

Abstract
Refractive errors, including myopia, are the most frequent eye disorders worldwide and an increasingly common cause of blindness. This genome-wide association meta-analysis in 160,420 participants and replication in 95,505 participants, increased the established independent signals from 37 to 161 and revealed high genetic correlation between Europeans and Asians (>0.78). Expression experiments and comprehensive in silico analyses identified retinal cell physiology and light processing as prominent mechanisms, and functional contributions to refractive error development in all cell types of the neurosensory retina, retinal pigment epithelium, vascular endothelium and extracellular matrix. Newly identified genes elicited novel mechanisms such as rod and cone bipolar synaptic neurotransmission, anterior segment morphology, and angiogenesis. Thirty-one loci resided in or near regions transcribing small RNAs, suggesting a role for post-transcriptional regulation. Our results support the notion that refractive errors are caused by a light-dependent retina-to-sclera signaling cascade, and delineate potential pathobiological molecular drivers.
INTRODUCTION
Refractive errors are common optical aberrations determined by mismatches in the focusing power of the cornea, lens and axial length of the eye. Their distribution is rapidly shifting towards myopia, or nearsightedness, all over the world. The myopia boom is particularly prominent in urban East Asia where up to 95% of twenty-year-olds in cities such as Seoul and Singapore have this refractive error1–4. Myopia prevalence is also rising throughout Western Europe and the USA, affecting ~50% of young adults in these regions5,6. While refractive errors can be optically corrected, even at moderate values they carry significant risk of ocular complications with high economic burden7–9. One in three individuals with high myopia (−6 diopters or worse) will develop irreversible visual impairment or blindness, mostly due to myopic macular degeneration, retinal detachment, or glaucoma10,11. At the other extreme, high hyperopia predisposes to strabismus, amblyopia and angle-closure glaucoma10,12.
Refractive errors result from a complex interplay of lifestyle and genetic factors. The most established lifestyle factors for myopia are high education, lack of outdoor exposure, and excessive near work3. Recent research has identified many genetic variants for refractive errors, myopia, and axial length13–25. Two large studies, the international Consortium for Refractive Error and Myopia (CREAM)26 and the personal genomics company 23andMe, Inc.17,27 have provided the most comprehensive results.28
Given that only 3.6% of the variance of the refractive error trait was explained by the identified genetic variants26, we presumed a high missing heritability. We therefore combined CREAM and 23andMe, and expanded the study sample to 160,420 individuals from a mixed ancestry population with quantitative information on refraction for a genome-wide association (GWAS) meta-analysis. Index variants were tested for replication in an independent cohort consisting of 95,505 individuals from the UK Biobank. We conducted systematic comparisons to assess differences in genetic inheritance and distribution of risk variants between Europeans and Asians. Polygenic risk analyses were performed to evaluate the contribution of the identified variants to the risk of myopia and hyperopia. Finally, we integrated expression data and bioinformatics on the identified genes to gain insight into the possible mechanisms underlying the genetic associations.
RESULTS
Susceptibility loci for refractive error
We performed a GWAS meta-analysis on adult untransformed spherical equivalent (SphE) using summary statistics from 37 studies from CREAM and on age of diagnosis of myopia (AODM) from two cohorts from 23andMe (Supplementary Figure 1, Supplementary Table 1a)26,27. Analyses were based on ~11 million genetic variants (SNPs, insertions and deletions) genotyped or imputed to 1000 Genomes Project Phase I reference panel (version 3, March 2012 release29) that passed extensive quality control (Supplementary Figures 2–4, Supplementary Table 1b).
Meta-analyses were conducted in three stages: Stage 1 CREAM (CREAM-EUR, N=44,192; CREAM-ASN, N=11,935); Stage 2 23andMe (N=104,293; Online Methods); Stage 3 joint meta-analysis of Stage 1 and 2. As CREAM and 23andMe applied different phenotype measures, we used signed Z-scores as the mean per-allele effect size and assigned equal weights to CREAM and 23andMe. We identified 7,967 genome-wide significant genetic variants clustering in 140 loci (Figure 1a,b; Supplementary Figure 5–6, Supplementary Table 2-5, Supplementary Data 1-2), replicating all 37 previously discovered loci and finding 104 novel loci. We applied genomic control at each stage and checked for population stratification using LD score regression30 (Stage 1-2 inflation factors (λGC) <1.1 and LD score regression intercepts (LDSCintercept) 0.892-1.023; Supplementary Table 6; Supplementary Figure 6-7). At Stage 3, we observed a genomic inflation (λGC=1.129; Supplementary Figure 6), probably due to true polygenicity rather than population stratification or cryptic relatedness31. LDSCintercept remained undetermined due to mixed ancestry.
Figure 1. GWAS meta-analysis identifies 140 loci for refractive error (Stage 3).
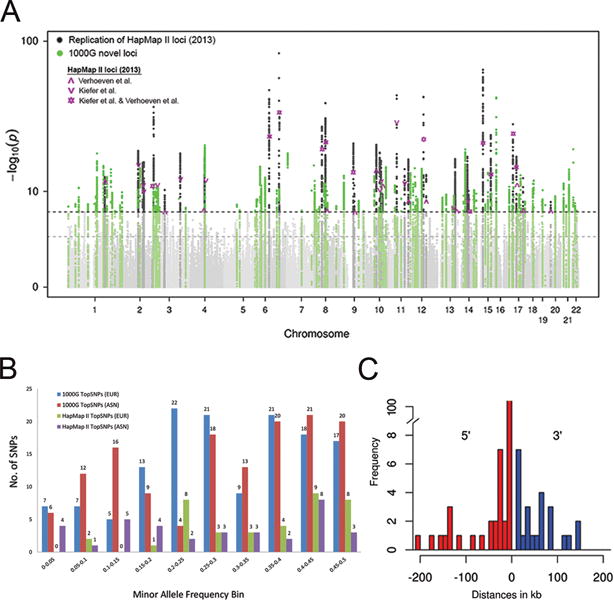
(a) We conducted a meta-analysis of genome-wide single-variant analyses for >10 million variants in 160,420 participants of CREAM and 23andMe (Stage 3). Shown is the Manhattan plot depicting P for association, highlighting new (P < 5 × 10−8 for the first time; green) and known (dark grey) refractive error loci previously found using HapMap II imputations from Kiefer et al.27 and Verhoeven et al.26 (Table 1). The horizontal lines indicate suggestive significance (P=1×10−5) or genome-wide significance (P=5×10−8). (b) We compared the minor allele frequencies of the 140 discovered index variants based on 1000G (blue: Europeans; red: Asians) to the minor allele frequencies of the previously found genetic variants based on HapMap II (green: Europeans; purple: Asians). Observed are an increase in genetic variants found across all minor allele frequency bins increase, including the lower minor allele frequency bins. (c) We annotated the 167 loci to genes using wANNOVAR. Shown are the distances between index variants from the nearest gene and its gene on the 5′ and/or 3′ site. The majority of index variants (84%) were at a distance of less than 50 kb up- or downstream from the annotated gene.
To detect the presence of multiple independent signals at the discovered loci, a stepwise conditional analysis was performed with GCTA-COJO32 on summary statistics from all European cohorts (N=148,485) using the Rotterdam Study I-III (RS I-III) as a reference panel for LD structure (NRSI-III=10,775). This analysis yielded 27 additional independent variants, resulting in a total of 167 loci (Supplementary Table 2).
We advanced these loci for replication in a GWAS of refractive error carried out by the UK Biobank Eye & Vision (UKEV) Consortium (N=95,505)33 (Online Methods). Six out of the 167 variants were not considered for replication analysis. One of these five variants (rs3138141, RDH5) was identified previously and therefore still considered as a refractive error risk variant26,27. The remaining 161 genetic variants were tested for replication. 86% (138/161) of the candidate variants replicated significantly: 104 (65%) replicated surpassing genome-wide significance and 34 replicated surpassing Bonferroni correction (P<3.0×10−4; 21.1%); another 12 showed nominal evidence for replication (0.05<P<3.0×10−4; 7.5%) and only 11 (7%) did not replicate at all (Table 1, Supplementary Table 2).
Table 1.
Results of the meta-analysis of CREAM and 23andMe for the previously-identified loci and a subset of the newly-identified loci, and replication in UK Biobank
| a Replication of the HapMap II index variants for refractive error per locus in the Stage 3 meta-analysis
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Position | Nearest Loci And Gene(s) |
Effect Allele |
Other Allele |
EAF EUR |
EAF ASN |
Z-score | Direction | P value | HetISq | HetPVal | Sample Size (N) |
HapMap II Discovery (2013) |
Category | P value Replication |
| rs10500355 | 16 | 7459347 | RBFOX1 | A | T | 0.354 | 0.133 | −13.73 | −− | 6.49E-43 | 9.1 | 2.93E-07 | 160,139 | Kiefer et al. & Verhoeven et al. | I | 2.50E-48 |
| rs11145465 | 9 | 71766593 | TJP2 | A | C | 0.212 | NA | −9.55 | −− | 1.35E-21 | 46.3 | 0.1722 | 153,174 | Kiefer et al. & Verhoeven et al. | I | 1.00E-10 |
| rs11178469 | 12 | 71275137 | PTPRR | T | C | 0.752 | 0.638 | −7.40 | −− | 1.33E-13 | 0 | 0.6989 | 160,139 | Verhoeven et al. | II (CREAM) | 2.60E-04 |
| rs11602008 | 11 | 40149305 | LRRC4C | A | T | 0.822 | 0.749 | 13.98 | ++ | 2.12E-44 | 22.5 | 1.56E-10 | 157,505 | Kiefer et al. | II (23andMe) | 2.90E-47 |
| rs12193446 | 6 | 129820038 | BC035400, LAMA2 | A | G | 0.906 | NA | −19.43 | −− | 4.21E-84 | 16.8 | 5.72E-15 | 150,269 | Kiefer et al. & Verhoeven et al. | I | 4.60E-106 |
| rs1550094 | 2 | 233385396 | CHRNG, PRSS56 | A | G | 0.701 | 0.705 | 12.74 | ++ | 3.64E-37 | 26.3 | 0.003 | 159,422 | Kiefer et al. & Verhoeven et al., Kiefer et al. | I | 4.10E-59 |
| rs1649068 | 10 | 60304864 | BICC1 | A | C | 0.475 | 0.504 | −9.44 | −− | 3.77E-21 | 0 | 0.712 | 160,144 | Verhoeven et al. | I | 7.50E-11 |
| rs17382981 | 10 | 94953258 | CYP26A1,MYOF | T | C | 0.417 | 0.190 | −6.31 | −− | 2.72E-10 | 67.9 | 0.077 | 155,332 | Verhoeven et al. | II (CREAM) | 4.10E-07 |
| rs17428076 | 2 | 172851936 | HAT1, METAP1D | C | G | 0.768 | 0.854 | −8.18 | −− | 2.77E-16 | 0 | 0.003 | 160,151 | Kiefer et al. | II (23andMe) | 7.50E-08 |
| rs1858001 | 1 | 207488004 | C4BPA,CD55 | C | G | 0.676 | 0.415 | 7.28 | ++ | 3.45E-13 | 59.6 | 0.020 | 160,149 | Verhoeven et al. | II (CREAM) | 6.70E-20 |
| rs1954761 | 11 | 105596885 | GRIA4 | T | C | 0.371 | 0.377 | −8.40 | −− | 4.57E-17 | 0 | 0.911 | 160,122 | Verhoeven et al. | I | 1.20E-16 |
| rs2155413 | 11 | 84634790 | DLG2 | A | C | 0.482 | 0.655 | −7.76 | −− | 8.85E-15 | 0 | 2.99E-04 | 159,504 | Kiefer et al. | II (23andMe) | 1.10E-17 |
| rs235770 | 20 | 6761765 | BMP2 | T | C | 0.372 | 0.388 | −5.93 | −− | 3.11E-09 | 0 | 0.547 | 157,521 | Verhoeven et al. | II (23andMe) | 4.80E-11 |
| rs2573081 | 2 | 178828507 | PDE11A | C | G | 0.524 | 0.538 | 8.21 | ++ | 2.18E-16 | 47.6 | 0.167 | 160,126 | Kiefer et al. | II (23andMe) | 1.60E-29 |
| rs2753462 | 14 | 60850703 | JB175233, C14orf39 | C | G | 0.296 | 0.568 | −6.49 | −− | 8.37E-11 | 73.9 | 0.050 | 157,352 | Verhoeven et al. | II (CREAM) | 2.00E-15 |
| rs2855530 | 14 | 54421917 | BMP4 | C | G | 0.507 | 0.474 | −8.58 | −− | 9.87E-18 | 41.7 | 0.190 | 160,092 | Kiefer et al. | I | 4.80E-22 |
| rs2908972 | 17 | 11407259 | SHISA6 | A | T | 0.415 | 0.484 | −11.13 | −− | 9.46E-29 | 23 | 0.254 | 160,123 | Kiefer et al. & Verhoeven et al. | I | 6.10E-29 |
| rs3138141 | 12 | 56115778 | BLOC1S1-RDH5,RDH5 | A | C | 0.214 | 0.147 | 13.80 | ++ | 2.46E-43 | 3.2 | 5.05E-07 | 157,531 | Kiefer et al. & Verhoeven et al. | I | 2.30E-56 |
| rs4687586 | 3 | 53837971 | CACNA1D | C | G | 0.691 | NA | −6.55 | −− | 5.86E-11 | 0 | 0.605 | 150,217 | Verhoeven et al. | III | 1.60E-08 |
| rs4793501 | 17 | 68718734 | KCNJ2, BC039327 | T | C | 0.575 | 0.444 | −7.21 | −− | 5.53E-13 | 0 | 0.592 | 160,150 | Verhoeven et al. | II (CREAM) | 3.70E-12 |
| rs524952 | 15 | 35005886 | GOLGA8B, GJD2 | A | T | 0.475 | 0.507 | −17.08 | −− | 2.28E-65 | 67.2 | 0.015 | 160,150 | Kiefer et al. & Verhoeven et al. | I | 1.60E-103 |
| rs56075542 | 2 | 146882415 | BC040861, PABPC1P2 | T | G | 0.552 | 0.472 | −8.99 | −− | 2.39E-19 | 13.9 | 0.001 | 159,478 | Kiefer et al. | II (23andMe) | 1.30E-18 |
| rs62070229 | 17 | 31227593 | MYO1D, TMEM98 | A | G | 0.807 | 0.874 | 8.58 | ++ | 9.64E-18 | 0 | 0.416 | 156,570 | Verhoeven et al. | I | 1.30E-18 |
| rs6495367 | 15 | 79375347 | RASGRF1 | A | G | 0.408 | 0.399 | −10.20 | −− | 1.95E-24 | 0 | 0.667 | 160,144 | Kiefer et al. & Verhoeven et al. | I | 7.20E-37 |
| rs7042950 | 9 | 77149837 | RORB | A | G | 0.732 | 0.392 | 6.80 | ++ | 1.07E-11 | 0 | 0.912 | 160,153 | Verhoeven et al. | III | 2.90E-18 |
| rs72621438 | 8 | 60178580 | SNORA51, CA8 | C | G | 0.642 | 0.609 | −13.14 | −− | 2.03E-39 | 38.4 | 0.006 | 160,128 | Kiefer et al. & Verhoeven et al. | I | 1.80E-49 |
| rs745480 | 10 | 85986554 | LRIT2,LRIT1 | C | G | 0.511 | 0.418 | 8.31 | ++ | 9.26E-17 | 67.3 | 0.081 | 159,504 | Kiefer et al. | II (23andMe) | 8.20E-18 |
| rs7624084 | 3 | 141093285 | ZBTB38 | T | C | 0.568 | 0.633 | −8.81 | −− | 1.24E-18 | 18.5 | 0.018 | 160,151 | Kiefer et al. | II (23andMe) | 6.50E-17 |
| rs7662551 | 4 | 80537638 | LOC100506035, PCAT4 | A | G | 0.723 | 0.558 | 8.53 | ++ | 1.47E-17 | 19.4 | 0.265 | 160,147 | Verhoeven et al. | I | 6.00E-12 |
| rs7692381 | 4 | 81903049 | C4orf22, BMP3 | A | G | 0.763 | 0.630 | 9.40 | ++ | 5.55E-21 | 0 | 0.013 | 160,134 | Kiefer et al. | I | 7.50E-13 |
| rs7744813 | 6 | 73643289 | KCNQ5 | A | C | 0.591 | 0.602 | −14.56 | −− | 5.43E-48 | 35 | 0.001 | 160,091 | Kiefer et al. & Verhoeven et al. | I | 1.00E-75 |
| rs7829127 | 8 | 40726394 | ZMAT4 | A | G | 0.792 | 0.897 | −10.91 | −− | 1.02E-27 | 15.9 | 2.77E-04 | 160,132 | Kiefer et al. & Verhoeven et al. | II (23andMe) | 3.10E-22 |
| rs7895108 | 10 | 79061458 | KCNMA1 | T | G | 0.351 | 0.118 | −8.87 | −− | 7.56E-19 | 32.8 | 0.021 | 160,140 | Kiefer et al. | II (23andMe) | 1.10E-27 |
| rs79266634 | 16 | 7309047 | RBFOX1 | C | G | 0.093 | 0.115 | −5.93 | −− | 3.00E-09 | 0 | 0.561 | 156,268 | Kiefer et al. & Verhoeven et al. | III | 1.50E-08 |
| rs837323 | 13 | 101175664 | PCCA | T | C | 0.512 | 0.762 | 6.32 | ++ | 2.65E-10 | 35.6 | 0.213 | 160,142 | Verhoeven et al. | II (23andMe) | 5.30E-16 |
| rs9517964 | 13 | 100717833 | ZIC2,PCCA | T | C | 0.589 | 0.786 | 8.42 | ++ | 3.68E-17 | 0 | 0.020 | 160,121 | Kiefer et al. | II (23andMe) | 3.40E-20 |
| b Subset of the new loci harboring the smallest p-values for refractive error in the Stage 3 meta-analysis | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Position | Nearest Loci And Gene(s) |
Effect Allele |
Other Allele |
EAF EUR |
EAF ASN | Z-score | Direction | P value | HetISq | HetPVal | Sample Size (N) |
Category | P value Replication |
| rs36024104 | 14 | 42294993 | LRFN5 | A | G | 0.823 | NA | 9.09 | ++ | 9.86E-20 | 15.9 | 0.01414 | 152,585 | II (23andMe) | 2.20E-12 |
| rs7456039 | 7 | 6901710 | CCZ1B, LOC100131257 | C | G | 0.183 | NA | 8.82 | ++ | 1.18E-18 | 42.1 | 3.79E-12 | 121,337 | II (23andMe) | 6.50E-01 |
| rs1556867 | 1 | 164213686 | 5S_rRNA, PBX1 | T | C | 0.264 | 0.494 | −8.81 | −− | 1.29E-18 | 71.1 | 0.06266 | 160,155 | II (23andMe) | 4.20E-17 |
| rs12667032 | 7 | 154406581 | DPP6 | A | G | 0.152 | 0.317 | 7.99 | ++ | 1.31E-15 | 82.3 | 1.02E-11 | 130,790 | II (23andMe) | 2.10E-01 |
| rs2225986 | 1 | 200311910 | LINC00862 | A | T | 0.381 | 0.169 | −7.96 | −− | 1.68E-15 | 40.2 | 0.196 | 160,152 | II (23andMe) | 7.50E-17 |
| rs1207782 | 6 | 22059967 | LINC00340 | T | C | 0.577 | 0.265 | −7.92 | −− | 2.47E-15 | 0 | 0.8946 | 160,149 | I | 4.90E-13 |
| rs72826094 | 10 | 114801488 | TCF7L2 | A | T | 0.799 | 0.838 | 7.88 | ++ | 3.20E-15 | 64.5 | 0.09323 | 156,825 | II (23andMe) | 4.90E-02 |
| rs297593 | 2 | 157363743 | GPD2 | T | C | 0.286 | 0.257 | −7.82 | −− | 5.45E-15 | 0 | 0.5285 | 159,461 | II (23andMe) | 7.80E-11 |
| rs5442 | 12 | 6954864 | GNB3 | A | G | 0.068 | NA | −7.82 | −− | 5.48E-15 | 8.8 | 0.03693 | 146,217 | II (23andMe) | 1.20E-33 |
| rs10880855 | 12 | 46144855 | ARID2 | T | C | 0.507 | 0.464 | −7.78 | −− | 7.35E-15 | 0 | 0.9683 | 160,144 | I | 4.80E-08 |
| rs12405776 | 1 | 242431557 | PLD5 | T | C | 0.220 | 0.521 | 7.75 | ++ | 9.52E-15 | 64.9 | 3.56E-10 | 153,784 | II (23andMe) | 1.50E-01 |
| rs2150458 | 21 | 47377296 | PCBP3, COL6A1 | A | G | 0.455 | 0.641 | 7.74 | ++ | 1.04E-14 | 55.7 | 0.1329 | 160,151 | II (23andMe) | 1.80E-13 |
| rs12898755 | 15 | 63574641 | APH1B | A | G | 0.245 | 0.456 | 7.53 | ++ | 4.98E-14 | 7.9 | 0.2974 | 159,506 | II (23andMe) | 1.40E-16 |
| rs7122817 | 11 | 117657679 | DSCAML1 | A | G | 0.507 | 0.662 | 7.51 | ++ | 5.73E-14 | 73.8 | 0.05077 | 160,147 | II (23andMe) | 1.10E-10 |
| rs10511652 | 9 | 18362865 | SH3GL2, ADAMTSL1 | A | G | 0.416 | 0.445 | 7.36 | ++ | 1.91E-13 | 44.8 | 0.1782 | 160,149 | II (23andMe) | 3.50E-18 |
| rs11101263 | 10 | 49414181 | FRMPD2 | T | C | 0.258 | 0.105 | −7.33 | −− | 2.33E-13 | 0 | 0.3477 | 160,155 | II (23andMe) | 2.20E-13 |
| rs11118367 | 1 | 219790221 | LYPLAL1 | T | C | 0.482 | 0.630 | −7.29 | −− | 3.16E-13 | 0 | 0.8576 | 160,141 | III | 1.20E-13 |
| rs9395623 | 6 | 50757699 | TFAP2D, TFAP2B | A | T | 0.315 | 0.381 | 7.25 | ++ | 4.16E-13 | 0 | 0.9579 | 160,151 | III | 2.20E-10 |
| rs284816 | 8 | 53362145 | ST18, FAM150A | A | G | 0.163 | 0.198 | −7.21 | −− | 5.52E-13 | 0 | 0.9242 | 160,140 | III | 1.60E-08 |
| rs12965607 | 18 | 47391025 | MYO5B | T | G | 0.857 | 0.923 | 7.07 | ++ | 1.52E-12 | 20.8 | 0.01674 | 157,604 | II (23andMe) | 8.10E-16 |
| rs7747 | 4 | 80827062 | ANTXR2 | T | C | 0.202 | 0.093 | 7.03 | ++ | 2.05E-12 | 5.4 | 0.01267 | 150,327 | II (23andMe) | 7.70E-16 |
| rs12451582 | 17 | 54734643 | NOG, C17orf67 | A | G | 0.369 | 0.308 | 7.02 | ++ | 2.22E-12 | 0 | 0.5925 | 160,155 | II (23andMe) | 8.80E-18 |
| rs80253120 | 17 | 14138507 | CDRT15 | T | C | 0.626 | 0.723 | 6.97 | ++ | 3.25E-12 | 58.6 | 0.12 | 156,054 | II (23andMe) | 7.20E-11 |
| 22:23069851:I | 22 | 23069851 | DKFZp667J0810 | ATG | A | 0.084 | 0.1582 | 6.95 | −+ | 3.56E-12 | 98.5 | 4.80E-16 | 120,481 | II (23andMe) | 9.30E-01 |
| rs7968679 | 12 | 9313304 | PZP | A | G | 0.700 | 0.894 | 6.95 | ++ | 3.65E-12 | 0 | 0.01951 | 160,076 | II (23andMe) | 4.20E-10 |
| rs11202736 | 10 | 90142203 | RNLS | A | T | 0.717 | 0.762 | −6.92 | −− | 4.53E-12 | 0 | 0.4007 | 160,150 | II (23andMe) | 9.40E-07 |
| rs11088317 | 21 | 16574122 | NRIP1, USP25 | T | C | 0.287 | 0.299 | −6.90 | −− | 5.38E-12 | 72.5 | 0.05657 | 160,116 | II (23andMe) | 6.50E-06 |
| rs10853531 | 18 | 42824449 | SLC14A2 | A | G | 0.200 | 0.182 | 6.88 | ++ | 5.89E-12 | 0 | 0.6755 | 160,104 | III | 2.60E-10 |
| rs72655575 | 8 | 60556509 | SNORA51, CA8 | A | C | 0.201 | 0.124 | 6.87 | ++ | 6.54E-12 | 0 | 0.8811 | 156,566 | I | 7.10E-07 |
| rs12998513 | 2 | 242879499 | CXXC11,AK097934 | A | G | 0.880 | 0.676 | −6.86 | +− | 7.15E-12 | 65.2 | 4.51E-14 | 117,611 | II (23andMe) | 7.80E-01 |
| rs1790165 | 11 | 131928971 | NTM | A | C | 0.411 | 0.283 | 6.85 | ++ | 7.17E-12 | 0 | 0.003708 | 160,131 | II (23andMe) | 1.80E-10 |
| rs511217 | 11 | 30029948 | METTL15, KCNA4 | A | T | 0.738 | 0.729 | −6.79 | −− | 1.10E-11 | 0 | 0.3626 | 160,143 | II (23andMe) | 1.40E-17 |
| rs1150687 | 6 | 28162469 | ZNF192P1, TRNA_Ser | T | C | 0.619 | 0.504 | 6.78 | ++ | 1.17E-11 | 56.2 | 0.131 | 159,448 | II (23andMe) | 3.10E-10 |
| rs56055503 | 16 | 80532694 | MAF, DYNLRB2 | A | G | 0.751 | 0.539 | −6.72 | −− | 1.83E-11 | 0 | 0.8407 | 160,145 | II (23andMe) | 8.00E-06 |
| rs9681162 | 3 | 8194734 | AK124857, LMCD1-AS1 | T | C | 0.680 | 0.437 | 6.70 | ++ | 2.10E-11 | 63 | 0.1002 | 160,152 | II (23andMe) | 6.30E-13 |
| rs11589487 | 1 | 61342229 | AK097193, BC030753 | A | G | 0.445 | 0.089 | 6.67 | ++ | 2.64E-11 | 34.6 | 0.2163 | 160,143 | II (23andMe) | 2.20E-10 |
We identified 140 loci for refractive error with genome-wide significance (P < 5 × 10−8) on the basis the meta-analyses of the genome-wide single-variant linear regressions performed in 160,420 participants of mixed ancestries (CREAM-ASN, CREAM-EUR and 23andMe). Shown are the replication of the previously found loci from HapMap II and a subset of the new loci harboring the smallest P values. For each locus, represented by an index variant (the variant with smallest p-value in that locus), Effect Allele, Other Allele, effect allele frequencies per ancestry (EAF AZN and EAF EUR), effect size (Z-score), direction of the effect (Direction), the P value, heterogeneity I square (HetISq), heterogeneity P value (HetPval), Sample Size (N), Category (I = both GWS in Stage 1 and 2, 2=one of two cohorts (CREAM or 23andMe) GWS, 3= both not GWS in Stage 1 or 2) and P value of the replication in UK Biobank are shown (Full table: Supplementary Table 2). Chr, chromosome; EAF, effect allele frequency; ASN, Asian; EUR, European; GWS, genome wide significant.
As CREAM and 23andMe employed different phenotypic outcomes, we evaluated consistency of genotypic effects by comparing marker-wise additive genetic effect sizes (in units diopters per risk allele variant) for SphE from CREAM-EUR against those (in units log(HR) per risk allele variant) for AODM from 23andMe. All variants strongly associated with either outcome (P<0.001) were concordant in direction-of-effect, and had highly correlated effect sizes (Figure 2a,b; Supplementary Figure 8). For these variants a 10% decrease in log(HR) for AODM, indicating an earlier age-at-myopia onset, was associated with a decrease of 0.15 diopters in SphE. A quantitative analysis for all common SNPs (MAF>0.01; HapMap3) using LD score regression yielded a genetic correlation of 0.93 (95% CI 0.86-0.99; P=2.1×10−159), confirming that effect sizes for both phenotypic outcomes were closely related.
Figure 2. Correlation of statistical significance and effect size of SNPs based on spherical equivalent (SphE) in diopters and age of diagnosis of myopia (AODM) in years.
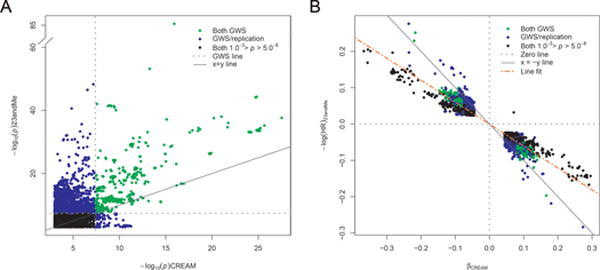
(a) P comparison of all genetic variants with P < 1.0 × 10−3 (n=7249) between CREAM meta-analysis (Stage 1) and 23andMe (Stage 2) meta-analysis. Shown is the overlap (red) and the difference (green) in P signals per cohort for genetic variants. Green genetic variants are only genome wide significant in either CREAM or 23andMe. Blue: genetic variants with P between 5.0 × 10−8 and 1.0 × 10−3 in both CREAM and 23andMe. (b) Comparison of effects (SphE and logHR of AODM in years; P < 1.0 × 10−3; n=7249) between CREAM and 23andMe. Same color code was applied as in (a). The effects were concordant in their direction of effect on refractive error. We performed a simple linear regression between the effects of CREAM and 23andMe; the regression slope is -0.15 diopters per logHR of AODM in years.
Gene annotation of susceptibility loci
We annotated all genetic variants with wANNOVAR using the University of California Santa Cruz (UCSC) Known Gene database34,35. The identified 139 genetic loci were annotated to 208 genes and known transcribed RNA genes (Table 1, Supplementary Table 2, Online Methods). The physical positions of the lead genetic variants relative to protein-coding genes are shown in Figure 1c. 86% of the identified variants were either intragenic or less than 50 kb from the 5′or 3′ end of the transcription start site. We found seven exonic variants (Supplementary Table 7) of which two had MAF≤0.05: rs5442 (GNB3) and rs17400325 (PDE11A). The index SNP in the GNB3 locus with MAF 0.05 in Europeans is a highly conserved missense variant (G272S) predicted to be damaging by PolyPhen-236 and SIFT37. PDE11A is presumed to play a role in tumorigenesis, brain function, and inflammation38. The index SNP in the PDE11A locus with MAF 0.03 in Europeans is also a highly conserved missense variant (Y727C); this variant was predicted to be damaging by PolyPhen36, SIFT39 and align GVGD40,41.The other exonic variants, rs1064583 (COL10A1), rs807037 (KAZALD1), rs1550094 (PRSS56), rs35337422 (RD3L), and rs6420484 (TSPAN10) were not predicted to be damaging.
The most significant variant (Stage 3; rs12193446, P=4.21×10−84) resides on chromosome 6 within a non-coding RNA, BC035400, in an intron of the LAMA2 gene. This locus had been identified previously, but our current fine mapping redefined the most associated variant. The function and potential downstream target sites for BC035400 are currently unknown. The previously most strongly associated variant, rs524952 on chromosome 15 near GJD2, was the second most significant variant (P=2.28×10−65).
Post-GWAS analyses
We performed two gene-based tests, fastBAT42 and EUGENE43, and applied a functional enrichment approach using fgwas44 (Online Methods). With fastBAT, we identified 13 genes at P <2.0×10−6, one of which (CHD7) had been identified previously26,27. Using EUGENE, we found 7 genes at P <2.0×10−6 after incorporation of blood eQTLs. With fgwas, we identified 6 loci, which could be annotated to 9 genes, at posterior probability >0.9. Two genes (HMGN4 and TLX1) showed significant associations in two or more approaches. Taken together, these post-GWAS approaches resulted in a total of 22 additional candidate loci for refractive error, annotated to 25 genes (Supplementary Table 8). This increases the overall number of significant genetic associations to 161 candidate loci.
Polygenic risk scores
We calculated polygenic risk scores (PGRS)45 per individual at various P thresholds (Online Methods) for Rotterdam Study I-III (RS I-III; N=10,792) after recalculating P and Z-scores of variants from Stage 3 excluding RS I-III. We found the highest fraction of phenotypic variance (7.8%) explained with 7,307 variants at P value threshold 0.005 (Supplementary Table 9). A PGRS based on these variants distinguished well between individuals with hyperopia and myopia at the lower and higher deciles (Figure 3); those in the highest decile had a 40-fold increased risk of myopia. When the PGRS was stratified for the median age (<63 or >63+ yrs), we found a significant difference in the variance explained (<63 yrs 8.9%; 63+ yrs 7.4%; P 0.0038). The variance explained by PGRS was not significantly different between males and females (8.3% vs 7.5%; P 0.13). The predictive value (area under the receiver operating characteristic curve, AUC) of the PGRS for myopia versus hyperopia adjusted for age and gender was 0.77 (95% CI=0.75–0.79), a 10% increase compared to previous estimations46.
Figure 3. Risk of refractive error per decile of polygenic risk score (Rotterdam Study I-III, N=10,792).
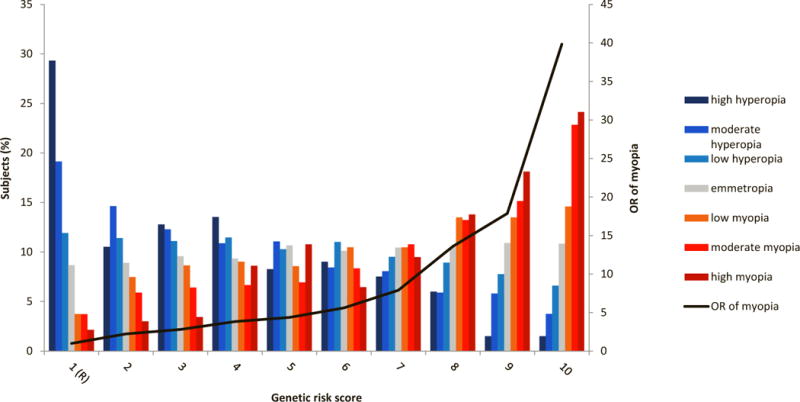
Distribution of refractive error in subjects from Rotterdam Study I–III (N=10,792) as a function of the optimal polygenic risk score (including 7,303 variants at P ≤ 0.005 explaining 7.8% of the variance of SphE; Supplementary Table 9). Mean OR of myopia (black line) was calculated per polygenic risk score category using the lowest category as a reference. High myopia (SphE ≤-6 D), moderate myopia (SphE >-6 D & ≤ −3 D), low myopia (SphE > −3 D & <-1.5 D), emmetropia (SphE ≥ −1.5 D and ≤ 1.5 D), low hyperopia (SphE > 1.5 D & < 3 D), moderate hyperopia (SphE ≥ 3 D & 6 D), high hyperopia (SphE ≥ 6 D).
Trans-ethnic comparison of genotypic effects
To explore potential ancestry differences in the identified refractive error loci, we calculated the heritability explained by common genetic variants (SNP-h2) for Europeans and Asians using LD score regression47. SNP-h2 was 0.214 (95% CI 0.185- 0.243) and 0.172 (95% CI 0.154- 0.190) in the European samples (CREAM-EUR and 23andMe), while it was only 0.053 (95% CI -0.025- 0.131) in the Asian sample (CREAM-EAS). Next, we estimated the genetic correlation between Europeans and Asians by comparing variant effect size for common variants using Popcorn48 (Online Methods). Two genetic correlation metrics were calculated T; First, a genetic effect correlation (ρge) that quantifies the correlation in SNP effect sizes between Europeans and Asians without taking into account ancestry-related differences in allele frequency; and second, a genetic impact correlation (ρgi) that estimates the correlation in variance-normalized SNP effect sizes between the two ancestry groups (Table 2). Estimates of ρge were high between Europeans and Asians, but significantly different from 1 (0.79 and 0.80, respectively at P <1.9×10−6; Table 2), indicating a clear genetic overlap but a difference in per allele effect size. Estimates of ρgi were similarly high (>0.8), but not significantly different from 1 for the correlation between CREAM-EUR and CREAM-ASN (P=0.065), indicating that the genetic impact of these alleles may still be similar.
Table 2.
Genetic correlation for refractive error between Europeans and East Asians
| Sample 1 | Sample 2 | Genetic effect correlation (pge) a | Standard error pge | P value pge | Genetic impact correlation (pgi) a | Standard error pgi | P value pgi |
|---|---|---|---|---|---|---|---|
| EUR CREAM | EAS CREAM | 0.804 | 0.041 | 1.83E-06 | 0.888 | 0.061 | 0.065 |
|
| |||||||
| EUR 23andMe | EAS CREAM | 0.788 | 0.041 | 2.48E-07 | 0.865 | 0.054 | 0.014 |
Abbreviations: EUR, European; EAS, East Asian.
P-value relates to a test of the null hypothesis that pge=1 or pgi=1.
We calculated the genetic correlation of effect (pge) and impact (pgi) using Popcorn to compare the genetic associations between Europeans (CREAM-EUR, N= 44,192; 23andMe, N=104,292) and East Asians (CREAM-ASN, N= 9,826). Reference panels for Popcorn were constructed using genotype data for 503 EUR and 504 EAS individuals sequenced as part of the 1000 Genomes Project. SNPs used had a MAF of at least 5% in both populations, resulting in a final set of 3,625,602SNPs for the analyses using the 23andMe GWAS sample and 3,642,928 SNPs for those using the CREAM-EUR sample. These findings support a largely common genetic predisposition to refractive error and myopia in Europeans and Asians, although ancestry-specific risk alleles may exist.
In silico pathway analysis
We used an array of bioinformatics tools to investigate potential functions and pathways of the associated genes. We first employed DEPICT49 to perform a gene set enrichment analysis, a tissue type enrichment, and a gene prioritization analysis, on all variants with P <1.00×10−5 from Stage 3. The gene set enrichment analysis resulted in 66 reconstituted gene sets, of which 55 (83%) were eye-related. To reduce redundancies between pathways, we clustered the significant pathways into 13 meta gene sets (false discovery rate (FDR) <5% and a P <0.05) (Supplementary Note 2, Figure 4, Supplementary Table 10). The most significant gene set was the ‘abnormal photoreceptor inner segment morphology’ (MP:0003730; P=1.79×10−7). The eye-related meta gene sets consisted of the ‘thin retinal outer nuclear layer’ (MP:0008515; 27 (55%) gene sets), ‘detection of light stimulus’ (GO:0009583; 13 (24%) gene sets), ‘nonmotile primary cilium’ (GO:0031513; 4 (6%) gene sets), and ‘abnormal anterior eye segment morphology’ (MP:0005193; 4 (6%) gene sets). The first three meta gene sets had a Pearson’s correlation > 0.6. Interestingly, RGR, RP1L1, RORB and GNB3 were present in all of these meta gene sets. Retina was the most significant tissue of expression according to the tissue enrichment analysis (P=1.11 × 10−4, FDR <0.01). From the gene prioritization according to DEPICT, 7 genes were highlighted as the most likely causal genes at P<7.62×10−6 and FDR<0.05: ANO2, RP1L1, GNB3, EDN2, RORB and CABP4.
Figure 4. Visualization of the DEPICT gene-set enrichment analysis based on loci associated with refractive error and the correlation between the (meta)gene sets.
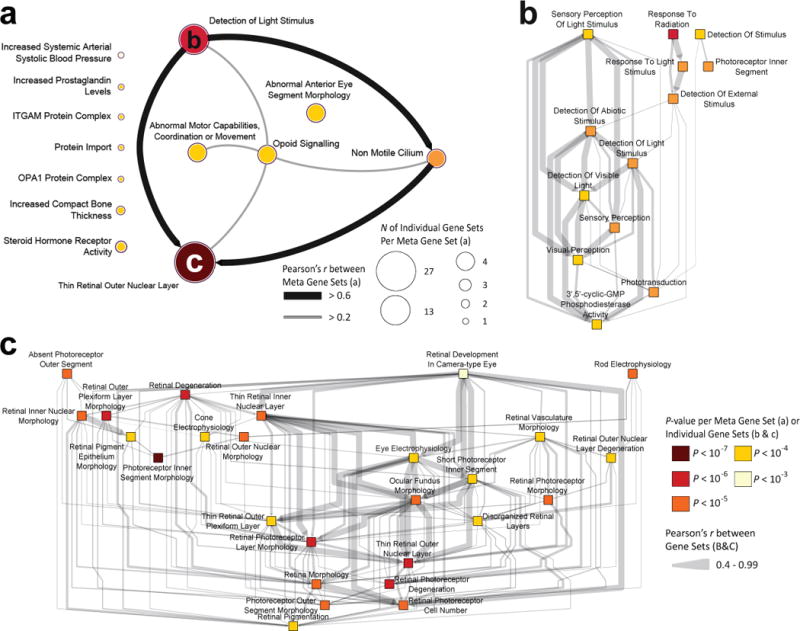
(a) Shown are the 66 significantly enriched reconstituted gene sets clustered into thirteen meta gene sets based on the gene set enrichment analysis of DEPICT (pairwise Pearson correlations; P < 0.05). All genetic variants with a P < 1 × 10−5 in the GWAS meta-analysis of stage 3 (n=21,073) and an FDR < 0.05 were considered. (b) Visualization of the interconnectivity between gene sets (n=13; pairwise Pearson correlations; P < 0.05) of the meta gene set ‘Detection of Light Stimulus’ (GO:0009583). (c) Visualization of the interconnectivity between gene sets (n=27; pairwise Pearson correlations; P < 0.05) of the largest meta gene set ‘Thin Retinal Outer Nuclear Layer’ (MP:0008515). In all panels, (meta)gene sets are represented by nodes colored according to statistical significance, and similarities between them are indicated by edges scaled according to their correlation; Pearson’s r ≥ 0.2 are shown in panel (a) and Pearson’s r ≥ 0.4 are shown in panel (b,c).
Next, we performed a canonical pathway analysis on all genes annotated to the variants of Stage 3 using Ingenuity Pathway Analysis (See URLs). All genes were run against the IPA database incorporating functional biological evidence on genomic and proteomic expression based on regulation or binding studies. IPA identified “Glutamate Receptor Signaling” with central player Nf-kB gene as the most significant pathway after correction for multiple testing (ratio of the number of molecules 8.8% and Fisher’s Exact test P=1.56×10−4; Supplementary Figure 9).
From disease-associated loci to biological mechanisms
We adapted the scoring scheme designed by Fritsche et al.50 to highlight genes for which there is biological plausibility for a role in eye growth50. We used 10 equally rated categories (Online Methods; Figure 5; Supplementary Table 11; Supplementary Note 2). One-hundred-and-nine index variants replicated in two or more individual cohorts; we found evidence for seven genetic variants with eQTL effects in multiple tissue types; nine exonic variants, of which seven predicted protein-alterations (Supplementary Table 7); 31 RNA genes, five located in the 3′ or 5′UTR (Supplementary Table 12, Supplementary Figure 10), 84 genes resulting in an ocular phenotype in humans (Supplementary Table 13) and 36 in mice (Supplementary Table 14); 172/212 (81%) genes expressed in human ocular tissue (Supplementary Note 2, Supplementary Table 15); 41 genes identified by DEPICT at P <5.4×10−4 and FDR<0.05 and 45 genes contributed to the most significant canonical pathways of IPA. Notably, 48 of the associated genes encode known drug targets (Supplementary Table 16).
Figure 5. Genes ranked according to biological and statistical evidence.
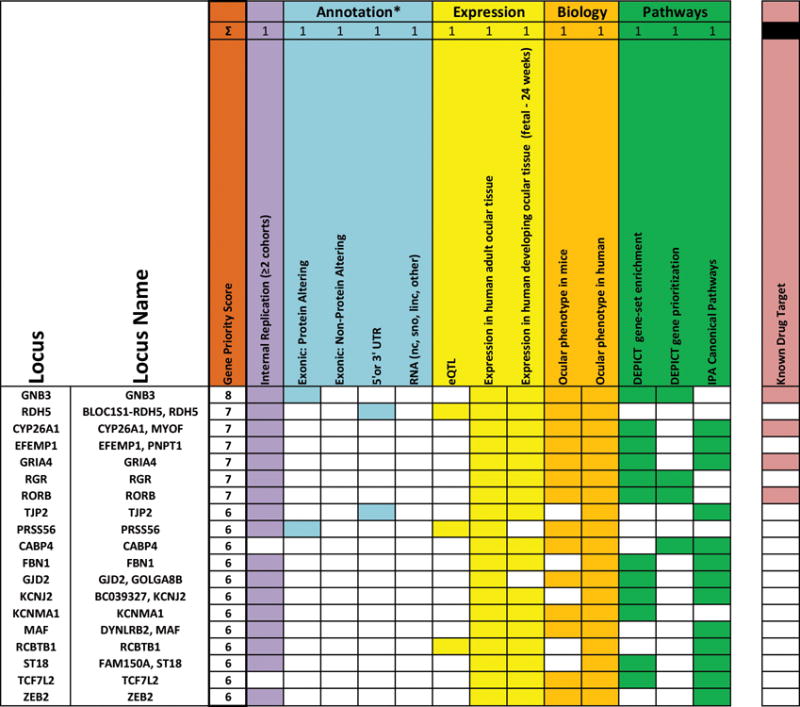
Genes were ranked (orange) based on 10 equal categories which can be divided in four categories: internal replication of genetic variant in more than two cohorts (purple; CREAM-EUR, CREAM-ASN and/or 23andMe), annotation (light blue; genetic variant harboring an exonic protein altering variant or non-protein altering variant, genetic variant residing in a 5′ or 3′ UTR region of a gene or transcribing an RNA structure), expression (yellow; eQTL, expression in adult human ocular tissue, expression in developing ocular tissue), biology (dark yellow; ocular phenotype in mice, ocular phenotype in humans), pathways (green; DEPICT gene-set enrichtment, DEPICT gene prioritization analysis and canonical pathway analysis of IPA). We assessed genes harboring drug targets (salmon red), but did not assign a scoring point to this category.
*Only one point can be assigned in the category ‘ANNOTATION’, even though it has four columns (i.e. a genetic variant is located in only 1 of these four categories).
The gene with the highest biological plausibility score (score=8) was GNB3, a highly conserved gene encoding a guanine nucleotide-binding protein expressed in rod and cone photoreceptors and ON-bipolar cells51. GNB3 participates in signal transduction through G-protein-coupled receptors and enhances the temporal accuracy of phototransduction and ON-center signaling in the retina51. As described above, the index SNP harbors a missense variant associated with refractive errors. Non-synonymous mutations within GNB3 are known to cause syndromic congenital stationary night blindness52 in humans, progressive retinopathy and globe enlargement in chickens51, and abnormal development of the photoreceptor-bipolar synapse in knock-out mice53,54.
Other genes highly ranked (score=7) include CYP26A1, GRIA4, RDH5, RORB, and RGR, all previously associated with refractive error, and one newly identified gene: EFEMP1. EFEMP1 encodes a member of the fibulin family of extracellular matrix glycoproteins, and is found pan-ocularly including in the inner nuclear layer and Bruch’s membrane. Mutations in this gene lead to specific macular dystrophies55, while variants have also been shown to co-segregate with primary open-angle glaucoma56 and associate with optic disc cup area57.
Several other genes are noteworthy for their function. CABP4, a calcium-binding protein expressed in cone and rod photoreceptor cells, mediates Ca2+-influx and glutamate release in the photoreceptor-bipolar synapse58. Mutations in this gene have been described in congenital cone-rod synaptic disorder59, a retinal dystrophy associated with nystagmus, photophobia, and, remarkably, high hyperopia. KCNMA1 encodes pore-forming alpha subunits of Ca2+-activated K+ (BK) channels. These channels regulate synaptic transmission exclusively in the rod pathway60. ANO2 is a Ca2+-activated Cl− channel recently reported to regulate retinal pigment epithelial (RPE) cell volume in a light-dependent manner64. EDN2 is a potent vasoconstrictor that binds to two G-protein-coupled receptors, EDNRA, which resides on bipolar dendrites, and EDNRB, which is present on Mueller and horizontal cells. Both receptors are also present on choroidal vessels65, implying that the choroid as well as retinal cells are target sites for this gene. RP1L1 is expressed in cone and rod photoreceptors where it is involved in the maintenance of microtubules in the connecting cilium66. Mutations in this gene cause dominant macular dystrophy and retinitis pigmentosa67. We replicated two genes known to cause myopia in family studies. FBN1 harbors mutations causing with Marfan (OMIM #154700) and Weil Marchesani (OMIM #608328) syndrome; PTPRR was one of the candidates in the MYP3 locus, which was found by linkage in families with high myopia68.
The location of rs7449443 (P=3.58×10−8) is notable as it resides in between DRD1 and FLJ16171. DRD1 encodes dopamine receptor 1 and is known to modulate dopamine receptor 2-mediated events69,70. The dopamine pathway has been implicated in myopia pathogenesis in many studies69,71. SNPs in and near other genes involved in the dopamine pathway (dopamine receptors, synthesis, degradation, and transporters)72–74 did not reveal genome-wide significant associations (Supplementary Note 3, Supplementary Table 17; Supplementary Figure 11).
There were 31 genetic variants in or near DNA structures transcribing RNA genes (non coding RNA, linc RNAs, tRNAs, snoRNas, rRNAs). Notably, five were in the transcription region and thirteen were in the vicinity (>0 kb and ≤50 kb) of start or end of the RNA gene transcription region. They received low scores, since many have no reported function or disease association to date (Figure 5, Supplementary Figure 10, Supplementary Table 12). Our ranking of genes based on functional information existing in the public domain does not necessarily represent the true order of importance for refractive error pathogenesis. The observation that genes with strong statistical association were distributed over all scores supports this concept. Nevertheless, this list may help to select genes for subsequent functional studies.
Finally, integration of all our findings, supported by literature, allowed us to annotate a large number of genes to ocular cell types (Figure 6). Remarkably, all cell types of the retina harbored refractive error genes, as well as the RPE, vascular endothelium, and extracellular matrix.
Figure 6. Schematic representation of the human eye, retinal cell types, and functional sites of associated genes.
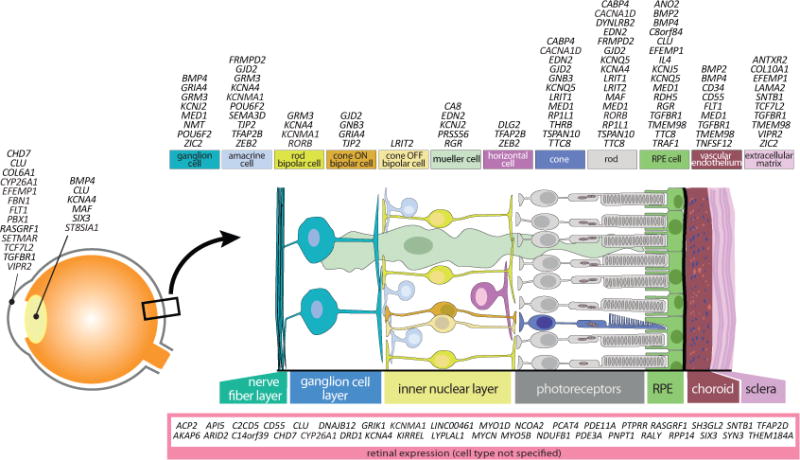
We assessed gene expression sites and/or functional target cells in the eye for all genes using our expression data and literature and data present in the public domain. The genes appear to be distributed across virtually all cell types in the neurosensory retina, in the RPE, vascular endothelium and extracellular matrix; i.e., the route of the myopic retina-to-sclera signalling cascade.
Genetic pleiotropy
We performed a GWAS catalogue look up using FUMA to investigate overlap of genes with other common traits (Supplementary Figure 12)75. Refractive error and hyperopia were replicated significantly after correcting for multiple testing (adjusted P value=1.44×10−52 and 9.34×10−9, respectively). We found significant overlap with 74 other traits, of which height (adjusted P value=1.11×10−10), obesity (adjusted P=1.38×10−10), and BMI (adjusted P=4.05×10−7) were most important. Ocular diseases significantly associated were glaucoma (optic cup area, intraocular pressure; adjusted P=2.69×10−5 and 3.01×10−5, respectively) and age-related macular degeneration (adjusted P value=1.27×10−3).
DISCUSSION
Myopia may become the leading cause of world blindness in the near future, a grim outlook for which current counteractions are still insufficient11,76. To improve understanding of the genetic landscape and biology of refractive error, we conducted a large GWAS meta-analysis in 160,420 participants of mixed ancestry and replicated in 95,505 participants. This led to the identification of 139 independent susceptibility loci by single variant analysis and 22 additional loci through post-GWAS methods, a four-fold increase in refractive error genes. The majority of annotated genes were found to be expressed in the human posterior segment of the eye. Using in silico analysis, we identified significant biological pathways, of which retinal cell physiology, light processing, and, specifically, glutamate receptor signaling were the most prominent mechanisms. Our integrated bio-informatic approach highlighted known ocular functionality for many genes.
To ensure robustness of our genetic associations, we included studies of various designs and populations, sought replication in an independent cohort of significant sample size, and stringently accounted for population stratification by performing genomic control at all stages of the meta-analysis77. We combined studies with outcomes based on actual refractive error measurements as well as on self-reported age-of-myopia-onset, and found the direction-of-effect of the associated variants, as well as their effect size, to be remarkably consistent. Combining two different outcome measures may appear unconventional, but age of onset and refractive error have been shown to be very tightly correlated11,28,78,79. Moreover, the high genetic correlation (93%) of common SNPs between the two phenotypes underscores their similarity. Most compelling evidence was provided by replication of 86% of the discovered variants in the independent UKEV which also used conventional refractive error measurements. This robustness indicates that both phenotypic outcomes can be used to capture a shared source of genetic variation. In addition, we found trans-ethnic replication of significant loci, and a high correlation of genetic effects of common variants in the Europeans and Asians. Our findings support a largely shared genetic predisposition to refractive error and myopia in the two ethnicities, although ancestry-specific allelic effects may exist. The low heritability estimate in Asians may, in part, be explained by the low representation of this ethnicity in our study sample. Alternatively, it may imply that environmental factors explain a greater proportion of the phenotypic risk and recent rise in myopia prevalence in this ancestry group80.
Limitations of our study were the possibility of false negative findings due to genomic control, and underrepresentation of studies with Asian ancestry. Heterogeneity of observed effect estimates was large for several associated variants, but not unexpected, given the large number of collaborating studies with varying methodology.
Although neurotransmission was previously suggested pathway26,27, our current pathway analyses provide more in-depth insights into the retinal circuitry driving refractive error. DEPICT identified ‘thin retinal outer nuclear layer’, ‘detection of light stimulus’, and ‘nonmotile primary cilium’ as the most important meta-gene sets. These are the main characteristics of photoreceptors, which are located in the outer retina and contain cilia. These photosensitive cells drive the phototransduction cascade in response to light, which in turn induces visual information processing. IPA pointed towards glutamate receptor signaling as the most significant pathway. Glutamate is released by photoreceptors and determines conductance of retinal signaling to the ON and OFF bipolar cells81. Our functional gene look ups provide evidence that rod (CLU) as well as cone (GNB3) bipolar cells play a role. Taken together, these findings strongly suggest that light response and light processing in the retina are initiating factors leading to refractive error.
The genetic association with light-dependent pathways may also link to the well-established protective effect of outdoor exposure on myopia. We found suggestive evidence for a genetic association with DRD1. The dopaminergic pathway has been studied extensively in animal models for its role in controlling eye growth in response to light69,71,82–91. DRD1 was found to be a mediator in this process, as bright light increased DRD1 activity in the bipolar ON-pathway, and diminished form-deprivation myopia in mice. Blockage of DRD1 reversed this inhibitory effect92. We did not find evidence for direct involvement of other genes in the dopamine pathway, but GNB3 may be an indirect modifier as it is a downstream signaling molecule of dopamine and has been shown to influence availability of the dopamine transporter DAT93. Although a promising target for therapy, further evidence of DRD1 in human myopiagenesis is warranted.
Novel pathways elicited by the newly identified genes are anterior segment morphology (TCF7L2, VIPR2, MAF) and angiogenesis (FLT1). In addition, the high number of variants residing near small RNA genes suggests that post-transcriptional regulation is an important mechanism, as these RNAs are known to play a distinct and central regulatory role in cells94. These findings will serve as leads for future studies performing detailed mapping of cellular networks, and functional studies on genes implicated in ocular phenotypes, harboring protein-altering variants, and proven drug targets.
Our evaluation of shared genetics between refractive error and other disease-relevant phenotypes highlighted overlap with anthropometric traits such as height, obesity, and body mass index. This could give valuable additional clues as to the phenotypic outcomes of perturbations of some of the networks identified.
Our genetic observations add credence to the current notion that refractive errors are caused by a retina-to-sclera signaling cascade that induces scleral remodeling in response to light stimuli. The concept of this cascade originates from various animal models showing that form deprivation, retinal defocus and contrast, ambient light, and wavelength can influence eye growth in young animals95–97. Cell-specific moieties in this putative signaling cascade in humans were largely unknown, although animal models implicated GABA, dopamine, all-trans-retinoic acid and TGF-β69,91,98,99. Our study provides a large number of new molecular candidates for this cascade, and clearly shows that a wide range of neuronal cell types in the retina, the RPE, the vascular endothelium, as well as components of the extracellular matrix are implicated. The many interprotein relationships exemplify the complexity of eye growth, and provide a challenge to develop strategies to prevent pathological eye elongation.
In conclusion, by using a cross-ancestry design in the largest study population on common refractive errors to date, we uncovered numerous novel loci and pathways involved in eye growth. Our multi-disciplinary approach incorporating GWAS data with in silico analyses and expression experiments provides an example for the design of future genetic studies for complex traits. Additional genetic insights into refractive errors will be gained by increasing sample size and genotyping depth, by performing family studies to identify rare alleles of large effects, and by evaluating population extremes. Our list of plausible genes and pathways provide a plethora of data for future studies focusing on gene-environment interaction, and on translation of GWAS findings into starting points for therapy.
ONLINE METHODS
Ethics Statement
All human research was approved by the relevant institutional review boards and conducted according to the Declaration of Helsinki. All CREAM participants provided written informed consent; all 23andMe applicants provided informed consent online, and answered surveys according to 23andMe’s human subjects protocol, which was reviewed and approved by Ethical & Independent Review Services, an AAHRPP-accredited institutional review board. The UK Biobank received ethical approval from the National Health Service National Research Ethics Service (reference 11/NW/0382).
Study data
The study populations were participants of the Consortium for Refractive Error and Myopia (CREAM) comprising of 41,793 individuals with European ancestry from 26 cohorts (CREAM-EUR) and 11,935 individuals with Asian ancestry from 8 studies (CREAM-ASN); and customers of the 23andMe genetic testing company who gave informed consent for inclusion in research studies consisting of 104,293 individuals (2 cohorts of individuals with European ancestry, N = 12,128 and N= 92,165, respectively). All participants included in this analysis from CREAM and 23andMe were aged 25 years or older. Participants with conditions that could alter refraction, such as cataract surgery, laser refractive procedures, retinal detachment surgery, keratoconus as well as ocular or systemic syndromes were excluded from the analyses. Recruitment and ascertainment strategies varied per study (Supplementary Table 1a,b, and Supplementary Note 4). Refractive error represented by measurements of refraction and analyzed as spherical equivalent (SphE =spherical refractive error + 1/2 cylinder refractive error) was the outcome variable for CREAM; myopic refractive error represented by self-reported age of diagnosis of myopia (AODM) for 23andMe27.
Genotype calling and imputation
Samples were genotyped on different platforms and study specific quality control measures of the genotyped variants were implemented before association analysis (Supplementary Table 1b). Genotypes were imputed using the appropriate ancestry-matched reference panel for all cohorts from the 1000 Genomes Project (Phase I version 3, March 2012 release) with either minimac100 or IMPUTE101. The metrics for pre-imputation quality control varied amongst studies, but genotype call rate thresholds were set at high level (≥0.95 for both CREAM and 23andMe). These metrics were similar to our previous GWAS analyses26,27; details per cohort can be found in Supplementary Table 1b.
GWAS per study
For each CREAM cohort, a single marker analysis for the SphE (in diopters) phenotype was carried out using linear regression adjusting for age, sex and up to the first five principal components. All non-family-based cohorts removed one of each pair of relatives (after detection using either GCTA or IBS/IBD analysis). In family-based cohorts, a score test-based association was used to adjust for within-family relatedness102. For the 23andMe participants, Cox proportional hazards analysis testing AODM as the dependent variable were performed as previously described27, with P calculated using a likelihood ratio test for the single marker genotype term. We used an additive SNP allelic effect model for all analyses.
Centralized quality control per study
After individual GWAS, all studies underwent a second round of quality control (QC). Quantile-quantile, effect allele frequency, P – Z test, standard error – sample size, and genomic control inflation factor plots were generated for each individual cohort using EasyQC103 (Supplementary Figure 2.1 (Supplementary Figure 2.1.1 and 2.1.2), 2.2 (Supplementary Figure 2.2.1 – 2.2.5), 2.3 (Supplementary Figure 2.3.1 and 2.3.2). All analytical issues discovered during this QC step were resolved per individual cohort.
GWAS meta-analyses
The GWAS meta-analyses were performed in three stages (Supplementary Figure 1). In Stage 1, European (CREAM-EUR, N=44,192) and Asian (CREAM-ASN, N=11,935) participants from the CREAM cohort were meta-analysed separately. Subsequently, all CREAM cohorts (CREAM-ALL) were meta-analysed. Variants with MAF < 1% or imputation quality score < 0.3 (info metric of IMPUTE) or Rsq < 0.3 (minimac) were excluded. A fixed effects inverse variance-weighted meta-analysis was performed using METAL104. 1,063 variants clustering in 24 loci (Supplementary Table 2) were genome-wide significant (P=5.0×10−8). All 37 loci that were previously found by CREAM and 23andMe using genotype data imputed to the HapMap II reference panel were replicated (pBonferroni 1.85×10−3), and 36 of the 37 were genome-wide significant (Supplementary Table 2)26,27. In Stage 2, a meta-analysis of the two 23andMe cohorts (N23andMe_V2=12,128; N23andMe_V3=92,165) was performed, using similar filtering but a lower MAF threshold (< 0.5%). A total of 5,205 genome-wide significant variants clustered in 112 loci (Supplementary Table 2).
In Stage 3, CREAM-ALL and 23andMe samples were combined using a fixed effects meta-analysis based on P value and direction of effect. In all stages, each genetic variant had to be represented by at least half of the entire study population and at least represented by 13 cohorts in CREAM and one cohort in 23andMe. For SNPs with high heterogeneity (at P < 0.05), we also performed a random effects meta-analysis using METASOFT50. We chose a different weighting scheme due to the differences in effect size scaling; 23andMe used a less accurate phenotype variable (AODM); i.e. the effective sample size of the 23andMe was approximately equivalent to the effective sample size of CREAM-ALL (Figure 2b), thus weighting by (1/√neffective) yielded a final weighting ratio of 1:1105. Genome-wide statistical significance was defined at P < 5.0 × 10−8 106.
All three meta-analysis stages were performed under genomic control. Study specific and meta-analysis lambda (λ) estimates are shown in Supplementary Figure 6; to check for confounding biases (e.g. cryptic relatedness and population stratification), LD score intercepts from LD score regressions per ancestry were constructed (Supplementary Figure 7)30. To check the robustness of signals, we performed a conventional random effects models using METASOFT, fixed effects models weighted on sample size and on weights estimated from standard error per allele tested using METAL (Supplementary Table 2 and Supplementary Table 3).
Manhattan (modified version of package ‘qqman’), regional, box, and forest plots were made using R version 3.2.3 and LocusZoom107. An overview of the Hardy Weinberg P of all index variants per cohort can be found in Supplementary Table 4. The comparison between refractive error and age-of-onset was performed using the LDSC program30.
Population stratification and heritability calculations
Each study assessed the degree of genetic admixture and stratification in their study participants through the use of principal components. Homogeneity of participants was assured by removal of all individuals whose ancestry did not match the prevailing ancestral group. We used genomic inflation factors to control for admixture and stratification, and performed genomic-controlled meta-analysis to account for the effects of any residual heterogeneity. To further distinguish between inflation from a true polygenic signal and population stratification, we examined the relationship between test statistics and linkage disequilibrium (LD) with LDSC. CREAM-EUR, CREAM-ASN and 23andMe were evaluated separately; variants not present in HapMap3 and MAF < 1% were excluded. SNP heritability estimates were calculated using LDSC for the same set of genetic variants.
Locus definition and annotation
All study effect size estimates were oriented to the positive strand of the NCBI Build 37 reference sequence of the human genome. The index variant of a locus was defined as the variant with the lowest P in a region spanning a 100 kb window of the most outer genome-wide significant variant of that same region. We annotated all index variants using the web-based version of ANNOVAR108 based on UCSC Known Gene Database35. For variants within the coding sequence or 5′ or 3′ untranslated regions of a gene, that gene was assigned to the index variant (note that this led to more than 1 gene being assigned to variants located within the transcription units of multiple, overlapping genes). For variants in intergenic regions, the nearest 5′ gene and the nearest 3′ gene were assigned to the variant. Index variants were annotated to functional RNA elements when described as such in the UCSC Known Gene Database. We used conservation (PhyloP109) and prediction tools (SIFT39, Mutation Taster110, align GVGD40,41, PolyPhen-236) to predict the pathogenicity of protein-altering exonic variants.
Conditional signal analysis
We performed conditional analysis to identify additional independent signals nearby the index variant at each locus, using GCTA-COJO32. We transformed the Z-scores of the summary statistics to beta’s using the following formula: . We performed the GCTA-COJO analysis32, utilizing summary-level statistics from the meta-analysis on all cohorts. Linkage disequilibrium (LD) between variants was estimated from the Rotterdam Study I-III.
Replication in UK Biobank
The UK Biobank Eye & Vision (UKEV) Consortium performed a GWAS of refractive error in 95,505 participants of European ancestry aged 37-73 year with no history of eye disorders33. Refractive error was measured using an autorefractor; SphE was calculated per eye and averaged between the two eyes. To account for relatedness a mixed model analysis with BOLT-LMM was used111, including age, gender, genotyping array, and the first 10 principal components as covariates. Analysis was restricted to markers present on the HRC reference panel112. We performed lookups for all independent genetic variants identified in our Stage 3 meta-analysis and conditional analysis. For 16 variants not present in UKEV, we performed lookups for a surrogate variant in high LD (r2 >0.8). When more than one potential surrogate variant was available, the variant in strongest LD with the index variant was selected. Six variants were not available for replication: one variant (rs188159083) was not present on the array nor was a surrogate available in UKEV and five variants showed evidence of departure from HWE (HWE exact test P<3.0×10−4).
Post-GWAS analyses
We performed two gene-based tests to identify additional significant genes not found in the single variant analysis. First, we applied the gene-based test implemented in fastBAT42 to the per-variant summary statistics of the meta-analysis of all European cohorts (23andMe and CREAM-EUR). We used the default parameters (all variants in or within 50kb of a gene) and focused on variants with a gene-based P <2 × 10−6 (Bonferroni correction based on 25,000 genes) and the per-variant P >5 × 10−8. Secondly, we applied another gene-based test in EUGENE43 which only includes variants which are eQTLs (GTex, blood113). EUGENE tests an hypothesis predicated on eQTLs as key drivers of the association signal. eQTLs within 50kb of a gene were included in the test. Genes with EUGENE P <2 × 10−6 (and not found in the single variant analysis) were considered to be significant. Finally, we used functional annotation information from genome-wide significant loci to reweigh results using fgwas (version 0.3.6444). Fgwas incorporates functional annotation (e.g. DNase I-hypersensitive sites in various tissues and 3′UTR regions) to reweight data from GWAS, and uses a Bayesian model to calculate a posterior probability of association. This approach is able to identify risk loci that otherwise might not reach the genome-wide significance threshold in standard GWAS. Details about this approach can be found in Supplementary Note 5.
Refractive errors and myopia risk prediction
To assess the risk of the entire range of refractive errors, we computed polygenic risk scores (PGRS) for the population-based Rotterdam Studies (RS) I, RS-II and RS-III using the P and Z scores from a meta-analysis on CREAM-ALL and 23andMe, excluding the RS I-III cohorts. Only variants with high imputation quality (IMPUTE info score > 0.5 or minimac Rsq > 0.8) and MAF > 1% were considered. P-based clumping was performed with PLINK114, using an r2 threshold of 0.2 and a physical distance threshold of 500 kb, excluding the MHC region. This resulted in a total of 243,938 variants. For each individual in RS-I, RS-II and RS-III (N = 10,792), PGRS were calculated using the –score command in PLINK across strata of P thresholds: 5.0 × 10−8, 5.0 × 10−7, 5.0 × 10−6, 5.0 × 10−5, 5.0 × 10−4, 0.005, 0.01, 0.05, 0.1, 0.5, 0.8 and 1.0. The proportion of variance explained by each PGRS model was calculated as the difference in the R2 between two regression models; one where SphE was regressed on age, sex, the first five principal components, and the other also including the PGRS as an additional covariate. Subsequently, AUCs were calculated for myopia (SphE ≤ -3 SD) versus hyperopia (SphE ≥ +3 SD).
Genetic correlation between ancestries
We used Popcorn48 to investigate ancestry-related differences in the genetic architecture of refractive error and myopia. Popcorn takes summary GWAS statistics from two populations and LD information from ancestry-matched reference panels, and computes genetic correlations by implementing a weighted likelihood function that accounts for the inflation of Z scores due to LD. Pairwise analyses were carried out using the GWAS summary statistics from 23andMe (N = 104,292), CREAM-EUR (N = 44,192) and CREAM-EAS (N = 9,826) meta-analyses. Only SNPs with MAF ≥ 5% were included, resulting in a final set of 3,625,602 SNPs for analyses involving 23andMe and 3,642,928 SNPs for the CREAM-EUR versus CREAM-EAS analysis. Reference panels were constructed using genotype data from 503 European and 504 East Asian individuals sequenced as part of the 1000 Genomes Project (release 2013-05-02 downloaded from: ftp.1000genomes.ebi.ac.uk). The reference panel VCF files were filtered using PLINK114 to remove indels, strand-ambiguous variants, variants without an “rs” id prefix, and variants located in the MHC region on chromosome 6 (chr6:25,000,000-33,500,000; Build 37).
Analysis between phenotypes
To evaluate consistency of genotypic effects across studies that employed different phenotype definitions, we compared effect sizes from GWAS studies of either SphE or AODM in Europeans, i.e. CREAM-EUR (N = 44,192) or 23andMe (N = 104,293) respectively. Marker-wise additive genetic effect sizes (in units diopters per copy of the risk allele) for SphE were compared against those (in units log(HR) per copy of the risk allele) for AODM. Data was visualised using R. Genetic correlation between the two phenotypes SphE and AODM was calculated using LD score regression. This analysis included all common SNPs (MAF > 0.01) present in HapMap3.
Evidence for functional involvement
In order to rank genes according to biological plausibility, we scored annotated genes based on our own findings and published reports for a potential functional role in refractive error. Points were assigned for each gene on the basis of 10 categories (details on the methodology per category are provided in Supplementary Note 4): internal replication of index genetic variants in the individual cohort GWAS analyses through Bonferroni corrections (CREAM-ASN, CREAM-EUR and 23andMe; pBonferroni 1.19 × 10−4), evidence for eQTL using the FUMA32 and extensive look-ups in GtEx, evidence of expression in the eye in developmental and adult ocular tissues, presence of an eye phenotype in knock-out mice (MGI and IMPC database), presence of an eye phenotype in humans (OMIM; see URLs, DisGeNET115), location in a functional region of a gene (wANNOVAR; see URLs), presence of the gene in a significant enriched functional pathway with false discovery rate < 0.05 (DEPICT49), presence of the gene in the gene priority analysis of DEPICT with false discovery rate < 0.05 and the presence of the gene in the canonical pathway analysis of Ingenuity Pathway Analysis (IPA; See URLs). Furthermore, we performed a systematic search for each gene to assess its potential as a drug target (SuperTarget116, STITCH117, DrugBank118, PharmaGkb119). All information derived from this study and literature were used to annotate genes to retinal cell types.
Genetic pleiotropy
To investigate overlap of genes with other common traits, we performed a look-up in the GWAS catalog using FUMA. Multiple testing correction (i.e. Benjamini-Hochberg) was performed. Traits were significantly associated when adjusted P ≤ 0.05 and the number of genes that overlap with the GWAS catalog gene sets was ≥ 2.
Data availability statement
The summary statistics of the Stage 3 meta-analysis are included in Supplementary Data 3 of this published article. In order to protect the privacy of the participants in our cohorts, further summary statistics of Stage 1 (CREAM) and Stage 2 (23andMe) will be available upon request. Please contact c.c.w.klaver@erasmusmc.nl (CREAM) and/or apply.research@23andMe.com (23andMe) for more information and to access the data.
Supplementary Material
Acknowledgments
We gratefully thank the invaluable contributions of all study participants, their relatives and staff at the recruitment centers. We thank all contributors to the CREAM Consortium, 23andMe, and UKEV for their generosity in sharing data and help in the production of this publication. Funding for this particular GWAS mega-analysis was provided by European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant 648268), Netherlands Organisation for Scientific Research (NWO, grant 91815655) and the National Eye Institute (grant R01EY020483). Funding agencies which facilitated the execution of the individual studies are acknowledged in Supplementary Note 1.
Footnotes
URLs. LDSC https://github.com/bulik/ldsc; Popcorn https://github.com/brielin/Popcorn; OMIM http://omim.org; wANNOVAR http://wannovar.wglab.org/; Polyphen http://genetics.bwh.harvard.edu/pph2/; SIFT http://sift.jcvi.org/www/SIFT_aligned_seqs_submit.html; Mutation Taster http://www.mutationtaster.org/; IPA http://www.ingenuity.com/index.html;
Competing interests
N.A.F., N.E., J.Y.T., and the 23andMe Research Team are current or former employees of 23andMe, Inc., and hold stock or stock options in 23andMe. J.B. J. is a patent holder with Biocompatibles UK Ltd. (Franham, Surrey, UK) (Title: Treatment of eye diseases using encapsulated cells encoding and secreting neuroprotective factor and/or anti-angiogenic factor; Patent number: 20120263794), and patent application with University of Heidelberg (Heidelberg, Germany) (Title: Agents for use in the therapeutic or prophylactic treatment of myopia or hyperopia; European Patent Number: 3 070 101). The other authors declare no competing financial interests.
Author contributions
M.S.T., V.J.M.V., S.M., J.A.G., A.I.I.G., R.W., P.G.H., A.I.I.G., and E.M.v.L. performed the analyses. C.C.W.K., V.J.M.V., M.S.T., R.W., J.A.G., and S.M. drafted the manuscript, and C.J.H., P.G.H., A.P.K., C.M.v.D., D.S, E.M.v.L., J.E.B.W., J.Y.T., N.A.F., Q.F., S.M.S., and V.V. critically reviewed the manuscript. A.N., A.P.K., A.T., C.B., C.Gi., C.L.S., C.Y.C., G.Bi., G.C., I.R., J.E.B.W., J.E.H., J.S.Ri., J.W., J.X., K.M.W., K.Y., M.P.C., M.S.H., M.S.T., N.A.F., N.E., P.C., P.Gh., P.K.J., Q.F., R.Ho., R.L.S., R.P.I., R.W., T.H., T.H.S.A., T.Z., V.V., W.Y.S., W.Z., X.L.S., Y.C.H., Y.S., and Y.Y.T. performed data analysis for the individual studies; and A.D.P., A.G.U., A.T., A.W.H., B.E.K.K., C.C.W.K., C.D., C.Gr., C.H., C.J.H., C.W., C.Y.C., D.A.M., F.R., G.Be., H.M.H., J.A.G., J.B.J., J.E.B.W., J.E.C., J.F.W., J.H.L., J.R.V., J.S.Ra., J.S.Ri., J.Y.T., K.Y., M.A.M.S., N.G.M., N.P., O.Po., O.Pa., O.T.R., P.Gu., P.J.F., P.M., P.N.B., R.K., S.K.I., S.M.S., T.L., T.M., W.Z., Y.C.H., and Y.X.W. contributed to data assembly. A.A.B.B., A.W., C.Gr., D.S., K.N.W., S.W.J.T., and T.Y. performed expression experiments, and M.S.T., A.A.B.B., P.J.v.d.S., and R.Ha. performed in silico pathway analyses. C.C.W.K. and C.J.H. conceived and designed the outline of the current report, and jointly with A.M., A.H., A.W.H., C.D., C.H., C.J.H., C.M.v.D., C.W., C.Y.C., D.A.M., D.S., E.S.T., F.M., G.Bi., I.R., J.A.G., J.B.J., J.E.B.W., J.E.C., J.F.W., J.H.L., J.R.V., J.Y.T., N.A., N.A.F., N.P., O.Pa., O.T.R., P.J.F., P.N.B., S.K.I., S.M.S., T.L., T.Y.W., T.Y., V.V., Y.X.W., and Y.Y.T. supervised conduction of experiments and analyses.
References for main text
- 1.Pan CW, Ramamurthy D, Saw SM. Worldwide prevalence and risk factors for myopia. Ophthalmic Physiol Opt. 2012;32:3–16. doi: 10.1111/j.1475-1313.2011.00884.x. [DOI] [PubMed] [Google Scholar]
- 2.Morgan IG. What Public Policies Should Be Developed to Deal with the Epidemic of Myopia? Optom Vis Sci. 2016;93:1058–60. doi: 10.1097/OPX.0000000000000980. [DOI] [PubMed] [Google Scholar]
- 3.Morgan I, Rose K. How genetic is school myopia? Prog Retin Eye Res. 2005;24:1–38. doi: 10.1016/j.preteyeres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Morgan IG, Ohno-Matsui K, Saw SM. Myopia. Lancet. 2012;379:1739–48. doi: 10.1016/S0140-6736(12)60272-4. [DOI] [PubMed] [Google Scholar]
- 5.Williams KM, et al. Increasing Prevalence of Myopia in Europe and the Impact of Education. Ophthalmology. 2015;122:1489–97. doi: 10.1016/j.ophtha.2015.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams KM, et al. Prevalence of refractive error in Europe: the European Eye Epidemiology (E(3)) Consortium. Eur J Epidemiol. 2015;30:305–15. doi: 10.1007/s10654-015-0010-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vongphanit J, Mitchell P, Wang JJ. Prevalence and progression of myopic retinopathy in an older population. Ophthalmology. 2002;109:704–11. doi: 10.1016/s0161-6420(01)01024-7. [DOI] [PubMed] [Google Scholar]
- 8.Seet B, et al. Myopia in Singapore: taking a public health approach. Br J Ophthalmol. 2001;85:521–6. doi: 10.1136/bjo.85.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith TS, Frick KD, Holden BA, Fricke TR, Naidoo KS. Potential lost productivity resulting from the global burden of uncorrected refractive error. Bull World Health Organ. 2009;87:431–7. doi: 10.2471/BLT.08.055673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verhoeven VJ, et al. Visual consequences of refractive errors in the general population. Ophthalmology. 2015;122:101–9. doi: 10.1016/j.ophtha.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 11.Tideman JW, et al. Association of Axial Length With Risk of Uncorrectable Visual Impairment for Europeans With Myopia. JAMA Ophthalmol. 2016;134:1355–1363. doi: 10.1001/jamaophthalmol.2016.4009. [DOI] [PubMed] [Google Scholar]
- 12.Flitcroft DI. The complex interactions of retinal, optical and environmental factors in myopia aetiology. Prog Retin Eye Res. 2012;31:622–60. doi: 10.1016/j.preteyeres.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Nakanishi H, et al. A genome-wide association analysis identified a novel susceptible locus for pathological myopia at 11q24.1. PLoS Genet. 2009;5:e1000660. doi: 10.1371/journal.pgen.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lam CY, et al. A genome-wide scan maps a novel high myopia locus to 5p15. Invest Ophthalmol Vis Sci. 2008;49:3768–78. doi: 10.1167/iovs.07-1126. [DOI] [PubMed] [Google Scholar]
- 15.Stambolian D, et al. Meta-analysis of genome-wide association studies in five cohorts reveals common variants in RBFOX1, a regulator of tissue-specific splicing, associated with refractive error. Hum Mol Genet. 2013;22:2754–64. doi: 10.1093/hmg/ddt116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan Q, et al. Genetic variants on chromosome 1q41 influence ocular axial length and high myopia. PLoS Genet. 2012;8:e1002753. doi: 10.1371/journal.pgen.1002753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan Q, et al. Meta-analysis of gene-environment-wide association scans accounting for education level identifies additional loci for refractive error. Nat Commun. 2016;7:11008. doi: 10.1038/ncomms11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng CY, et al. Nine Loci for Ocular Axial Length Identified through Genome-wide Association Studies, Including Shared Loci with Refractive Error. Am J Hum Genet. 2013;93:264–77. doi: 10.1016/j.ajhg.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, et al. Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet. 2011;7:e1002084. doi: 10.1371/journal.pgen.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi Y, et al. Genetic variants at 13q12.12 are associated with high myopia in the Han Chinese population. Am J Hum Genet. 2011;88:805–13. doi: 10.1016/j.ajhg.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li YJ, et al. Genome-wide association studies reveal genetic variants in CTNND2 for high myopia in Singapore Chinese. Ophthalmology. 2011;118:368–75. doi: 10.1016/j.ophtha.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z, et al. A genome-wide association study reveals association between common variants in an intergenic region of 4q25 and high-grade myopia in the Chinese Han population. Hum Mol Genet. 2011;20:2861–8. doi: 10.1093/hmg/ddr169. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Zhang HX. Polymorphism in the 11q24.1 genomic region is associated with myopia: a comprehensive genetic study in Chinese and Japanese populations. Mol Vis. 2014;20:352–8. [PMC free article] [PubMed] [Google Scholar]
- 24.Tran-Viet KN, et al. Mutations in SCO2 are associated with autosomal-dominant high-grade myopia. Am J Hum Genet. 2013;92:820–6. doi: 10.1016/j.ajhg.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldahmesh MA, et al. Mutations in LRPAP1 are associated with severe myopia in humans. Am J Hum Genet. 2013;93:313–20. doi: 10.1016/j.ajhg.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verhoeven VJ, et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat Genet. 2013 doi: 10.1038/ng.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiefer AK, et al. Genome-wide analysis points to roles for extracellular matrix remodeling, the visual cycle, and neuronal development in myopia. PLoS Genet. 2013;9:e1003299. doi: 10.1371/journal.pgen.1003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wojciechowski R, Hysi PG. Focusing in on the complex genetics of myopia. PLoS Genet. 2013;9:e1003442. doi: 10.1371/journal.pgen.1003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genomes Project, C et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19:807–12. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. FUMA: Functional mapping and annotation of genetic associations. bioRxiv. 2017 doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plotnikov D, Guggenheim J. Is a large eye size a risk factor for myopia? A Mendelian randomization study. bioRxiv. 2017 [Google Scholar]
- 34.UCSC Genome Browser.
- 35.Hsu F, et al. The UCSC Known Genes. Bioinformatics. 2006;22:1036–46. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 36.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly MP. Does phosphodiesterase 11A (PDE11A) hold promise as a future therapeutic target? Curr Pharm Des. 2015;21:389–416. doi: 10.2174/1381612820666140826114941. [DOI] [PubMed] [Google Scholar]
- 39.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 40.Mathe E, et al. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–25. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tavtigian SV, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bakshi A, et al. Fast set-based association analysis using summary data from GWAS identifies novel gene loci for human complex traits. Sci Rep. 2016;6:32894. doi: 10.1038/srep32894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferreira MA, et al. Gene-based analysis of regulatory variants identifies 4 putative novel asthma risk genes related to nucleotide synthesis and signaling. J Allergy Clin Immunol. 2016 doi: 10.1016/j.jaci.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pickrell JK. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am J Hum Genet. 2014;94:559–73. doi: 10.1016/j.ajhg.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.International Schizophrenia, C et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verhoeven VJ, et al. Large scale international replication and meta-analysis study confirms association of the 15q14 locus with myopia. The CREAM consortium. Hum Genet. 2012;131:1467–80. doi: 10.1007/s00439-012-1176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Finucane HK, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–35. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown BC, Asian Genetic Epidemiology Network Type 2 Diabetes C. Ye CJ, Price AL, Zaitlen N. Transethnic Genetic-Correlation Estimates from Summary Statistics. Am J Hum Genet. 2016;99:76–88. doi: 10.1016/j.ajhg.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pers TH, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun. 2015;6:5890. doi: 10.1038/ncomms6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fritsche LG, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48:134–43. doi: 10.1038/ng.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchey ER, et al. Vision-guided ocular growth in a mutant chicken model with diminished visual acuity. Exp Eye Res. 2012;102:59–69. doi: 10.1016/j.exer.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vincent A, et al. Biallelic Mutations in GNB3 Cause a Unique Form of Autosomal-Recessive Congenital Stationary Night Blindness. Am J Hum Genet. 2016;98:1011–9. doi: 10.1016/j.ajhg.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blake JA, et al. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res. 2017;45:D723–D729. doi: 10.1093/nar/gkw1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nikonov SS, et al. Cones respond to light in the absence of transducin beta subunit. J Neurosci. 2013;33:5182–94. doi: 10.1523/JNEUROSCI.5204-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stone EM, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- 56.Mackay DS, Bennett TM, Shiels A. Exome Sequencing Identifies a Missense Variant in EFEMP1 Co-Segregating in a Family with Autosomal Dominant Primary Open-Angle Glaucoma. PLoS One. 2015;10:e0132529. doi: 10.1371/journal.pone.0132529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Springelkamp H, et al. ARHGEF12 influences the risk of glaucoma by increasing intraocular pressure. Hum Mol Genet. 2015;24:2689–99. doi: 10.1093/hmg/ddv027. [DOI] [PubMed] [Google Scholar]
- 58.Haeseleer F, et al. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci. 2004;7:1079–87. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Littink KW, et al. A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder. Invest Ophthalmol Vis Sci. 2009;50:2344–50. doi: 10.1167/iovs.08-2553. [DOI] [PubMed] [Google Scholar]
- 60.Grimes WN, Li W, Chavez AE, Diamond JS. BK channels modulate pre- and postsynaptic signaling at reciprocal synapses in retina. Nat Neurosci. 2009;12:585–92. doi: 10.1038/nn.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parker RO, Crouch RK. Retinol dehydrogenases (RDHs) in the visual cycle. Exp Eye Res. 2010;91:788–92. doi: 10.1016/j.exer.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Radu RA, et al. Retinal pigment epithelium-retinal G protein receptor-opsin mediates light-dependent translocation of all-trans-retinyl esters for synthesis of visual chromophore in retinal pigment epithelial cells. J Biol Chem. 2008;283:19730–8. doi: 10.1074/jbc.M801288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo T, Sakai Y, Wagner E, Drager UC. Retinoids, eye development, and maturation of visual function. J Neurobiol. 2006;66:677–86. doi: 10.1002/neu.20239. [DOI] [PubMed] [Google Scholar]
- 64.Keckeis S, Reichhart N, Roubeix C, Strauss O. Anoctamin2 (TMEM16B) forms the Ca2+-activated Cl- channel in the retinal pigment epithelium. Exp Eye Res. 2016;154:139–150. doi: 10.1016/j.exer.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 65.Prasanna G, Narayan S, Krishnamoorthy RR, Yorio T. Eyeing endothelins: a cellular perspective. Mol Cell Biochem. 2003;253:71–88. doi: 10.1023/a:1026005418874. [DOI] [PubMed] [Google Scholar]
- 66.Yamashita T, et al. Essential and synergistic roles of RP1 and RP1L1 in rod photoreceptor axoneme and retinitis pigmentosa. J Neurosci. 2009;29:9748–60. doi: 10.1523/JNEUROSCI.5854-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davidson AE, et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat. 2013;34:506–14. doi: 10.1002/humu.22264. [DOI] [PubMed] [Google Scholar]
- 68.Hawthorne F, et al. Association mapping of the high-grade myopia MYP3 locus reveals novel candidates UHRF1BP1L, PTPRR, and PPFIA2. Invest Ophthalmol Vis Sci. 2013;54:2076–86. doi: 10.1167/iovs.12-11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feldkaemper M, Schaeffel F. An updated view on the role of dopamine in myopia. Exp Eye Res. 2013;114:106–19. doi: 10.1016/j.exer.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 70.Paul ML, Graybiel AM, David JC, Robertson HA. D1-like and D2-like dopamine receptors synergistically activate rotation and c-fos expression in the dopamine-depleted striatum in a rat model of Parkinson’s disease. J Neurosci. 1992;12:3729–42. doi: 10.1523/JNEUROSCI.12-10-03729.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stone RA, Lin T, Laties AM, Iuvone PM. Retinal dopamine and form-deprivation myopia. Proc Natl Acad Sci U S A. 1989;86:704–6. doi: 10.1073/pnas.86.2.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gardner M, Bertranpetit J, Comas D. Worldwide genetic variation in dopamine and serotonin pathway genes: implications for association studies. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1070–5. doi: 10.1002/ajmg.b.30717. [DOI] [PubMed] [Google Scholar]
- 73.D’Souza UM, Craig IW. Functional polymorphisms in dopamine and serotonin pathway genes. Hum Mutat. 2006;27:1–13. doi: 10.1002/humu.20278. [DOI] [PubMed] [Google Scholar]
- 74.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 75.MacArthur J, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Res. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holden BA, et al. Global Prevalence of Myopia and High Myopia and Temporal Trends from 2000 through 2050. Ophthalmology. 2016;123:1036–42. doi: 10.1016/j.ophtha.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 77.Cardon LR, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;361:598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- 78.Chua SY, et al. Age of onset of myopia predicts risk of high myopia in later childhood in myopic Singapore children. Ophthalmic Physiol Opt. 2016;36:388–94. doi: 10.1111/opo.12305. [DOI] [PubMed] [Google Scholar]
- 79.Williams KM, et al. Age of myopia onset in a British population-based twin cohort. Ophthalmic Physiol Opt. 2013;33:339–45. doi: 10.1111/opo.12042. [DOI] [PubMed] [Google Scholar]
- 80.Dolgin E. The myopia boom. Nature. 2015;519:276–8. doi: 10.1038/519276a. [DOI] [PubMed] [Google Scholar]
- 81.Connaughton V. Glutamate and Glutamate Receptors in the Vertebrate Retina. In: Kolb H, Fernandez E, Nelson R, editors. Webvision: The Organization of the Retina and Visual System. Salt Lake City (UT): 1995. [Google Scholar]
- 82.Hung GK, Mahadas K, Mohammad F. Eye growth and myopia development: Unifying theory and Matlab model. Comput Biol Med. 2016;70:106–18. doi: 10.1016/j.compbiomed.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 83.Norton TT. What Do Animal Studies Tell Us about the Mechanism of Myopia-Protection by Light? Optom Vis Sci. 2016;93:1049–51. doi: 10.1097/OPX.0000000000000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weiss S, Schaeffel F. Diurnal growth rhythms in the chicken eye: relation to myopia development and retinal dopamine levels. J Comp Physiol A. 1993;172:263–70. doi: 10.1007/BF00216608. [DOI] [PubMed] [Google Scholar]
- 85.Stone RA, Lin T, Iuvone PM, Laties AM. Postnatal control of ocular growth: dopaminergic mechanisms. Ciba Found Symp. 155:45–57. doi: 10.1002/9780470514023.ch4. discussion 57-62 (1990) [DOI] [PubMed] [Google Scholar]
- 86.Morgan IG. The biological basis of myopic refractive error. Clin Exp Optom. 2003;86:276–88. doi: 10.1111/j.1444-0938.2003.tb03123.x. [DOI] [PubMed] [Google Scholar]
- 87.Li XX, Schaeffel F, Kohler K, Zrenner E. Dose-dependent effects of 6-hydroxy dopamine on deprivation myopia, electroretinograms, and dopaminergic amacrine cells in chickens. Vis Neurosci. 1992;9:483–92. doi: 10.1017/s0952523800011287. [DOI] [PubMed] [Google Scholar]
- 88.Iuvone PM, Tigges M, Stone RA, Lambert S, Laties AM. Effects of apomorphine, a dopamine receptor agonist, on ocular refraction and axial elongation in a primate model of myopia. Invest Ophthalmol Vis Sci. 1991;32:1674–7. [PubMed] [Google Scholar]
- 89.Ashby R, McCarthy CS, Maleszka R, Megaw P, Morgan IG. A muscarinic cholinergic antagonist and a dopamine agonist rapidly increase ZENK mRNA expression in the form-deprived chicken retina. Exp Eye Res. 2007;85:15–22. doi: 10.1016/j.exer.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 90.Ashby R. Animal Studies and the Mechanism of Myopia-Protection by Light? Optom Vis Sci. 2016;93:1052–4. doi: 10.1097/OPX.0000000000000978. [DOI] [PubMed] [Google Scholar]
- 91.Rymer J, Wildsoet CF. The role of the retinal pigment epithelium in eye growth regulation and myopia: a review. Vis Neurosci. 2005;22:251–61. doi: 10.1017/S0952523805223015. [DOI] [PubMed] [Google Scholar]
- 92.Chen S, et al. Bright Light Suppresses Form-Deprivation Myopia Development With Activation of Dopamine D1 Receptor Signaling in the ON Pathway in Retina. Invest Ophthalmol Vis Sci. 2017;58:2306–2316. doi: 10.1167/iovs.16-20402. [DOI] [PubMed] [Google Scholar]
- 93.Chen PS, et al. Effects of C825T polymorphism of the GNB3 gene on availability of dopamine transporter in healthy volunteers–a SPECT study. Neuroimage. 2011;56:1526–30. doi: 10.1016/j.neuroimage.2010.10.082. [DOI] [PubMed] [Google Scholar]
- 94.Scott MS, Ono M. From snoRNA to miRNA: Dual function regulatory non-coding RNAs. Biochimie. 2011;93:1987–92. doi: 10.1016/j.biochi.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McFadden SA. Understanding and Treating Myopia: What More We Need to Know and Future Research Priorities. Optom Vis Sci. 2016;93:1061–3. doi: 10.1097/OPX.0000000000000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smith EL, 3rd, Hung LF, Arumugam B. Visual regulation of refractive development: insights from animal studies. Eye (Lond) 2014;28:180–8. doi: 10.1038/eye.2013.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Y, Wildsoet CF. RPE and Choroid Mechanisms Underlying Ocular Growth and Myopia. Prog Mol Biol Transl Sci. 2015;134:221–40. doi: 10.1016/bs.pmbts.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harper AR, Summers JA. The dynamic sclera: extracellular matrix remodeling in normal ocular growth and myopia development. Exp Eye Res. 2015;133:100–11. doi: 10.1016/j.exer.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Summers JA. The choroid as a sclera growth regulator. Exp Eye Res. 2013 doi: 10.1016/j.exer.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–9. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 102.Chen WM, Abecasis GR. Family-based association tests for genomewide association scans. Am J Hum Genet. 2007;81:913–26. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Winkler TW, et al. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc. 2014;9:1192–212. doi: 10.1038/nprot.2014.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zaykin DV. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J Evol Biol. 2011;24:1836–41. doi: 10.1111/j.1420-9101.2011.02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol. 2008;32:227–34. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pruim RJ, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–66. doi: 10.1038/nprot.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cooper GM, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 111.Loh PR, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47:284–90. doi: 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McCarthy S, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bauer-Mehren A, Rautschka M, Sanz F, Furlong LI. DisGeNET: a Cytoscape plugin to visualize, integrate, search and analyze gene-disease networks. Bioinformatics. 2010;26:2924–6. doi: 10.1093/bioinformatics/btq538. [DOI] [PubMed] [Google Scholar]
- 116.Gunther S, et al. SuperTarget and Matador: resources for exploring drug-target relationships. Nucleic Acids Res. 2008;36:D919–22. doi: 10.1093/nar/gkm862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kuhn M, et al. STITCH 4: integration of protein-chemical interactions with user data. Nucleic Acids Res. 2014;42:D401–7. doi: 10.1093/nar/gkt1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wishart DS, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–72. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Whirl-Carrillo M, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92:414–7. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The summary statistics of the Stage 3 meta-analysis are included in Supplementary Data 3 of this published article. In order to protect the privacy of the participants in our cohorts, further summary statistics of Stage 1 (CREAM) and Stage 2 (23andMe) will be available upon request. Please contact c.c.w.klaver@erasmusmc.nl (CREAM) and/or apply.research@23andMe.com (23andMe) for more information and to access the data.