

Abstract
Background
Submandibular glands (SMGs) are specialized epithelial structures which generate saliva necessary for mastication and digestion. Loss of SMGs can lead to inflammation, oral lesions, fungal infections, problems with chewing/swallowing, and tooth decay. Understanding the development of the SMG is important for developing therapeutic options for patients with impaired SMG function. Recent studies have suggested Sonic hedgehog (Shh) signaling in the epithelium plays an integral role in SMG development; however, the mechanism by which Shh influences gland development remains nebulous.
Results
Using the Kif3af/f;Wnt1-Cre ciliopathic mouse model to prevent Shh signal transduction via the loss of primary cilia in neural crest cells, we report that mesenchymal Shh activity is necessary for gland development. Furthermore, using a variety of murine transgenic lines with aberrant mesenchymal Shh signal transduction, we determine that loss of Shh activity, via loss of the Gli activator, rather than gain of Gli repressor, is sufficient to cause the SMG aplasia. Finally, we determine that loss of the SMG correlates with reduced Neuregulin1 (Nrg1) expression and lack of innervation of the SMG epithelium.
Conclusion
Together, these data suggest a novel mechanistic role for mesenchymal Shh signaling during SMG development.
Keywords: Primary cilia, submandibular salivary glands, Gli, Hedgehog
Introduction
Submandibular gland (SMG) development requires integration of many cell types and signaling pathways to coordinate organogenesis (Patel et al., 2006; Knosp et al., 2012; Mattingly et al., 2015). In particular, proper timing and communication between the ectodermally derived-epithelium and the underlying neural crest cell (NCC) derived-mesenchyme is required (Jaskoll et al., 2002; Rothova et al., 2012). For example, it was previously reported that during early development of the SMG, NCC-derived mesenchyme provides signaling cues such as Fibroblast growth factors (Fgf), to promote epithelial proliferation and branching (Jaskoll et al., 2005; Wells et al., 2013). While the role of some signaling pathways, like Fgf, have been well studied in SMG development (De Moerlooze et al., 2000; Koyama et al., 2012; Wells et al., 2013; Teshima et al., 2016; Chatzeli et al., 2017), our overall understanding of the signaling interactions that occur between the SMG epithelium and NCC-derived mesenchyme remains rudimentary.
The Sonic hedgehog (Shh) pathway is a well characterized signaling pathway involved in several developmental events (Riddle et al., 1993; Dassule et al., 2000; Hebrok et al., 2000; Wijgerde et al., 2002; Cobourne and Green, 2012). Briefly, Shh signal transduction is initiated upon a Shh ligand binding to its membrane-bound receptor, Patched (Ptc). Shh-Ptc binding alleviates Ptc-mediated repression of the pathway effector Smoothened (Smo). Activated Smo then promotes the processing of the transcription factors of the pathway, the Gli factors, into activators (GliA). GliA then moves into the nucleus and activates target gene expression. In the absence of the Shh ligand, Smo is repressed by Ptc, and Gli is processed into a truncated repressor (GliR) which will enter the nucleus to repress Gli target genes (reviewed in (Robbins et al., 2012). The role of Shh activity in the development of many organ systems, including limb (Riddle et al., 1993), neural tube (Belloni et al., 1996; Chiang et al., 1996; Roessler et al., 1996; Nanni et al., 1999; Wijgerde et al., 2002; Ybot-Gonzalez et al., 2002; Vokes et al., 2007; Chamberlain et al., 2008), and many craniofacial components (Muenke et al., 1994; Hu and Helms, 1999; Morava et al., 2003; Schimmenti et al., 2003; Garavelli et al., 2004) is well documented and understood.
A handful of studies have specifically examined a role for Shh signaling within the developing SMG (Jaskoll et al., 2004; Hashizume and Hieda, 2006; Mizukoshi et al., 2016). These studies observed expression of several components of the Shh pathway in the epithelium of the developing SMG, and found that Shh-null mice presented with ‘paedomorphic’ SMGs, which fail to progress past the pseudoglandular stage (Jaskoll et al., 2004). Together, these data suggested that epithelial Shh signaling was intrinsically required to maintain epithelial progenitors during branching morphogenesis (Jaskoll et al., 2004; Mizukoshi et al., 2016), and to promote epithelial proliferation and lumen formation via epithelial polarization (Hashizume and Hieda, 2006; Patel et al., 2006). While there has been some data suggesting Shh induces these processes by activating members of the Epidermal growth factor (Egf) family (Mizukoshi et al., 2016), by in large, the detailed mechanism and downstream targets of Shh signaling in the developing SMG remain unclear.
In the last few decades, several studies have convincingly shown that Shh signal transduction requires primary cilia: microtubule-based extensions present on almost all vertebrate cells not actively undergoing mitosis (Sorokin, 1962; Huangfu et al., 2003; Corbit et al., 2005; Huangfu and Anderson, 2005; Rohatgi et al., 2007). Loss of primary cilia disrupts Shh signaling, as functional primary cilia are required for proper processing of both GliA and GliR (Huangfu and Anderson, 2005). Our previous studies have focused on understanding the role of cilia-dependent Shh signaling during craniofacial development (Brugmann et al., 2010; Chang et al., 2016; Millington et al., 2017). To address this, we perturbed primary cilia on NCC-derived mesenchyme within the developing craniofacial complex, by conditionally deleting the kinesin-2 motor protein subunit Kif3a with the Wnt1-Cre driver (Kif3af/f;Wnt1-Cre). Herein, we examine a previously uncharacterized SMG phenotype of Kif3af/f;Wnt1-Cre mutant mice. In pursuit of the molecular mechanism for this SMG phenotype, we also uncovered novel details regarding the role Shh plays during SMG development and hypothesized that primary cilia-dependent Shh signaling in NCCs was necessary for SMG development.
Results
Conditional ablation of primary cilia in neural crest cells resulted in SMG aplasia
We previously characterized midfacial and glossal phenotypes which resulted from a loss of primary cilia in NCCs using the Kif3af/f;Wnt1-Cre conditional mouse model (Brugmann et al., 2010; Chang et al., 2016; Millington et al., 2017). In addition to these previously described facial phenotypes, we recently observed a loss of SMGs in Kif3af/f;Wnt1-Cre embryos (Fig. 1). To characterize SMG aplasia in Kif3af/f;Wnt1-Cre mutants, we first histologically examined SMG initiation and development. SMG initiation begins at e11.5 (prebud stage) with the formation of epithelial thickenings (primitive knots) bilaterally adjacent to the developing tongue (Fig. 1A, A’). At e12.5 (initial bud stage) the epithelia elongate into a condensed NCC-derived mesenchyme forming a solid stalk which terminates in a bulb (Fig. 1C). In Kif3af/f;Wnt1-Cre mutants, the primitive knot and thickened epithelium were observed at e11.5 (Fig. 1B-B’); however, epithelial elongation was stunted and failed to progress past the initial bud stage (Fig. 1D). These data suggested that albeit severely compromised, the earliest stages of SMG initiation occurred when primary cilia were lost on NCCs.
Figure 1. Conditional loss of primary cilia on NCCs results in aplasia of the SMG.

(A, B) H&E on e11.5 frontal sections through wild-type and Kif3af/f;Wnt1-Cre SMG prebud epithelium (dotted black lines and black arrows). (A’, B’) High-magnification images of A, B. (C, D) H&E on e12.5 frontal sections through wild-type and Kif3af/f;Wnt1-Cre initial bud SMG (dotted black lines). (E, F) H&E on e14.5 frontal sections through wild-type and Kif3af/f;Wnt1-Cre mutant embryos showing the presence and absence of SMGs (dotted black lines), respectively. (E’, F’) High-magnification images of E, F. (G-J) Anti-Pdgfrβ (green) and Anti-Krt8 (red) immunostaining on e14.5 wild-type and Kif3af/f;Wnt1-Cre mutant SMGs. The (G, I) SMG or (H, J) mutant mesenchymal capsule are indicated with dotted white lines. Nuclei are counterstained with Hoechst. (K, L) Anti-Sox9 (red) immunostaining on e11.5 wild-type and Kif3af/f;Wnt1-Cre mutant embryos, showing the presence (dotted white lines) or absence (white asterisks) of Sox9 expression in the prebud epithelium. Nuclei are counterstained with Hoechst. (M, N) Anti-Arl13b (green) immunostaining on e11.5 wild-type and Kif3af/f;Wnt1-Cre developing SMGs (dotted white lines). Nuclei are counterstained with Hoechst. (O) Quantification of CC3-positive cells in the e11.5 prebud epithelium and mesenchyme of wild-type and Kif3af/f;Wnt1-Cre mutants. (P) Quantification of PHH3-positive cells in the e11.5 prebud epithelium and mesenchyme of wild-type and Kif3af/f;Wnt1-Cre mutants. (Q) X-gal staining on a frontal section through the developing SMG of an e14.5 R26R;Wnt1-Cre embryo. The SMG is indicated with a dotted black line. (R) Frontal section through the developing SMG of a ROSAmT/mG;Wnt1-Cre embryo at e14.5. NCCs express EGFP (green). Non-NCCs express Td Tomato (red). Scale bars= (A-D, E’, F’, G-J) 100μm; (E, F) 1mm; (K, L) 20μm; (M, N, R) 200μm; (Q) 500μm.
By e14.5, H&E staining on frontal sections of wild-type embryos confirmed the presence of pseudoglandular stage SMGs, which contain a branched epithelium surrounded by condensed NCC-derived mesenchyme (Fig. 1E, E’). Relative to wild-type embryos, Kif3af/f;Wnt1-Cre mutants lacked SMG epithelial structures and only possessed a condensed mesenchymal capsule (Fig. 1F, F’). To confirm our initial histological characterization of SMG aplasia in Kif3af/f;Wnt1-Cre mutants, we next performed immunostaining with molecular markers for SMG mesenchyme and epithelium. In wild-type embryos, SMG mesenchyme and epithelium were present as evident by positive staining for Platelet-derived growth factor receptor beta (Pdgfrβ) (Yamamoto et al., 2008; Nelson et al., 2013; Kwon and Larsen, 2015)(Fig. 1G) and Cytokeratin 8 (Krt8) (Rebustini et al., 2007)(Fig. 1I), respectively. Kif3af/f;Wnt1-Cre embryos retained a Pdgfrβ-positive mesenchymal capsule (Fig. 1H), but did not contain any Krt8-positive epithelial cells (Fig. 1J). We further sought to characterize the Kif3af/f;Wnt1-Cre prebud epithelium by examining Sox9 expression. Sox9 expression, recently determined to be dispensable for SMG initiation (Chatzeli et al., 2017), was detected in wild-type prebud epithelium (Fig. 1K), but lost in the prebud epithelium of Kif3af/f;Wnt1-Cre mutants (Fig. 1L). Finally, to confirm the SMG phenotype was attributed to NCC-specific loss of primary cilia, we performed immunostaining for the primary cilia marker ADP ribosylation factor like GTPase 13B (Arl13b) at the prebud stage. In wild-type embryos, both prebud epithelium and NCC-derived mesenchyme extended Arl13b-positive primary cilia (Fig. 1M). Kif3af/f;Wnt1-Cre embryos retained epithelial primary cilia; however, no Arl13b-positive primary cilia were detected in the NCC-derived mesenchyme (Fig. 1N).
To further characterize early SMG development in Kif3af/f;Wnt1-Cre mutant embryos, we examined cell death and proliferation using immunostaining Cleaved caspase 3 (CC3) and Phosphohistone H3 (PHH3), respectively. While we did not observe a significant difference in cell death in either the epithelium or mesenchyme of wild-type and Kif3af/f;Wnt1-Cre mutants (Fig. 1O, p>0.05; n=8 wild-type, n=4 Kif3af/f;Wnt1-Cre), Kif3af/f;Wnt1-Cre mutants display significantly less proliferation in the prebud epithelium compared to wild-type embryos (Fig. 1P, p=0.7453; n=8 wild-type, n=4 Kif3af/f;Wnt1-Cre). No significant difference was detected in proliferation of the mesenchymal cells between wild-type and Kif3af/f;Wnt1-Cre mutants (Fig. 1P, p=0.0104; n=8 wild-type, n=4 Kif3af/f;Wnt1-Cre,). Together, these data were consistent with our characterization of the Kif3af/f;Wnt1-Cre phenotype primarily affecting the developing SMG epithelium, despite the loss of cilia in the NCC-derived mesenchyme.
To confirm the mesenchyme of the SMG was NCC-derived, we utilized two distinct transgenic reporter lines to visualize tissue of origin. We first utilized R26R-LacZ reporter mice (Soriano, 1999) and generated R26R;Wnt1-Cre embryos. X-gal staining within the mesenchyme surrounding the developing SMG epithelium suggested the mesenchyme was of NCC origin (Fig. 1Q). We also utilized the ROSAmT/mG dual reporter, which expressed membrane tdTomato in cells which do not express Cre recombinase, and expressed membrane EGFP upon exposure to Cre-recombinase (Muzumdar et al., 2007). We observed EGFP-positive mesenchyme surrounding tdTomato-positive branched epithelium in ROSAmT/mG;Wnt1-Cre embryos (Fig. 1R). Together, these data suggested that the mesenchymal cells of the SMG were NCC-derived and that loss of primary cilia on NCCs exerted an extrinsic effect on the developing SMG epithelium. We next investigated how loss of primary cilia on NCCs affected SMG development, relative to known mechanisms of SMG organogenesis.
SMG initiation and Fgf expression are maintained when primary cilia are removed from NCCs
It is well established that Fgf signaling plays an essential role in SMG development (De Moerlooze et al., 2000; Jaskoll et al., 2002; Wells et al., 2013). Specifically, Fgf10/Fgf2rb signaling is essential for the branching and histodifferentiation of the SMG epithelium, but not the earliest initial bud formation (Jaskoll et al., 2005). Since the SMGs of Kif3af/f;Wnt1-Cre embryos appear to undergo early initiation, but fail to progress through epithelial branching and differentiation, we hypothesized that impaired Fgf signaling could be associated with ciliopathic SMG aplasia. To determine if SMG aplasia observed in our ciliopathic mutant was due to a loss of Fgf signaling, we next examined Fgf expression by performing sectioned in situ hybridization for Fgfr2 and Fgf10 at e11.5 in wild-type and Kif3af/f;Wnt1-Cre embryos. Using a probe that detected both the epithelial isoform (Fgfr2b) and the mesenchymal isoform (Fgfr2c) of Fgfr2 (Knosp et al., 2012; Goetz and Mohammadi, 2013), we observed Fgfr2 was expressed within the wild-type oral epithelium, including the SMG primitive knot and some NCC-derived mesenchyme (Fig. 2A). Fgf10 was expressed in the NCC-derived SMG mesenchyme surrounding the SMG primitive knot (Fig. 2C). Interestingly, Fgfr2 and Fgf10 expression were maintained in the Kif3af/f;Wnt1-Cre mutants (Fig. 2B, D). To determine if Fgf signaling was impaired, we next examined expression of three Fgf targets, Etv5, Spry1, and Spry2 by in situ hybridization at e11.5. Similar to Fgfr2, and Fgf10 expression, Etv5, Spry1, and Spry2 expression was maintained in Kif3af/f;Wnt1-Cre mutants compared to wild-type embryos (Fig. 2E-J). We next confirmed and quantified expression of Fgf pathway members via RNA-sequencing and RT-qPCR. Compared to wild-type mandibular prominences, there was no significant difference in the expression of Fgfr2, Fgf10, Etv5, Spry1, or Spry2 in e11.5 Kif3af/f;Wnt1-Cre mandibular prominences by RNA-sequencing (Fig. 2K). Additionally, RT-qPCR revealed no significant difference in expression of Fgf10 and Fgfr2b between e11.5 wild-type and Kif3af/f;Wnt1-Cre mutants (Fig. 2L; p=0.6248 and p=0.7953; n=3). These data suggested that SMG aplasia in Kif3af/f;Wnt1-Cre embryos was not due to loss of Fgf expression or activity.
Figure 2. Fgf signaling is maintained in Kif3af/f;Wnt1-Cre mutants.
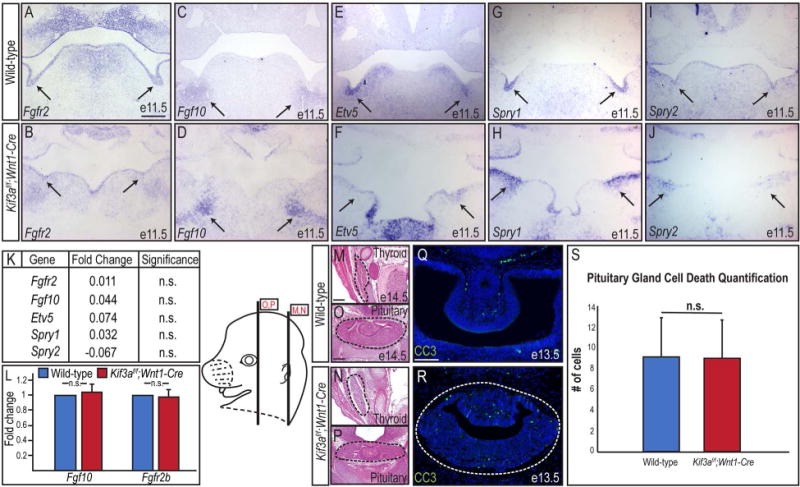
(A-J) Sectioned in situ hybridization for Fgf10, Fgfr2, Etv5, Spry1, and Spry2 in SMG epithelium (black arrows) of e11.5 wild-type and Kif3af/f;Wnt1-Cre mutant embryos. (K) Fold change in expression levels of Fgf10, Fgfr2, Etv5, Spry1, and Spry2 by RNA-Sequencing in e11.5 Kif3af/f;Wnt1-Cre mutant mandibular prominences compared to e11.5 wild-type mandibular prominences. (L) RT-qPCR for Fgf10 and Fgfr2b transcript levels in wild-type SMGs and Kif3af/f;Wnt1-Cre mutant mesenchymal capsules. (M-P) H&E on frontal sections through the thyroid and pituitary glands (dotted black lines) of e14.5 wild-type and Kif3af/f;Wnt1-Cre embryos. (Q, R) Anti-CC3 immunostaining (green) on e13.5 wild-type and Kif3af/f;Wnt1-Cre pituitary glands (dotted white lines). Nuclei counterstained with Hoechst. (S) Quantification of Q and R. Scale bars= (A-J, M-P) 200μm; (Q, R) 100μm.
To confirm that loss of Fgf activity was not responsible for SMG agenesis in Kif3af/f;Wnt1-Cre mutants, we next compared the development of additional glands known to require Fgf input for organogenesis. Loss of Fgf10 in NCCs (Fgf10f/f;Wnt1-Cre) affected the development of the thyroid gland, but did not severely impact pituitary gland development (Teshima et al., 2016). Contrary to Fgf10f/f;Wnt1-Cre embryos, Kif3af/f;Wnt1-Cre mutants did not display an obvious thyroid phenotype (Fig. 2M, N); however, the pituitary gland was dysmorphic (Fig. 2O, P). Furthermore, unlike Fgf10f/f;Wnt1-Cre mutants, Kif3af/f;Wnt1-Cre mutants did not have any observable changes in cell death in developing pituitary gland (Fig. 2Q-S; p=0.8551, n=28 wild-type, n=41 Kif3af/f;Wnt1-Cre). These data, taken together with our analysis of Fgf10, Fgfr2, Etv5, Spry1, and Spry2 expression, strongly suggested that loss of Fgf signaling was not causal for the SMG phenotype in Kif3af/f;Wnt1-Cre mutants.
The Hedgehog pathway is active in early SMG development
Several studies have suggested a role for the Shh pathway during SMG development. These suggestions were based on reported expression of pathway components within the developing gland, a hypoplastic SMG phenotype in Shh-null mice, and a reported role for Shh in generation of SMG endbud number (Jaskoll et al., 2004; Melnick et al., 2009; Häärä et al., 2011; Mizukoshi et al., 2016). Despite these studies suggesting an important role for Shh in SMG development, the mechanism by which Shh functions during the process is still poorly understood. Since the Kif3af/f;Wnt1-Cre SMG phenotype is caused by the loss of primary cilia, and the primary cilium has been intricately linked to the transduction of the Shh pathway (Huangfu et al., 2003; Corbit et al., 2005; Huangfu and Anderson, 2005; Rohatgi et al., 2007), we next examined if the SMG aplasia observed in Kif3af/f;Wnt1-Cre embryos was due to aberrant Shh activity.
To further understand the role of Shh signaling during SMG development we first analyzed Shh-expression and Shh-responsiveness (Fig. 3). We utilized the Shh-GFP reporter line (Harfe et al., 2004) to confirm the presence of Shh expression in the initial SMG bud at e12.5. GFP was robustly expressed throughout the SMG epithelium and in the mesenchyme adjacent to the epithelium at initial bud stage (Fig. 3A). We next confirmed the presence of Shh in the developing SMG via immunostaining, confirming antibody specificity in the e12.5 ventral neural tube (Fig. 3B). Using the anti-Shh antibody, we detected Shh expression in wild-type SMG epithelium and adjacent mesenchyme from prebud through pseudoglandular stages (Fig. 3C-E). In Kif3af/f;Wnt1-Cre embryos Shh expression was maintained at e11.5 (Fig. 3F) and e12.5 (Fig. 3G), but was almost completely absent by e14.5, concordant with the loss of a branched SMG epithelium (Fig. 3H). Together, these data suggested that a loss of Shh expression was not causal for SMG agenesis in Kif3af/f;Wnt1-Cre mutants.
Figure 3. Shh pathway is active during early SMG development.
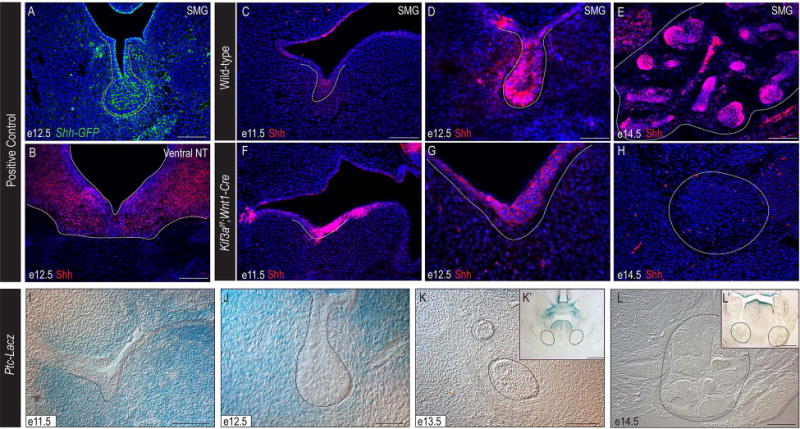
(A) Anti-GFP immunostaining (green) on e12.5 Shh-GFP SMG (dotted white line). (B) Anti-Shh immunostaining (red) on e12.5 ventral neural tube (dotted white line). (C-H) Anti-Shh immunostaining (red) on e11.5, e12.5, and e14.5 wild-type and Kif3af/f;Wnt1-Cre mutant SMGs (dotted white lines). Nuclei counterstained with Hoechst. (I-L) X-gal staining on frontal sections of Ptc-LacZ embryos. Dotted black lines indicate developing SMGs. (K’, L’) High-magnification images of K and L. (A, B, C, E, F, H, I) 100μm; (D, G, J) 50μm; (K, L) 500μm; (K’, L’) 200μm.
Finally, we assayed for Shh-responsive cells in the developing gland using the Patched-LacZ reporter line (Goodrich et al., 1996). At both e11.5 and e12.5, we observed X-gal staining throughout the NCC-derived mesenchyme surrounding the initial SMG bud (Fig. 3I, J). Although it has been hypothesized in the literature that Shh later promotes branching morphogenesis within the SMG (Jaskoll et al., 2004; Mizukoshi et al., 2016), we did not observe Shh-responsive cells within the early pseudoglandular epithelium at e13.5 or within the mid-stage pseudoglandular epithelium at e14.5, during branching morphogenesis (Fig. 3K-L’). These data suggest that Shh, originating from the initial bud, signals to the developing NCC-derived mesenchyme.
Hedgehog activity, via the Gli activator, is required for epithelial development in the SMG
To further explore the role of Shh activity in the NCC-derived SMG mesenchyme and determine if impaired Shh signaling in the NCC was responsible for the SMG phenotype observed in Kif3af/f;Wnt1-Cre embryos, we analyzed SMG development in several transgenic mouse lines with Shh pathway components conditionally knocked-out or compromised in the NCC-derived mesenchyme. We first examined Smof/f;Wnt1-Cre mutant mice, in which Smo, a transmembrane-protein required for intracellular signal transduction of the Shh pathway, was conditionally ablated from NCCs. Compared to e14.5 wild-type SMGs, which expressed Pdgfrβ in the SMG mesenchyme and Krt8 in the SMG epithelium (Fig. 4A-C), e14.5 Smof/f;Wnt1-Cre mutant embryos phenocopied the SMG aplasia observed in Kif3af/f;Wnt1-Cre embryos. Both Kif3af/f;Wnt1-Cre and Smof/f;Wnt1-Cre mutant embryos had mesenchymal capsules devoid of SMG epithelium (Fig. 4D-I). Perhaps the only obvious difference between Kif3af/f;Wnt1-Cre and Smof/f;Wnt1-Cre mutants was that the mesenchymal capsules of the Smof/f;Wnt1-Cre mutants were located more medially. We next compared the Kif3af/f;Wnt1-Cre SMG phenotype to that generated when the transcription factors of the Shh pathway, Gli2 and Gli3, were genetically ablated from the NCC-derived mesenchyme. Gli2f/f;Gli3f/f;Wnt1-Cre also phenocopied Kif3af/f;Wnt1-Cre mutants in that there was a Pdgfrβ-positive mesenchymal capsule present and a complete loss of Krt8 positive epithelium (4J-L); however the mesenchymal capsules of Gli2f/f;Gli3f/f;Wnt1-Cre mutants appeared to have a higher cell density than Kif3af/f;Wnt1-Cre or Smof/f;Wnt1-Cre mutants. Thus, two separate transgenic lines with impaired Shh activity in the NCC-derived mesenchyme phenocopied the Kif3af/f;Wnt1-Cre SMG aplasia. Taken together, these data suggested that Shh activity in the NCC-derived mesenchyme was essential for SMG development and supported our hypothesis that the loss of Shh activity in the NCC-derived mesenchyme could be responsible for SMG agenesis in Kif3af/f;Wnt1-Cre embryos.
Figure 4. Shh and GliA activity are required in NCCs for SMG development.
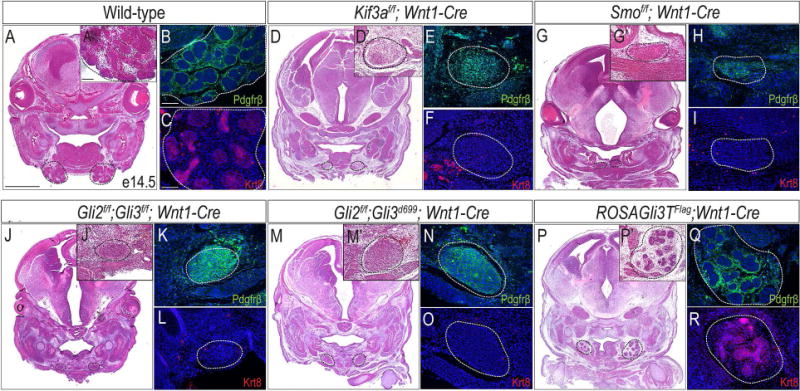
(A, D, G, J, M, P) H&E staining on frontal sections of wild-type, Kif3af/f;Wnt1-Cre, Smof/f;Wnt1-Cre, Gli2f/f;Gli3f/f;Wnt1-Cre, Gli2f/f;Gli3d699;Wnt1-Cre, and ROSAGli3TFlag;Wnt1-Cre embryos. Dotted black lines indicate developing and mutant SMGs. (A’, D’, G’, J’, M’, P’) High-magnification images of A, D, G, J, M, P. (B, E, H, K, N, Q) Anti-Pdgfrβ immunostaining (green) on wild-type, Kif3af/f;Wnt1-Cre, Smof/f;Wnt1-Cre, Gli2f/f;Gli3f/f;Wnt1-Cre, Gli2f/f;Gli3d699;Wnt1-Cre, and ROSAGli3TFlag;Wnt1-Cre SMGs or mutant mesenchymal capsules (dotted white lines). (C, F, I, L, O, R) Anti-Krt8 immunostaining (red) on wild-type, Kif3af/f;Wnt1-Cre, Smof/f;Wnt1-Cre, Gli2f/f;Gli3f/f;Wnt1-Cre, Gli2f/f;Gli3d699;Wnt1-Cre, and ROSAGli3TFlag;Wnt1-Cre SMGs or mutant mesenchymal capsules (dotted white lines). Nuclei counterstained with Hoechst. Scale bars= (A, D, G, J, M, P) 1mm; (A’, B, C, D’, E, F, G’, H, I, J’, K, L, M’, N, O, P’, Q, R) 100μm.
As the effectors of the Shh pathway, the Gli transcription factors can act as both activators or repressors (Hui and Angers, 2011). As such, Shh pathway activity can be reduced by means of either losing Gli activator (GliA) activity or gaining Gli repressor (GliR) activity. Our previous studies confirmed that GliA and GliR activity were impaired in Kif3af/f;Wnt1-Cre mutant embryos (Chang et al., 2016; Millington et al., 2017), thus to gain insight into a possible mechanism for SMG aplasia in Kif3af/f;Wnt1-Cre embryos, we analyzed additional transgenic mouse lines in which either GliA or GliR expression was compromised. We first genetically deleted GliA from NCC cells, by generating Gli2f/f;Gli3d699;Wnt1-Cre embryos, in which we conditionally deleted Gli2 (the predominant activator of the pathway) in NCC cells and introduced the Gli3d699 allele (Millington et al., 2017). Gli3d699 murine mutants only express the truncated N-terminal repressor domain of Gli3, Gli3R (Bose et al., 2002). Similar to Kif3af/f;Wnt1-Cre and Smof/f;Wnt1-Cre embryos, Gli2f/f;Gli3d699;Wnt1-Cre embryos only had a Pdgfrβ-positive mesenchymal capsule which lacked epithelialized SMG structures (Fig. 4M-O). Interestingly, similar to Gli2f/f;Gli3f/f;Wnt1-Cre embryos, the mesenchymal capsules of Gli2f/f;Gli3d699;Wnt1-Cre mutants appeared to have a higher cell density than Kif3af/f;Wnt1-Cre or Smof/f;Wnt1-Cre mutants. Thus, loss of GliA activity alone in NCCs, was sufficient to phenocopy the SMG aplasia present in Kif3af/f;Wnt1-Cre embryos.
To investigate an alternative method by which Shh signaling could be lost, we next tested if reducing Shh pathway activity via overexpression of GliR, would impact SMG development. To test this hypothesis we used the ROSAGli3TFlag transgenic line (Vokes et al., 2008) to conditionally overexpress Gli3R in NCCs (ROSAGli3TFlag;Wnt1-Cre) (Millington et al., 2017). Interestingly, ROSAGli3TFlag;Wnt1-Cre embryos still developed SMGs, albeit slightly hypoplastic relative to wild-type embryos (Fig. 4P-R, dotted black lines). ROSAGli3TFlag;Wnt1-Cre SMGs had both a Pdgfrβ-positive mesenchyme and Krt8-positive epithelia (Fig. 4Q, R), suggesting that despite being smaller in size, these SMGs were comprised of mesenchyme and epithelium similar to wild-type embryos. These data, taken together with those from other transgenic lines, suggested that the loss of GliA activity, rather than a gain of GliR activity or disruptions in the GliA:GliR ratio, was sufficient to cause SMG aplasia.
Egf/ErbB ligand expression is lost in both ciliopathic and Shh mutants with SMG aplasia
Previous hypotheses suggested Shh was required for branching morphogenesis (Jaskoll et al., 2004; Mizukoshi et al., 2016); however, our Ptc-LacZ reporter analysis (Fig. 3) did not indicate epithelial cells were responsive to a Shh signal during branching. To address this contradiction, we hypothesized that Shh signaling induced an intermediate factor which promoted epithelial branching. Ex vivo studies utilizing SMG explants suggested that Shh signaling induced Epidermal growth factor (Egf) ligand/Egf receptor (ErbB) signaling, which was required for epithelial proliferation and branching morphogenesis in the SMG (Mizukoshi et al., 2016). Furthermore, loss of Egf/ErbB signaling resulted in reduced epithelial cell proliferation and structures (Haara et al., 2009), similar to that observed in both Kif3af/f;Wnt1-Cre (Fig. 1P) and various Hh mutants (Fig. 4). To examine the possibility that cilia-dependent Shh signaling in NCCs decreased Egf/ErbB signaling, we performed RT-qPCR for several ErbB ligands on e14.5 wild-type and Kif3af/f;Wnt1-Cre mutant SMGs. While no significant difference was detected in Egf expression (Fig. 5A; p=0.7479; n=6), expression of several Neuregulin (Nrg) family members, including Nrg1, Nrg2 and Nrg3, was significantly decreased in Kif3af/f;Wnt1-Cre mutant SMGs (Fig. 5A; p<0.0001; n=6). We confirmed our qPCR data by performing immunostaining for Nrg1, an Nrg family member previously implicated in SMG development (Miyazaki et al., 2004; Nedvetsky et al., 2014). Nrg1 expression was detected in both NCC-derived mesenchyme and SMG epithelial cells in wild-type embryos (Fig. 5B), and significantly reduced in Kif3af/f;Wnt1-Cre mutant SMGs capsules (Fig. 5C). Although Nrg expression was significantly reduced in mutant embryos via both qPCR and immunostaining, the caveat for this experiment was that we examined expression after the onset of the phenotype. To determine if loss of Nrg ligand expression was potentially causal for ciliopathic SMG aplasia, we next analyzed expression of one Nrg ligand prior to the onset of the aplastic phenotype.
Figure 5. Nrg1 expression is reduced in ciliopathic and GliA mutants.
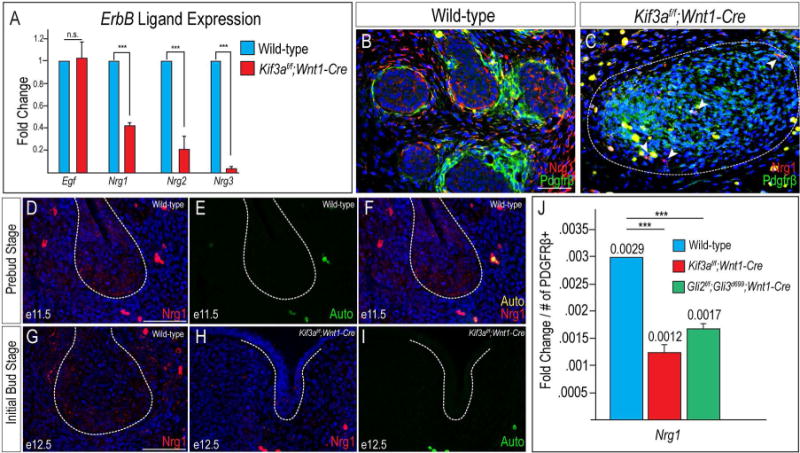
(A) RT-qPCR for Egf, Nrg1, Nrg2, and Nrg3 transcript levels in wild-type SMGs and Kif3af/f;Wnt1-Cre mutant mesenchymal capsules. (B, C) Anti-Nrg1 (red) and anti-Pdgfrβ (green) co-immunostaining on e14.5 wild-type SMGs and Kif3af/f;Wnt1-Cre mutant mesenchymal capsules (dotted white lines). White arrow heads in C indicate a small number of Nrg1-positive cells in the Kif3af/f;Wnt1-Cre mutant mesenchymal capsules. (D) Anti-Nrg1 immunostaining (red) on frontal section of a wild-type e11.5 SMG (dotted white lines). (E) Auto-fluorescence of red blood cells from the 488 filter in D. (F) Merged images from D and E. (G, H) Anti-Nrg1 immunostaining (red) on frontal sections of e12.5 wild-type and Kif3af/f;Wnt1-Cre SMGs (dotted white line). Nuclei counterstained with Hoechst. (I) Auto-fluorescence of red blood cells from the 488 filter in H. (J) RT-qPCR for Nrg1 transcript levels in wild-type, Kif3af/f;Wnt1-Cre, and Gli2f/f;Gli3d699;Wnt1-Cre SMGs or mutant mesenchymal capsules. Fold change of transcript levels were normalized to Pdgfrβ+ cells. ***=p<0.001 Scale bars= (B-I) 50μm.
Since Nrg1 has previously been detected in the SMG and implicated in epithelial branching (Miyazaki et al., 2004; Nedvetsky et al., 2014), we focused our subsequent analysis on Nrg1 expression during SMG development in both wild-type and mutant embryos. To test the hypothesis that reduced Nrg1 could contribute to the loss of SMG epithelial structures in Kif3af/f;Wnt1-Cre mutants, we first determined the spatiotemporal expression of Nrg1. Previous studies examining the e14.0 SMG using a pan-Nrg1 antibody reported that expression was confined to the mesenchyme (Miyazaki et al., 2004); however, at e11.5, we observed punctate Nrg1 expression in both the epithelium and NCC-derived mesenchyme of wild-type embryos (Fig. 5D). Auto-fluorescent red blood cells were also observed in this tissue (Fig. 5E), but the nonspecific emission artifacts were confirmed via merging these images and observing their contribution to multiple wavelengths (Fig. 5F). At e12.5, during the initial bud stage, Nrg1 expression was observed in NCC-derived mesenchymal cells surrounding the growing epithelial bud and within the periphery of the bud itself (Fig. 5G). In Kif3af/f;Wnt1-Cre mutants, Nrg1 expression is noticeably absent in the epithelium of the hypoplastic initial bud, and reduced in the surrounding NCC-derived mesenchyme (Fig. 5H), distinct from auto-fluorescence red blood cells (Fig. 5I). Thus, the spatiotemporal expression of Nrg1, and subsequent loss of expression in Kif3af/f;Wnt1-Cre mutants supported our hypothesis that loss of cilia-dependent Shh signaling in NCCs could impair expression of intermediate factors, perhaps Nrg ligands including Nrg1, necessary for epithelial branching.
Loss of primary cilia impairs Gli processing (Huangfu et al., 2003; Huangfu and Anderson, 2005). Our previous work with Kif3af/f;Wnt1-Cre mutants suggested that despite having an increased amount of GliA protein present in the developing facial prominences, the lack of ciliary processing rendered the GliA protein inactive (Chang et al., 2016; Millington et al., 2017). Since the SMG phenotype of Kif3af/f;Wnt1-Cre mutants phenocopies the Gli2f/f;Gli3d699;Wnt1-Cre mutant that lacks GliA, we wondered if Nrg1 levels were also reduced in Gli2f/f;Gli3d699;Wnt1-Cre mutants. RT-qPCR on SMGs from wild-type embryos and the mesenchymal capsule of Gli2f/f;Gli3d699;Wnt1-Cre mutant embryos revealed there was a significant decrease in Nrg1 expression in mutants compared to wild-type embryos (Fig. 5J; p<0.0001; n=6). These data supported the hypothesis that SMG aplasia present in both ciliopathic and GliA mutants could be due to a loss of Egf/ErbB signaling in the SMG mesenchyme.
Loss of primary cilia on neural crest cells does not impair neural crest cell differentiation into cranial nerves, but does impair innervation of the developing SMG
Previous studies correlated SMG development with sympathetic and parasympathetic innervation (Knox et al., 2010; Knox et al., 2013; Nedvetsky et al., 2014; Knosp et al., 2015). Specifically, it was reported that epithelial progenitors required signals from parasympathetic ganglia for maintenance and proliferation (Knox et al., 2010; Knosp et al., 2015). Furthermore, mice that lack Nrg1 were reported to have depleted innervation of the SMG (Nedvetsky et al., 2014). Since the autonomic ganglia, including all parasympathetic nerves, are NCC-derived (Le Douarin and Teillet, 1974; D'Amico-Martel and Noden, 1983; Ferreira and Hoffman, 2013), we next asked if loss of cilia on NCCs impaired neuronal development and/or innervation of the developing SMG. To investigate this hypothesis, we harvested e12.5 wild-type and Kif3af/f;Wnt1-Cre mutant embryos and performed immunostaining for ß-III Tubulin (Tubb3) on frontal sections to mark neurons. Tubb3-positive neurons were observed near the primary duct of the e12.5 initial bud of wild-type SMGs, as previously described (Knox et al., 2010) (Fig. 6A). Although Tubb3-positive neurons were detected in Kif3af/f;Wnt1-Cre mutant embryos, they were never observed coalescing near the epithelium (Fig. 6B), suggesting subsequent innervation on the gland could be impaired. We next sought to examine if the parasympathetic ganglia were associated with the developing SMG in wild-type and mutant embryos. Whole mount co-immunostaining for Tubb3 and Krt8 was performed on e14.5 wild-type and Kif3af/f;Wnt1-Cre mutant embryos to mark neurons and secretory epithelium, respectively. Tubb3-positive neurons were observed throughout the craniofacial complex and associated with the Krt8-positive epithelial branches of the developing SMG in wild-type embryos (Fig. 6C). Interestingly, Tubb3-positive neurons were not only present, but appeared to have increased axonemal branching in Kif3af/f;Wnt1-Cre embryos, but were unable to coalesce with epithelial structures because they were not present (Fig. 6D). Sections through the developing SMG confirmed our whole mount observations. Tubb3 and E-cadherin (Ecad) immunostaining revealed neurons surrounding SMG epithelium in e14.5 wild-type embryos (Fig. 6E, G). Despite Tubb3-positive neurons in the area surrounding the SMG mesenchymal capsule, there was a complete lack of SMG epithelial structures in the Kif3af/f;Wnt1-Cre mutant embryos (Fig. 6F, H), and thus a lack of any innervation. These data suggested that SMG agenesis in Kif3af/f;Wnt1-Cre mutant embryos was not due to the loss of NCC-derived cranial nerves, but rather a defect in the ability of the existing NCC-derived neurons to coalesce and innervate the SMG epithelium.
Figure 6. Loss of primary cilia on NCCs does not impair differentiation into cranial nerves, but does impair innervation.
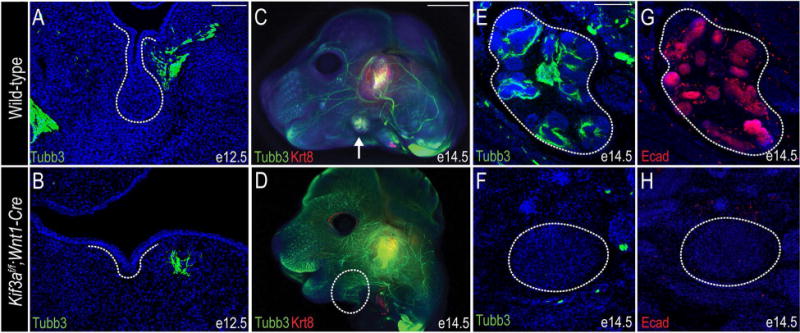
(A, B) Anti-βIII-tubulin (Tubb3) immunostaining (green) on e12.5 wild-type and Kif3af/f;Wnt1-Cre mutant SMGs (dotted white lines). (C, D) Whole mount co-immunostaining for anti-Tubb3 (green) and anti-Krt8 (red) on wild-type and Kif3af/f;Wnt1-Cre e14.5 heads. White arrow in C indicates SMG; dotted white line in B indicates remaining SMG mesenchymal capsule. (E, F) Anti-Tubb3 immunostaining (green) on frontal sections through e14.5 wild-type and Kif3af/f;Wnt1-Cre SMGs (dotted white line). (G, H) Anti-E-cadherin immunostaining (red) on frontal sections through e14.5 wild-type and Kif3af/f;Wnt1-Cre SMGs (dotted white line). Nuclei are counterstained with Hoechst. Scale bars= (A, B, E-H) 100μm; (C, D) 2mm.
Based on the data presented herein, we propose a role for cilia-dependent Shh activity in the NCC-derived mesenchyme in prebud and initial bud stage development of the SMG. We hypothesized that in wild-type SMGs, primary cilia on the NCC-derived mesenchyme surrounding the initial bud stage SMG epithelium were required for transducing a Shh signal from the SMG epithelium. In response to this Shh signal, primary cilia on the NCCs processed full-length Gli (GliFL) into a fully functional GliA. We hypothesize that GliA, either directly or indirectly, then induced expression of downstream genes that impact development of the gland epithelium, like Nrg1, in the NCC-derived mesenchyme. Secreted Nrg1 becomes bound to ErbB receptors on SMG epithelial cells, culminating in proliferation, and eventual branching morphogenesis and proper innervation (Fig. 7A). When primary cilia were lost on NCCs (Kif3af/f;Wnt1-Cre mutants), the Shh signal was not transduced in NCCs and a functional GliA was not produced (Chang et al., 2016; Millington et al., 2017). Decreased GliA activity, either directly or indirectly, prevented Nrg1 expression, and thus the development of the SMG epithelium was arrested at prebud stage and SMG aplasia occurred (Fig. 7B). We were able to support this hypothesis by examining the Gli2f/f;Gli3d699;Wnt1-Cre mutants, which completely lack Gli2A and express a truncated version of Gli3 that can only be converted to Gli3R. Gli2f/f;Gli3d699;Wnt1-Cre mutants had reduced levels of GliA, and downstream genes such as Nrg1 were not transcribed. As a result, SMG development was arrested and SMG aplasia occurred (Fig. 7C). Interestingly, when GliR was overexpressed in NCC-derived SMG mesenchyme (ROSAGli3TFlag;Wnt1-Cre), endogenous GliFL was still processed into GliA, however the ratio of GliA:GliR was likely reduced. Under these conditions, SMG epithelium still underwent branching morphogenesis and developed–possibly due to meeting a threshold of functional, ciliary-processed GliA promoting Nrg1 expression (Fig. 7D). Although we have highlighted Nrg1 in our study and in our hypothesized model, we acknowledge that impaired expression of other factors (any GliA target that impacts proliferation, branching morphogenesis or proper innervation) could fit into this model and contribute to this phenotype.
Figure 7. Hypothesized model for primary cilia and GliA-dependent SMG development.
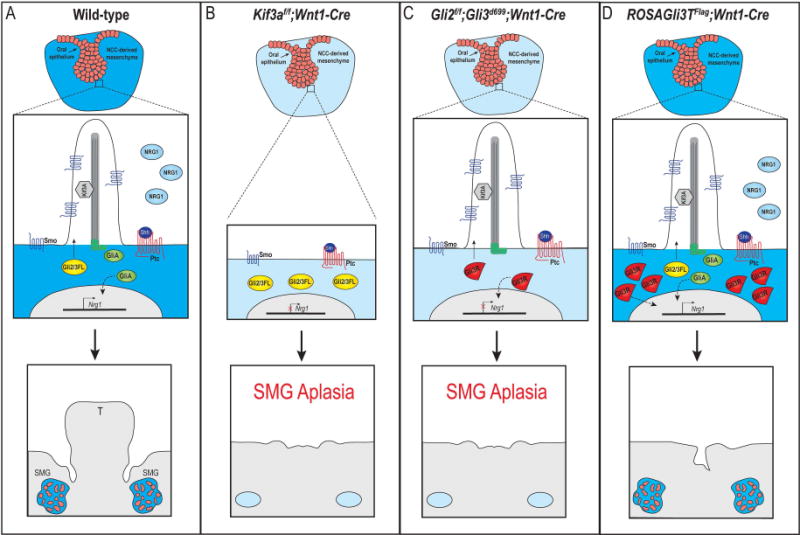
(A) In response to Shh binding to Ptc in wild-type SMG mesenchymal cells, Gli2/3FL is shuttled through the primary cilia and processed into GliA. GliA moves to the nucleus to induce transcription of downstream genes (either directly or indirectly; dotted black line), possibly including Nrg1. SMG organogenesis occurs. (B) In Kif3af/f;Wnt1-Cre mutant embryos, cilia are lost on the NCC-derived mesenchyme and Gli2/3FL cannot be processed into GliA. Nrg1 expression is not induced and SMG aplasia occurs. (C) In Gli2f/f;Gli3f/f;Wnt1-Cre mutant embryo, primary cilia remain, but functional GliA is not produced. Nrg1 expression is not induced and SMG aplasia occurs. (D) In ROSAGli3TFlag;Wnt1-Cre mutant embryos, primary cilia remain, but an excessive amount of GliR are produced. Functional GliA is still produced and moves to the nucleus to induce transcription of downstream genes (either directly or indirectly; doted black line), possibly including Nrg1. SMG organogenesis occurs. T, tongue.
Discussion
We previously reported the requirement of primary cilia-dependent Shh signaling in the development of several craniofacial components (Brugmann et al., 2010; Chang et al., 2016; Millington et al., 2017). Herein, we reported that the loss of primary cilia on NCC-derived SMG mesenchymal cells caused SMG aplasia. We confirmed this phenotype was not due to a lack of gland initiation, reduced Fgf signaling or a lack of NCC differentiation into parasympathetic nerves: three previously reported mechanisms of impaired SMG formation. We confirmed previous reports that Shh ligand was expressed in gland epithelium (Jaskoll et al., 2004) (Fig. 3) and additionally reported that the NCC-mesenchyme surrounding the gland epithelium was responsive to a Shh signal at the initial bud stage (Fig. 3). We further reported that the loss of Shh activity in the NCC-gland mesenchyme, via loss of GliA, phenocopied the Kif3af/f;Wnt1-Cre SMG phenotype (Fig. 4). Finally, we observed that reduced expression of Nrg ligands (Fig. 5) of the EGF/ErbB pathway, correlated with lack of SMG innervation and SMG aplasia (Fig. 6) when ciliogenesis or Shh signaling was impaired in the NCC-derived mesenchyme.
Based on these data, we hypothesize a mechanism by which primary cilia on NCC-derived SMG mesenchyme are required for proper SMG development. First, a Shh ligand from the prebud epithelium signals to the adjacent NCC-derived mesenchyme. The NCC-derived mesenchyme then receives and transduces the signal via the primary cilia. We then hypothesize that the Shh signal is transduced in the NCC-derived mesenchyme via cilia-dependent processing of the Gli transcription factors. GliA in the mesenchyme then induces the expression of downstream genes necessary for proliferation and branching of the SMG epithelium. Although we do not have direct evidence, we suggest that one of these genes could be the ErbB ligand, Nrg1. We further hypothesize that when primary cilia are lost on NCCs, the epithelium will still produce a Shh signal; however, the NCC-derived SMG mesenchymal cells, which lack primary cilia, will be incapable of transducing the Shh signal. Without transduction of a Shh signal, functional GliA protein is not produced and downstream genes like Nrg1 are not transcribed. Subsequently, the prebud epithelium does not receive the necessary signals to induce proliferation and branching, and innervation does not occur.
The role of NCC-derived mesenchyme in SMG development
Previous reports have shown that components of the Shh pathway were expressed in SMG epithelium during development (Jaskoll et al., 2002; Jaskoll et al., 2004; Mizukoshi et al., 2016), and that loss of Shh resulted in aplastic glands (Jaskoll et al., 2004; Mizukoshi et al., 2016). Based on our experimental design (loss of cilia in NCC-derived mesenchyme), we chose to further examine the role NCC-derived mesenchymal cells played in SMG organogenesis. NCC-derived mesenchyme is important for maintaining tissue-tissue interactions during development of several craniofacial components. Notably, the tongue, tooth, and whisker all require competent NCCs adjacent to either ectodermal or mesodermal derivatives to sustain a developmental program (Bitgood and McMahon, 1995; Chiang et al., 1999; Dassule et al., 2000; Jeong et al., 2004; Millington et al., 2017). It has previously been shown that via a Fgf-dependent mechanism, SMG mesenchyme was able to illicit branching when recombined with non-glandular epithelium (Wells et al., 2013). Our data further support an instructive role for the NCC-derived mesenchyme, and provide new evidence that an additional role of the mesenchyme is to transduce a Shh signal. These data suggest that whereas Shh signaling initiates in the epithelium, the mesenchyme must properly transduce the signal to induce expression of downstream genes that communicate back to the epithelium. Despite our data suggesting an important role for Shh signaling in the SMG mesenchyme, we concede that there must be other pathways active in the mesenchyme playing a role in this process because the loss of cilia on NCCs elicit a more severe phenotype (SMG aplasia) than that reported in Shh-null embryos (gland arrest at an early pseudoglandular stage) (Jaskoll et al., 2004). Since the primary cilia function as molecular signaling hubs, it is likely that loss of the cilia impacts other signaling pathway that are also required for SMG organogenesis. Understanding the participating pathways and their interplay will be an essential step in gaining insight into this process.
A glossal requirement for SMG development?
While the tongue and SMGs are distinct structures, they develop adjacent to one another, both spatially and temporally. Glossal development begins at e10.5 with two lateral lingual swellings comprised of NCC-derived mesenchyme arising from the floor of the mandible. These swellings then fuse and tissue-tissue interactions between the NCC-derived mesenchyme and mesodermally-derived muscle precursors facilitate the survival and proliferation of the glossal musculature (Noden and Trainor, 2005; Parada et al., 2012; Millington et al., 2017). SMG development has been tightly associated with glossal development (Knosp et al., 2012), as these structures develop bilaterally adjacent to the tongue in approximately the same temporal window. In addition to their spatial and temporal commonalities, the SMGs and the tongue also utilize several of the same signaling pathways, including Fgf, Wnt, and Hh for normal development (Han et al., 2012; Parada et al., 2012; Mattingly et al., 2015; Millington et al., 2017). While many aspects of these structures’ development are obviously linked, and gland development is thought to be dependent upon establishment of a proper oral cavity (Tucker, 2007); our data questions the extent of the requirement of the tongue for SMG development. While we presented instances where both structures are completely lost (Smof/f;Wnt1-Cre, Gli2f/f;Gli3f/f;Wnt1-Cre, Gli2f/f;Gli3d699;Wnt1-Cre), we also presented an example of severe hypoglossia/aglossia in which SMGs were maintained (ROSAGli3TFlag;Wnt1-Cre). ROSAGli3TFlag;Wnt1-Cre mutants have normal SMG tissue with expected epithelial and mesenchymal contributions, but development of glossal tissue is severely impacted. Observations such as this challenge the notion that SMG development is reliant upon tongue development. Understanding the molecular requirements for the development of each of these structures is a topic of ongoing study in our lab.
Requirement of Gli activator in SMG development
Transduction of a Shh signal requires Gli transcription factor activity. Shh-target gene activation or repression is dependent on the balance between GliA and GliR isoforms; however, several methods exist to achieve activation or repression of said targets (Aberger and Ruiz, 2014; Falkenstein and Vokes, 2014). The threshold activation model suggests that the presence or absence of a threshold level of GliA defines a transcriptional response (Falkenstein and Vokes, 2014). Alternatively, the threshold repression model suggests that the presence or absence of a threshold level of repression by GliR dictates a transcriptional response (Falkenstein and Vokes, 2014). Finally, the ratio sensing model suggests that it is not a threshold level of GliA or GliR that is important, rather it is the relative ratio between GliA and GliR that drives a specific response (Falkenstein and Vokes, 2014). We examined mutants in which either GliA and GliR isoforms were perturbed to reveal a particular sensitivity of the SMG to the loss of a threshold of GliA activity. In the instance of the Gli2f/f;Gli3d699;Wnt1-Cre mutant embryos, GliA levels were significantly reduced and SMGs failed to develop properly. Alternatively, when we did not perturb endogenous expression of GliA and just overexpressed GliR (ROSAGli3TFlag;Wnt1-Cre), thus altering the ratio of GliA:GliR, SMGs developed. Given these results we favor the threshold activation model for Gli-mediated SMG development. Our future work will focus on identifying direct targets of GliA that are necessary for SMG and mandibular development.
We also investigated the spatiotemporal details of Shh signaling during SMG development. In agreement with previous reports (Jaskoll et al., 2004), we observed Shh expression at both the initial bud and pseudoglandular phases, however we detected Shh-responsive cells only at the initial bud stage (Fig. 3). The lack of Shh-responsive cells at the pseudoglandular stage suggested that while Shh may be responsible for initiating the process of branching morphogenesis, a Shh-independent signal may be necessary to maintain this process. Understanding how different pathways coordinate and interact with each other to promote SMG development will be essential for gaining a deeper understanding of SMG organogenesis.
Neuregulin-1 potentially mediates mesenchymal signaling back to the epithelium
While we have further supported the hypothesis that SMG mesenchyme is indispensable for epithelial development, the exact mechanism by which this occurs has not been fully deciphered. Egf/ErbB signaling was previously shown to be required for epithelial proliferation in the SMG (Miyazaki et al., 2004; Mizukoshi et al., 2016), and ex vivo experiments have shown that Shh ligand can increase ErbB ligand expression (Mizukoshi et al., 2016). We examined one ErbB ligand, Nrg1. We characterized Nrg1 expression during SMG development, and found its expression coincides with our hypothesized role for the protein as a mesenchymal factor that exerts an extrinsic effect on the SMG epithelium. While our current studies are only correlative, together they support a hypothesized mechanism by which primary cilia-dependent GliA activity could induce targets like Nrg1 in the mesenchyme to promote epithelial proliferation and branching. Our future studies will focus on determining if Nrg1 is a direct target of GliA in the developing mandible. Furthermore, since previous studies have reported that a loss of Wnt (Knosp et al., 2015) or neurturin (Knox et al., 2013) signaling can result in lack of innervation and impaired SMG development, it will be interesting to determine if other signaling pathways are also impaired in ciliopathic mutants.
In summary, our data suggest that primary cilia are necessary for SMG development. Patients with ciliopathies frequently present with phenotypes associated with aberrant tissue-tissue interactions like polydactyly, cranial midline disruptions, tongue defects, and kidney cysts (Waters and Beales, 2011). Currently, we are not aware of any reports of salivary gland-associated phenotypes in ciliopathy patients. Moving forward, it will be interesting to more closely examine human ciliopathy patients for SMG defects and identify additional molecular pathways which signal through the cilia during SMG organogenesis.
Experimental Procedures
Mouse strains
The Wnt1-Cre, ROSAmT/mG, ROSAGli3TFlag c/c, and Smof/f (Smotm2/Amc/J) mouse strains were purchased from Jackson Laboratory. R26R (B6;129S4-Gt(ROSA)26Sortm1Sor/J) and Ptc-LacZ (Ptch1tm1Mps/J) mouse strains were generously acquired from Dr. Rolf Stottmann at Cincinnati Children’s Hospital Medical Center. Kif3af/f mice were obtained from Dr. Bradley Yoder at University of Alabama at Birmingham, Gli2f/f were provided by Dr. Alexandra Joyner at Memorial Sloan-Kettering Cancer Center, Gli3d699 mice were acquired from Dr. Chi-Chung Hui at Hospital for Sick Kids, Canada, and ShhGFP/+ mice were a gift from Dr. Yu Lan at Cincinnati Children’s Hospital Medical Center. All mice were maintained on a CD1 background. All mouse usage was approved by the Institutional Animal Care and Use Committee (IACUC) and maintained by the Veterinary Services at Cincinnati Children’s Hospital Medical Center.
Embryo collection and tissue preparation
Timed mouse matings were performed and embryonic day (e) 0.5 was assigned the day a vaginal plug was discovered. Embryos were harvested between e11.5-15.5, collected in PBS, and fixed in 4% paraformaldehyde (PFA) overnight at 4°C, unless otherwise noted. For paraffin embedding, embryos were dehydrated through an ethanol series and stored in 70% ethanol before washing in xylene and embedding in paraffin. 8μm sections were cut on a microtome. For cryoembedding, embryos were equilibrated into 10% sucrose, then incubated in 30% sucrose overnight at 4°C, embedded in OCT, and snap frozen in a dry ice-ethanol slurry. Blocks were stored at −80°C and 10μm sections were cut on a cryostat.
LacZ detection
ß-galactosidase activity was detected by X-gal staining. Embryos were collected in cold PBS and fixed with 0.4% PFA for 2 hours at room temperature, equilibrated into 10% sucrose and then 30% sucrose overnight, embedded in OCT, and snap frozen in a dry ice-ethanol slurry. 10μm sections were cut on a cryostat and X-gal staining was performed at 37°C.
Histology and immunostaining
Hematoxylin and eosin (H&E) staining was performed using standard protocols. Sectioned immunostaining was performed using standard protocols, including antigen retrieval and incubation with primary antibody in 10% normal goat serum overnight at 4°C. All secondary antibodies were used at a 1:1000 dilution in 10% normal goat serum at room temperature for 1 hour. For whole mount immunostaining, dissected embryos were blocked at room temperature for 24 hours in 10% normal goat serum and 0.1% triton-X, then incubated with primary antibody for 2-4 days at room temperature, and finally incubated with secondary antibody at room temperature for 2 days. Stained embryos were cleared for ease of imaging using RIMS solution for 1-2 days and imaged in RIMS using Leica imaging software. Primary antibodies used: anti-Krt8 (TROMA-I, RRID: AB_531826, University of Iowa Developmental Studies Hybridoma Bank, 1:50), anti-beta III Tubulin (ab18207, Abcam, 1:1000), anti-Ecadherin (610182, BD Biosciences, 1:250), anti-Sox9 (ab71762, Abcam, 1:2000), anti-Cleaved caspase 3 (9661S, Cell Signaling Technology, 1:1000), anti-Pdgfrβ (ab32570, Abcam, 1:100), anti-Neuregulin-1 (MA5-12896, Life Sciences 1:50), and anti-Sonic Hedgehog (sc-9024, Santa Cruz).
In situ hybridization
Sectioned in situ hybridization was performed using a modified version of the Gallus gallus expression in situ hybridization analysis (GEISHA) protocol (Darnell et al., 2007). Digoxigenin-labeled anti-sense riboprobes for Fgf10, Fgf2, Etv5, Spry1 and Spry2 were generated using primers designed based on sequences from Gene Paint (www.genepaint.org) and mouse e11.5 cDNA generated from mouse facial prominences.
Quantitative RT-PCR
mRNA was prepared with TRIzol reagent (Thermo Fisher Scientific) from e14.5 wild-type, Kif3af/f;Wnt1-Cre, or Gli2f/f;Gli3d699;Wnt1-Cre mutant SMGs. cDNA was made through reverse transcription, and utilized for quantitative PCR. Using SsoAdvanced Universal SYBR Green Supermix, BioRad, expression of Egf, Nrg1, Nrg2, Nrg3 was examined and quantified in relation to GAPDH. The following primers were used: Egf forward: CCTCATATGATGGATACTGCCTCAA; Egf reverse: ACCAGTGCCACCATGCAGA; Nrg1 forward: CATCACAACCCTGCACATCA; Nrg1 reverse: CCTGGGTCGTTTCGTATTCC; Nrg2 forward: GGATGGCAAGGAACTCAACC, and Nrg2 reverse: TCGGCCTCACAGACGTACT; Nrg3 forward: TTACGCTGTAGCGACTGCATC; Nrg3 reverse: GCCTACCACGATCCATTTAAGC. Student’s t-test was used to determine significance. p-values less than or equal to 0.05 (95% confidence level) were considered as statistically significant differences.
Acknowledgments
This work was funded by NIH/NIDCR R01DE023804 awarded to S.A.B
References
- Aberger F, Ruiz IAA. Context-dependent signal integration by the GLI code: the oncogenic load, pathways, modifiers and implications for cancer therapy. Semin Cell Dev Biol. 2014;33:93–104. doi: 10.1016/j.semcdb.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell HF, Donis-Keller H, Helms C, Hing AV, Heng HH, Koop B, Martindale D, Rommens JM, Tsui LC, Scherer SW. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996;14:353–356. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- Bitgood MJ, McMahon AP. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev Biol. 1995;172:126–138. doi: 10.1006/dbio.1995.0010. [DOI] [PubMed] [Google Scholar]
- Bose J, Grotewold L, Ruther U. Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum Mol Genet. 2002;11:1129–1135. doi: 10.1093/hmg/11.9.1129. [DOI] [PubMed] [Google Scholar]
- Brugmann SA, Allen NC, James AW, Mekonnen Z, Madan E, Helms JA. A primary cilia-dependent etiology for midline facial disorders. Hum Mol Genet. 2010;19:1577–1592. doi: 10.1093/hmg/ddq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain CE, Jeong J, Guo C, Allen BL, McMahon AP. Notochord-derived Shh concentrates in close association with the apically positioned basal body in neural target cells and forms a dynamic gradient during neural patterning. Development. 2008;135:1097–1106. doi: 10.1242/dev.013086. [DOI] [PubMed] [Google Scholar]
- Chang CF, Chang YT, Millington G, Brugmann SA. Craniofacial Ciliopathies Reveal Specific Requirements for GLI Proteins during Development of the Facial Midline. PLoS Genet. 2016;12:e1006351. doi: 10.1371/journal.pgen.1006351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzeli L, Gaete M, Tucker AS. Fgf10-Sox9 are essential for establishment of distal progenitor cells during salivary gland development. Development. 2017 doi: 10.1242/dev.146019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- Chiang C, Swan RZ, Grachtchouk M, Bolinger M, Litingtung Y, Robertson EK, Cooper MK, Gaffield W, Westphal H, Beachy PA, Dlugosz AA. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol. 1999;205:1–9. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- Cobourne MT, Green JB. Hedgehog signalling in development of the secondary palate. Front Oral Biol. 2012;16:52–59. doi: 10.1159/000337543. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- D’Amico-Martel A, Noden DM. Contributions of placodal and neural crest cells to avian cranial peripheral ganglia. Am J Anat. 1983;166:445–468. doi: 10.1002/aja.1001660406. [DOI] [PubMed] [Google Scholar]
- Darnell DK, Kaur S, Stanislaw S, Davey S, Konieczka JH, Yatskievych TA, Antin PB. GEISHA: an in situ hybridization gene expression resource for the chicken embryo. Cytogenet Genome Res. 2007;117:30–35. doi: 10.1159/000103162. [DOI] [PubMed] [Google Scholar]
- Dassule HR, Lewis P, Bei M, Maas R, McMahon AP. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development. 2000;127:4775–4785. doi: 10.1242/dev.127.22.4775. [DOI] [PubMed] [Google Scholar]
- De Moerlooze L, Spencer-Dene B, Revest J, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- Falkenstein KN, Vokes SA. Transcriptional regulation of graded Hedgehog signaling. Semin Cell Dev Biol. 2014;33:73–80. doi: 10.1016/j.semcdb.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira JN, Hoffman MP. Interactions between developing nerves and salivary glands. Organogenesis. 2013;9:199–205. doi: 10.4161/org.25224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavelli L, Zanacca C, Caselli G, Banchini G, Dubourg C, David V, Odent S, Gurrieri F, Neri G. Solitary median maxillary central incisor syndrome: clinical case with a novel mutation of sonic hedgehog. Am J Med Genet A. 2004;127a:93–95. doi: 10.1002/ajmg.a.20685. [DOI] [PubMed] [Google Scholar]
- Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP. Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- Häärä O, Fujimori S, Schmidt-Ullrich R, Hartmann C, Thesleff I, Mikkola ML. Ectodysplasin and Wnt pathways are required for salivary gland branching morphogenesis. Development. 2011;138:2681. doi: 10.1242/dev.057711. [DOI] [PubMed] [Google Scholar]
- Haara O, Koivisto T, Miettinen PJ. EGF-receptor regulates salivary gland branching morphogenesis by supporting proliferation and maturation of epithelial cells and survival of mesenchymal cells. Differentiation. 2009;77:298–306. doi: 10.1016/j.diff.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Han D, Zhao H, Parada C, Hacia JG, Bringas P, Jr, Chai Y. A TGFbeta-Smad4-Fgf6 signaling cascade controls myogenic differentiation and myoblast fusion during tongue development. Development. 2012;139:1640–1650. doi: 10.1242/dev.076653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118:517–528. doi: 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Hashizume A, Hieda Y. Hedgehog peptide promotes cell polarization and lumen formation in developing mouse submandibular gland. Biochem Biophys Res Commun. 2006;339:996–1000. doi: 10.1016/j.bbrc.2005.11.106. [DOI] [PubMed] [Google Scholar]
- Hebrok M, Kim SK, St Jacques B, McMahon AP, Melton DA. Regulation of pancreas development by hedgehog signaling. Development. 2000;127:4905–4913. doi: 10.1242/dev.127.22.4905. [DOI] [PubMed] [Google Scholar]
- Hu D, Helms JA. The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development. 1999;126:4873–4884. doi: 10.1242/dev.126.21.4873. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–537. doi: 10.1146/annurev-cellbio-092910-154048. [DOI] [PubMed] [Google Scholar]
- Jaskoll T, Abichaker G, Witcher D, Sala FG, Bellusci S, Hajihosseini MK, Melnick M. FGF10/FGFR2b signaling plays essential roles during in vivo embryonic submandibular salivary gland morphogenesis. BMC Dev Biol. 2005;5:11. doi: 10.1186/1471-213X-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskoll T, Leo T, Witcher D, Ormestad M, Astorga J, Bringas P, Jr, Carlsson P, Melnick M. Sonic hedgehog signaling plays an essential role during embryonic salivary gland epithelial branching morphogenesis. Dev Dyn. 2004;229:722–732. doi: 10.1002/dvdy.10472. [DOI] [PubMed] [Google Scholar]
- Jaskoll T, Zhou YM, Chai Y, Makarenkova HP, Collinson JM, West JD, Hajihosseini MK, Lee J, Melnick M. Embryonic submandibular gland morphogenesis: stage-specific protein localization of FGFs, BMPs, Pax6 and Pax9 in normal mice and abnormal SMG phenotypes in FgfR2-IIIc(+/Delta), BMP7(−/−) and Pax6(−/−) mice. Cells Tissues Organs. 2002;170:83–98. doi: 10.1159/000046183. [DOI] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes & Development. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knosp WM, Knox SM, Hoffman MP. Salivary gland organogenesis. Wiley Interdiscip Rev Dev Biol. 2012;1:69–82. doi: 10.1002/wdev.4. [DOI] [PubMed] [Google Scholar]
- Knosp WM, Knox SM, Lombaert IM, Haddox CL, Patel VN, Hoffman MP. Submandibular parasympathetic gangliogenesis requires sprouty-dependent Wnt signals from epithelial progenitors. Dev Cell. 2015;32:667–677. doi: 10.1016/j.devcel.2015.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox SM, Lombaert IM, Haddox CL, Abrams SR, Cotrim A, Wilson AJ, Hoffman MP. Parasympathetic stimulation improves epithelial organ regeneration. Nat Commun. 2013;4:1494. doi: 10.1038/ncomms2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox SM, Lombaert IM, Reed X, Vitale-Cross L, Gutkind JS, Hoffman MP. Parasympathetic innervation maintains epithelial progenitor cells during salivary organogenesis. Science. 2010;329:1645–1647. doi: 10.1126/science.1192046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama N, Hayashi T, Mizukoshi K, Matsumoto T, Gresik EW, Kashimata M. Extracellular regulated kinase5 is expressed in fetal mouse submandibular glands and is phosphorylated in response to epidermal growth factor and other ligands of the ErbB family of receptors. Dev Growth Differ. 2012;54:801–808. doi: 10.1111/dgd.12008. [DOI] [PubMed] [Google Scholar]
- Kwon HR, Larsen M. The contribution of specific cell subpopulations to submandibular salivary gland branching morphogenesis. Curr Opin Genet Dev. 2015;32:47–54. doi: 10.1016/j.gde.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin NM, Teillet MA. Experimental analysis of the migration and differentiation of neuroblasts of the autonomic nervous system and of neurectodermal mesenchymal derivatives, using a biological cell marking technique. Dev Biol. 1974;41:162–184. doi: 10.1016/0012-1606(74)90291-7. [DOI] [PubMed] [Google Scholar]
- Mattingly A, Finley JK, Knox SM. Salivary gland development and disease. Wiley Interdiscip Rev Dev Biol. 2015;4:573–590. doi: 10.1002/wdev.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick M, Phair RD, Lapidot SA, Jaskoll T. Salivary gland branching morphogenesis: a quantitative systems analysis of the Eda/Edar/NFkappaB paradigm. BMC Dev Biol. 2009;9:32. doi: 10.1186/1471-213X-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millington G, Elliott KH, Chang YT, Chang CF, Dlugosz A, Brugmann SA. Cilia-dependent GLI processing in neural crest cells is required for tongue development. Dev Biol. 2017;424:124–137. doi: 10.1016/j.ydbio.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki Y, Nakanishi Y, Hieda Y. Tissue interaction mediated by neuregulin-1 and ErbB receptors regulates epithelial morphogenesis of mouse embryonic submandibular gland. Dev Dyn. 2004;230:591–596. doi: 10.1002/dvdy.20078. [DOI] [PubMed] [Google Scholar]
- Mizukoshi K, Koyama N, Hayashi T, Zheng L, Matsuura S, Kashimata M. Shh/Ptch and EGF/ErbB cooperatively regulate branching morphogenesis of fetal mouse submandibular glands. Dev Biol. 2016;412:278–287. doi: 10.1016/j.ydbio.2016.02.018. [DOI] [PubMed] [Google Scholar]
- Morava E, Bartsch O, Czako M, Frensel A, Kalscheuer V, Karteszi J, Kosztolanyi G. Small inherited terminal duplication of 7q with hydrocephalus, cleft palate, joint contractures, and severe hypotonia. Clin Dysmorphol. 2003;12:123–127. doi: 10.1097/01.mcd.0000059768.40218.9b. [DOI] [PubMed] [Google Scholar]
- Muenke M, Gurrieri F, Bay C, Yi DH, Collins AL, Johnson VP, Hennekam RC, Schaefer GB, Weik L, Lubinsky MS, et al. Linkage of a human brain malformation, familial holoprosencephaly, to chromosome 7 and evidence for genetic heterogeneity. Proc Natl Acad Sci U S A. 1994;91:8102–8106. doi: 10.1073/pnas.91.17.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nanni L, Ming JE, Bocian M, Steinhaus K, Bianchi DW, Die-Smulders C, Giannotti A, Imaizumi K, Jones KL, Campo MD, Martin RA, Meinecke P, Pierpont ME, Robin NH, Young ID, Roessler E, Muenke M. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet. 1999;8:2479–2488. doi: 10.1093/hmg/8.13.2479. [DOI] [PubMed] [Google Scholar]
- Nedvetsky PI, Emmerson E, Finley JK, Ettinger A, Cruz-Pacheco N, Prochazka J, Haddox CL, Northrup E, Hodges C, Mostov KE, Hoffman MP, Knox SM. Parasympathetic innervation regulates tubulogenesis in the developing salivary gland. Dev Cell. 2014;30:449–462. doi: 10.1016/j.devcel.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DA, Manhardt C, Kamath V, Sui Y, Santamaria-Pang A, Can A, Bello M, Corwin A, Dinn SR, Lazare M, Gervais EM, Sequeira SJ, Peters SB, Ginty F, Gerdes MJ, Larsen M. Quantitative single cell analysis of cell population dynamics during submandibular salivary gland development and differentiation. Biol Open. 2013;2:439–447. doi: 10.1242/bio.20134309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005;207:575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada C, Han D, Chai Y. Molecular and cellular regulatory mechanisms of tongue myogenesis. J Dent Res. 2012;91:528–535. doi: 10.1177/0022034511434055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel VN, Rebustini IT, Hoffman MP. Salivary gland branching morphogenesis. Differentiation. 2006;74:349–364. doi: 10.1111/j.1432-0436.2006.00088.x. [DOI] [PubMed] [Google Scholar]
- Rebustini IT, Patel VN, Stewart JS, Layvey A, Georges-Labouesse E, Miner JH, Hoffman MP. Laminin alpha5 is necessary for submandibular gland epithelial morphogenesis and influences FGFR expression through beta1 integrin signaling. Dev Biol. 2007;308:15–29. doi: 10.1016/j.ydbio.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal. 2012;5:re6. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Rothova M, Thompson H, Lickert H, Tucker AS. Lineage tracing of the endoderm during oral development. Dev Dyn. 2012;241:1183–1191. doi: 10.1002/dvdy.23804. [DOI] [PubMed] [Google Scholar]
- Schimmenti LA, de la Cruz J, Lewis RA, Karkera JD, Manligas GS, Roessler E, Muenke M. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am J Med Genet A. 2003;116a:215–221. doi: 10.1002/ajmg.a.10884. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol. 1962;15:363–377. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teshima THN, Lourenco SV, Tucker AS. Multiple Cranial Organ Defects after Conditionally Knocking Out Fgf10 in the Neural Crest. Frontiers in Physiology. 2016;7:488. doi: 10.3389/fphys.2016.00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker AS. Salivary gland development. Semin Cell Dev Biol. 2007;18:237–244. doi: 10.1016/j.semcdb.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Vokes SA, Ji H, McCuine S, Tenzen T, Giles S, Zhong S, Longabaugh WJ, Davidson EH, Wong WH, McMahon AP. Genomic characterization of Gli-activator targets in sonic hedgehog-mediated neural patterning. Development. 2007;134:1977–1989. doi: 10.1242/dev.001966. [DOI] [PubMed] [Google Scholar]
- Vokes SA, Ji H, Wong WH, McMahon AP. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 2008;22:2651–2663. doi: 10.1101/gad.1693008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011;26:1039–1056. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells KL, Gaete M, Matalova E, Deutsch D, Rice D, Tucker AS. Dynamic relationship of the epithelium and mesenchyme during salivary gland initiation: the role of Fgf10. Biol Open. 2013;2:981–989. doi: 10.1242/bio.20135306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijgerde M, McMahon JA, Rule M, McMahon AP. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002;16:2849–2864. doi: 10.1101/gad.1025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Fukumoto E, Yoshizaki K, Iwamoto T, Yamada A, Tanaka K, Suzuki H, Aizawa S, Arakaki M, Yuasa K, Oka K, Chai Y, Nonaka K, Fukumoto S. Platelet-derived growth factor receptor regulates salivary gland morphogenesis via fibroblast growth factor expression. J Biol Chem. 2008;283:23139–23149. doi: 10.1074/jbc.M710308200. [DOI] [PubMed] [Google Scholar]
- Ybot-Gonzalez P, Cogram P, Gerrelli D, Copp AJ. Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development. 2002;129:2507–2517. doi: 10.1242/dev.129.10.2507. [DOI] [PubMed] [Google Scholar]