

Abstract
The attribution of incentive salience to reward-predictive stimuli has been shown to be associated with substance abuse-like behavior such as increased drug taking. Evidence suggests that glutamate neurotransmission and sequential N-methyl-D-aspartate (NMDA) activation are involved in the attribution of incentive salience. Here we further explore the role of second-by-second glutamate neurotransmission in the attribution of incentive salience to reward-predictive stimuli by measuring sign-tracking behavior during a Pavlovian conditioned approach procedure using ceramic-based microelectrode arrays configured for sensitive measures of extracellular glutamate in awake behaving Sprague Dawley rats. Specifically, we show that there is an increase in extracellular glutamate levels in the prelimbic cortex (PrL) and the nucleus accumbens core (NAcC) during sign-tracking behavior to a food-predictive conditioned stimulus (CS+) compared to the presentation of a non-predictive conditioned stimulus (CS−). Further, the results indicate greater increases in extracellular glutamate levels in the PrL compared to NAcC in response to the CS+, including differences in glutamate release and signal decay. Taken together, the present research suggests that there is differential glutamate signaling in the NAcC and PrL during sign-tracking behavior to a food-predictive CS+.
Keywords: Glutamate, Incentive Salience, Nucleus Accumbens, Pavlovian Conditioned Approach, Prelimbic Cortex, Sign-tracking
Graphical Abstract
The attribution of incentive salience to reward-predictive stimuli has been shown to be associated with substance abuse-like behavior such as increased drug taking. We use a novel electrochemical technology to show that differential glutamate signaling occurs in the nucleus accumbens core and prelimbic cortex in freely-moving rats exhibiting sign-tracking behavior in a Pavlovian conditioned approach procedure. Overall, our data suggest that glutamate signaling in the nucleus accumbens core and prelimbic cortex are important in incentive salience, providing more evidence supporting the importance of glutamatergic signaling underlying behavior associated with substance use. Abbreviations: conditioned stimulus plus (CS+), conditioned stimulus minus (CS−).
INTRODUCTION
Through associative learning, stimuli paired with drugs of abuse can come to influence substance use (Hogarth et al., 2010). Interestingly, not all reward-paired stimuli function equivalently; for example, discrete localizable stimuli are more likely to be attributed with value relative to diffuse stimuli (Robinson et al., 2014). Further, the propensity of a reward-predictive stimulus to be attributed with incentive value has been shown to predict how quickly animals acquire drug self-administration and their sensitivity to reinstatement of drug-seeking behavior (Saunders and Robinson 2010; Beckmann et al., 2011). Thus, understanding the neurobehavioral mechanisms that mediate incentive value attribution to reward-related stimuli may help elucidate the mechanisms of substance use disorder.
A number of studies have used a Pavlovian conditioned approach (PCA) procedure to assess how stimuli predictive of rewards are specifically attributed with incentive value. (Boakes, 1977, Tomie et al., 2012, Robinson et al., 2014). In this procedure, a lever next to a food receptacle consistently predicts a food reward, and over time this pairing comes to elicit sign- or goal-tracking behavior in response to the lever alone (Robinson et al., 2014). Sign-tracking responses to the lever (i.e. approach and interaction with the lever) are thought to represent “incentive salience” or value attributed to the lever above and beyond its predictive relationship with food delivery (Robinson et al., 2014). Conversely, goal-tracking responses to the food receptacle (i.e. approach to the place of the forthcoming food pellet in the presence of the lever) are thought to represent the simple predictive relationship of the stimulus (lever) and the reward (Robinson et al., 2014). Recent evidence suggests that independent but parallel valuation systems in the brain may drive sign- or goal-tracking behavior (Clark et al., 2012). In fact, research shows that sign- and goal-tracking responses differentially engage the dopamine system, with sign-tracking being more sensitive to changes in dopamine function than goal-tracking (Flagel et al., 2011; Saunders and Robinson, 2013; Chow et al., 2016). That being said, the neurobehavioral mechanisms for sign- and goal-tracking behavior are still largely unknown.
In the past two decades, some research has begun to focus on the role of glutamate in promoting and maintaining drug-taking behavior (Kalivas, 2009). For example, evidence suggests that glutamate signaling may be disrupted by chronic exposure to cocaine and that this disruption may lead to the reinstatement of drug seeking after a period of extinction (McFarland et al., 2003; Park et al., 2002). Specifically, disruptions in glutamate signaling in glutamate projections from the prelimbic cortex (PrL) to the nucleus accumbens core (NAcC) seem to be particularly important in promoting drug-seeking behavior (Kalivas et al., 2005; McFarland et al., 2003). Further, evidence suggests that pharmacotherapies that manipulate the glutamate system can normalize glutamate signaling in the aforementioned brain areas and reduce cue-induced drug seeking and sign-tracking behavior (Di Ciano et al., 2001; Peters and Kalivas, 2006; Zhou and Kalivas, 2007). While there is some evidence to suggest that glutamate signaling may be involved (Di Ciano et al., 2001; Fitzpatrick and Morrow, 2016), the role of glutamatergic signaling in the attribution of incentive salience remains unclear.
In this study, we explore the role of glutamate in incentive salience using novel ceramic-based microelectrode arrays configured for second-by-second measures of extracellular glutamate in awake, behaving animals (Hascup et al., 2007; Hascup et al., 2012; Stephens et al., 2014). We measured phasic glutamate release and signal decay in the PrL and NAcC during the attribution of incentive salience in feely-moving rats performing under a PCA discrimination procedure in order to assess glutamate dynamics during this process. Our results suggest that differential glutamate signaling occurs in the PrL and NAcC during sign-tracking responses. Together, these results support that glutamatergic signaling is involved in sign-tracking behavior to a food-predictive CS+.
MATERIALS AND METHODS
Experimental Subjects
Ten adult male Sprague Dawley rats (Harlan Inc.; Indianapolis, USA; RRID:RDG_5508397) were housed separately in a temperature-controlled environment on a 12:12-hour light/dark cycle (lights on at 0700 hours) with ad libitum access to food and water. All animals were allowed to acclimate to the colony environment and were handled daily one week prior to experimentation. All experimentation occurred during the light phase. All animal care and procedures were approved by the Institutional Animal Care and Use Committee at the University of Kentucky, which is AAALACI approved; the ethical approval reference number is 2011-0885. Please note that this study was not preregistered.
Apparatus
Experiments were conducted in an operant conditioning chamber (ENV-008, Med Associates) housed within a sound-attenuating compartment (ENV-018M, Med Associates). The operant chamber was modified for low noise second-by-second measures of glutamate during the behavioral events and synchronized to the glutamate recording system and its digital I/O lines. Each chamber was connected to a microcomputer (SG-502, Med Associates) controlled by MED-PC software. The operant chamber contained a 5.1 cm × 5.1 cm recessed food receptacle (ENV-200R2MA) on the front response panel with two retractable levers on either side (ENV-122CM). Above each lever was one white cue light (ENV-221M). Located above the top left cue light was a Sonalert tone (ENV-223 AM) and located above the top right cue light was another Sonalert tone (ENV-223 HAM). A house light (ENV-227M) was placed 17 cm above the metal floor in the middle of the back wall. Two nosepoke response receptacles (ENV-114BM) were located on both sides of the back wall across from the front response levers. Sucrose-based food pellets (45 mg, Dustless Precision Pellets, #F0021; Bio Serv) were delivered via a dispenser (ENV-203M-45) placed behind the food receptacle.
Glutamate Biosensor Preparation
Microelectrode arrays (MEAs, S2 configuration; CenMeT, University of Kentucky) consisting of four platinum recording sites (15 μm × 333 μm) arranged in dual pairs were first built into an implantable headcap as previously described (Rutherford et al., 2007). Briefly, both ends of an ~2.5 cm long varnished 30 AWG copper wire (Radioshack, Fort Worth, TX) were scraped to expose ~ 0.25 cm of copper wire and fluxed (#186 Rosin flux type RMA, Kester). One end of the wire was soldered (~200 °C) to a gold-plated socket (Ginder Scientific, Nepean, ON). The other end of the wire was soldered into the paddle portion of the MEA. This was done four times in order to wire up all four measuring sites. The four wires containing gold-plated sockets were then inserted into four holes in a nine-hole ABS plug and the wires were wrapped around the plug. A Teflon coated, 5 cm long 200 μm O.D. Ag wire (A-M Systems, Carlberg, WA), which was electroplated to form an Ag/AgCl wire to serve as a reference electrode once in contact with CSF containing Cl-, was then scraped (~0.25 cm), fluxed, soldered into a gold-plated socket, and placed in the ABS plug. The assembly was then covered with a heavy layer of marine quality epoxy and allowed to cure for at least 48 hours to ensure a waterproof seal (Rutherford et al., 2007).
MEAs were then configured for selective measures of glutamate. Specifically, a solution of 1% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO), 0.125% glutaraldehyde (Sigma-Aldrich), and 1% glutamate oxidase (GluOX; US Biological, Salem, MA) was coated on the bottom two recording sites of the MEA by syringe application (3 coats) to allow for conversion of glutamate into α-ketoglutarate and the reporter molecule, peroxide (Burmeister and Gerhardt, 2001; Day et al., 2006). The top two recording sites were coated with the BSA/glutaraldehyde matrix (without GluOX) to allow for background current subtraction thus resulting in a self-referenced glutamate signal (Burmeister and Gerhardt, 2001). After the MEAs were configured with enzymes to measure glutamate they were allowed to cure for at least 72 hours and then all four recording sites were electroplated with m-phenylenediamine (mPD; Acros, Fisher Scientific, Waltham, MA). mPD is a size exclusion layer that eliminates signals from interferent molecules such as ascorbic acid and dopamine thus allowing for more selective glutamate measurements (Miller et al., 2015). Electrodes were allowed to sit for at least 24 hours before implantation.
In Vitro Glutamate Calibration and Electrode Characterization
Amperometric recordings were collected at 4 Hz using the FAST16mkIII electrochemical recording system (Fast Analytical Sensing Technology, Quanteon, LLC, Nicholasville, KY). Immediately before in vivo implantation, all electrodes underwent an in vitro calibration to determine sensitivity (slope, nA/μM), selectivity (glutamate vs. ascorbic acid sensitivity), and limit of detection (in μM, signal-to-noise = 3). Note that a flow injection analysis system was used to accurately determine the time it took for the MEA current to rise from baseline to 90% of the maximum (T90 response time) using six additional MEAs. The T90 response time of the MEAs is 0.17 ± 0.07 seconds.
In Vivo Glutamate Biosensor Implantation and Electrochemical Recordings
Immediately before surgery all rats were given subcutaneous injections of carprofen (Rimadyl©, Pfizer, NYC) at a dose of 10mg/kg and 1 mL of 0.9% sterile saline in order to decrease animal suffering. All rats were anesthetized using 4% isoflurane (Isothesia, Henry Schein, Melville, NY). Once anesthetized, animals were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA) and maintained at an isoflurane level of 1–3%. Body temperature was maintained at 37°C using a circulating water bath attached to a water pad (Gaymar Industries, Orchard Park, NY). Artificial tears (Rugby Laboratories, Inc., Duluth, GA) were then applied to the rat’s eyes, the rat’s head was shaved, and Providone scrub (The Butler Co., Columbus, OH) and 70% ethanol were used to disinfect the surgery site. The skin overlying the skull was then reflected. A craniotomy was performed exposing the left hemisphere of either the PrL (AP: +3.2; ML: ±0.8; DV: −3.5) or NAcC (AP: +1.3; ML: ±2.1; DV: −7.0) (Paxinos and Watson, 2009). A small burr hole was made contralateral to the site of the craniotomy for implantation of the small Ag/AgCl reference electrode. Three screws (Amazon supply, part No. B00FN0K02) were then screwed into the skull. The dura was then reflected where the craniotomy was performed and the MEA was implanted in the respective brain region and the Ag/AgCl reference was placed into the contralateral burr hole epidurally. Anterior-posterior and medial-lateral coordinates for MEA implantation were calculated relative to bregma and dorsal-ventral coordinates from the brain surface. The MEA headcap was then set in place on the rat’s skull using dental acrylic (Ortho Jet Powder and Jet Acrylic Liquid, Lang Dental Manufacturing Co., Wheeling, IL). The animals were allowed to recover for 3 days before the recordings began; they were given carprofen (10 mg/kg) and 1 mL 0.9% sterile saline subcutaneously during this period in order to minimize animal suffering. Glutamate measurements were performed using the FAST-16 mkIII recording system using a low noise 4-channel Rat Hat amplifier (Quanteon, LLC, Nicholasville, KY) system connected through a low-noise commutator (Plastics One, Inc., Roanoke, VA) for the length of the PCA session. Each animal was first ran through the PCA procedure at +0.7 V potential vs. Ag/AgCl reference (able to oxidize the recorder molecule, peroxide [i.e. able to detect glutamate]), allowed to sit in their home cage for ~2–3 hours, and then ran through the PCA procedure again at +0.2 V potential vs. Ag/AgCl reference (unable to oxidize the recorder molecule, peroxide [i.e. unable to detect glutamate]). Taking measurements at both +0.7 V and +0.2 V potential within a single animal served as an internal control in that any release events seen during the PCA procedure at the +0.7 V potential should disappear at the +0.2 V potential due to the fact that the reporter molecule from MEA detection of glutamate, peroxide, cannot be measured at the +0.2 V potential. Note that the blinding of the experimenters did not take place in this study. However, animals were pseudo-randomly assigned to different experimental groups. This was accomplished by placing every other electrode in a different brain region. In this case, we started by placing the first electrode in the NAcC thus the second electrode was placed in the PrL and so on until equal groups of animals had electrodes placed in both brain regions. Note that this method did not create differences in behavioral performance between groups. Upon completion of the MEA recordings animals were euthanatized using both CO2 and decapitation, the brains were extracted, flash frozen, and 40 μm slices were prepared using a cryostat. The slices were stained using Cresyl Violet (Sigma-Aldrich, St. Louis, MO) and were visualized to confirm MEA placements into NAcC or PrL (Figures 1A and 1B, respectively).
Figure 1. Glutamate electrode placement.

(A) Triangles represent the placement of the tip of the glutamate biosensor in the NAcC (n = 5). (B) Triangles represents the placement of the tip of the glutamate biosensor in the PrL (n = 5).
CS+/CS− PCA Discrimination Procedure with In Vivo Glutamate Recordings
All rats (n = 5/brain region) were first magazine shaped for two consecutive days by releasing food pellets into the food receptacle one time every minute for 15 minutes (Beckmann et al., 2011). Rats then began training on the PCA CS+/CS− procedure (Flagel et al., 2007; Beckmann et al., 2011) where one lever was the food-predictive conditioned stimulus (CS+) and the other the non-predictive conditioned stimulus (CS−) (counterbalanced). Each lever was presented (8 seconds) in pseudo-random order (no more than two presentations) and a food pellet was non-contingently delivered after the presentation of the CS+. A 90- second variable time (VT) intertrial interval (ITI), a time where no manipulandum were present and all stimuli were off, followed each trial. Each lever was presented 16 times for a total of 32 trials; each session lasted ~50 minutes. Sign-tracking responses were measured via lever depression and goal-tracking responses as breaks in the food receptacle photobeam during CS+/− presentation. Head receptacle entries during the 8-second period preceding stimulus presentations (8-second pre-CS) were also recorded. After stable responding (~14 sessions), rats were implanted with the glutamate biosensor (see above) and then phasic glutamate measurements were taken as the animal performed under the PCA discrimination procedure. For a timeline of experimental procedures, see Figure 2.
Figure 2. Timeline of experimental procedures.

Animals were first acclimated to the colony room for 7 days and then trained on the PCA procedure. After stable performance rats were implanted with chronic MEA implants in the NAcC or PrL. After post-surgery care glutamate data was collected as animals performed in the PCA procedure. After glutamate data collection the animals were euthanized.
Data Analyses
Data were only excluded if there were electrical failures in the electrode, amplifier, or commutator tethering the animal during a given session. Failures only occurred in six rats thus behavioral and electrochemical data were only analyzed for 10/16 rats. For behavioral measures, the sign-tracking and goal-tracking response rates were defined as the number of responses that happened during the presentation of the 8-second CS+/CS−. The amperometric data collected were analyzed using custom MATLAB-based software (Quanteon LLC, Nicholasville, KY). The parameters analyzed were the amplitude of glutamate release (μM) to the CS+ and CS−, the time to rise (Trise; sec) of the glutamate release event, and the area under the curve (AUC) of the glutamate release event from the maximum amplitude to baseline (Hascup et al., 2007; Hinzman et al., 2010). Statistical analyses were conducted using JMP Pro 12.1.0. Behavioral and glutamate release amplitude data were analyzed using linear mixed-effects modeling (Gelman and Hill, 2006), with subject as a random factor. Akaike information criterion (AIC) values were used to compare models; only statistics from the models that were most likely to describe the data are presented. Further, we also calculated differences in AIC values (ΔAICs) in order to calculate the relative difference of information loss of all the other models compared to the best model. From the ΔAICs we then calculated and presented evidence ratios for the best model relative to the second-best model (Burnham & Anderson, 2002; Burnham et al., 2011). The evidence ratios indicate the relative strength of the preferred model to the second-best model. Thus, by using evidence ratios we can say that the evidence for the preferred model is ‘x’ times stronger than that of the other model. The Trise and decay (AUC) of the glutamate release events to the CS+ between the NAcC and PrL were analyzed using unpaired t-tests. Note that essentially no glutamate release was observed to the presentation of the CS− (see below), and consequently there were no release rise or decay to analyze; thus, we only assessed the rise and decay of release events associated with the CS+. The Cohen’s d calculation was performed in order to determine the size of our effect for the rise and decay data during the +0.7 V applied potential. Note that no sample size calculation was performed before the beginning of the study. However, the evidence ratio and effect size calculations suggest that our N size was large enough to detect meaningful differences in the data (see below). All interactions were probed using the Tukey HSD, and statistical significance was defined a p < 0.05 for all analyses.
RESULTS
Sign-Tracking and Goal-Tracking Responses during the PCA Procedure
Figures 3A–3D illustrate the response rates to the CS+ and CS− for both sign-tracking and goal-tracking responses for animals with MEAs implanted in the NAcC and PrL, respectively. The model that had the lowest AIC value included only the stimulus (CS+/CS−) as a predictor of sign-tracking and goal-tracking behavior and the evidence ratio suggests that the evidence for this model is 2,428 times stronger than that for the full model which included stimulus and brain region as predictors. Higher sign-tracking response rates were seen to the CS+ compared to the CS− in animals with the MEA implanted in both brain regions [F(1, 9) = 43.30, p = 0.0001]. No significant differences were seen with goal-tracking behavior [F(1, 9) = 0.77, p = 0.40].
Figure 3. Sign-tracking and goal-tracking response rates to CS+/CS− during the PCA procedure.

(A/B) There was a significant main effect of stimulus for sign-tracking behavior with more sign-tracking seen to the CS+ compared to the CS− in rats with glutamate measurements taken from the NAcC or PrL [F(1, 9) = 43.30, *p < 0.05]. (C/D) No statistically significant differences were observed to the CS+ or CS− for goal-tracking behavior in rats with measurements taken from the NAcC or PrL [F(1, 9) = 0.77, *p < 0.05]. Data are presented as mean ± SEM; n = 5/brain region.
Glutamate Overflow Dynamics between the NAc Core and PrL Cortex during the PCA Procedure
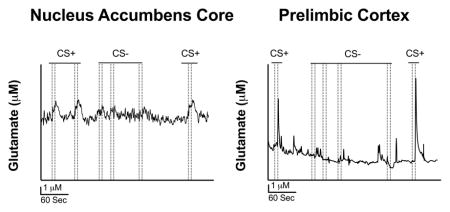
Figures 4A and 4B show representative traces of glutamate overflow (nA) to the CS+ and CS− in the NAcC and PrL before sentinel site subtraction, respectively. The CS+ was seen to produce detectable and reproducible changes in extracellular glutamate in the NAcC and PrL that were not observed during the CS−. Figures 4C and 4D illustrate representative traces of glutamate overflow (μM) to the CS+ and CS− in the NAcC and PrL, after sentinel site subtraction, respectively. Notice the difference in release, decay, and peak amplitude concentration to the CS+ and CS− between the two brain regions (Figures 4C and 4D). Also, note how the glutamate release events to the CS+ and CS− considerably decrease in both brain regions at the +0.2 V potential where glutamate is unable to be detected (Figures 5A and 5B), further confirming the selectivity of the MEA measures for extracellular glutamate. Figures 5C and 5D show the concentration of the glutamate release events to the CS+ and CS− when the MEA data was collected at +0.7 V and +0.2 V potential in animals with electrodes implanted in the NAcC and PrL, respectively. This was carried out to further confirm that the MEAs were responding to the reporter molecule, peroxide, and not to another detected electrode active species such as dopamine. The full model including stimulus, brain region, and electrical potential as predictors of glutamate release concentration had the lowest AIC value and had an evidence ratio of 982 compared to the model including only stimulus and potential as predictors. A higher concentration of glutamate release was seen in the NAcC and the PrL to the CS+ at the +0.7 V potential compared all other potentials and stimulus types between the two brain regions [F(1, 8) = 14.24, p = 0.005]. Release concentration in the PrL to the CS+ at the +0.7 V potential was significantly greater than release concentration in the NAcC to the CS+ at the +0.7 V potential [F(1, 8) = 14.24, p = 0.005]. All of these differences can be clearly seen in the traces presented in Figures 4C–4D and 5A–5B. Overall, this supports that extracellular glutamate concentration is greater to the presentation of the CS+ compared to the CS− in both brain regions at the +0.7 V potential and also that release concentration to the CS+ at the +0.7 V potential is greater in the PrL compared to the NAcC.
Figure 4. Representative glutamate traces from the NAc core and PrL cortex.

(A) Representative trace of glutamate release (nA) in the NAcC before sentinel site subtraction. (B) Representative trace of glutamate release (nA) in the PrL before sentinel site subtraction. (C) Representative trace of the concentration of glutamate release (μM) in the NAcC after sentinel site subtraction. (D) Representative trace of the concentration of glutamate release (μM) in the PrL after sentinel site subtraction. Note that the dotted lines represent lever extension and retraction, respectively.
Figure 5. Glutamate release in the NAc core and PrL cortex and the effects of applied potential on MEA responses.

(A) Representative trace at the +0.2 V potential (unable to detect glutamate) in the NAcC. (B) Representative trace at the +0.2 V potential (unable to detect glutamate) in the PrL. Notice how glutamate events are absent in response to the stimuli; however, there is some noise in the signal which is contributing to the increase seen in the bar graph at +0.2 V potential. Dashed lines represent lever onset and offset, respectively. (C/D) There was a significant stimulus × potential × brain region interaction with a greater concentration of glutamate release seen in the PrL and NAcC to the CS+ at the +0.7 V potential compared to all other stimuli presentations, brain regions, and potentials [F(1,8) = 14.24, *p < 0.05]. Note that release in the PrL to the CS+ at the +0.7 V potential was significantly greater than release in the NAcC to the CS+ at the +0.7 V potential (Tukey HSD, *p < 0.05). Data are presented as mean ± SEM; n = 5/brain region.
Additional analyses were carried out on the temporal properties of the glutamate signals seen during the CS+. Figure 6A shows a layover trace of a glutamate overflow event to the CS+ from the NAcC and PrL, respectively. Notice the difference in rise time and decay in the signal between the two brain regions. Specifically, the glutamate signal in the NAcC began at CS+ onset and peaked with CS+ offset and pellet delivery whereas the glutamate signal in the PrL only occurred to CS+ offset and pellet delivery. These differences are further seen in that Trise measures were longer in time course in the NAcC compared to the PrL [t(8) = 2.54, p = 0.03]; this is clearly seen in Figures 6A–6B. Figure 6C illustrates the average AUC of the signal from the maximum amplitude of the elicited glutamate peaks in the NAcC and PrL to the presentation of the CS+. Specifically, the AUC of decay was larger in the NAcC compared to the PrL [t(8) = 2.36, p = 0.04]. Considering the response time of the MEAs were 0.17 seconds (i.e. faster than the glutamate signal), it is unlikely that the response time of our sensors distorted our glutamate measurements. Note that the effect size was also calculated for both the Trise (d = 1.61) and the AUC (d = 1.49) data and was found to be large for both measures. Taken together with our prior studies on glutamate overflow in these two brain structures (Thomas et al., 2009; Mattinson et al., 2011; Miller at al., 2015), these data suggest that glutamate release is a longer lasting event in the NAcC to the CS+ and that glutamate signal decay is slower in the NAcC following the CS+.
Figure 6. Average time course data of CS+-induced glutamate signals in the NAc core and PrL cortex.

(A) Representative layover trace of CS+ elicited glutamate release in the NAcC and PrL. Notice the difference in release rise and decay. Dotted lines represent stimulus onset and offset. (B) Trise measures were significantly longer in time course in the NAcC compared to the PrL [t(8) = 2.54, p < 0.05]. (C) AUC measures from the peak amplitude were significantly larger in the NAcC compared to the PrL [t(8) = 2.36, *p < 0.05]. Data are presented as mean ± SEM; n = 5/brain region.
DISCUSSION
The results from the experiments presented here show that glutamate signaling in the NAcC and the PrL are differentially involved in sign-tracking performance. Specifically, release concentrations to the presentation of the CS+ were greater in both brain regions compared to presentation of the CS−. Glutamate release concentrations to the CS+ were also greater in the PrL compared to the NAcC. We also showed the reliability of our measure in that glutamate events occurred only at the +0.7 V potential that is able to measure glutamate from the detection of the enzyme reporter molecule and not due to other electroactive species or electrical anomalies. We also showed temporal differences in the glutamate overflow events to the CS+. Specifically, release was longer lasting in the NAcC compared to the PrL and glutamate signal decay was slower in the NAcC compared to the PrL.
Previous work has shown that glutamate neurotransmission in the NAcC and the PrL are involved in drug seeking and relapse (Kalivas et al., 2005; McFarland et al., 2003). Specifically, evidence suggests that the glutamate projections from the PrL to the NAcC and ventral tagmental area (VTA) are especially important in promoting drug seeking and cue-induced relapse (Kalivas, 2004). For example, pharmacological inhibition of the prefrontal cortex prevents cue-induced reinstatement (McClaughlin & See, 2003). Further, glutamate projections from the PrL to the NAcC have been shown to be important for encoding the meaning of cues (Kelley, 2004). Thus, considering that a hallmark of incentive salience is value attribution to a cue (CS) above and beyond its predictive relationship, it follows that glutamate signaling events in the PrL and NAcC are important in the attribution of incentive salience. Further, recent evidence showed that NMDA inhibition during PCA acquisition specifically blocked sign-tracking behavior to a lever CS+ suggesting that glutamate signaling and NMDA activation are critical in the attribution of incentive salience (Chow & Beckmann, 2017). To our knowledge, we are the first to show differential glutamate overflow in the PrL and NAcC during the attribution of incentive salience; thus, it leaves us to speculate as to why the peak concentration of glutamate release was larger in the PrL compared to the NAcC during sign-tracking behavior. One possible explanation for our results could be due to differential glutamatergic inputs between the PrL and NAcC. Evidence suggests that glutamate projections to the PrL arise primarily from the mediodorsal thalamus whereas glutamate inputs to the NAcC arise primarily from the PrL (Phillipson & Griffiths, 1985; Romanides et al., 1999). Further, evidence shows that dopamine projections can directly or indirectly affect the aforementioned glutamate projections (Miller et al., 2013). Considering that dopamine release in the NAcC is essential for the attribution of incentive salience (Berridge & Robinson, 1998), it is possible that differential dopamine tone on these glutamate projections is responsible for the glutamate concentrations observed in the PrL and NAcC. Further, evidence from the fear conditioning literature suggests that CS− invoked fear responses are related to an increase glutamatergic neuronal activity in the PrL and a decrease in glutamate release in the NAc (Saul’skaya & Marsden, 1997; Burgos-Robles et al., 2009). Here we found an increase in glutamate concentrations to CS+ offset and food pellet delivery in the PrL, and an increase in glutamate concentrations to CS+ onset in the NAcC. Our results are somewhat at odds with findings from the fear conditioning literature; however, this may be due to a difference in how the glutamate systems in these brain regions encode appetitive vs. aversive stimuli. However, more research will need to be conducted to further explore how glutamate dynamics in the PrL and NAcC specifically encode appetitive and aversive stimuli.
We also found that glutamate concentrations rose more slowly in the NAcC. It is not clear as to why glutamate release had a longer lasting rise time in the NAcC compared to the PrL. However, there is evidence that NAc neurons fire more to a food-paired CS+ than to the reinforcer itself and that this neuronal excitation to the CS+ may be due to glutamatergic inputs from cortical and limbic structures (Pennartz et al., 1994; Day & Carelli, 2007; Stuber et al., 2008). Therefore, the physiological nature of the aforementioned glutamate neurons may account for the slower rising Trise results presented here in the NAcC because the glutamate events began shortly after the presentation of the CS+ and peaked with the delivery of the reinforcer. However, in the PrL the rise in glutamate concentrations was seen more prominently to lever retraction and receipt of the food pellet. Thus, eating of the food pellet may have caused the increase in glutamate overflow in the PrL. In fact, evidence suggests that the PrL is involved in conditioned feeding as seen by decreased CS+ elicited feeding behavior in animals with PrL lesions (Holland & Petrovich, 2005). Thus, our glutamate measures map on to what has been observed in the NAcC and PrL in electrophysiological and lesion studies (see Figure 6A–6B; Holland & Petrovich, 2005; Day et al., 2006), which may explain why our Trise measures are different in the NAcC compared to the PrL. Further, it is unlikely that the rise time differences are due to differences in brain diffusion considering that tortuosity is similar across all brain regions in the healthy brain (Skyova & Nichols, 2008; Nicholson & Hrabetova, 2017
We also found that glutamate signal decay was slower in the NAcC compared to the PrL. This difference in decay is likely due to a difference in uptake between the PrL and NAcC, especially considering the fact that diffusion in the healthy brain is roughly the same across brain regions (Nicholson & Hrabetova, 2017) and thus should not contribute to the AUC measure from peak amplitude. It is possible that the differential glutamate uptake in the NAcC may be due to a change in GLT-1 expression. Trantham-Davidson et al. (2012) suggests that cocaine affects GLT-1 in the NAcC such that glutamate uptake is decreased. Herein, animals were never given cocaine; however, there is evidence that animals that sign-track are more susceptible to the rewarding properties of drugs of abuse (Tomie et al., 2008; Beckmann et al., 2011; Robinson et al., 2014). Thus, it is possible that the decreased decay in the glutamate signal seen here in the NAcC could be due to a natural decrease in GLT-1 that occurs during the acquisition of incentive salience and that this decrease predisposes animals to have a heightened reward sensitivity to drugs of abuse. This is especially so considering that GLT-1 expression is roughly the same in the PrL and NAcC in naïve animals (Vollbrecht et al., 2014; Knackstedt et al., 2010). However, more experiments will need to be conducted to support this possibility. Note that all glutamate measurements in these experiments were taken from the left hemisphere of both brain regions. This is a potential weakness of the study. However, others (e.g. McFarland et al., 2003; LaLumiere et al., 2008) have conducted similar experiments and have collected their data by either counterbalancing hemispheres or collapsing data from both hemispheres into a single data point. Both of the aforementioned ways suggest that robust hemispheric differences in glutamate signaling do not exist in the PrL and NAcC at least in relation to appetitive behavior. Thus, only having left hemisphere data in this study likely did not skew our results. Further, the response time of our biosensors was found to be 0.17 seconds, which is faster than the average glutamate signal. Thus, our temporal results are also likely not distorted by the response time of our MEAs.
It is worth noting that in this experiment we had very few animals that goal-tracked to the lever CS+. This could be for several reasons. One reason could be that most research studying sign- and goal-tracking behavior have used a large group of animals initially trained on a single-lever PCA procedure (Robinson et al., 2014). Specifically, in the aforementioned procedure, animals that predominantly sign-track during initial training are added to the “sign-trackers” cohort, those that predominantly goal-track are added to the “goal-trackers” cohort, and those that both sign- and goal-track are added to the “intermediate” cohort for future experimentation (Meyer et al., 2012). However, there is evidence that even when running large groups of animals that the response distributions on the single-lever PCA procedure are not uniform, with a heavy bias toward sign-tracking responses (Holland et al., 2014). There is some evidence that this difference in behavioral allocation could be due to a difference in animal vendors or even breeding colonies within vendors (Fitzpatrick et al., 2013). Thus, it is possible we did not observe robust goal-tracking behavior here due to the fact that we did not run a large cohort of animals or because of the vendor/breeding colony where the animals were purchased. Another reason for the lack of goal-tracking behavior observed here could be that the cohort studies use a retractable lever that is illuminated from behind whereas here we used a retractable lever that is not illuminated. There is evidence that illuminated nose pokes can promote goal-tracking behavior (Beckmann and Chow, 2015); thus, it is possible that the illuminated lever used in the cohort designs is picking up different selective associations made between the illuminated lever and food leading to a difference in the expression of sign- and goal-tracking behavior across animals. It is worth noting that others (Chang and Holland, 2013; Chang, 2014; Holland et al., 2014) have also observed a bias towards sign-tracking responses in a lever CS+/CS− procedure. Thus, our results are not unlike what others have observed.
In conclusion, here we present results showing that there is differential glutamate signaling in the PrL and NAcC during sign-tracking behavior. Specifically, we showed that glutamate overflow is greater to the CS+ compared to the CS− in both brain regions, with essentially no detected glutamate change from the CS−. Further, we showed that glutamate overflow to the CS+ was greater in the PrL compared to the NAcC. Lastly, we showed that the time course of the glutamate signal was different between the two brain regions with longer rise times and slower glutamate decay in the NAcC. Collectively, these data suggest that glutamatergic signaling in the NAcC and PrL is involved in incentive salience. Thus, a deeper understanding in the role of glutamate signaling and pharmacological approaches to modulate glutamate signaling during sign-tracking behavior may lead to the development of pharmacotherapies that decrease abuse-like behavior.
Acknowledgments
The authors would like to thank Josh Lavy for his technical assistance.
Abbreviations
- AIC
Akaike Information Criterion
- ΔAIC
delta AIC
- AUC
Area Under the Curve
- BSA
Bovine Serum Albumin
- GluOX
Glutamate Oxidase
- ITI
Intertrial Interval
- mPD
m-phenylenediamine
- MEA
Microelectrode Array
- CS−
Non-predictive Conditioned Stimulus
- PCA
Pavlovian Conditioned Approach
- CS+
Predictive Conditioned Stimulus
- PrL
Prelimbic cortex
- NAcC
Nucleus Accumbens Core
- Trise
Time to Peak Signal Rise
- T90
Time to 90% Peak Signal Rise
- VT
Variable Time
- RRID
Research Resource Identifier
Footnotes
Conflicts of interest: GAG is the principal owner of Quanteon, LLC that manufactures the FAST recording system. The remaining authors have nothing to disclose.
FUNDING AND DISCLOSURE
This research was funded by NIDA grant DA033373. GAG is the principal owner of Quanteon, LLC that manufactures the FAST recording system. The remaining authors have nothing to disclose.
References
- Beckmann JS, Chow JJ. Isolating the incentive salience of reward-associated stimuli: value, choice, and persistence. Learn Mem. 2015;22:116–127. doi: 10.1101/lm.037382.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann JS, Marusich JA, Gipson CD, Bardo MT. Novelty seeking, incentive salience and acquisition of cocaine self-administration in the rat. Behavioural brain research. 2011;216:159–165. doi: 10.1016/j.bbr.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Rev. 1998;28:309–369. doi: 10.1016/s0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- Boakes RA. Performance on learning to associate a stimulus with positive reinforcement. In: Davis H, Hurwitz H, editors. Operant-Pavlovian interactions. Earlbaum; Hillsdale, NJ: 1977. pp. 67–97. [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Quirk GJ. Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:8474–8482. doi: 10.1523/JNEUROSCI.0378-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister JJ, Gerhardt GA. Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Anal Chem. 2001;73:1037–1042. doi: 10.1021/ac0010429. [DOI] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR. Model selection and multimodal inference. 2. Springer-Verlag; New York, NY: 2002. [Google Scholar]
- Burnham KP, Anderson DR, Huyvaert KP. AIC model selection and multimodal inference in behavioral ecology: some background, observations, and comparisions. Behav Ecol Sociobiol. 2011;65:23–35. [Google Scholar]
- Chang SE. Effects of orbitofrontal cortex lesions on autoshaped lever pressing and reversal learning. Behavioural brain research. 2014;273:52–56. doi: 10.1016/j.bbr.2014.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SE, Holland PC. Effects of nucleus accumbens core and shell lesions on autoshaped lever-pressing. Behavioural brain research. 2013;256:36–42. doi: 10.1016/j.bbr.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JJ, Nickell JR, Darna M, Beckmann JS. Toward isolating the role of dopamine in the acquisition of incentive salience attribution. Neuropharmacology. 2016;109:320–331. doi: 10.1016/j.neuropharm.2016.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JJ, Beckmann JS. NMDA receptor blockade specifically impedes the acquisition of incentive salience attribution. Behavioural Brain Res. 2017;338:40–46. doi: 10.1016/j.bbr.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JJ, Hollon NG, Phillips PE. Pavlovian valuation systems in learning and decision making. Curr Opin Neurobiol. 2012;22:1054–1061. doi: 10.1016/j.conb.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day BK, Pomerleau F, Burmeister JJ, Huettl P, Gerhardt GA. Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. Journal of neurochemistry. 2006;96:1626–1635. doi: 10.1111/j.1471-4159.2006.03673.x. [DOI] [PubMed] [Google Scholar]
- Day JJ, Carelli RM. The nucleus accumbens and Pavlovian reward learning. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. 2007;13:148–159. doi: 10.1177/1073858406295854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Ciano P, Cardinal RN, Cowell RA, Little SJ, Everitt BJ. Differential involvement of NMDA, AMPA/kainate, dopamine receptors in the nucleus accubmens core in the acquisition and performance of pavlovian approach behavior. J Neuroscience. 21:9471–9477. doi: 10.1523/JNEUROSCI.21-23-09471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick CJ, Gopalakrishnan S, Cogan ES, et al. Variation in the form of pavlovian conditioned approach behavior among outbred male sprague-dawley rats from different vendors and colonies: sign-tracking vs. goal-tracking. Plos One. 2013;8:e75042. doi: 10.1371/journal.pone.0075042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick CJ, Morrow JD. Subanesthetic ketamine decreases the incentive-motivational value of reward-related cues. J Psychopharmacol. 2017;31:67–74. doi: 10.1177/0269881116667709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagel SB, Watson SJ, Robinson TE, Akil H. Individual differences in the propensity to approach signals vs goals promote different adaptations in the dopamine system of rats. Psychopharmacology. 2007;191:599–607. doi: 10.1007/s00213-006-0535-8. [DOI] [PubMed] [Google Scholar]
- Flagel SB, Clark JJ, Robinson TE, et al. A selective role for dopamine in stimulus-reward learning. Nature. 2011;469:53–57. doi: 10.1038/nature09588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A, Hill J. Data analysis using regression and multilevel/hierarchical models. Cambridge University Press; New York: 2006. [Google Scholar]
- Hascup KN, Rutherford EC, Quintero JE, et al. Second-by-second measures of L-glutamate and other neurotransmitter systems using enzyme based microelectrode arrays. In: Michael AC, Borland LM, editors. Electrochemical Methods for Neuroscience. Boca Raton (FL): CRC Press/Taylor & Francis; 2007. [PubMed] [Google Scholar]
- Hascup ER, Hascup KN, Pomerleau F, Huettl P, Hajos-Korcsok E, Kehr J, Gerhardt GA. An allosteric modulator of metabotropic glutamate receptors (mGluR(2)), (+)-TFMPIP, inhibits restraint stress-induced phasic glutamate release in rat prefrontal cortex. Journal of neurochemistry. 2012;122:619–627. doi: 10.1111/j.1471-4159.2012.07784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman JM, Thomas TC, Burmeister JJ, Quintero JE, Huettl P, Pomerleau F, Gerhardt GA, Lifshitz J. Diffuse brain injury elevates tonic glutamate levels and potassium-evoked glutamate release in discrete brain regions at two days post-injury: an enzyme-based microelectrode array study. J Neurotrauma. 2010;27:889–899. doi: 10.1089/neu.2009.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth L, Dickinson A, Duka T. The associative basis of cue-elicited drug taking in humans. Psychopharmacology. 2010;208:337–351. doi: 10.1007/s00213-009-1735-9. [DOI] [PubMed] [Google Scholar]
- Holland PC, Petrovich GD. The neural systems analysis of the potentiation of feeding by conditioned stimuli. Physiology of Behavior. 2005;86:747–761. doi: 10.1016/j.physbeh.2005.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland PC, Asem JS, Galvin CP, Keeney CH, Hsu M, Miller A, Zhou V. Blocking in autoshaped lever-pressing procedures with rats. Learn Behav. 2014;42:1–21. doi: 10.3758/s13420-013-0120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. Glutamate systems in cocaine addiction. Curr Opin Pharmacol. 2004;4:23–29. doi: 10.1016/j.coph.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nature reviews Neuroscience. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–650. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, Melendez RI, Kalivas PW. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine-seeking. Biol Psychiatry. 2010;67:81–84. doi: 10.1016/j.biopsych.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neuroscience. 2008;28:3170–3177. doi: 10.1523/JNEUROSCI.5129-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neuroscience. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EM, Thomas TC, Gerhardt GA, Glaser PEA. Dopamine and Glutamate Interactions in ADHD: Implications for the Future Neuropharmacology of ADHD. In: Banerjee Somnath., Dr, editor. Attention Deficit Hyperactivity Disorder in Children and Adolescents. InTech; 2013. [DOI] [Google Scholar]
- Mattinson CE, Burmeister JJ, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA. Tonic and phasic release of glutamate and acetylcholine neurotransmission in sub-regions of the rat prefrontal cortex using enzyme-based microelectrode arrays. Journal of neuroscience methods. 2011;202:199–208. doi: 10.1016/j.jneumeth.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, See RE. Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology. 2003;168:57–65. doi: 10.1007/s00213-002-1196-x. [DOI] [PubMed] [Google Scholar]
- Meyer PJ, Lovic V, Saunders BT, Yager LM, Flagel SB, Morrow JD, Robinson TE. Quantifying individual variation in the propensity to attribute incentive salience to reward cues. PloS one. 2012;7:e38987. doi: 10.1371/journal.pone.0038987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EM, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA, Glaser PE. Simultaneous glutamate recordings in the frontal cortex network with multisite biomorphic microelectrodes: New tools for ADHD research. Journal of neuroscience methods. 2015 doi: 10.1016/j.jneumeth.2015.01.018. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Hrabetova S. Brain Extracellular Space: The Final Frontier of Neuroscience. Biophysical journal. 2017 doi: 10.1016/j.bpj.2017.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, Pierce RC. Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neuroscience. 2002;22:2916–2925. doi: 10.1523/JNEUROSCI.22-07-02916.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Boston: AcademicPress/Elsevier; 2009. [Google Scholar]
- Pennartz C, Groenewegen H, Lopesdasilva F. The nucleus accumbens as a complex of functionally distinct neuronal ensembles: An integration of behavioural, electrophysiological and anatomical data. Progress in neurobiology. 1994;42:719–761. doi: 10.1016/0301-0082(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Peters J, Kalivas PW. The group II metabotropic glutamate receptor agonist, LY379268, inhibits both cocaine- and food-seeking behavior in rats. Psychopharmacology. 2006;186:143–149. doi: 10.1007/s00213-006-0372-9. [DOI] [PubMed] [Google Scholar]
- Phillipson OT, Griffiths AC. The topographic order of inputs to the nucleus accumbens in the rat. Neuroscience. 1985;16:275–296. doi: 10.1016/0306-4522(85)90002-8. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Yager LM, Cogan ES, Saunders BT. On the motivational properties of reward cues: Individual differences. Neuropharmacology. 2014;76(Pt B):450–459. doi: 10.1016/j.neuropharm.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanides AJ, Duffy P, Kalivas PW. Glutamatergic and dopaminergic afferents to the prefrontal cortex regulates spatial working memory in rats. Neuroscience. 1999;92:97–106. doi: 10.1016/s0306-4522(98)00747-7. [DOI] [PubMed] [Google Scholar]
- Rutherford EC, Pomerleau F, Huettl P, Stromberg I, Gerhardt GA. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. Journal of neurochemistry. 2007;102:712–722. doi: 10.1111/j.1471-4159.2007.04596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul’skaya NB, Marsden CA. Glutamate levels in the nucleus accumbens in conditioned emotional response. Neuroscience and Behavioral Physiology. 1997:548–551. doi: 10.1007/BF02463899. [DOI] [PubMed] [Google Scholar]
- Saunders BT, Robinson TE. A cocaine cue acts as an incentive stimulus in some but not others: implications for addiction. Biological psychiatry. 2010;67:730–736. doi: 10.1016/j.biopsych.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders BT, Robinson TE. Individual variation in resisting temptation: implications for addiction. Neuroscience and biobehavioral reviews. 2013;37:1955–1975. doi: 10.1016/j.neubiorev.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens ML, Williamson A, Deel ME, Bensalem-Owen M, Davis VA, Slevin J, Pomerleau F, Huettl P, Gerhardt GA. Tonic glutamate in CA1 of aging rats correlates with phasic glutamate dysregulation during seizure. Epilepsia. 2014;55:1817–1825. doi: 10.1111/epi.12797. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Klanker M, de Ridder B, Bowers MS, Joosten RN, Feenstra MG, Bonci A. Reward-predictive cues enhance excitatory synaptic strength onto midbrain dopamine neurons. Science. 2008;321:1690–1692. doi: 10.1126/science.1160873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykova E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev. 2008;88:1277–1340. doi: 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas TC, Grandy DK, Gerhardt GA, Glaser PE. Decreased dopamine D4 receptor expression increases extracellular glutamate and alters its regulation in mouse striatum. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2009;34:436–445. doi: 10.1038/npp.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomie A, Grimes KL, Pohorecky LA. Behavioral characteristics and neurobiological substrates shared by Pavlovian sign-tracking and drug abuse. Brain research reviews. 2008;58:121–135. doi: 10.1016/j.brainresrev.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomie A, Lincks M, Nadarajah SD, Pohorecky LA, Yu L. Pairings of lever and food induce Pavlovian conditioned approach of sign-tracking and goal-tracking in C57BL/6 mice. Behavioural brain research. 2012;226:571–578. doi: 10.1016/j.bbr.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trantham-Davidson H, LaLumiere RT, Reissner KJ, Kalivas PW, Knackstedt LA. Ceftriaxone normalizes nucleus accumbens synaptic transmission, glutamate transport, and export following cocaine self-administration and extinction training. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:12406–12410. doi: 10.1523/JNEUROSCI.1976-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollbrecht PJ, Simmler LD, Blakely RD, Deutch AY. Dopamine denervation of the prefrontal cortex increases expression astrocytic glutamate transporter GLT-1. J Neurochemistry. 2014;130:109–114. doi: 10.1111/jnc.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Kalivas PW. N-acetylcysteine reduces extinction responding and induces enduring reductions in cue- and heroin-induced drug-seeking. Biological psychiatry. 2008;63:338–340. doi: 10.1016/j.biopsych.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]