (WT‐AT1 receptor) and active state (N111G‐AT1 receptor). The ARBs are shown as sticks with red (candesartan), magenta (telmisartan) and blue (eprosartan) carbons. Side‐chain positions of residues investigated in this report are located within a 10 Å pocket for each ARB. In each ARB‐bound model, single side‐chain mutations affecting binding with a >3‐fold change in Ki are indicated by a thick green colour and bold label, both in the ground state (WT‐AT1 receptor) and active state (N111G‐AT1 receptor). A red residue label denotes a significant effect on inverse agonism for IP formation in WT‐ and N111G‐AT1 receptors. Highlighted residues shown as yellow spheres denote a unique influence on inverse agonism for IP formation in the specified state of AT1 receptors for the particular ARB.
As shown in Figure 5 and the recently solved AT1 receptor structure (Zhang et al., 2015), the residues in TM helices I–VII, as well as extracellular loop 2 (ECL2), comprise the canonical ARB binding pocket. The residues Tyr113TM3, Phe182ECL2, Tyr184ECL2 and His256TM6 in the AT1 receptor ligand‐binding pocket may interact hydrophobically with all three ARBs. The candesartan docking model shows that the tetrazole group forms hydrogen bonds (H‐bonds) or salt bridges with Arg167ECL2 and is predicted to form a salt bridge with Lys199TM5, while the carboxyl group of the benzimidazole moiety forms additional salt bridges with Arg167ECL2. In addition, the benzimidazole ring of candesartan interacts with the floor of the ligand‐binding pocket, including residues Tyr292TM7 and Asn295TM7, while the biphenyl rings of candesartan interact with Val108TM3 and Ser109TM3, as well as with Trp253TM6 and Gln257TM6. The telmisartan docking model shows that the carboxyl group forms salt bridges with Arg167ECL2 and Lys199TM5. The two consecutive benzimidazole groups of telmisartan interact with the floor of the ligand pocket, including residues Tyr292TM7 and Asn295TM7. In addition, of the two consecutive benzimidazole groups of telmisartan, the proximal group forms a H‐bond with Tyr35TM1 and forms π–π contacts with Trp84TM2, while the distal group extends to Tyr92ECL1, forming additional hydrophobic and π–π contacts. The biphenyl rings of telmisartan interact with Val108TM3 and Ser109TM3, as well as with Trp253TM6 and Gln257TM6. The eprosartan docking model indicates that the two carboxyl groups individually form salt bridges with Arg167, while the thiophen ring exhibits hydrophobic interactions with Pro285TM7 and Ile288TM7 and reaches towards Met284TM7. The phenyl ring of eprosartan interacts with Val108TM3 and Ser109TM3, as well as with Trp253TM6 and Gln257TM6. The flexible side chain of Lys199TM5 provides some conformational heterogeneity in AT1 receptors (Kellici et al., 2016a,b); the amino group of this residue may reach the carboxyl group of eprosartan by forming salt bridges or may interact through water‐mediated interactions with the phenyl scaffold, which may explain the reduced binding affinity and inverse agonist activity of eprosartan upon mutation of Lys199TM5 (Takezako et al., 2015; Zhang et al., 2015; Takezako et al., 2017). The residues Tyr35TM1, Trp84TM2, Arg167ECL2, Met284TM7, Pro285TM7 and Ile288TM7 were not targeted in this study and are therefore not shown in Figure 5.
Effect of mutations on binding and inverse agonism in the superimposed ARB docking models
Molecular modelling of the active state of N111G‐ AT1 receptors is difficult, as the long timescale required for molecular dynamics simulations of GPCRs is untenable (Manglik and Kobilka, 2014). Simulations on shorter timescales showed subtle changes in the binding pocket of AT1 receptors with low P values (Takezako et al., 2015). In addition, comparisons between multiple active and inactive crystal structures of GPCRs have been reported and demonstrated only subtle conformational differences in the ligand‐binding pockets of the ground and active states (Katritch et al., 2013). Therefore, our approach does not involve modelling N111G‐ AT1 receptors. The docking of N111G‐ AT1 receptors is shown in identical pose with WT‐ AT1 receptors. Colour‐highlighted residues shown in Figure 5B–D are based on the experimental data (effect of mutated residues on the binding affinity and inverse agonist activity) for each ARB in the ground and active states.
Superposition of the experimental data for WT‐ and N111G‐ AT1 receptors is shown in Figure 5B–D. Different residues affect both the binding affinities and inverse agonist activities of the ARBs, suggesting subtle movement of the TM helices and extracellular loop regions in N111G‐ AT1 receptors. The candesartan/WT‐AT1 receptor docking model suggests that Ser109TM3, Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 are essential for candesartan's inverse agonist activity, while the telmisartan/WT‐ AT1 receptor docking model shows that Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 are essential for telmisartan's inverse agonist activity, and the eprosartan/WT‐AT1 receptor model shows that Ser109TM3, Glu173ECL2, Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 are essential for eprosartan's inverse agonist activity. On the other hand, in the N111G‐ AT1 receptor docking model, different sets of interactions mediate the inverse agonism. The candesartan/N111G‐ AT1 receptor docking model suggests that Sre109TM3, Phe182ECL2 and Gln257TM6 are essential for candesartan's inverse agonist activity and that Ala163TM4, Lys199TM5, Tyr292TM7 and Asn295TM7 influence this activity. The telmisartan/N111G docking model shows that Val108TM3, Ala163TM4, Phe182ECL2, Lys199TM5, Gln257TM6, Tyr292TM7 and Asn295TM7 influence telmisartan's inverse agonist activity, while the eprosartan/N111G‐ AT1 receptor docking model suggests that Ser109TM3, Glu173ECL2, Lys199TM5, His256TM6, Gln257TM6 and Tyr292TM7 are essential for eprosartan's inverse agonist activity.
Discussion
Although ARBs that exhibit robust inverse agonist activity for the active state of AT1 receptors could potentially demonstrate enhanced therapeutic effects for diseases such as hypertension, renal allograft rejection, primary aldosteronism and systemic sclerosis, such an ARB has not yet been discovered. Here, we have identified the non‐biphenyl‐tetrazole ARB eprosartan as a robust inverse agonist for not only the ground state but also the active state of the AT1 receptor and proposed a possible molecular mechanism for this activity.
Mutagenesis studies, along with the recently solved AT1 receptor crystal structure, elucidated that the H‐bond between Asn111TM3 and Asn295TM7 stabilizes AT1 receptors in an inactive conformation (Noda et al., 1996; Balmforth et al., 1997; Groblewski et al., 1997; Zhang et al., 2015). The Asn111TM3–Asn295TM7 H‐bond network involves additional residues, namely, Asn46TM1, Asp74TM2, Trp253TM6, Phe77TM2, Val108TM3, Ile288TM7, Tyr292TM7 and Asn298TM7, and this network of residues is likely to convey the conformational changes in the ligand‐binding pocket to the cytoplasmic domain coupling to the G proteins during the activation process of AT1 receptors (Takezako et al., 2015; Takezako et al., 2017). This network of residues may also affect the inter‐helical interactions required for the binding and inverse agonist activity of ARBs. Experimental data and results of the candesartan/WT‐ AT1 receptor docking model suggest that interaction of candesartan with Ser109TM3, Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 results in inverse agonism for the ground state of AT1 receptors, confirming the recently proposed common mechanism of inverse agonist activity of the biphenyl‐tetrazole ARBs (Takezako et al., 2015; Takezako et al., 2017). On the other hand, experimental data and results of the telmisartan/WT‐ AT1 receptor docking model suggest that interaction of telmisartan with Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 results in inverse agonism for the ground state of AT1 receptors. Although the docking model shows that telmisartan interacts with Ser109TM3, experimental data demonstrate that Ser109TM3 is not necessary for telmisartan's inverse agonist activity. In contrast, eprosartan exhibits a considerably different structure compared with the biphenyl‐tetrazole ARBs and telmisartan. However, experimental data and results of the eprosartan/WT‐ AT1 receptor docking model suggest that interaction of eprosartan with residues Ser109TM3, Glu173ECL2, Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 results in inverse agonism for the ground state of AT1 receptors. Thus, these results suggest that residues Ser109TM3, Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 are essential for the inverse agonist activity of not only the biphenyl‐tetrazole ARBs but also telmisartan and eprosartan. We propose that interaction of ARBs with residues Ser109TM3, Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7 stabilizes the above‐mentioned Asn111–Asn295 H‐bond network in the inactive conformation, thereby leading to stabilization of AT1 receptors in an inactive state, which results in robust inverse agonism in the ground state. On the other hand, as Glu173 is essential for the inverse agonist activity of eprosartan, as well as losartan and EXP3174, but not for other ARBs (Takezako et al., 2015) (Figure 3), interaction with this residue is therefore not a common feature of the inverse agonist activity of ARBs.
Previous structure–function studies using a substituted cysteine accessibility method proposed that both rotational and translational motion of TM2, TM3, TM5, TM6 and TM7 occurred in N111G‐ AT1 receptors, which mimics the active state of AT1 receptors (Boucard et al., 2003; Martin et al., 2004; Martin et al., 2007; Domazet et al., 2009). In the active state of AT1 receptors, an Asn46–Asp74–Asn295 H‐bond network is proposed to form, which involves additional interacting residues around Asn111TM3 and Asn295TM7 (Cabana et al., 2013). The active state of AT1 receptors was also proposed to hydrate the hydrophobic core and facilitate the interaction of the ‘toggle switch’ residue, Trp253TM6, with Ala291TM7 and Leu112TM3 (Cabana et al., 2013). The ARBs may destabilize the Asn46–Asp74–Asn295 H‐bond network, as well as the additional interacting residues around Asn111TM3 and Asn295TM7, and reduce hydration of the TM core via their hydrophobic properties.
In the active state (i.e. N111G‐ AT1 receptors), the H‐bond network and the residues contributing to inverse agonism are different from those of the ground state (i.e. WT‐ AT1 receptors). Mutation of Ser109TM3, Ala163TM4, Phe182ECL2, Lys199TM5, Gln257TM6, Tyr292TM7 and Asn295TM7 affects the inverse agonist activity of candesartan, while mutation of Val108TM3, Ala163TM4, Phe182ECL2, Lys199TM5, Gln257TM6, Tyr292TM7 and Asn295TM7 affects the inverse agonist activity of telmisartan, and mutation of Val108TM3, Glu173ECL2, Lys199TM5, His256TM6, Gln257TM6 and Tyr292TM7 affects the inverse agonist activity of eprosartan. Interaction with TM3, TM4, ECL2, TM5, TM6 and TM7 residues participates in the inverse agonist activity of candesartan in the active state, while interaction with TM3, ECL2, TM6 and TM7 residues participates in the inverse agonist activity of candesartan in the ground state. Interaction with TM3, TM4, ECL2, TM5, TM6 and TM7 residues participates in the inverse agonist activity of telmisartan in the active state, while interaction with ECL2, TM6 and TM7 residues participates in the inverse agonist activity of telmisartan in the ground state. Finally, interactions with TM3, ECL2, TM5, TM6 and TM7 residues participate in the inverse agonist activity of eprosartan in the active state, while interactions with TM3, ECL2, TM6 and TM7 residues participate in the inverse agonist activity of eprosartan in the ground state. These comparisons suggest that ‘leaning’ of the three ARBs on TM helices and ECL2 changes upon transition of AT1 receptors from the ground to active state, similar to the effects observed for losartan, EXP3174, valsartan and irbesartan (Takezako et al., 2015). This change in ‘leaning’ on TM helices and ECL2 is significant for candesartan and telmisartan, as well as for losartan, EXP3174, valsartan and irbesartan (Takezako et al., 2015). Therefore, destabilization of the Asn46–Asp74–Asn295 H‐bond network and reduction in hydration of the TM core by the biphenyl‐tetrazole ARBs and telmisartan may be weak, which may explain the decreased inverse agonist activity for the active state of AT1 receptors. In contrast, as the change in ‘leaning’ of eprosartan on TM helices and ECL2 seems to be subtle, eprosartan can strongly destabilize the Asn46–Asp74–Asn295 H‐bond network and reduce the hydration of the TM core, which may explain the robust inverse agonist activity observed for eprosartan for the active state of AT1 receptors.
We observed that mutation of all or some of the residues Val108TM3, Ala163TM4, Phe182ECL2, Gln257TM6 and Asn295TM7 switched the activity of candesartan and telmisartan from inverse agonism towards agonism in N111G‐ but not in WT‐ AT1 receptors (Figure 4), consistent with results observed for losartan, EXP3174, valsartan and irbesartan. Although the exact mechanism for the change in ligand‐dependent function of the receptor is unclear, we recently suggested possible mechanisms for this phenomenon (Takezako et al., 2015; Takezako et al., 2017). Briefly, bulky substitution of Val108TM3 and Ala163TM4 may result in steric hindrance in the ARB‐induced inactive AT1 receptor conformation, while removal of the side chains of Gln257TM6, Asn295TM7 and Phe182ECL2 may weaken interactions with the ARBs, which may hydrate the hydrophobic core and stabilize the Asn46–Asp74–Asn295 H‐bond network. Although elucidation of the precise mechanism of such a transformation of pharmacological activity of ARBs requires additional biophysical analysis, such as a comparison of bound water molecules in the active and inactive states, current resolution of the AT1 receptor structure is inadequate to perform this kind of analysis. Although saturation mutagenesis for the above residues, combined with ligand binding and receptor functional assays, are indirect methods, this type of study alternatively may clarify the potential mechanism of this phenomenon.
Prior to the present study, the biphenyl‐tetrazole ARBs have been known to exhibit robust inverse agonist activity for the ground state of AT1 receptors, but the inverse agonist activity of these ARBs is strongly decreased upon transition of AT1 receptors to the active state. Novel finding of the present study is that the non‐biphenyl‐tetrazole ARB eprosartan causes robust inverse agonist activity for both the ground and active states of AT1 receptors. Although N111G mutation, mechanical stress and autoantibody activate the AT1 receptor, conformation of N111G mutant, mechanically activated AT1 receptors and autoantibody‐activated AT1 receptors may not be identical. However, eprosartan remarkably shows robust inverse agonist activity for both WT and N111G mutant receptors, suggesting that eprosartan can stabilize various conformational states of AT1 receptors between ground and active state in an inactive state to similar degrees. Therefore, we suppose that eprosartan can show potent inverse agonist activity for various activated state of AT1 receptors in living cells. Transition of AT1 receptors to the active state largely changes the ligand–receptor interactions for the biphenyl‐tetrazole ARBs and telmisartan, which decreases their respective inverse agonist activities for the active state of AT1 receptors. However, active‐state transition of AT1 receptors causes only subtle changes in ligand–receptor interactions for eprosartan. Thus, eprosartan is able to conserve the robust inverse agonist activity for the active state of AT1 receptors. Subtle changes in ligand–receptor interactions for eprosartan may explain the observation that eprosartan activity does not switch from inverse agonism towards agonism upon mutation of residues Val108TM3, Ala163TM4, Phe182ECL2, Gln257TM6 and Asn295TM7 in the active state of AT1 receptors. As eprosartan may exhibit enhanced therapeutic effects for diseases caused by agonist‐independent activation of AT1 receptors, such as hypertension, cardiac hypertrophy, renal transplantation, primary aldosteronism and systemic sclerosis, additional experimental and clinical studies need to be performed to compare the effects of eprosartan with those of other ARB.
As we have focused on inverse agonism of the ARBs on Gq‐protein signalling pathway, the present study has limitations. The AT1 receptor has Gq‐protein‐independent signalling pathways such as β‐arrestin‐mediated pathway as well as Gq‐protein signalling pathway. Effects of AT1 receptor‐mediated β‐arrestin signalling varies depending on the organ. For example, in the heart, AT1 receptor‐mediated β‐arrestin signalling has been shown to cause effects opposite to those of Gq‐protein‐mediated signalling. The β‐arrestin signalling causes protective effect for cardiac hypertrophy (Teixeira et al., 2017) and heart failure (Ryba et al., 2017), whereas Gq‐mediated signalling causes cardiac hypertrophy (Nakayama et al., 2010; Matsushita et al., 2014) and exacerbates heart failure (Matsushita et al., 2014). On the other hand, in the adrenal grand, AT1 receptor‐mediated Gq‐protein signalling and β‐arrestin signalling have shown to have the same effect. Recent study showed that all commercially available ARBs prevent Ang II‐stimulated β‐arrestin signalling (Dabul et al., 2015). In addition, although effect of the ARBs on β‐arrestin signalling in the N111G‐ AT1 receptors has not been examined, recent studies have shown that losartan and telmisartan prevent β‐arrestin signalling by the mechanically activated AT1 receptors (Rakesh et al., 2010; Tang et al., 2014), indicating that these ARBs are inverse agonists for β‐arrestin signalling as well as Gq protein signalling in active state of AT1 receptors. Therefore, pharmacological activity of the ARBs on β‐arrestin signalling needs to be examined in a future study.
Author contributions
T.T. conceived and coordinated the study; T.T. and H.U. performed the research; T.T., H.U. and S.S.K. analysed the data; and T.T., H.U., S.S.K. and K.N. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Conflict of interest
K.N. was financially supported by contributions from Merck & Co., Inc., Shionogi & Co., Ltd., and Novartis Pharma K.K.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by a Grant‐in‐Aid for Research Activity Start‐up (18890141) to T.T. from the Ministry of Education, Culture, Sports, Science and Technology in Japan. This work was supported in part by a National Institutes of Health RO1 Grant (HL57470) to S.S.K. and a Grant T32 (HL007914) to H.U.
Takezako, T. , Unal, H. , Karnik, S. S. , and Node, K. (2018) The non‐biphenyl‐tetrazole angiotensin AT1 receptor antagonist eprosartan is a unique and robust inverse agonist of the active state of the AT1 receptor. British Journal of Pharmacology, 175: 2454–2469. doi: 10.1111/bph.14213.
References
- Abagyan RA, Totrov MM (1997). Contact area difference (CAD): a robust measure to evaluate accuracy of protein models. J Mol Biol 268: 678–685. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmforth AJ, Lee AJ, Warburton P, Donnelly D, Ball SG (1997). The conformational change responsible for AT1 receptor activation is dependent upon two juxtaposed asparagine residues on transmembrane helices III and VII. J Biol Chem 272: 4245–4251. [DOI] [PubMed] [Google Scholar]
- Boucard AA, Roy M, Beaulieu ME, Lavigne P, Escher E, Guillemette G et al (2003). Constitutive activation of the angiotensin II type 1 receptor alters the spatial proximity of transmembrane 7 to the ligand‐binding pocket. J Biol Chem 278: 36628–36636. [DOI] [PubMed] [Google Scholar]
- Cabana J, Holleran B, Beaulieu ME, Leduc R, Escher E, Guillemette G et al (2013). Critical hydrogen bond formation for activation of the angiotensin II type 1 receptor. J Biol Chem 288: 2593–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabul S, Bathgate‐Siryk A, Valero TR, Jafferjee M, Sturchler E, McDonald P et al (2015). Suppression of adrenal βarrestin1‐dependent aldosterone production by ARBs: head‐to‐head comparison. Sci Rep 5: 8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domazet I, Martin SS, Holleran BJ, Morin ME, Lacasse P, Lavigne P et al (2009). The fifth transmembrane domain of angiotensin II type 1 receptor participates in the formation of the ligand‐binding pocket and undergoes a counterclockwise rotation upon receptor activation. J Biol Chem 284: 31953–31961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierens FL, Vanderheyden PM, De Backer JP, Vauquelin G (1999). Insurmountable angiotensin AT1 receptor antagonists: the role of tight antagonist binding. Eur J Pharmacol 372: 199–206. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB (2003). The G‐protein‐coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63: 1256–1272. [DOI] [PubMed] [Google Scholar]
- Garland SL (2013). Are GPCRs still a source of new targets? J Biomol Screen 18: 947–966. [DOI] [PubMed] [Google Scholar]
- Groblewski T, Maigret B, Larguier R, Lombard C, Bonnafous JC, Marie J (1997). Mutation of Asn111 in the third transmembrane domain of the AT1A angiotensin II receptor induces its constitutive activation. J Biol Chem 272: 1822–1826. [DOI] [PubMed] [Google Scholar]
- Gunther J, Kill A, Becker MO, Heidecke H, Rademacher J, Siegert E et al (2014). Angiotensin receptor type 1 and endothelin receptor type A on immune cells mediate migration and the expression of IL‐8 and CCL18 when stimulated by autoantibodies from systemic sclerosis patients. Arthritis Res Ther 16: R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren TA (1995). Potential energy functions. Curr Opin Struct Biol 5: 205–210. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC (2013). Structure–function of the G protein‐coupled receptor superfamily. Annu Rev Pharmacol Toxicol 53: 531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellici TF, Ntountaniotis D, Kritsi E, Zervou M, Zoumpoulakis P, Potamitis C et al (2016a). Leveraging NMR and X‐ray data of the free ligands to build better drugs targeting angiotensin II type 1 G‐protein coupled receptor. Curr Med Chem 23: 36–59. [DOI] [PubMed] [Google Scholar]
- Kellici TF, Ntountaniotis D, Liapakis G, Tzakos A, Mavromoustakos T (2016b). The dynamic properties of angiotensin II type 1 receptor inverse agonists in solution and in the receptor site. Arab J Chem. https://doi.org/10.1016/j.arabjc.2016.11.014. [Google Scholar]
- Khan BV (2011). The effect of amlodipine besylate, losartan potassium, olmesartan medoxomil, and other antihypertensives on central aortic blood pressure and biomarkers of vascular function. Ther Adv Cardiovasc Dis 5: 241–273. [DOI] [PubMed] [Google Scholar]
- Le MT, Pugsley MK, Vauquelin G, Van Liefde I (2007). Molecular characterisation of the interactions between olmesartan and telmisartan and the human angiotensin II AT1 receptor. Br J Pharmacol 151: 952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Saver JL, Hong KS, Hao Q, Chow J, Ovbiagele B (2012). Renin–angiotensin system modulators modestly reduce vascular risk in persons with prior stroke. Stroke 43: 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Yu X, Cicala MV, Mantero F, Benbrook A, Veitla V et al (2015). Prevalence of angiotensin II type 1 receptor (AT1R)‐activating autoantibodies in primary aldosteronism. J Am Soc Hypertens 9: 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kobilka B (2014). The role of protein dynamics in GPCR function: insights from the β2AR and rhodopsin. Curr Opin Cell Biol 27: 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SS, Boucard AA, Clement M, Escher E, Leduc R, Guillemette G (2004). Analysis of the third transmembrane domain of the human type 1 angiotensin II receptor by cysteine scanning mutagenesis. J Biol Chem 279: 51415–51423. [DOI] [PubMed] [Google Scholar]
- Martin SS, Holleran BJ, Escher E, Guillemette G, Leduc R (2007). Activation of the angiotensin II type 1 receptor leads to movement of the sixth transmembrane domain: analysis by the substituted cysteine accessibility method. Mol Pharmacol 72: 182–190. [DOI] [PubMed] [Google Scholar]
- Matsushita N, Kashihara T, Shimojo H, Suzuki S, Nakada T, Takeishi Y et al (2014). Cardiac overexpression of constitutively active Gαq causes angiotensin II type1 receptor activation, leading to progressive heart failure and ventricular arrhythmias in transgenic mice. PLoS One 9: e106354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederos y Schnitzler M, Storch U, Gudermann T (2011). AT1 receptors as mechanosensors. Curr Opin Pharmacol 11: 112–116. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier TL, Mikoshiba K et al (2010). The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res 107: 659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda K, Feng YH, Liu XP, Saad Y, Husain A, Karnik SS (1996). The active state of the AT1 angiotensin receptor is generated by angiotensin II induction. Biochemistry 35: 16435–16442. [DOI] [PubMed] [Google Scholar]
- Noda K, Saad Y, Kinoshita A, Boyle TP, Graham RM, Husain A et al (1995). Tetrazole and carboxylate groups of angiotensin receptor antagonists bind to the same subsite by different mechanisms. J Biol Chem 270: 2284–2289. [DOI] [PubMed] [Google Scholar]
- Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA (2010). β‐Arrestin‐biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal 3: ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemekasten G, Philippe A, Nather M, Slowinski T, Muller DN, Heidecke H et al (2011). Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann Rheum Dis 70: 530–536. [DOI] [PubMed] [Google Scholar]
- Rossitto G, Regolisti G, Rossi E, Negro A, Nicoli D, Casali B et al (2013). Elevation of angiotensin‐II type‐1‐receptor autoantibodies titer in primary aldosteronism as a result of aldosterone‐producing adenoma. Hypertension 61: 526–533. [DOI] [PubMed] [Google Scholar]
- Ryba DM, Li J, Cowan CL, Russell B, Wolska BM, Solaro RJ (2017). Long‐term biased β‐arrestin signaling improves cardiac structure and function in dilated cardiomyopathy. Circulation 135: 1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch U, Mederos y Schnitzler M, Gudermann T (2012). G protein‐mediated stretch reception. Am J Physiol Heart Circ Physiol 302: H1241–H1249. [DOI] [PubMed] [Google Scholar]
- Takezako T, Gogonea C, Saad Y, Noda K, Karnik SS (2004). “Network leaning” as a mechanism of insurmountable antagonism of the angiotensin II type 1 receptor by non‐peptide antagonists. J Biol Chem 279: 15248–15257. [DOI] [PubMed] [Google Scholar]
- Takezako T, Unal H, Karnik SS, Node K (2015). Structure–function basis of attenuated inverse agonism of angiotensin II type 1 receptor blockers for active‐state angiotensin II type 1 receptor. Mol Pharmacol 88: 488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezako T, Unal H, Karnik SS, Node K (2017). Current topics in angiotensin II type 1 receptor research: focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol Res 123: 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Strachan RT, Lefkowitz RJ, Rockman HA (2014). Allosteric modulation of β‐arrestin‐biased angiotensin II type 1 receptor signaling by membrane stretch. J Biol Chem 289: 28271–28283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira LB, Parreiras ESLT, Bruder‐Nascimento T, Duarte DA, Simoes SC, Costa RM et al (2017). Ang‐(1‐7) is an endogenous β‐arrestin‐biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci Rep 7: 11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans PB (1999). Pharmacological properties of angiotensin II receptor antagonists. Can J Cardiol 15 (Suppl F): 26F–28F. [PubMed] [Google Scholar]
- Unal H, Jagannathan R, Bhatnagar A, Tirupula K, Desnoyer R, Karnik SS (2013). Long range effect of mutations on specific conformational changes in the extracellular loop 2 of angiotensin II type 1 receptor. J Biol Chem 288: 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal H, Jagannathan R, Karnik SS (2012). Mechanism of GPCR‐directed autoantibodies in diseases. Adv Exp Med Biol 749: 187–199. [DOI] [PubMed] [Google Scholar]
- Vejakama P, Thakkinstian A, Lertrattananon D, Ingsathit A, Ngarmukos C, Attia J (2012). Reno‐protective effects of renin–angiotensin system blockade in type 2 diabetic patients: a systematic review and network meta‐analysis. Diabetologia 55: 566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraghavan K, Deedwania P (2011). Renin–angiotensin–aldosterone blockade for cardiovascular disease prevention. Cardiol Clin 29: 137–156. [DOI] [PubMed] [Google Scholar]
- Wallukat G, Schimke I (2014). Agonistic autoantibodies directed against G‐protein‐coupled receptors and their relationship to cardiovascular diseases. Semin Immunopathol 36: 351–363. [DOI] [PubMed] [Google Scholar]
- Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA et al (2015). Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 161: 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]