

Abstract
Allostery, which is regulation from distant sites, plays a major role in biology. While traditional allostery is described in terms of conformational change upon ligand binding as an underlying principle, it is possible to have allosteric regulations without significant conformational change through modulating the conformational dynamics by altering the local effective elastic modulus of the protein upon ligand binding. Pin1 utilizes this dynamic allostery to regulate its function. It is a modular protein containing a WW domain and a larger peptidyl prolyl isomerase domain (PPIase) that isomerizes phospho-serine/threonine-proline (pS/TP) motifs, The WW domain serves as a docking module, whereas catalysis solely takes place within the PPIase. Here, we analyze the change in dynamic flexibility profile of PPIase domain upon ligand binding to the WW domain. Substrate binding to the WW domain induces formation of new rigid hinge site around the interface of two domain and loosens flexibility of a rigid site existing in the Apo form around the catalytic site. This hinge shift mechanism enhances the dynamic coupling of the catalytic positions with the PPIase domain, where the rest of the domain can cooperatively respond to the local conformational changes around the catalytic site, leading to increase in catalytic efficiency.
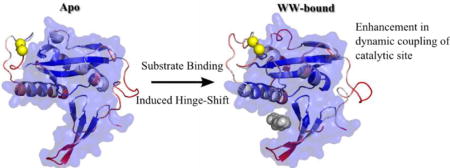
TOC image
Introduction
The biological function of any given enzyme is directly associated with its catalytic/active site. While changes at these sites, such as binding of a substrate or mutations, will directly affect the function of the protein, it is also possible to indirectly affect protein active sites through a process known as allostery. A classical definition of allostery involves a ligand binding event to a receptor distal to the active site yet having an effect on the active site, such as altering the active site’s binding affinity or overall rate of enzymatic activity1,2. This can come as a result of a change in the global conformation of the protein or local changes at the active site upon binding. However, conformational changes are not necessary for the observance of allosteric effects. More recently, the concept of dynamic allostery has been proposed in which allosteric communication proceeds even in the absence of structural changes3–8. It follows that a binding event at a site distal to an active site can have an allosteric effect on the active site in the absence of structural change by altering the protein’s internal communication network, a phenomenon investigated further through computational studies9–13.
The protein Pin1 provides an excellent test case to examine potential mechanisms underlying allostery without a large global change in conformation. Particularly the allosteric regulations of Pin1 have garnered interest within the scientific community as its up-regulation and down-regulation have been associated with various cancers and Alzheimer’s disease, respectively14–18. Pin1 is a modular protein with two flexibly linked binding domains: (i) the enzymatically active PPIase domain and (ii) an inactive WW domain, distantly located from the PPIase active site19,20(Figure 1). While both domains bind to substrates with phospho-Ser/Thr-Pro (pS/T-P) motifs, only the PPIase domain catalyzes the cis–trans isomerization of the substrates. Experimental and computational studies have shown that PPIase activity is enhanced when the pS/T-P substrate binds to the WW domain21,22. Furthermore, experimental studies comparing the full length Pin1 to the isolated PPIase domain have shown that the binding affinity and catalytic rate of the PPIase domain differs when the WW domain is present19–25. NMR studies have shown substrate binding induces changes in side chain flexibility, suggesting the role of dynamic allosteric regulation in Pin1 through transient contact formation and dynamic communications22,22,26–29. While PPIase binding events lead to stiffening of conserved hydrophobic residues between the PPIase binding sites and the PPIase active sites23,30, substrate binding to the WW domain changes the intra/interdomain mobility, thereby altering substrate activity in the distal PPIase domain catalytic site21–23. Furthermore, previous work by the Zhou group has proposed the existence of two allosteric pathways of communication between the WW and PPIase domains, one of which is dormant in the apo (unbound) structure and becomes activated when the second pathway is formed in the FFpSPR (WW-bound) structure21. This detailed MD analysis provided mechanistic insights of allosteric regulation of PPIase activity upon WW domain binding. WW domain binding events induce quenching of fast local motions and lead to an enhancement in communication pathways between the two domains, and generate a decrease in flexibility of three PPIase catalytic loops, thus decreasing the entropic cost of substrate binding in the PPIase domain21.
Figure 1.
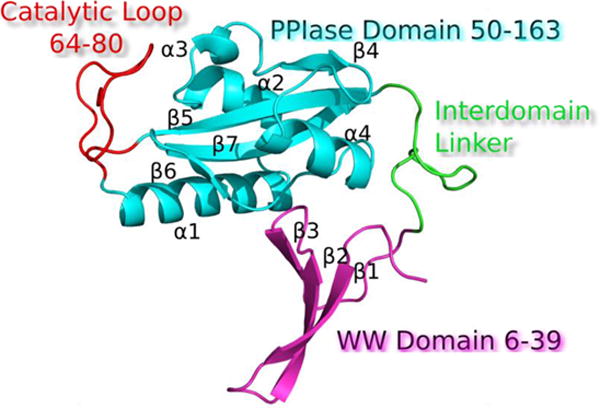
Ribbon representation of Pin1 modular organization (PDB: 1PIN) N-terminal WW domain (magenta), flexible linker (green), and a C-terminal PPIase domain (cyan). Functional loops are annotated in red.
Here we attempt to further elucidate the mechanisms of allosteric communication in Pin1. Specifically, how active site interactions and active site communication with the rest of the structure changes upon binding of a substrate to the WW domain and whether these changes differ from changes upon PPIase binding. First, we analyzed the allosteric effects of ligand binding events through changes in flexibility of the structure by implementing a metric known as the dynamic flexibility index (DFI), a site specific metric that measures each position’s resilience to perturbations within the network of interactions in native equilibrium dynamics11,31. These analyses show that substrate binding to the WW domain significantly alters the dynamic flexibility profile of the PPIase domain. A low DFI site, referred to as a hinge, forms at the interstitial region of the two domains central to the structure while another hinge around the catalytic region loosens. This observed hinge shift mechanism may underlie the critical change in dynamics that enhances catalytic efficiency of the PPIase domain. On the other hand, substrate binding to the PPIase domain when the WW domain is unbound does not induce a significant change in the flexibility profile of WW domain, suggesting that there is unidirectional control through WW domain binding in agreement with prior work21,28. Additionally, we investigated changes in the internal network of dynamic communication upon substrate binding to the WW domain by constructing pathways of force propagation from the binding site of the PPIase domain to that of the WW domain using perturbation response scanning (PRS)11. This analysis showed that a WW domain binding event increases the number of these pathways, where most of them travel through the PPIase core to the interdomain interface. We also generated force propagation pathways from the PPIase active site residues to all other residues within the protein to analyze changes in the network of interactions of the active site to the rest of the structure upon substrate binding. We observed that there is a significant increase in pathways from the active site to the PPIase binding domain upon substrate binding to the WW domain. This suggests that the catalytic site becomes more dynamically coupled to the active PPIase domain. Particularly, enhancement in dynamic coupling of catalytic site residues with the distal sites of PPIase domain may increase overall catalytic efficiency since the catalytic reaction of cis-trans isomerization involves the association and disassociation of substrate which utilizes the collective dynamics of the full PPIase domain where distal sites also cooperatively move with the catalytic sites. To further explore this, we analyzed the changes in coupling strength between the PPIase active site, the PPIase binding domain and the WW binding domain using the dynamic coupling index (DCI). In agreement with the pathway analysis, we observed that WW-bound Pin1 exhibits a global increase in dynamic coupling of the active site to PPIase domain binding residues.
Methods
Dynamic Flexibility Index (DFI)
DFI measures the response of a residue upon perturbations to the rest of the protein. The dynamic response profile of a given position within a protein is explored by the Perturbation Response Scanning technique (PRS) that combines equilibrium dynamics with linear response theory11,31. The original PRS approach uses the Elastic Network Model (ENM) to obtain correlated dynamics of positions in native equilibrium32. ENM is a coarse-grained model in which the nodes are represented by Cα atoms11,31,33,34 and the pairwise potential between each atom is given as the potential of a harmonic spring. A small perturbation in the form of a random Brownian kick is applied sequentially to each Cα atom in the elastic network. As a first order approximation, this perturbation mimics the forces exerted by an approaching protein or a ligand in a crowded cellular environment. The perturbations on a single residue result in a cascade of perturbations to all other atoms in the network, inducing a global response. The fluctuation response profile of the positions upon perturbation of a single residue is obtained using linear response theory and given by the equation11,31,
| (1) |
where H is the hessian, a 3N×3N matrix composed of the second order derivatives of the harmonic potential with respect to the components of the position vectors for a chain of length N, giving the position co-variance of all residue pairs in equilibrium. F is the external force vector applied at N residues in the protein and ΔR is the response of the force. The force is applied in all directions at each residue and the magnitude of the response profile is averaged to give an isotropic measure of response.
However, in order to more accurately model changes in all networks of interaction upon ligand binding, we replace the inverse of Hessian with the covariance matrices obtained from molecular dynamics simulations.
| (2) |
Here, G is the covariance matrix containing the dynamic properties of the system. The covariance matrix contains the data for long range interactions, solvation effects and biochemical specificities of all types of interactions. In this work, the covariance matrices of Apo and bound complexes were constructed using previously obtained 100 ns all-atom explicit water molecular dynamics simulations. (See ref 21 for details). Structural models of the Apo Pin1 and all Pin1-ligand complexes were generated as averaged structures from previous equilibrium molecular dynamics simulation data21.
DFI is calculated by applying unit isotropic perturbations to each individual residue, one at a time, and obtaining the residue fluctuation response profile of each position upon perturbing a specific position using equation 2, repeating this process until we obtain the perturbation response matrix that contains the residue response profiles for all positions in a protein.
| (3) |
Where, is the magnitude of response at site ‘i’ due to the perturbation at site ‘j’.
DFI of a position ‘i’ is defined as the total fluctuation response of that position normalized with the net displacement of the entire protein when all the residues are perturbed, i.e.
| (4) |
Thus, a higher DFI score of a residue position ‘i’ implies a more flexible site and a low score implies a rigid site with lower response to perturbations in the protein.
Dynamic Coupling Index (DCI)
We also have extended our DFI approach to identify dynamic coupling between any given residue and functionally critical residues such us binding sites or catalytic sites through computing a new metric called the dynamic coupling index (DCI). The DCI metric can identify sites that are distal to functional sites but impact active site dynamics through dynamic allosteric coupling35–37. These distal sites, called dynamic allosteric Residue coupling (DARC) spots, play vital roles in function. Our previous analysis shows that genetic disease mutations observed in human enzymes that leads a change in function are usually observed at DARC spots36,37.
As defined, DCI is the ratio of the sum of the mean square fluctuation response of the residue ‘i’ upon functional site perturbations (i.e., catalytic residues) to the response of residue ‘i’ upon perturbations on all residues,
| (5) |
Where |∆Rj|i is the response fluctuation profile of residue ‘i’ upon perturbation of residue ‘j’. The numerator is the average mean square fluctuation response obtained over the perturbation of the functionally critical residues Nfunctional and the denominator is the average mean square fluctuation response over all residues.
Force Propagation Pathways through Perturbation Response Analysis
Through PRS we measure the direction and magnitude of a response upon exerting a random Brownian kick to one position11. This can be utilized to create force propagation pathways used to establish networks of communication internal to a given protein. Here, a residue ‘i’ is perturbed by a unit force F averaged over multiple directions to create an isotropic perturbation. The displacement response vector ri of the resultant fluctuations of both ‘i’ and nearby residues ‘j’ (within 10 angstroms of Cα Euclidean distance) with a sequence separation of three or greater (j i+3) is then recorded. Residues j meeting these criteria are considered linked to residue ‘i’ if they respond with a perturbation response directionality of 0.98 or greater to that of residue ‘i’, as evaluated by the cosine of the angle between response vectors. The cutoff of 0.98 ensures almost completely concerted motion while allowing for a small amount of deviation, where the same relative behavior in shortest distance pathway analysis was observed for cutoffs of 0.95 to 0.98 in 0.01 increments.
| (6) |
In order to efficiently enumerate through all possible pathways, Dijkstra’s algorithm is applied and the shortest pathway that connects beginning and ending is ensured38.
Results and Discussion
Substrate binding to WW domain induces a hinge shift mechanism
We first explored changes in conformational dynamics of Pin1 in different bound forms compared to the unbound form through our dynamic flexibility index (DFI). Here we constructed DFI profiles for three conformations of Pin1; Apo, WW-FFpSPR bound and PPI-cis bound structures (Figure 1). DFI measures the fluctuation response of a given position to the perturbations that occur at different parts of the protein using linear response theory, capturing the multi-dimensional effects of its conformational landscape when the protein structure is displaced out of equilibrium11,31. Thus, DFI enables us to probe the conformational space of a protein at the residue level and provides a measure for the local vibrational entropy for a given position. It also allows us to identify and map flexible and rigid positions in the structure11,39. DFI has been applied to several protein families to provide mechanistic insights about emergence of new functions40, explain the molecular basis of single-nucleotide polymorphisms associated with genetic-diseases11,36,37,39 and to shed light onto induced cooperativity of butyrylcholinesterase upon mutations distal from the catalytic site mutations35.
Low DFI sites, called hinges, are notably important for coordinating functionally critical conformational dynamics (i.e. like joints in a skeleton). Hinge sites do not exhibit a high fluctuation response, yet transfer the perturbation efficiently to the rest of chain in a controlled fashion. On the other hand, high DFI sites, flexible regions, play critical roles in substrate recognition and ligand binding11,36. Here we explore how substrate binding to the WW domain induces a new DFI profile on distal sites of Pin1, particularly within the PPIase domain. Interestingly we observe a hinge-shift mechanism (i.e. losing hinges and gaining hinges in a distal region) as seen in the evolution of red fluorescence proteins from their ancestors, green fluorescence proteins (GFP)40. The 13 substitutions distributed throughout the 3-D structure of the beta-barrel induce the rigid hinge region near the chromophore to be shifted across the beta-barrel in response to accommodate the required flexibility for photoconversion of the red chromophore. Likewise, binding of a substrate to the WW domain induces formation of a new hinge around D136-R142, near the alpha-4 helix at the domain interface, and losing a hinge around the catalytic loop region S72-Q75. Thus, in agreement with previous computational work,41 our analysis suggests that the hinge shift mechanism upon substrate binding to the WW domain induces new dynamics to accommodate substrate binding and catalysis at the PPIase domain (Figure 2).
Figure 2.
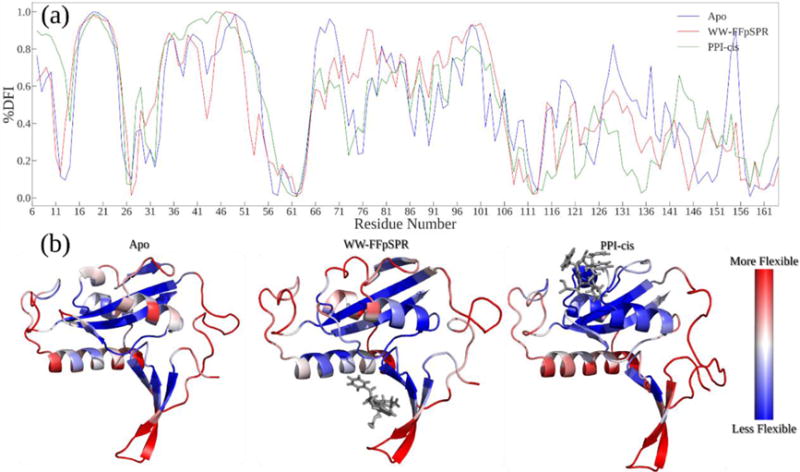
(A) DFI profile comparison of Apo to WW-FFpSPR complex and PPI-cis complex. A hinge shift mechanism, where the hinge near catalytic loop S72-75Q of the apo form becomes flexible upon substrate binding and a hinge forms with residues D136-R142 at the domain interface upon substrate binding to WW domain. Substrate binding event of PPIase domain slightly alters DFI profile of WW domain, only around loop 1 region (B) Structures colored by DFI values from more flexible (red) to less flexible (blue).
Additionally, the WW-bound complex of Pin1 exhibits decreased flexibility compared to the WW-unbound counterparts around the loop region directly surrounding the proline binding pocket of the PPIase domain. In conjunction with the pre-existing hinges present in the beta2-beta3 linker region of the WW-domain, we see that the presence of a ligand bound to the WW domain dampens the allowed degrees of freedom within regions of the alpha-1 helix. Overall, besides the aforementioned hinge-shift, our DFI analysis is in agreement with previous experimental work showing that WW binding produces chemical shift changes in residues between 115-14022,23,30. Furthermore, they also agree with the current proposed mechanism for an increased binding affinity at the PPIase domain as a result of lower conformational entropy in specific regions surrounding the PPIase binding pocket21. Finally, DFI analysis of a cis inhibitor bound to the PPIase domain presents a decrease in the DFI profile of interdomain sites. On the other hand, the DFI profile of the WW domain changes only slightly (a minor decrease of flexibility of loop 1) in accordance with the previous results indicating that changes in dynamics and allosteric communication are unidirectional21,28.
Mapping the Changes in Internal Networking via Force Propagation Pathways reveals enhancement in dynamic networking of catalytic site in WW-bound complexes
Changes in interdomain communication have been long-cited as a means for Pin1 allosteric regulation; namely, how communication from the WW domain to the PPIase domain changes in the presence of a WW-bound ligand. To further understand the important routes of internal communication between these two binding domains we constructed force propagation pathways from signals originating at the PPIase binding domain traveling to the WW domain (Figure 2 shows the shortest distance pathways). Here we perturb one of the residues from the selected starting sites using an isotropic Brownian kick and measure the response of surrounding residues in both magnitude and direction using linear response theory. If a residue follows a direction similar to the perturbed residue (based on the cosine of the angle between residue displacement vectors) and is within 10 angstroms of the perturbed residue, a link is built between these two residues. The newly linked residue is then perturbed and this process is repeated until all possible paths between all starting residues and the target residues have been constructed.
When FFpSPR is bound only to the WW domain, pathways leading from the PPIase binding pocket to the WW domain binding residues become more diverse, with the pathways of WW-FFpSPR bound complex predominately traveling through the interdomain backside and deeper into the WW binding pocket. This again suggests unidirectional communication21,28 and that WW domain binding triggers new dynamics that accommodate an efficient substrate search for the PPIase domain22,42. (Figure 3).
Figure 3.
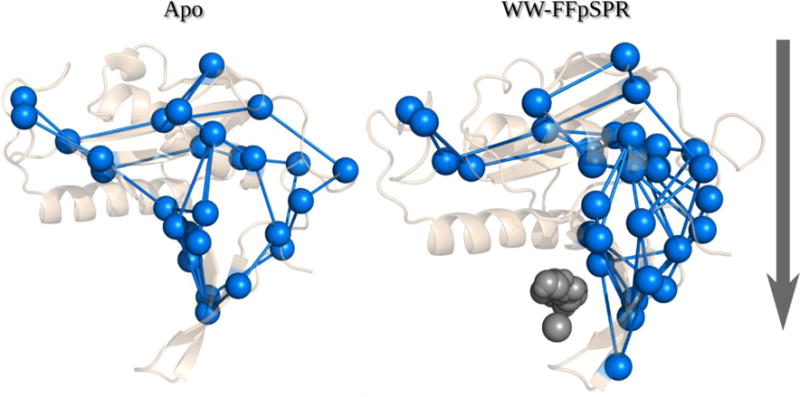
Shortest distance force propagation pathways from signals originating at the PPIase binding domain traveling to the WW domain. The Apo structure exhibits fewer pathways than the WW-FFpSPR structure which tend to pass through the core of the structure and deeper into the WW domain through the interstitial domain backside.
To further explore how a hinge-shift mechanism upon substrate binding to the WW domain affects the catalytic site, we also enumerated all force propagation pathways out from each catalytic site residue to every other residue within the protein for the Apo and WW-FFpSPR complexes. As summarized in Table 1, WW domain binding leads to a drastic increase in the number of pathways from catalytic site residues to the PPIase domain binding pocket., clearly indicating enhanced catalytic site networking.
Table 1.
Statistics for shortest distance pathways originating at the PPIase active site residues out to all other residues within the protein.
| PPIase active residue i.d. | Apo Pin1 | FFpSPR-WW domain bound complex |
|---|---|---|
| K63 | 5 | 33 |
| R68 | 5 | 7 |
| R69 | 6 | 9 |
| M130 | 6 | 28 |
| Total | 22 | 77 |
Substrate binding decreases the dynamic coupling of WW domain binding sites with the PPIase catalytic domain
As noted above, a WW-domain binding event cannot be characterized by a global increase or decrease in structural flexibility but by changes that must be described as site-specific. In the WW-bound case, we found that much of the alpha-1 helix becomes less flexible and also observed formation of new hinges in the WW-bound structure while losing existing hinges found in the Apo structure. Because, hinges, much like joints in a skeleton, are crucial regions for controlling the movement and subsequent communication between connected regions, this hinge shift mechanism upon substrate binding to the WW domain opens up the potential for a new communication pathway between the active site and the rest of the chain through dynamic fluctuations as also suggested by force propagation analysis. To further investigate this, we built a dynamic coupling index (DCI), a measure of dynamic coupling for a given position with a specific group of functionally critical positions through a dynamic network of interactions (see Methods for details) (Figure 3). We first explore how substrate binding impacts the dynamic coupling of Pin1, particularly the PPIase domain with the WW domain binding pocket. DCI analysis evaluates site-specific changes in dynamic coupling to the WW binding sites upon substrate binding to the WW domain using the co-variance matrices of the Apo and WW-FFpSPR forms by perturbing WW domain binding pocket residues. High %DCI indicates the positions which respond more strongly than the average to the perturbations at the WW binding sites. Interestingly, substrate binding to the WW domain does not induce significant changes in dynamic coupling between WW domain binding sites and residues located within the WW domain (Figure 4). On the other hand, we observe a decrease in DCI profiles of the catalytic loop, suggesting that the catalytic loop becomes more independent upon substrate binding to the WW domain, in agreement with a recent NMR analysis suggesting negatively controlled allosteric regulation as a result of WW domain binding reduces the interdomain contact as compared to the Apo state, thus allowing the PPIase domain to freely search for a distinct pS/T-P substrate42.
Figure 4.
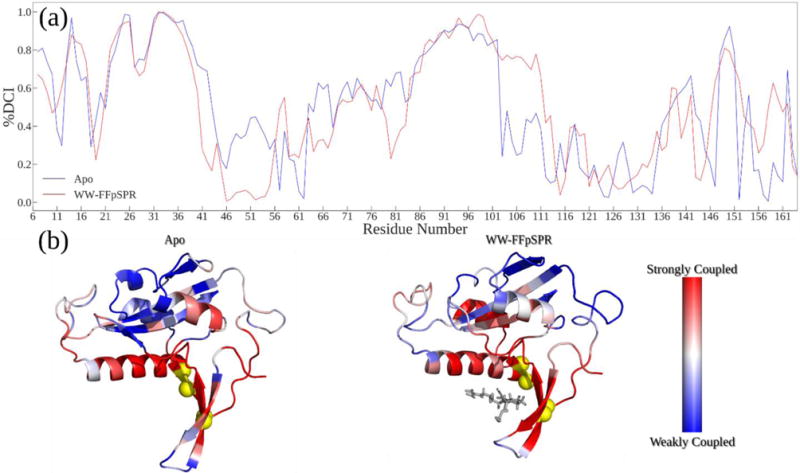
(A) %DCI values (coupling strength) between WW domain binding sites and the rest of the structure. The WW-FFpSPR structure exhibits similar coupling strength to the WW domain binding pocket, but a notable decrease in coupling to catalytic loop regions and the backside interstitial region between the WW and PPIase domains. (B) Structures colored by %DCI values.
WW binding events increase dynamic coupling of the active site with the PPIase domain
We also explore how substrate binding to the WW domain alters dynamic coupling of catalytic residues with the rest of Pin1. Particularly, we investigate whether WW domain binding increases the dynamic coupling between the PPIase binding domain and two very critical catalytic residues R68 and R69, as mutations those site have been shown to lead to complete functional loss42. Strikingly, our results are in complete agreement with the proposed mechanism of Peng et al42 where upon ligand binding to the WW domain, catalytic residues completely lose coupling with the WW domain, becoming free to search for substrates (Figure 5). Moreover, we also see enhancement of dynamic coupling of the catalytic site with the PPIase domain, particularly the PPIase binding pocket and regions surrounding the catalytic loop. This enhanced dynamic coupling could accommodate cooperative, necessary dynamics of the PPIase domain as an entity for substrate binding, catalysis and product release, leading to the increase in the efficiency of catalysis.
Figure 5.
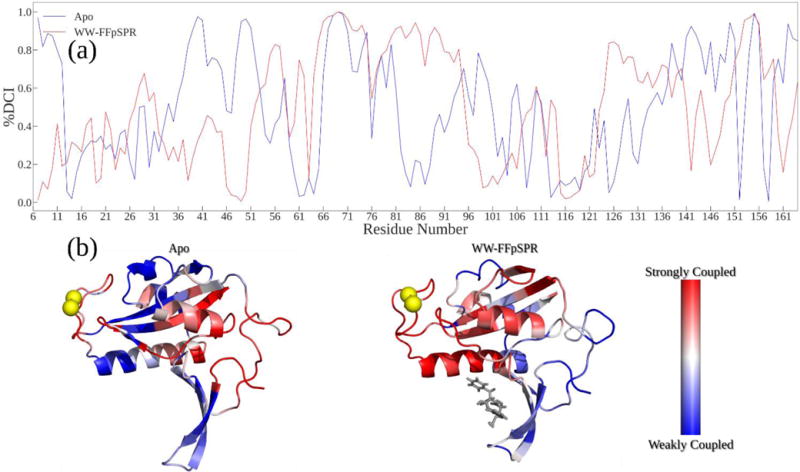
(A) %DCI values (coupling strength) between the active site and the rest of the structure. The WW bound structure shows an increase in this active site coupling with other regions of the PPIase domain, including the catalytic loops and PPIase binding residues. (B) Structures colored by %DCI values indicates active site residues are more strongly coupled to the interstitial alpha helix and the loop around the proline binding pocket.
Conclusions
Through NMR and all atom MD simulations of Pin1, it has been shown that substrate binding to the WW domain regulates PPIase activity without significant structural change between Apo and bound states, indicating the role of dynamic allostery in modulating enzymatic activity. Here we investigated how substrate binding alters conformational dynamics of Pin1 by comparing the flexibility profiles of the Pin1 structure in different bound conformations to that of the Apo form through our dynamic flexibility index (DFI) analysis. In agreement with previous MD studies, differences in DFI profiles are unidirectional, where major changes in these flexibility profiles can only be seen when the WW domain is in its bound form. Substrate binding to the WW domain induces a hinge shift mechanism by shifting hinge from a region near catalytic loop to the interstitial region around the alpha 4 helix and core domain of the PPIase region. This hinge shift enhances the dynamic coupling of catalytic residues with the PPIase domain, allowing the rest of the PPIase domain to move cooperatively with the catalytic site for association, catalysis and disassociation of the substrate. Overall, our results also agree with the proposed mechanism of recent NMR results of interface mutations of Pin1 where substrate binding to the WW domain enhances the conformational sampling of the PPIase domain, increasing its affinity for the substrate.
Acknowledgments
Support from NSF-MCB Award 1715591 and Scialog Fellow Award by RCS and the Gordon & Betty Moore Foundation is gratefully acknowledged by SBO. HXZ is supported in part by National Institutes of Health Grant GM118091.
References
- 1.Monod J, Wyman J, Changeux J-P. On the nature of allosteric transitions: A plausible model. Journal of Molecular Biology. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 2.Fischer S, Olsen KW, Nam K, Karplus M. Unsuspected pathway of the allosteric transition in hemoglobin. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5608–5613. doi: 10.1073/pnas.1011995108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper A, Dryden DTF. Allostery without conformational change. Eur Biophys J. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 4.Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL. Hidden dynamic allostery in a PDZ domain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18249–18254. doi: 10.1073/pnas.0904492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLeish TCB, Cann MJ, Rodgers TL. Dynamic Transmission of Protein Allostery without Structural Change: Spatial Pathways or Global Modes? Biophysical journal. 2015;109:1240–1250. doi: 10.1016/j.bpj.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 7.Nussinov R, Ma B, Tsai C-J. Multiple conformational selection and induced fit events take place in allosteric propagation. Biophysical chemistry. 2014;186:22–30. doi: 10.1016/j.bpc.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nussinov R, Tsai C-J. Allostery without a conformational change? Revisiting the paradigm. Current opinion in structural biology. 2015;30:17–24. doi: 10.1016/j.sbi.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Bowman GR, Geissler PL. Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11681–11686. doi: 10.1073/pnas.1209309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dima RI, Thirumalai D. Determination of network of residues that regulate allostery in protein families using sequence analysis. Protein science : a publication of the Protein Society. 2006;15:258–268. doi: 10.1110/ps.051767306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerek ZN, Ozkan SB. Change in allosteric network affects binding affinities of PDZ domains: Analysis through perturbation response scanning. PLoS computational biology. 2011;7:e1002154. doi: 10.1371/journal.pcbi.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchenberg S, Sittel F, Stock G. Time-resolved observation of protein allosteric communication. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E6804–E6811. doi: 10.1073/pnas.1707694114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 15.Lu KP, Zhou XZ. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nature reviews. Molecular cell biology. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 16.Lu KP. Pinning down cell signaling, cancer and Alzheimer’s disease. Trends in biochemical sciences. 2004;29:200–209. doi: 10.1016/j.tibs.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. The EMBO journal. 2001;20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou XZ, Lu KP. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nature reviews. Cancer. 2016;16:463–478. doi: 10.1038/nrc.2016.49. [DOI] [PubMed] [Google Scholar]
- 19.Lu P. Function of WW Domains as Phosphoserine- or Phosphothreonine-Binding Modules. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]
- 20.Zhou XZ, Kops O, Werner A, Lu P-J, Shen M, Stoller G, Küllertz G, Stark M, Fischer G, Lu KP. Pin1-Dependent Prolyl Isomerization Regulates Dephosphorylation of Cdc25C and Tau Proteins. Molecular Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 21.Guo J, Pang X, Zhou H-X. Two pathways mediate interdomain allosteric regulation in pin1. Structure (London, England : 1993) 2015;23:237–247. doi: 10.1016/j.str.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng JW. Investigating Dynamic Interdomain Allostery in Pin1. Biophysical reviews. 2015;7:239–249. doi: 10.1007/s12551-015-0171-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Namanja AT, Wang XJ, Xu B, Mercedes-Camacho AY, Wilson KA, Etzkorn FA, Peng JW. Stereospecific gating of functional motions in Pin1. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12289–12294. doi: 10.1073/pnas.1019382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng JW, Wilson BD, Namanja AT. Mapping the dynamics of ligand reorganization via 13CH3 and 13CH2 relaxation dispersion at natural abundance. Journal of biomolecular NMR. 2009;45:171–183. doi: 10.1007/s10858-009-9349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nature structural biology. 2000;7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 26.Jacobs DM, Saxena K, Vogtherr M, Bernado P, Pons M, Fiebig KM. Peptide binding induces large scale changes in inter-domain mobility in human Pin1. The Journal of biological chemistry. 2003;278:26174–26182. doi: 10.1074/jbc.M300796200. [DOI] [PubMed] [Google Scholar]
- 27.Labeikovsky W, Eisenmesser EZ, Bosco DA, Kern D. Structure and dynamics of pin1 during catalysis by NMR. Journal of Molecular Biology. 2007;367:1370–1381. doi: 10.1016/j.jmb.2007.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo J, Zhou H-X. Protein Allostery and Conformational Dynamics. Chemical reviews. 2016;116:6503–6515. doi: 10.1021/acs.chemrev.5b00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barman A, Hamelberg D. Coupled Dynamics and Entropic Contribution to the Allosteric Mechanism of Pin1. The journal of physical chemistry B. 2016;120:8405–8415. doi: 10.1021/acs.jpcb.6b02123. [DOI] [PubMed] [Google Scholar]
- 30.Namanja AT, Peng T, Zintsmaster JS, Elson AC, Shakour MG, Peng JW. Substrate recognition reduces side-chain flexibility for conserved hydrophobic residues in human Pin1. Structure (London, England : 1993) 2007;15:313–327. doi: 10.1016/j.str.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Nevin Gerek Z, Kumar S, Banu Ozkan S. Structural dynamics flexibility informs function and evolution at a proteome scale. Evolutionary applications. 2013;6:423–433. doi: 10.1111/eva.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atilgan C, Gerek ZN, Ozkan SB, Atilgan AR. Manipulation of conformational change in proteins by single-residue perturbations. Biophysical journal. 2010;99:933–943. doi: 10.1016/j.bpj.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of Fluctuation Dynamics of Proteins with an Elastic Network Model. Biophysical journal. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bahar I, Lezon TR, Yang L-W, Eyal E. Global dynamics of proteins: Bridging between structure and function. Annual review of biophysics. 2010;39:23–42. doi: 10.1146/annurev.biophys.093008.131258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larrimore KE, Kazan IC, Kannan L, Kendle RP, Jamal T, Barcus M, Bolia A, Brimijoin S, Zhan C-G, Ozkan SB, et al. Plant-expressed cocaine hydrolase variants of butyrylcholinesterase exhibit altered allosteric effects of cholinesterase activity and increased inhibitor sensitivity. Scientific reports. 2017;7:10419. doi: 10.1038/s41598-017-10571-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar A, Glembo TJ, Ozkan SB. The Role of Conformational Dynamics and Allostery in the Disease Development of Human Ferritin. Biophysical journal. 2015;109:1273–1281. doi: 10.1016/j.bpj.2015.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar A, Butler BM, Kumar S, Ozkan SB. Integration of structural dynamics and molecular evolution via protein interaction networks: A new era in genomic medicine. Current opinion in structural biology. 2015;35:135–142. doi: 10.1016/j.sbi.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dijkstra EW. A note on two problems in connexion with graphs. Numer Math. 1959;1:269–271. [Google Scholar]
- 39.Butler BM, Gerek ZN, Kumar S, Ozkan SB. Conformational dynamics of nonsynonymous variants at protein interfaces reveals disease association. Proteins. 2015;83:428–435. doi: 10.1002/prot.24748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim H, Zou T, Modi C, Dörner K, Grunkemeyer TJ, Chen L, Fromme R, Matz MV, Ozkan SB, Wachter RM. A hinge migration mechanism unlocks the evolution of green-to-red photoconversion in GFP-like proteins. Structure (London, England : 1993) 2015;23:34–43. doi: 10.1016/j.str.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo J, Zhou H-X. Dynamically Driven Protein Allostery Exhibits Disparate Responses for Fast and Slow Motions. Biophysical journal. 2015;108:2771–2774. doi: 10.1016/j.bpj.2015.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Mahoney BJ, Zhang M, Zintsmaster JS, Peng JW. Negative Regulation of Peptidyl-Prolyl Isomerase Activity by Interdomain Contact in Human Pin1. Structure (London, England : 1993) 2015;23:2224–2233. doi: 10.1016/j.str.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]