

Abstract
Background
A comprehensive report on the clinical course of the three major genotypes of the long QT syndrome (LQTS) in a large U.S. patient cohort is lacking.
Methods
Our study consisted of 1,923 U.S. subjects from the Rochester‐based LQTS Registry with genotype‐positive LQT1 (n = 879), LQT2 (n = 807), and LQT3 (n = 237). We evaluated the risk of a first cardiac event (syncope, aborted cardiac arrest, or sudden cardiac death, whichever occurred first) from birth through age 50 years. Cox proportional hazards regression models incorporating clinical covariates were used to assess genotype‐specific risk of cardiac events.
Results
For all three genotypes, the cumulative probability of a first cardiac event increased most markedly during adolescence. Multivariate analysis identified proband status and QTc > 500 ms as predictors of cardiac events in all three genotypes, and males <14 years and females >14 years as predictors of cardiac events in LQT1 and LQT2 only. Beta‐blockers significantly reduced the risk of cardiac events in LQT1 (HR: 0.49, p = .002) and LQT2 patients (HR: 0.48, p = .001). A trend toward beta‐blocker benefit in reducing cardiac events was found in LQT3 females (HR: 0.32, p = .078), but not in LQT3 males (HR: 1.37, p = .611).
Conclusion
Risk factors and outcomes in LQTS patients varied by genotype. In all three genotypes, proband status and prolonged QTc were risk factors for cardiac events. Younger males and older females experienced increased risk in LQT1 and LQT2 only. Beta‐blockers were most effective in reducing cardiac events in LQT1 and LQT2, with a potential benefit in LQT3 females.
Keywords: Long QT syndrome, outcomes, risk factors
1. INTRODUCTION
The long QT syndrome was first described by Jervell and Lange‐Nielsen in 1957 and involved family members with congenital deafness, QT prolongation on the ECG, and sudden death (Jervell & Lange‐Nielsen, 1957). In 1958, Levine and Woodworth reported a similar family (Levine & Woodworth, 1958). A few years later, QT prolongation in children with syncope and sudden death but without deafness was reported (Romano, Gemme, & Pongiglione, 1963; Ward, 1964). Several isolated case reports of LQTS without deafness were published in the next few years.
In 1968, the University of Rochester cardiology and neurosurgical groups developed the first effective treatment for refractory long QT syndrome (LQTS) with the introduction of left cervicothoracic sympathetic ganglionectomy to prevent recurrent ventricular tachycardia and ventricular fibrillation in a 39‐year‐old female with marked QTc prolongation. After a 3‐year asymptomatic follow‐up following the sympathectomy, the clinical results in this individual patient were reported (Moss & McDonald, 1971). The patient is still alive and doing well at age 85 years. Following the publication in 1971 (Moss & McDonald, 1971), the University of Rochester received numerous referrals of patients with LQTS, and in 1975 a Rochester‐based LQTS Registry was established. With the introduction of beta‐blockers for the treatment of LQTS later in the 1970s, the LQTS Registry expanded and involved patient referrals from more centers in the United States as well as from clinical centers in Europe (Moss, Schwartz, Crampton, Locati, & Carleen, 1985; Moss et al., 1991).
The published clinical literature on LQTS expanded during the next 20 years, (Ackerman, 1998; Priori et al., 1997; Schwartz, Moss, Vincent, & Crampton, 1993) and in the early 1990s the genetic mutations associated with several forms of LQTS were identified (Keating et al., 1991; Wang et al., 1996). Subsequently, clinical, genetic, and basic science aspects of LQTS expanded dramatically with increasing focus on the molecular genetics of this disorder (Moss et al., 2007; Shimizu et al., 2009; Wilde et al., 2016; Zareba et al., 1998). During this period of time, our Rochester‐based LQTS Registry involving exclusively U.S. patients progressively enrolled almost 2,000 genetically confirmed LQT1, LQT2, and LQT3 patients who have been followed on a regular basis for their clinical course, their cardiac events, and for their time‐dependent therapies related to beta‐blockers, pacemakers, implantable defibrillators, and sympathectomy that were prescribed. The findings from this updated LQTS Registry are the subject of this report.
2. METHODS
2.1. Study population
Our Rochester‐based LQTS Registry involving exclusively U.S. subjects progressively enrolled 879 genetically confirmed LQT1 patients (due to mutational reduction of function in the KCNQ1 channel gene that encodes for the IKs current), 807 genetically confirmed LQT2 patients (due to mutational reduction of function in the KCNH2 channel gene that encodes for the IKr current), and 237 genetically confirmed LQT3 patients (due to mutational gain of function in the SCN5A alpha subunit of the Na channel gene that encodes for the INa current). Patients with more than one LQTS mutation and those having the Jervell and Lange‐Nielsen syndrome were not included in this study. Only a limited number of patients were tested for LQT4‐LQT14 genotypes; they make up only a very small fraction of the LQTS syndrome in the Registry, and patients with these mutations were also excluded from this study. Patients were enrolled in the Registry from birth through age 50 years when they were clinically identified with LQTS, with genotype determined at experienced academic laboratories or from professional commercial laboratories. This research study was approved by the University of Rochester Medical Center Research Subject Review Board, and all subjects gave written informed consent.
Once a patient with a positive LQT1, LQT2, or LQT3 genotype was enrolled in the Registry, the clinical history dating back to birth was obtained with specific information on date and type of cardiac events (syncope, aborted cardiac arrest, or sudden cardiac death) and on therapy rendered including beta‐blockers, and other relevant drug therapies involving pacemakers, sympathectomy, and implanted cardioverter defibrillators. After formal enrollment in the LQTS Registry, similar information was also collected every 2 years on all the patients in the Registry, with a few patients having incomplete clinical data. If a family member had died suddenly up to age 50 who was not initially in the LQTS Registry, the patient was included in this study if the pre‐ or postmortem testing was positive for LQT1, LQT2, or LQT3 genotype.
2.2. ECG parameters
The first recorded ECG was used to determine the heart rate, the QT interval, and the Bazett‐corrected QTc interval. For infants who survived the first year of life, we used ECGs during subsequent years, usually beginning at age 3–5 years of age for baseline QT measurements.
2.3. Cardiac events
Cardiac events were defined as syncope, aborted cardiac arrest (requiring defibrillation), or sudden cardiac death. Cardiac events were identified by history before initiation of any therapy and after start of relevant LQTS‐related therapies.
To limit the potential confounding effect of coronary artery disease on the outcome of studied patients, only cardiac events occurring until age 50 years were considered. We also analyzed the frequency by genotype of recurrent cardiac events including atrial fibrillation and syncope. An independent end point committee categorized all identified cardiac events in the LQTS registry.
2.4. Statistical analysis
The clinical characteristics of study patients were compared by genotype using the chi‐square test for categorical variables and the nonparametric Wilcoxon rank‐sum test for continuous variables.
The cumulative probability of first cardiac event stratified by genotype was estimated using the Kaplan–Meier method. Stratified Cox multivariable proportional hazards regression models were used to estimate hazard ratios of the standard risk factors potentially associated with time to first cardiac event within each genotype.
All models were stratified by the decade in which study patients were born to account for changes in the baseline hazard function for different calendar time periods. To account for the lack of proportionality of the sex covariate, the models estimated separate hazard ratios for sex before and after 14 years of age. Beta‐blocker use was modeled as a time‐dependent variable in the regression models.
The statistical software used for the analyses involved SAS version 9.4 (SAS Institute Inc., Cary, NC, USA). All tests were two‐sided, and a p‐value <.05 was taken as statistically significant.
3. RESULTS
3.1. Study population
The clinical characteristics of the study population involving the LQT1, LQT2, and LQT3 genotypes are presented in Table 1. LQT1 made up 46% (879/1,923), LQT2 42% (807/1,923), and LQT3 12% (237/1,923) of the study population. There were many similarities in the clinical makeup of the three genotypes including age at enrollment into the Registry, median duration of follow‐up, and QTc duration. However, there were more beta‐blocker prescriptions in LQT1 and LQT2 patients, and more ICDs for treatment of LQT3 patients.
Table 1.
Clinical characteristics of the study population
| Clinical characteristics | LQT1 | LQT2 | LQT3 | p‐value |
|---|---|---|---|---|
| # of patients | 879 | 807 | 237 | |
| Male, # (%) | 378 (43) | 362 (45) | 99 (42) | .617 |
| Proband # (%) | 136 (15) | 163 (20) | 33 (14) | .013 |
| Age at 1st ECG, years | 26 ± 21 | 25 ± 21 | 26 ± 20 | .727 |
| Median duration of follow‐up, years | 40.6 ± 15.8 | 39.0 ± 16.1 | 40.1 ± 16.4 | .749 |
| ECG, mean ± SD | ||||
| RR, ms | 831 ± 216 | 820 ± 247 | 840 ± 248 | .625 |
| QRS, ms | 80 ± 15 | 81 ± 16 | 82 ± 14 | .043 |
| QTc, ms | 480 ± 48 | 480 ± 53 | 474 ± 54 | .164 |
| Treatment, # (%) | ||||
| Beta‐blocker use during follow‐up | 478 (54) | 518 (64) | 124 (52) | <.001 |
| LCSD during follow‐up | 8 (1) | 14 (2) | 1 (0) | .209 |
| Pacemaker during follow‐up | 20 (2) | 64 (8) | 14 (6) | <.001 |
| ICD during follow‐up | 95 (11) | 171 (21) | 67 (28) | <.001 |
| Syncope: pts. with 1 or more episodes, # (%) | 318 (36) | 299 (37) | 54 (23) | <.001 |
| Cardiac events, # (%) | ||||
| ACA during follow‐up | 26 (3) | 40 (5) | 13 (5) | .062 |
| Sudden cardiac death during follow‐up | 19 (2) | 28 (3) | 15 (6) | .005 |
| Appropriate ICD shock during follow‐up | 13 (1) | 31 (4) | 8 (3) | .009 |
ACA, aborted cardiac arrest; AF, atrial fibrillation; ICD, implantable cardioverter defibrillator; LCSD, left cardiac sympathetic denervation.
3.2. Kaplan–Meier graphs of cardiac events
The time dependence of a first cardiac event (syncope, aborted cardiac arrest, or sudden cardiac death), aborted cardiac arrest or sudden death, and sudden cardiac death by genotype are presented in Figure 1a–c.
Figure 1.
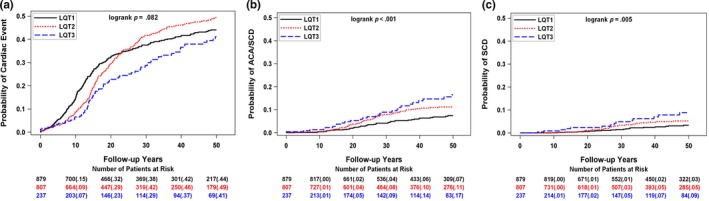
Frequency of (a) first cardiac event (syncope, aborted cardiac arrest, or sudden cardiac death), (b) first aborted cardiac arrest or sudden cardiac death (ACA/SCD), and (c) sudden cardiac death (SCD) by genotype
In each genotype, the rate of increase in cardiac events was most marked during adolescence, but the distribution among the genotypes was variable depending on the end point. The cumulative probability of a first cardiac event was initially greatest for LQT1, but highest for LQT2 patients in the latter years. The probability of aborted cardiac arrest or sudden cardiac death was somewhat more frequent in LQT2 and LQT3 relative to LQT1 patients.
The probability of a first cardiac event for three levels of QTc (<480, 480–500, and >500 ms) for each genotype is presented in Figure 2a–c. In LQT1 and LQT2, QTc > 500 ms is associated with a higher cumulative frequency of cardiac events, but no QTc dominance was observed in LQT3 for these levels of QTc. Similar findings were revealed for aborted cardiac arrest or sudden cardiac death (data not shown).
Figure 2.
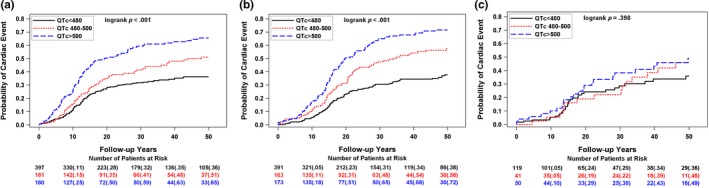
Frequency of a first cardiac event for three levels of QTc (<480, 480–500, and >500) for (a) LQT1, (b) LQT2, (c) LQT3 patients
The cumulative probability of a first cardiac event by gender for each genotype is presented in Figure 3a–c. Females had significantly more frequent events than males in LQT2 and LQT3. A similar pattern was evident when the end point was limited to aborted cardiac arrest or sudden cardiac death, whichever came first (data not shown).
Figure 3.
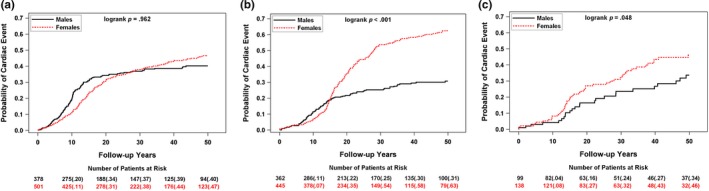
Frequency of a first cardiac event by gender for (a) LQT1, (b) LQT2, (c) LQT3 patients
3.3. Risk factors for cardiac events by LQTS genotype
The relevant factors together with hazard ratios associated with first cardiac events for syncope, aborted cardiac arrest, or sudden cardiac death, whichever came first, by genotype are presented in Table 2. In multivariable analyses, probands and subjects with QTc > 500 ms had significantly greater risk of cardiac events for all three genotypes. Young males and older females were also at significant risk for cardiac events in LQT1 and LQT2, but not for LQT3. Beta‐blockers reduced the risk for cardiac events in LQT1 and LQT2, but not in LQT3. For female LQT3 patients, however, beta‐blockers were associated with a trend toward reduction in cardiac events, an effect not found in male LQT3 subjects.
Table 2.
Hazard ratios for factors associated with a first cardiac event (syncope, aborted cardiac arrest, or sudden cardiac death)
| Risk factor | LQT1: Hazard ratio 95% confidence int. (p‐value) | LQT2: Hazard ratio 95% confidence int. (p‐value) | LQT3: Hazard ratio 95% confidence int. (p‐value) |
|---|---|---|---|
| Proband vs. family member |
3.21 2.47–4.16 (p < .001) |
2.48 1.91–3.22 (p < .001) |
2.83 1.62–4.95 (p < .001) |
| QTc > 500 ms vs. <500 ms |
1.90 1.44–2.50 (p < .001) |
2.02 1.50–2.71 (p < .001) |
1.93 1.05–3.52 (p = .033) |
| Male <age 14 years |
2.15 1.59–2.90 (p < .001) |
1.69 1.15–2.48 (p = .007) |
0.54 0.23–1.28 (p = .161) |
| Female ≥age 14 years |
1.76 1.08–2.84 (p = .023) |
3.15 2.14–4.64 (p < .001) |
1.68 0.85–3.31 (p = .137) |
| Beta‐blockers vs. no beta‐blockers |
0.49 0.31–0.76 (p = .002) |
0.48 0.31–0.74 (p = .001) |
0.63a 0.30–1.32 (p = .219) |
Hazard ratios were adjusted for decade of birth.
Number of cardiac events utilized in each genotype analysis: LQT1 = 303; LQT2 = 297; LQT3 = 69. Significant hazard ratios are highlighted in bold.
LQT3 gender‐specific effects were: females HR = 0.32, p = .078, males HR = 1.37, p = .611.
4. DISCUSSION
In the present study, we report the clinical course of the three major genotypes of LQTS syndrome from the U.S. Rochester‐based LQTS Registry, the largest database with genotyped LQTS probands and family members who were followed on a regular basis over the course of several decades. Risk factors and outcomes in LQTS patients varied by genotype, and probands were at significantly greater risk in all three genotypes. In LQT1 and LQT2 patients, prolonged QTc, younger males, and older females experienced an increased risk for cardiac events. Beta‐blockers were most effective in reducing cardiac events in LQT1 and LQT2. Among patients with LQT3, who made up only 13% of the LQTS study population, beta‐blockers were potentially beneficial for females but not males.
Our findings have several important clinical implications. Patients with LQT1 may need greater attention and advanced treatment early on during their lifetime, since they exhibited an increased probability of cardiac events during the first 20 years of life. On the contrary, LQT2 patients exhibited a high probability of cardiac events primarily following adolescence, predominantly in females. Such differences in the development of risk over time can be useful in clinically managing LQTS patients.
In LQT1, LQT2, and LQT3 genotypes, proband status consistently predicted increased cardiac events and aborted cardiac arrest/sudden cardiac death, likely due to the fact that the clinical phenotype is most striking in probands. The presence of QTc > 500 ms also predicted cardiac events in all three genotypes.
In LQT1 and LQT2 patients, there were a number of additional simple clinical predictors identifying risk for cardiac events, including younger males and older females. A greater risk of cardiac events in older females has been shown in prior studies (Locati et al., 1998; Zareba et al., 2003). History of syncope was related to increased risk of aborted cardiac arrest/syncope in LQT1 and LQT2 patients, and serves as an added marker of poor outcomes for these two genotypes.
Beta‐blocker treatment modalities are basic therapy in LQT1 and LQT2 patients (Abu‐Zeitone, Peterson, Polonsky, McNitt, & Moss, 2014; Priori et al., 2004), and this study provides further evidence from a large aggregate database on LQTS patients that beta‐blocker treatment reduces the risk of cardiac events with hazard ratios of 0.49 and 0.48 in LQT1 and LQT2, and dramatically reduced the risk of aborted cardiac arrest/sudden cardiac death even further in these genotypes. In our study, this beneficial effect of beta‐blockers was not evident in the total cohort of LQT3 patients. Similar to the Wilde analysis of 391 LQT3 patients, (Wilde et al., 2016) a trend toward reduction of cardiac events with beta‐blockers occurred in females, but not in males with LQT3. In LQT3 males, Wilde et al. showed a HR of 0.51 with a p‐value .308 for beta‐blocker effect to reduce ACA/SCD events, while in our study the HR was 1.37 for LQT3 males, with a p‐value of .611. Both studies however might be underpowered to detect the effects of beta‐blockers on survival in LQT3, due to the small number of patients and events. Therefore, further studies are warranted investigating beta‐blocker therapy in LQT3 patients, especially males.
What are the indications for ICD therapy in LQTS? As stated in the 2012 ACCF/AHA/HRS Guidelines for Device‐based Therapy, (Epstein et al., 2013) ICD implantation is recommended for selected LQTS patients with recurrent syncope despite beta‐blocker therapy and may be considered when there is a strong family history of SCD or when compliance or intolerance to drugs is a concern (Chatrath, Porter, & Ackerman, 2002; Goel, Berger, Pelech, & Dhala, 2004; Monnig et al., 2005; Zareba et al., 2003). In the present study, ICDs were implanted in 10% of LQT1, 21% of LQT2, and 26% of LQT3 patients. While these percentages seem high, and there is a concern about ICD overutilization in LQTS patients with potentially high frequency of adverse events with ICD therapy, (Schwartz et al., 2010) those at high risk for SCD should nevertheless be protected by the ICD. Furthermore, sympathectomy remains a therapeutic option for LQTS patients with refractory arrhythmia events.
This LQTS study has the inherent limitations of a registry database with lack of randomization. Randomized studies in rare genetic diseases are often not feasible. Since this is an observational study, there might have been unmeasured confounders influencing our study. However, we adjusted the models for major known risk factors. Specific genetic mutations within each genotype were not utilized for risk stratification or as the basis for therapy in this study since the field continues to change rapidly with new LQTS mutations being reported frequently with limited follow‐up.
5. CONCLUSIONS
In conclusion, we report from the Rochester‐based U.S. LQTS Registry a varying clinical course across the three major LQTS genotypes, and present specific risk models applicable to LQT1, LQT2, and LQT3 patients to predict major cardiac events including syncope, aborted cardiac arrest, and sudden cardiac death. Our report should help clinicians improve risk stratification of their LQTS patients and optimize treatment modalities that can improve outcome in high‐risk patients.
DISCLOSURES
All authors have nothing else to disclose.
ACKNOWLEDGEMENTS
The authors would like to dedicate this work to the late Dr. Arthur J. Moss, the founder of the Rochester‐based LQTS‐Registry. His insightful, dedicated work on LQTS syndrome led to many major scientific discoveries and development of treatment modalities, saving numerous lives with LQTS.
Kutyifa V, Daimee UA, McNitt S, et al. Clinical aspects of the three major genetic forms of long QT syndrome (LQT1, LQT2, LQT3). Ann Noninvasive Electrocardiol. 2018;23:e12537 10.1111/anec.12537
Funding information
Supported in‐part by research grants HL‐33853 (AJM), HL‐51618 (AJM), and HL‐123483 (AJM) from the National Institutes of Health, Bethesda, MD, USA and by a research grant from Gilead Sciences, Inc., Foster City, CA, USA (AJM).
[Correction Statement added on 15th March 2018, after first online publication. Initial “A” has been added to the author “Usama Daimee” in the author byline.]
REFERENCES
- Abu‐Zeitone, A. , Peterson, D. R. , Polonsky, B. , McNitt, S. , & Moss, A. J. (2014). Efficacy of different beta‐blockers in the treatment of long QT syndrome. Journal of the American College of Cardiology, 64, 1352–1358. 10.1016/j.jacc.2014.05.068 [DOI] [PubMed] [Google Scholar]
- Ackerman, M. J. (1998). The long QT syndrome. Pediatrics in Review, 19, 232–238. 10.1542/pir.19-7-232 [DOI] [PubMed] [Google Scholar]
- Chatrath, R. , Porter, C. B. , & Ackerman, M. J. (2002). Role of transvenous implantable cardioverter‐defibrillators in preventing sudden cardiac death in children, adolescents, and young adults. Mayo Clinic Proceedings, 77, 226–231. 10.4065/77.3.226 [DOI] [PubMed] [Google Scholar]
- Goel, A. K. , Berger, S. , Pelech, A. , & Dhala, A. (2004). Implantable cardioverter defibrillator therapy in children with long QT syndrome. Pediatric Cardiology, 25, 370–378. [DOI] [PubMed] [Google Scholar]
- Epstein, A. E. DiMarco, J. P. , Ellenbogen, K. A. , Estes, N. A. , Freedman, R. A. , Gettes, L. S. … Heart Rhythm Society (2013). 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device‐based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Journal of the American College of Cardiology, 61, e6–75. [DOI] [PubMed] [Google Scholar]
- Jervell, A. , & Lange‐Nielsen, F. (1957). Congenital deal‐mutism, functional heart disease with prolongation of the Q‐T interval and sudden death. American Heart Journal, 54, 59–68. 10.1016/0002-8703(57)90079-0 [DOI] [PubMed] [Google Scholar]
- Keating, M. , Dunn, C. , Atkinson, D. , Timothy, K. , Vincent, G. M. , & Leppert, M. (1991). Consistent linkage of the long‐QT syndrome to the Harvey ras‐1 locus on chromosome 11. American Journal of Human Genetics, 49, 1335–1339. [PMC free article] [PubMed] [Google Scholar]
- Levine, S. A. , & Woodworth, C. R. (1958). Congenital deaf‐mutism, prolonged QT interval, syncopal attacks and sudden death. New England Journal of Medicine, 259, 412–417. 10.1056/NEJM195808282590902 [DOI] [PubMed] [Google Scholar]
- Locati, E. H. , Zareba, W. , Moss, A. J. , Schwartz, P. J. , Vincent, G. M. , Lehmann, M. H. , … Andrews, M. (1998). Age‐ and sex‐related differences in clinical manifestations in patients with congenital long‐QT syndrome: Findings from the International LQTS Registry. Circulation, 97, 2237–2244. 10.1161/01.CIR.97.22.2237 [DOI] [PubMed] [Google Scholar]
- Monnig, G. , Kobe, J. , Loher, A. , Eckardt, L. , Wedekind, H. , Scheld, H. H. , … Böcker, D. (2005). Implantable cardioverter‐defibrillator therapy in patients with congenital long‐QT syndrome: A long‐term follow‐up. Heart Rhythm, 2, 497–504. 10.1016/j.hrthm.2005.02.008 [DOI] [PubMed] [Google Scholar]
- Moss, A. J. , & McDonald, J. (1971). Unilateral cervicothoracic sympathetic ganglionectomy for the treatment of long QT interval syndrome. New England Journal of Medicine, 285, 903–904. 10.1056/NEJM197110142851607 [DOI] [PubMed] [Google Scholar]
- Moss, A. J. , Schwartz, P. J. , Crampton, R. S. , Locati, E. , & Carleen, E. (1985). The long QT syndrome: A prospective international study. Circulation, 71, 17–21. 10.1161/01.CIR.71.1.17 [DOI] [PubMed] [Google Scholar]
- Moss, A. J. , Schwartz, P. J. , Crampton, R. S. , Tzivoni, D. , Locati, E. H. , MacCluer, J. , … Garson, A. (1991). The long QT syndrome. Prospective longitudinal study of 328 families. Circulation, 84, 1136–1144. 10.1161/01.CIR.84.3.1136 [DOI] [PubMed] [Google Scholar]
- Moss, A. J. , Shimizu, W. , Wilde, A. A. , Towbin, J. A. , Zareba, W. , Robinson, J. L. , … Hofman, N. (2007). Clinical aspects of type‐1 long‐QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation, 115, 2481–2489. 10.1161/CIRCULATIONAHA.106.665406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori, S. G. , Mortara, D. W. , Napolitano, C. , Diehl, L. , Paganini, V. , Cantù, F. , … Schwartz, P. J. (1997). Evaluation of the spatial aspects of T‐wave complexity in the long‐QT syndrome. Circulation, 96, 3006–3012. 10.1161/01.CIR.96.9.3006 [DOI] [PubMed] [Google Scholar]
- Priori, S. G. , Napolitano, C. , Schwartz, P. J. , Grillo, M. , Bloise, R. , Ronchetti, E. , … Nastoli, J. (2004). Association of long QT syndrome loci and cardiac events among patients treated with beta‐blockers. JAMA, 292, 1341–1344. 10.1001/jama.292.11.1341 [DOI] [PubMed] [Google Scholar]
- Romano, C. , Gemme, G. , & Pongiglione, R. (1963). [Rare cardiac arrythmias of the pediatric age. II. Syncopal attacks due to paroxysmal ventricular fibrillation. (Presentation of 1st case in Italian pediatric literature)]. La Clinica Pediatrica, 45, 656–683. [PubMed] [Google Scholar]
- Schwartz, P. J. , Moss, A. J. , Vincent, G. M. , & Crampton, R. S. (1993). Diagnostic criteria for the long QT syndrome. An update. Circulation, 88, 782–784. 10.1161/01.CIR.88.2.782 [DOI] [PubMed] [Google Scholar]
- Schwartz, P. J. , Spazzolini, C. , Priori, S. G. , Crotti, L. , Vicentini, A. , Landolina, M. , … Toivonen, L. (2010). Who are the long‐QT syndrome patients who receive an implantable cardioverter‐defibrillator and what happens to them? Data from the European long‐QT syndrome implantable cardioverter‐defibrillator (LQTS ICD) registry. Circulation, 122, 1272–1282. 10.1161/CIRCULATIONAHA.110.950147 [DOI] [PubMed] [Google Scholar]
- Shimizu, W. , Moss, A. J. , Wilde, A. A. , Towbin, J. A. , Ackerman, M. J. , January, C. T. , … Vincent, G. M. (2009). Genotype‐phenotype aspects of type 2 long QT syndrome. Journal of the American College of Cardiology, 54, 2052–2062. 10.1016/j.jacc.2009.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Curran, M. E. , Splawski, I. , Burn, T. C. , Millholland, J. M. , VanRaay, T. J. , … Schwartz, P. J. (1996). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nature Genetics, 12, 17–23. 10.1038/ng0196-17 [DOI] [PubMed] [Google Scholar]
- Ward, O. C. (1964). A new familial cardiac syndrome in children. Journal of the Irish Medical Association, 54, 103–106. [PubMed] [Google Scholar]
- Wilde, A. A. , Moss, A. J. , Kaufman, E. S. , Shimizu, W. , Peterson, D. R. , Benhorin, J. , … Ackerman, M. J. (2016). Clinical aspects of type 3 long‐QT syndrome: An International Multicenter Study. Circulation, 134, 872–882. 10.1161/CIRCULATIONAHA.116.021823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zareba, W. , Moss, A. J. , Daubert, J. P. , Hall, W. J. , Robinson, J. L. , & Andrews, M. (2003). Implantable cardioverter defibrillator in high‐risk long QT syndrome patients. Journal of Cardiovascular Electrophysiology, 14, 337–341. 10.1046/j.1540-8167.2003.02545.x [DOI] [PubMed] [Google Scholar]
- Zareba, W. , Moss, A. J. , Locati, E. H. , Lehmann, M. H. , Peterson, D. R. , Hall, W. J. , … Towbin, J. A. (2003). Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. Journal of the American College of Cardiology, 42, 103–109. 10.1016/S0735-1097(03)00554-0 [DOI] [PubMed] [Google Scholar]
- Zareba, W. , Moss, A. J. , Schwartz, P. J. , Vincent, G. M. , Robinson, J. L. , Priori, S. G. , … Lehmann, M. H. (1998). Influence of genotype on the clinical course of the long‐QT syndrome. New England Journal of Medicine, 339, 960–965. 10.1056/NEJM199810013391404 [DOI] [PubMed] [Google Scholar]