

Abstract
Hepatic ischemia/reperfusion injury (IRI) is an unavoidable consequence of liver transplantation that can lead to postoperative hepatic dysfunction. Myeloid cells that include Kupffer cells, monocytes, and neutrophils contribute to the inflammatory response and cellular injury observed during hepatic IRI. We hypothesize that overactivation of the nuclear erythroid 2 p45- related factor 2 (Nrf2)–antioxidant response element (ARE) pathway in myeloid cells leads to decreased cellular damage after hepatic IRI. We constructed transgenic mice with constitutively active nuclear erythroid 2 p45-related factor 2 (caNrf2) that over activates the Nrf2-ARE pathway in myeloid cells (lysozyme M cre recombinase [LysMcre]+/caNrf2−, n = 9), and their littermate controls lacking transgene expression (LysMcre+/caNrf2−, n = 11). The mice underwent either sham or partial hepatic ischemia surgery, with 60 minutes of ischemia followed by 6 hours of reperfusion. After IRI, LysMcre+/caN-rf2+ mice demonstrated significantly decreased serum alanine aminotransferase and decreased areas of necrosis. Immunohistochemistry and immunoblot of caspase 3 showed a significantly decreased cleaved to full-length caspase 3 ratio in LysMcre+/caNrf2+ animals. Lymphocyte antigen 6 complex locus G and CD68 staining demonstrated reduced inflammatory cell infiltration. LysMcre+/caNrf2+ animals also had significantly decreased gene expression of proinflammatory cytokines, including interleukin (IL) 1β, IL6, tumor necrosis factor α, chemokine (C-C motif) ligand 2, and chemokine (C-X-C motif) ligand 10, and significantly decreased levels of 8-isoprostanes. In our model, Nrf2 overactivation in myeloid cells leads to decreased hepatocellular damage, necrosis, apoptosis, inflammation, and oxidative stress. Pharmacologic targeting of the Nrf2-ARE pathway in myeloid cells may be a novel strategy to mitigate hepatic IRI.
Liver transplantation, the standard treatment for endstage liver disease, is limited by the significant donor organ shortage. To combat the organ shortage, many centers are using donor livers that are older, steatotic, or recovered from donation after circulatory death donors. These livers are more susceptible to ischemia/reperfusion injury (IRI) and suffer a higher percentage of allograft dysfunction and failure.(1)
Hepatic IRI is a complex, multifactorial process that involves several mechanisms of cellular injury including impaired oxidative metabolism, depletion of adenosine triphosphate, increased production of reactive oxygen species (ROS), and decreased expression of cytoprotective genes.(2) During the initial phase of IRI, ischemic stress leads to hepatocellular damage and the release of damage-associated molecular patterns that bind to Toll-like receptors on Kupffer cells (KCs), resulting in KC activation.(3) Upon activation, KCs respond by releasing a multitude of inflammatory cytokines including tumor necrosis factor α (TNF-α), interleukin (IL) 1, C-C and C-X-C family chemokines, and cytotoxic ROS that further potentiate the inflammatory response.(4) During the later phase, cytokine release from KCs results in the recruitment of additional myeloid cells (neutrophils and monocytes) into the liver that further enhance the inflammatory milieu and tissue injury. The abundance of ROS overcomes the intrinsic, antioxidant defense mechanisms of the cell, leading to lipid peroxidation, disruption of calcium homeostasis, and ultimately direct cellular injury.(4)
Nuclear erythroid 2 p45-related factor 2 (Nrf2) is a basic leucine zipper transcription factor with a cap ‘n’ collar structure that is key to the cellular antioxidative response. Nrf2 is normally bound in the cytoplasm to the kelch-like ECH-associated protein 1 (KEAP1) via the Nrf2-ECH homology (Neh2) domain at the N-terminus of Nrf2. KEAP1 acts as a negative regulator of Nrf2 by targeting Nrf2 toward ubiquitination and degradation.(5) Electrophilic compounds and in some cases ROS directly lead to modification of KEAP1’s cysteine thiols, resulting in a conformational change in KEAP1 and disruption of Nrf2 ubiquitination. This leads to translocation of newly synthesized Nrf2 into the nucleus.(6,7) Once in the nucleus, Nrf2 binds to antioxidant response elements (AREs), which control several antioxidative and other cytoprotective genes.(8)
Previous studies have demonstrated a protective role of Nrf2 in the setting of hepatic IRI. The global absence of Nrf2 results in increased cellular damage seen during warm hepatic IRI.(9,10) Overactivation of Nrf2 in hepatocytes through expression of a constitutively active mutant in these cells leads to a reduction of hepatocellular damage after warm hepatic IRI.(10) In addition, overactivation of Nrf2 in hepatocytes of donor livers using hepatocyte-specific KEAP1-knockdown mice leads to a reduction in cold preservation-IRI in a murine liver transplant model.(11) Despite these Nrf2-dependent protective effects, the selective impact of Nrf2 activation in nonparenchymal cells of the liver during IRI remains unclear. Because of the significant proinflammatory and oxidative effects of activated myeloid cells during hepatic IRI, we chose to investigate the Nrf2-ARE pathway in these cells during hepatic IRI. We hypothesized that induction of the Nrf2-ARE pathway in myeloid cells, which include KCs, neutrophils, and monocytes, may lead to a reduction in the oxidative stress and cellular damage that is seen during hepatic IRI. We used a novel transgenic mouse that specifically over activates the Nrf2-ARE pathway in myeloid cells to assess the protective impact in a model of hepatic IRI.
Materials and Methods
ANIMALS
Mice expressing a constitutively active nuclear erythroid 2 p45-related factor 2 (caNrf2) under the control of a cytomegalovirus promoter and β-actin enhancer(12) were mated with transgenic mice expressing cyclization recombination (Cre) recombinase in myeloid cells under control of the lysozyme2 (Lyz2) promoter (LysMcre mice; Jackson Labs, Bar Harbor, ME). Upon mating, the caNrf2 mice with mice expressing Cre under control of the Lyz2 promoter, Cre recombinase excises the STOP cassette, and caNrf2 is expressed specifically in myeloid cells. In previous studies where the caNrf2 mice were mated with mice expressing Cre in hepatocytes under the control of the albumin (Alb) promoter, the transgenic mice were shown to express the caNrf2 protein at similar levels as the endogenous protein in the liver.(13) Because the caNrf2 protein is constitutively present in the nucleus, it efficiently induces expression of Nrf2 target genes, resulting in their up-regulation.
Transgenic mice (age 6-12 weeks) with myeloid-specific expression of caNrf2 (LysMcre+/caNrf2+; n = 9) and littermate controls (LysMcre+/caNrf2−; n = 11) were subjected to either sham or partial hepatic ischemia surgery. Animals that underwent sham surgery had laparotomy performed, followed by abdominal closure. For animals that underwent partial hepatic ischemia surgery, a vascular clip was applied to the left pedicle of the portal triad to render 70% of the liver ischemic. Ischemia was verified by visual confirmation. After 60 minutes of partial hepatic ischemia, the vascular clip was removed to allow for reperfusion, followed by abdominal closure. Six hours after the procedure, animals were killed, and liver and serum samples were collected for analysis. All experiments were performed according to the ethical guidelines outlined in the Guide for Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee at the University of Wisconsin School of Medicine and Public Health.
KC AND HEPATOCYTE ISOLATION
To confirm the efficiency of genetic recombination of the transgene in the KCs of our mice, we isolated hepatocytes and KCs from LysMcre+/caNrf2+ (n = 4) and LysMcre+/caNrf2− (n = 4) mice. The transgenic mice were anesthetized, and midline laparotomy was performed. The liver was flushed with Hank’s balanced salt solution, followed by 2.50 mg/mL of Pronase (Roche Diagnostics, Indianapolis, IN) in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Mediatech, Manassas, VA) at 3.0 mL/minute, followed by 0.15 U/mL Liberase Tm (Roche Diagnostics) and 0.50 mg/mL of Pronase in DMEM/F12. The liver was resected and digested further in the previous Liberase TM/Pronase solution with additional 0.1 mg/mL of deoxyribonuclease I (DNaseI) (Roche Diagnostics) for 30 minutes at 37°C with agitation. The cell suspension was filtered through a 70-μm cell strainer, and viable cells were pelleted by centrifugation at 700g for 10 minutes. Hepatocytes were pelleted by suspending the cell pellet in DMEM/F12 and centrifuging at 50g for 2 minutes.
The supernatant containing the nonparenchymal cells (NPCs) was collected for further isolation. The NPCs were pelleted by centrifugation at 700g for 10 minutes. The pellet was then suspended in 70% Percoll in DMEM/F12. This suspension was layered at the bottom of a conical tube, followed by a layer of 50% Percoll, 26% Percoll, and finally a DMEM/F12 layer. The gradient was centrifuged for 30 minutes at 400g, 4°C, with no brake. The KC layer was isolated between the 50% and 26% Percoll gradient interface. This layer was further purified for KCs by using magnetic cell sorting (MACS) with positive selection with α-F4/80-Biotin in combination with α-Biotin MicroBeads, per commercial protocol (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany).
BONE MARROW (BM) NEUTROPHIL AND MONOCYTE ISOLATION
Deletion of the STOP cassette through Cre-mediated recombination and expression of the caNrf2 transgene was confirmed in myeloid cells of the BM by performing isolations of BM from LysMcre+/caNrf2+ (n = 3) and LysMcre+/caNrf2− (n = 3) mice. The mice were killed under isoflurane anesthesia using the cervical dislocation method. The skin was removed from the hind limbs, and hind limbs were resected from the hip joint. The epiphyses were cut off, and the long bones were flushed using a 25-gauge needle with DMEM supplemented with 10% fetal bovine serum and 2 mM ethylene diamine tetraacetic acid. The cell suspension was filtered through a 70-μm cell strainer. The bone epiphyses were cut into small pieces with a scalpel and pushed through the 70-μm strainer. Viable BM cells were pelleted by centrifugation at 1400 rpm for 7 minutes at 4°C. The cells were washed with perfusate and again pelleted by centrifugation at 1400 rpm for 7 minutes at 4°C. The isolated BM was then purified for neutrophils and monocytes by performing sequential MACS isolations with α-Biotin MicroBeads, per commercial protocol. Neutrophil isolation was performed using positive selection with α–lymphocyte antigen 6 complex locus G (Ly6-G)–Biotin; this was followed by selection with α-CD11b-Biotin for monocytes (Miltenyi Biotec GmbH).
POLYMERASE CHAIN REACTION (PCR)–BASED RECOMBINATION ASSAY
RNA was isolated from KCs, hepatocytes, BM neutrophils, and BM monocytes using TRIzol Reagent, according to the manufacturer’s protocol (Life Technologies, Carlsbad, CA). Following RNA isolation, single-stranded complementary DNA (cDNA) was reverse-transcribed (Promega, Madison, WI) with a specific reverse primer that hybridizes in the caNrf2 sequence downstream of the floxed transcription/translation STOP cassette. After cDNA synthesis of this specific region, a real-time polymerase chain reaction (RT-PCR)–based assay was employed using 2 primer sets. The first primer set confirmed the presence of the caNrf2 transgene by amplifying a 306 base pair (bp) segment in the caNrf2 sequence upstream of the cDNA synthesis site. The second primer set was designed to determine allele recombination. In nonrecombined alleles, this second primer set amplified a 1652 bp segment consisting of the β-actin promoter, floxed STOP cassette, and caNrf2 sequence. In recombined alleles, the floxed STOP cassette is excised by Cre recombinase, and the second primer set amplifies a 273 bp segment consisting only of the β-actin promoter and caNrf2 sequence. Following RT-PCR amplification, the products were run on an agarose gel and visualized in Syngene G:Box (Syngene, Frederick, MD). Hepatocytes were also isolated from previously characterized transgenic mice with hepatocyte-specific overactivation of Nrf2 (AlbCre+/caNrf2+) and control mice (AlbCre+/caNrf2−).(13,14) RNA isolated from these animals served as positive and negative controls, respectively (data not shown).
HISTOLOGICAL ANALYSIS
Formalin-fixed, paraffin-embedded livers were sectioned and hematoxylin-eosin (H & E)–stained slides were reviewed by 2 pathologists (Kristina A. Matkowskyj and Shelly Cook) to determine histopathological features. Immunohistochemistry (IHC) was performed, as previously described, using rabbit anti-cleaved caspase 3 (1:100; Epitomics, Burlingame, CA), rat anti-Ly6-G (1:100) (Biolegend, San Diego, CA), or rabbit anti-CD68 (1:500; Abcam, Cambridge, MA) primary antibodies.(14)
IMAGE ANALYSIS
The slides for each animal were visualized under light microscopy using Axiovert 200M microscope (Carl Zeiss, Gottingen, Germany). Four random high-powered fields of view were taken for each animal.
SERUM ANALYSIS
Whole blood was collected at the time of death. Blood was allowed to coagulate at room temperature for at least 30 minutes and centrifuged to collect serum. Serum alanine aminotransferase (ALT) was measured using the IDEXX VetTest Chemistry Analyzer (IDEXX, Westbrook, ME).
8-ISOPROSTANE
To assess the degree of lipid peroxidation and oxidative stress in damaged livers, we measured 8-isoprostane levels in the hepatic tissue using the 8-Isoprostane enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI). The assay was carried out as previously described.(14)
QUANTITATIVE RT-PCR
Quantitative RT-PCR was performed as previously described.(14) We investigated messenger RNA (mRNA) abundance of β-actin, nicotinamide adenine dinucleotide phosphate dehydrogenase, reduced form, quinone 1 (NQO1), glutathione S-transferase alpha 2 (GSTA2), glutathione S-transferase alpha 4 (GSTA4), glutathione S-transferase mu 1 (GSTM1), glutathione S-transferase mu 2 (GSTM2), glutamate-cysteine ligase, modifier subunit (GCLM), peroxiredoxin 1 (PRDX1), thioredoxin reductase 1 (TXNRD1), IL1β, IL6, TNF-α, chemokine (C-C motif) ligand 2 (CCL2), chemokine (C-X-C motif) ligand 10 (CXCL10), and intercellular cell adhesion molecule 1 (ICAM1), using the primer sequences listed in the Supporting Table.
WESTERN BLOT ANALYSIS
Three random sham animals and 4 random IRI animals from each experimental group were used for Western blot analysis. Western blotting technique was performed, as previously described, with primary antibodies for anti-caspase 3 (Cell Signaling, Danvers, MA), anti-cleaved caspase 3 (Cell Signaling), or anti-β-actin (Abcam).(14) Densitometry for bands of interest was performed using ImageJ.(15)
STATISTICS
All data in figures are presented as mean 6 standard error of the mean (SEM). Comparisons between the groups were performed with either 1-way analysis of variance followed by Fisher’s least significant difference post hoc test, or Student t test where appropriate. Log transformation was performed as appropriate as determined by Levene’s test for equality of variance. P < 0.05 was used for statistical significance. All statistical tests were performed using IBM SPSS Statistics, version 22 (IBM, Armonk, NY).
Results
EFFICIENT RECOMBINATION AT THE TRANSGENE LOCUS IN MYELOID CELLS FROM LysMcre+/caNrf2+ MICE, RESULTING IN INCREASED EXPRESSION OF Nrf2–DEPENDENT GENES
To determine the Cre-mediated recombination efficiency at the caNrf2 transgene locus in myeloid cells, a PCR-based recombination assay was performed using RNA isolated from hepatocytes, KCs, BM-derived neutrophils, and monocytes of LysMcre+/caNrf2+ mice and littermate controls. Representative agarose gels are depicted in Fig. 1A,B. A 306 bp band was present in all 4 cell populations isolated from LysMcre+/caNrf2+ mice, confirming the presence of the caNrf2 transgene. In addition, KCs from all 4 LysMcre+/caNrf2+ mice (Fig. 1A) and neutrophils and monocytes from all 3 LysMcre+/caNrf2+ mice (Fig. 1B) had a 273 bp band present, indicating a recombination efficiency of 100. None of the myeloid cell populations isolated from the LysMcre+/caNrf2− mice nor hepatocytes isolated from either mouse demonstrated a 273 bp band, thus confirming the selective recombination of caNrf2 in myeloid cells.
FIG. 1.
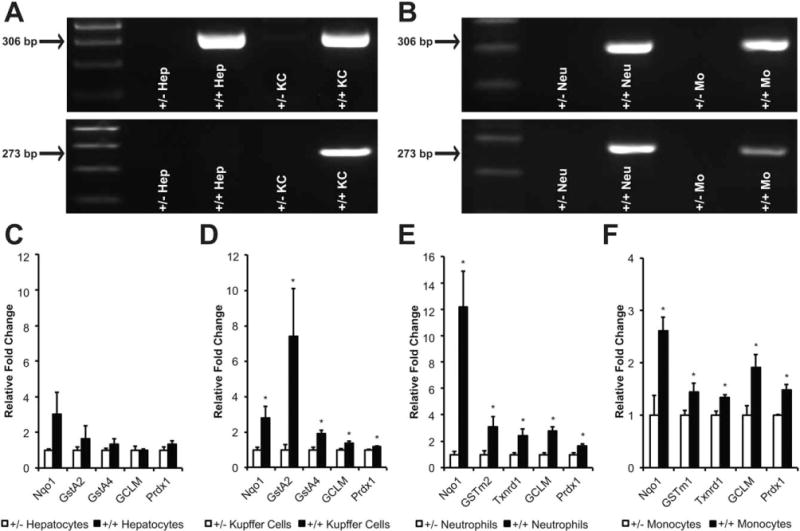
Cre-mediated recombination at the transgene locus and Nrf2 overactivation in LysMcre+/caNrf2+ mice. Hepatocytes and myeloid cells, including KC, neutrophils, and monocytes, were isolated from LysMcre+/caNrf2+ mice. (A, B) RT-PCR was performed on multiple different cell isolations from the transgenic mice and control littermates. The upper panels of A and B show amplification of a 306 bp band demonstrating the insertion of the caNrf2 transgene in caNrf2+ animals. The lower panels show the amplification of a 273 bp band demonstrating successful Cre-mediated recombination specifically in (A) KCs and (B) neutrophils and monocytes of LysMcre+/caNrf2+ animals, and not in (A) hepatocytes. Quantitative RT-PCR for Nrf2-dependent genes was performed on LysMcre+/caNrf2+ for (C) hepatocytes, (D) KCs, (E) neutrophils, and (F) monocytes. Mean ± SEM. * P < .05 as compared to LysMcre+/caNrf2− cells.
Quantitative RT-PCR was then performed on RNA isolated from hepatocytes and each group of myeloid cells to determine the expression of prototypical Nrf2-dependent genes. Messenger RNA abundance was measured for a panel of Nrf2-dependent genes (GCLM, GSTA2, GSTA4, GSTM1, GSTM2, NQO1, PRDX1, and/or TXNRD1). We did not observe any differences in the mRNA abundance of Nrf2-dependent genes in the hepatocytes (Fig. 1C). However, significant elevations in mRNA abundance of most of the genes were observed in each of the myeloid cells isolated from LysMcre+/caNrf2+ mice, as compared to littermate controls (Fig. 1D-F). These data confirmed the overactivation of the Nrf2-ARE pathway in the myeloid cells of our transgenic mice. We also measured mRNA expression of Nrf2 and caNrf2 in the myeloid cells of the LyzMcre+/caNrf2+ mice. The ratio of endogenous Nrf2:caNrf2 was 8.6:1 in KCs, 1.3:1 in neutrophils, and 5.3:1 in monocytes (data not shown). Even though the mRNA abundance of caNrf2 was less than endogenous Nrf2, we observed significant increases in the expression of Nrf2-dependent genes in the LyzMcre+/caNrf2+ mice, most likely due to the high stability of the caNrf2 mutant, which is no longer degraded via the KEAP1-ubiquitin-proteasome pathway and constitutively present in the nucleus.
LysMcre+/caNrf2+ MICE HAVE DECREASED SERUM ALT AFTER PARTIAL HEPATIC ISCHEMIA/REPERFUSION (IR)
After verifying the transgenic system, either partial hepatic IR or sham surgery was performed on the LysMcre+/caNrf2+ mice and their littermate controls. After IR, the LysMcre+/caNrf2− and LysMcre+/caNrf2+ mice had significantly increased serum ALT levels as compared to their sham counter-parts, indicative of hepatocellular damage from IRI. However, when serum ALT levels in the LysMcre+/caNrf2+ mice were compared to their littermate controls, there was a significant, almost 80% decrease in serum ALT levels, suggesting decreased hepatocellular damage from IRI in the transgenic mice (Fig. 2).
FIG. 2.
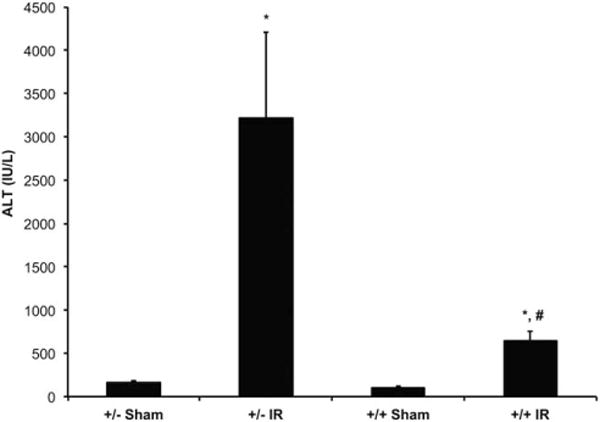
Serum ALT levels of LysMcre/caNrf2 animals after partial hepatic ischemia. LysMcre+/caNrf2+ and LysMcre+/caNrf2− animals were subjected to either sham or partial hepatic IR surgery (LysMcre+/caNrf2− sham, n = 4; LysMcre+/caNrf2− IR, n = 7; LysMcre+/caNrf2+ sham, n = 4; LysMcre+/caNrf2+ IR, n = 5). Animals were killed 6 hours after reperfusion, and their serum collected for analysis. Serum ALT demonstrates a significant increase after IR in the LysMcre+/caNrf2− animals. However, when comparing animals undergoing IR, LysMcre+/caNrf2+ animals have significantly decreased serum ALT as compared to LysMcre+/caNrf2−. Mean ± SEM. * P < 0.05 as compared to the sham group of the same genotype. # P < 0.05 as compared to +/− group of the same treatment.
LysMcre+/caNrf2+ MICE HAVE DECREASED AREAS OF NECROSIS AFTER PARTIAL HEPATIC IR
H & E–stained liver parenchyma was examined to determine the degree of liver damage after IR or sham surgery. As expected, animals that underwent sham surgery had minimal areas of liver damage. Among the animals that underwent IR, there were large areas of damage noted in the LysMcre+/caNrf2− animals. However, livers from the LysMcre+/caNrf2+ animals had minimal areas of cellular damage (Fig. 3A-D). To better quantify the degree of liver damage, 2 independent, blinded pathologists reviewed the H & E– stained slides. The slides were scored using the Suzuki scoring method that takes into account cellular congestion, vacuolization, and necrosis.(16) There were no differences in the degree of congestion, vacuolization, and overall Suzuki score between the LysMcre+/caNrf2+ and LysMcre+/caNrf2− mice. However, the LysMcre+/caNrf2− animals undergoing IR had a significantly higher Suzuki score for necrosis than the LysMcre+/caNrf2− mice undergoing sham surgery. In contrast, the LysMcre+/caNrf2+ animals undergoing IR had similar necrosis scores as compared to the sham animals. In addition, for those animals undergoing IR, the mean necrosis score was significantly lower in the LysMcre+/caNrf2+ mice compared to that seen in the littermate controls (Fig. 3E).
FIG. 3.
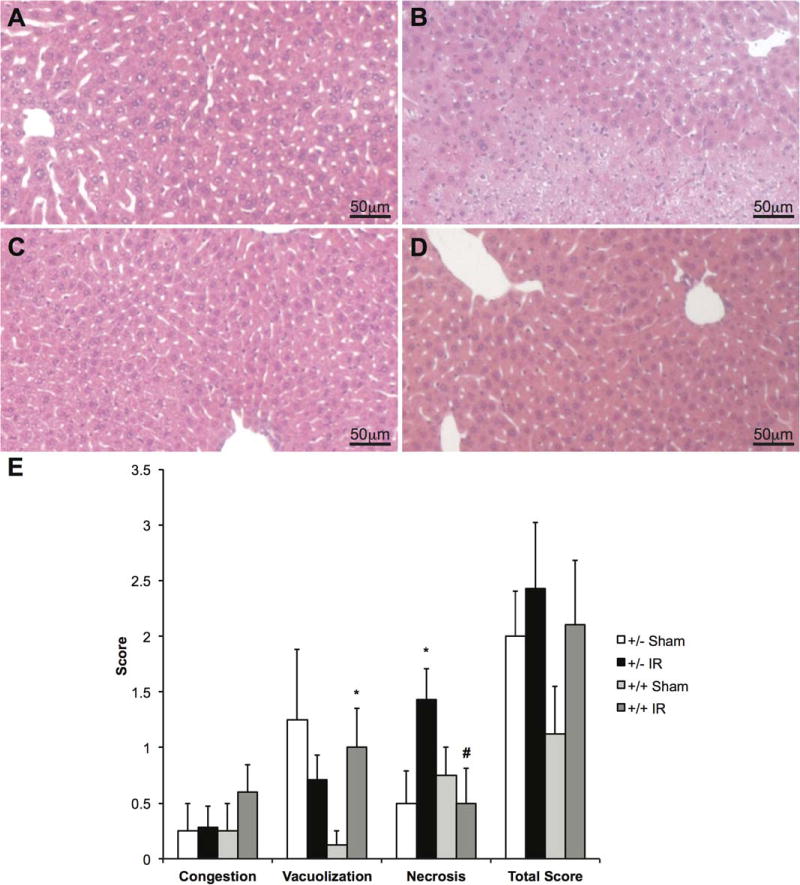
H & E staining of LysMcre/caNrf2 animals after partial hepatic ischemia. H & E staining of the LysMcre/caNrf2 animal livers was performed after either sham or partial hepatic ischemia surgery. Representative images for (A) LysMcre+/caNrf2− sham, (B) LysMcre+/caNrf2− IR, (C) LysMcre+/caNrf2+ sham, and (D) LysMcre+/caNrf2+ IR animals are shown. The sham group in both genotypes demonstrated normal liver histology. Although LysMcre+/caNrf2− animals after IR had large areas of necrosis, the LysMcre+/caNrf2+ animals after IR showed minimal necrosis. (E) The H & E slides were presented to 2 independent pathologists in a blinded manner, and Suzuki score was evaluated. On the basis of Suzuki scoring, the LysMcre+/caNrf2+ IR group demonstrated significantly decreased necrosis scores as compared to the LysMcre+/caNrf2− IR group. *P < 0.05 as compared to the sham group of the same genotype. # P < 0.05 as compared to +/− group of the same treatment.
LysMcre+/caNrf2+ MICE HAVE DECREASED APOPTOSIS AFTER PARTIAL HEPATIC IR
We performed IHC on liver tissue for cleaved caspase 3 to determine the effect of Nrf2 overactivation in myeloid cells on apoptosis. As expected, livers from sham animals had minimal cleaved caspase 3 staining (Fig. 4A,C). However, there was significantly more cleaved caspase 3 staining surrounding areas of necrosis in the LysMcre+/caNrf2− animals undergoing IR (Fig. 4B), as compared to the LysMcre+/caNrf2+ animals (Fig. 4D). On the basis of cellular morphology, the majority of cleaved caspase 3–positive cells appeared to be hepatocytes.
FIG. 4.
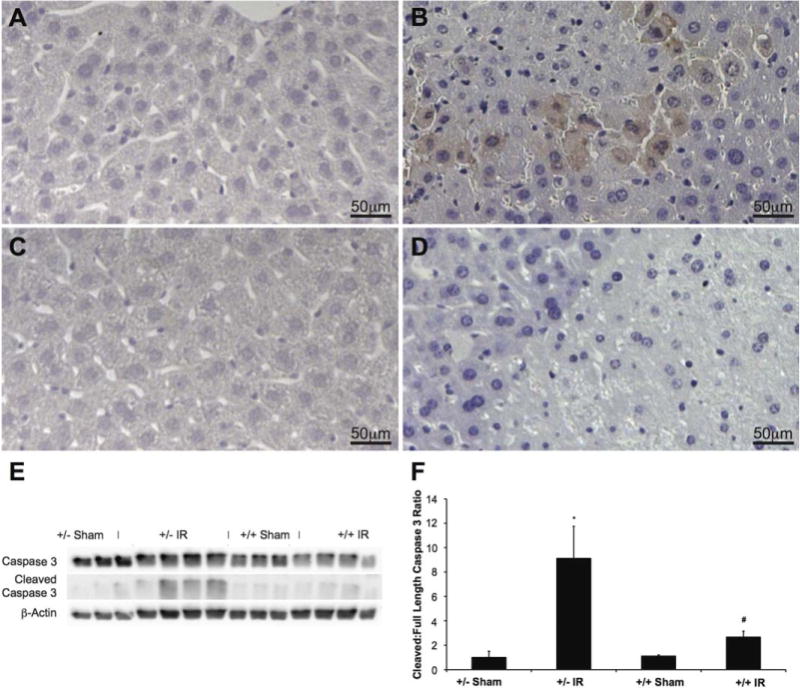
Cleaved caspase 3 IHC and Western blot after partial hepatic ischemia. Cleaved caspase 3 was examined in each of the different groups to assess for evidence of apoptosis. Representative immunohistochemical staining for cleaved caspase 3 is shown for (A) LysMcre+/caNrf2− sham, (B) LysMcre+/caNrf2− IR, (C) LysMcre+/caNrf2+ sham, and (D) LysMcre+/caNrf2+ IR mice. The sham group in both genotypes demonstrated no cleaved caspase 3 staining. LysMcre+/caNrf2− animals after IRI had high numbers of cells with cleaved caspase 3 staining surrounding areas of necrosis. In the LysMcre+/caNrf2+ animals after IRI, in comparable areas of necrosis, there is minimal, if any, cleaved caspase 3 staining. To further clarify apoptosis in our experimental groups, whole cell protein lysates of the livers were examined by Western blot. (E) Western blotting was performed for caspase 3, cleaved caspase 3, and β-actin. (F) Densitometry of the cleaved caspase 3 to full-length caspase 3 was quantified using ImageJ, and the ratio normalized to LysMcre+/caNrf2− sham group. The IR groups in both genotypes have a significantly increased cleaved to full-length caspase 3 ratio, as compared to sham animals of the same genotype. The LysMcre+/caNrf2+ IR group has a significantly decreased cleaved to full-length caspase 3 ratio as compared to the LysMcre+/caNrf2− IR group, demonstrating decreased apoptosis. * P < 0.05 as compared to the sham group of the same genotype. # P < 0.05 as compared to +/− group of the same treatment.
Western blot for cleaved caspase 3 was performed to assess the presence of apoptosis in the different experimental groups (Fig. 4E). The band densities for cleaved caspase 3 and full-length caspase 3 were quantified and analyzed (Fig. 4F). Consistent with the IHC findings, increased caspase 3 cleavage was observed only in the LysMcre+/caNrf2− mice undergoing IR, whereas the sham groups and the LysMcre+/caNrf2+ animals undergoing IR had minimal cleaved caspase 3. In comparison, LysMcre+/caNrf2+ animals undergoing IR had significantly decreased caspase 3 cleavage as compared to their littermate counterparts, suggesting decreased apoptosis in the mice with myeloid-specific Nrf2 overactivation.
LysMcre+/caNrf2+ MICE HAVE DECREASED INFLAMMATORY CELL INFILTRATE AFTER PARTIAL HEPATIC IR
To examine the extent of inflammation present in the livers after IR, we performed IHC with anti-Ly6-G antibody. Ly6-G, which is predominantly expressed on neutrophils, was used as a marker of neutrophilic infiltration. Animals undergoing sham surgery had minimal Ly6-G staining (Fig. 5A,C), whereas LysMcre+/caNrf2− animals undergoing IR had increased amounts of Ly6-G–positive cells (Fig. 5B). LysMcre+/caNrf2+ animals undergoing IR had minimal staining surrounding comparable areas of necrosis (Fig. 5D).
FIG. 5.
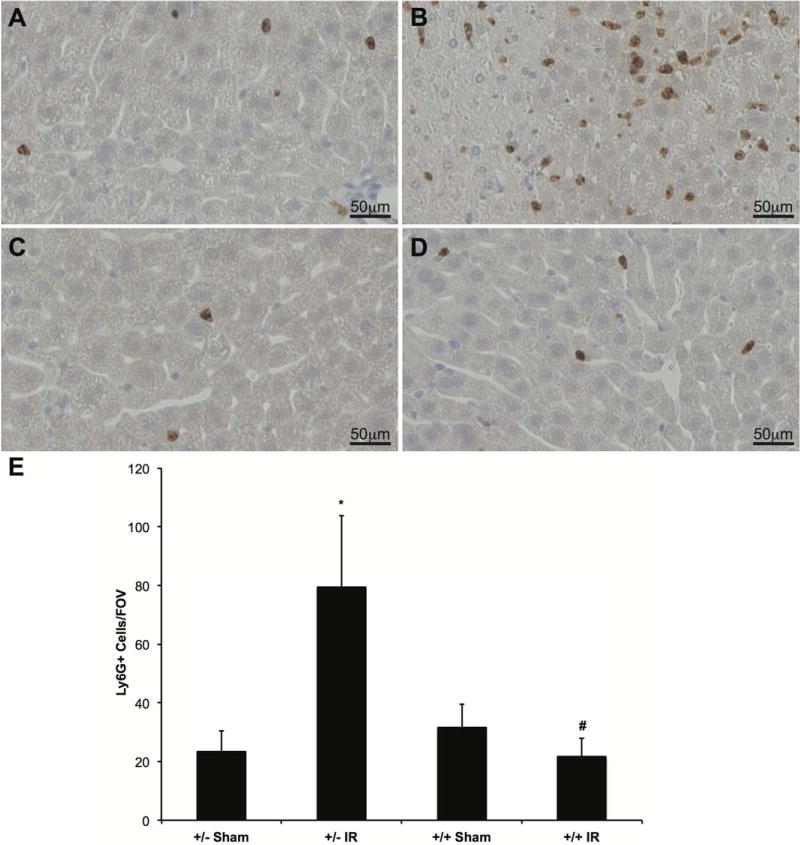
IHC for neutrophilic infiltration. To evaluate the extent of neutrophil infiltration in the different groups, IHC with a Ly6-G antibody was performed. Ly6-G is a protein expressed on neutrophils, and thus serves as a marker of neutrophil infiltration. Representative slides for (A) LysMcre+/caNrf2− sham (n = 4), (B) LysMcre+/caNrf2− IR (n = 7), (C) LysMcre+/caNrf2+ sham (n = 4), and (D) LysMcre+/caNrf2+ IR (n = 5) animals are shown. There is minimal neutrophilic infiltrate in the sham groups of both genotypes and the LysMcre+/caNrf2+ IR group. However, there is a marked increase in neutrophilic infiltrate in the LysMcre+/caNrf2− IR group. (E) The number of Ly6-G positive cells was quantified for each experimental group. Confirmatory to our visual findings, we see significantly increased numbers of Ly6-G staining cells in the LysMcre+/caNrf2− IR group as compared to its corresponding sham group. We also see significantly decreased numbers of Ly6-G positive cells in the LysMcre+/caNrf2+ IR group as compared to the LysMcre+/caNrf2− IR group. * P < 0.05 as compared to the sham group of the same genotype. # P < 0.05 as compared to +/− group of the same treatment.
To quantify neutrophilic infiltration, we counted the number of Ly6-G–positive cells. LysMcre+/caNrf2− mice undergoing IR had a significantly increased number of Ly6-G–positive cells as compared to their sham counterparts. In contrast, there were no differences in the number of Ly6-G–positive cells in the LysMcre+/caNrf2+ animals undergoing IR, as compared to the sham animals. In comparison, LysMcre+/caNrf2+ animals undergoing IR had significantly fewer Ly6-G–positive cells as compared to the LysMcre+/caNrf2− animals (Fig. 5E).
To examine monocytic infiltration and the number of KCs in the liver tissue, we performed IHC using anti-CD68 antibody. Representative staining is depicted in Fig. 6A-D. There was a significant increase in the number of CD68-positive cells in the livers of LysMcre+/caNrf2+ and LysMcre+/caNrf2− mice undergoing IRI compared to sham animals. There was a significant reduction in CD68-positive cells in the livers of LysMcre+/caNrf2+ mice compared to LysMcre+/caNrf2− mice undergoing IRI (Fig. 6E). Overactivation of the Nrf2 pathway in myeloid cells resulted in a decrease in inflammatory cells in the liver after IRI.
FIG. 6.
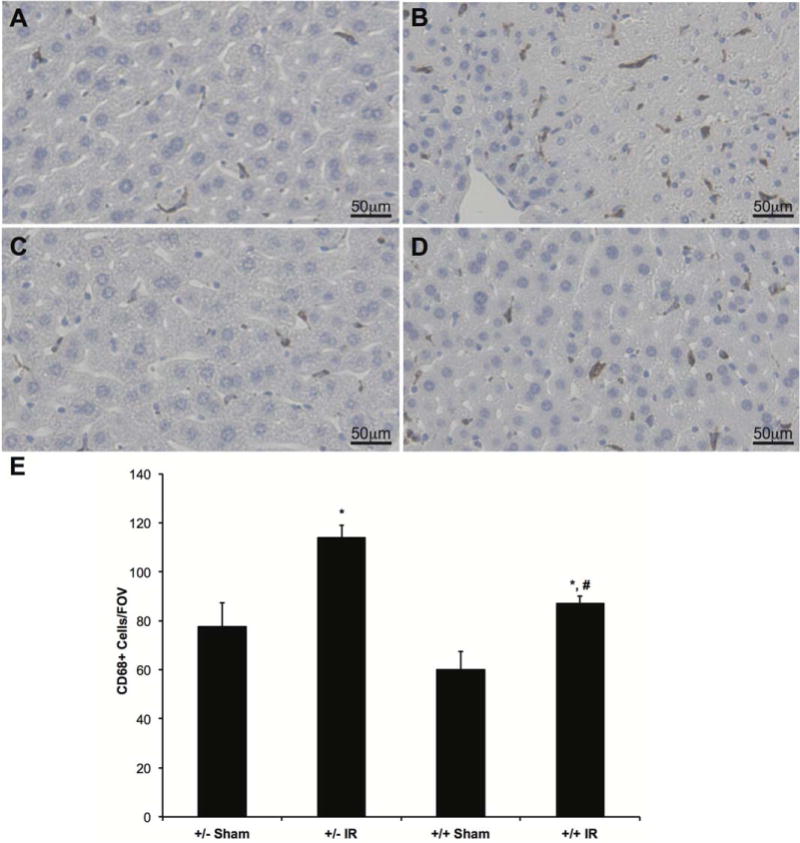
IHC for KCs and infiltrated monocytes. To evaluate the extent of monocyte infiltration and KC staining in the different groups, IHC with a CD68 antibody was performed. CD68 is a marker expressed on monocytes/immature macrophages, as well as KCs. Representative slides for (A) LysMcre+/caNrf2− sham (n = 4), (B) LysMcre+/caNrf2− IR (n = 7), (C) LysMcre+/caNrf2+ sham (n = 4), and (D) LysMcre+/caNrf2+ IR (n = 5) animals are shown. There is similar CD68 staining in the sham groups of both genotypes. After IR, there is a marked increase in CD68 staining in both IR groups, but when compared to LysMcre+/caNrf2−, CD68 staining is decreased in the LysMcre+/caNrf2+ IR group. (E) The number of CD68 staining cells was quantified for each experimental group. Similar to visual observation, there is a significantly increased number of CD68 positive cells in both IR groups, as compared to sham counterparts. We also see significantly decreased numbers of CD68 positive cells in the LysMcre+/caNrf2+ IR group as compared to the LysMcre+/caNrf2− IR group. * P < 0.05 as compared to sham group of the same genotype. # P < 0.05 as compared to +/− group of the same treatment.
LysMcre+/caNrf2+ MICE HAVE DECREASED PROINFLAMMATORY CYTOKINE EXPRESSION AFTER PARTIAL HEPATIC IR
Quantitative RT-PCR was performed to examine classically up-regulated inflammatory cytokines in the liver tissue to determine the inflammatory state of the different experimental groups (Fig. 7A). Although IRI in LysMcre+/caNrf2− mice led to increases in the mRNA abundance of the proinflammatory cytokines and chemokines IL6, TNF-α, and CCL2, there was only minimal change in the panel of these proinflammatory genes in the liver of LysMcre+/caNrf2+ animals after IR. In addition, we even observed a significantly decreased expression of IL1β, IL6, TNF-α, CCL2, and CXCL10 in the LysMcre+/caNrf2+ IRI group as compared to the littermate counterparts, suggestive of a decreased inflammatory state (Fig. 7A).
FIG. 7.
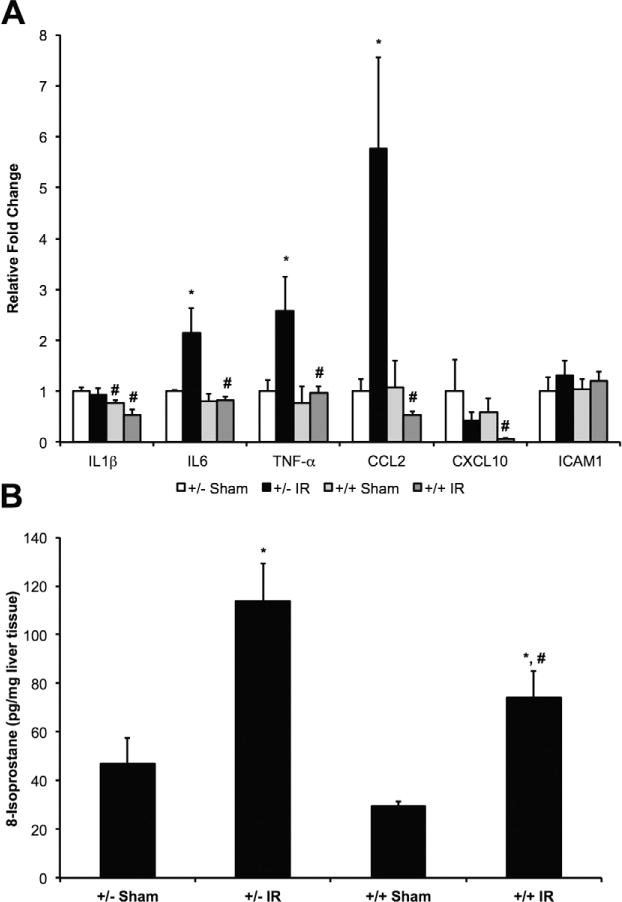
Inflammation and oxidative stress after partial hepatic IRI. (A) RNA was isolated from livers of experimental animals, and quantitative RT-PCR of some classical proinflammatory cytokines was performed. There were significant increases in mRNA abundance of IL6, TNF-α, and CCL2 in the LysMcre+/caNrf2− IR animals, as compared to its sham group. When comparing the LysMcre+/caNrf2+ IR group to the LysMcre+/caNrf2− IR group, there were significant decreases in IL1β, IL6, TNF-α, CCL2, and CXCL10. (B) To assess the degree of oxidative stress, 8-isoprostane levels in the livers were assessed. Hepatic 8-isoprostane levels were significantly elevated in IR groups of both genotypes as compared to their sham counterparts. Hepatic 8-isoprostane levels were significantly decreased in the LysMcre+/caNrf2+ IR group as compared to the LysMcre+/caNrf2− IR group. * P < 0.05 as compared to the sham group of the same genotype. # P < 0.05 as compared to +/− group of the same treatment.
LysMcre+/caNrf2+ MICE HAVE DECREASED HEPATIC OXIDATIVE STRESS AFTER PARTIAL HEPATIC IR
Finally, we determined the degree of oxidative stress in each experimental group by measuring the amount of 8-isoprostane present in the liver tissue (Fig. 7B). Isoprostanes are formed from oxidation of essential fatty acids by oxygen radicals and are reliable markers of lipid peroxidation and tissue oxidative stress. In animals that underwent IRI, liver parenchyma demonstrated elevated levels of 8-isoprostane as compared to their sham counterparts. However, the livers of the LysMcre+/caNrf2+ IRI animals had significantly decreased 8-isoprostane as compared to the LysMcre+/caNrf2− IRI animals, suggestive of decreased oxidative stress in the transgenic animals with myeloid-specific overactivation of the Nrf2 pathway.
Discussion
Nrf2 activation has been shown to protect the liver from several disease states. Our laboratory and others have previously shown that Nrf2 induction can lead to a reduction in the development of steatosis in dietary models of fatty liver disease.(14,17) Nrf2 activation also protects the liver from T cell–mediated hepatic inflammation(18) and acetaminophen toxicity.(19–21) Nrf2 is a positive regulator of human bile salt export pump expression, and its increased expression may benefit patients with cholestatic liver disease.(22) Each of these disease states involves components of oxidative damage and inflammation that appear to be attenuated by Nrf2 activation.
The protective effects of the Nrf2-ARE pathway have been previously described in IRI models of the kidney,(23) heart,(24) brain,(25,26) and liver.(9,27) All of these studies use Nrf2 (−/−) mice and demonstrate increased tissue damage in the absence of Nrf2. A recent study demonstrated that transgenic overexpression of Nrf2 in hepatocytes of lean donor livers can protect those livers from hepatocellular damage sustained after liver transplantation.(11) We have recently shown that overactivation of Nrf2 in hepatocytes leads to reduced cellular damage after IRI.(10) However, the study described here is the first to look at the role of direct, cell-specific overactivation of Nrf2 in nonparenchymal cells in a model of warm hepatic IRI. By using novel transgenic mice, we intended to more precisely decipher the cell-specific role of Nrf2 in hepatic IRI.
In this study, we used Cre-lox technology to over activate the Nrf2-ARE pathway in a cell specific manner. The LysMcre construct places Cre recombinase under control of the myeloid-specific lysozyme promoter.(28) This allows for the transcription and translation of our caNrf2 mutant to over activate the Nrf2-ARE pathway in myeloid cells. Upon differentiation, myeloid progenitor cells develop into multiple cell types including monocytes, neutrophils, and tissue macrophages. Although recent studies have shown that the vast majority of KCs are derived from erythromyeloid progenitor cells in the yolk sac before the emergence of adult hematopoietic stem cells,(29) we demonstrate that the LysMcre transgene is active in KCs, neutrophils, and monocytes. By using the LysMcre transgene, we were able to over activate the Nrf2-ARE pathway in these myeloid cells, as demonstrated in Fig. 1.
We also demonstrated that Nrf2-ARE pathway overactivation in myeloid cells decreased hepatic IRI with decreased tissue oxidative stress, cytokine production, necrosis, and apoptosis. It has been long established that myeloid cells play a prominent role in hepatic IRI. By up-regulating Nrf2 signaling in myeloid cells, we have demonstrated decreased gene expression of inflammatory cytokines within the liver. This is associated with decreased neutrophilic infiltration and a reduction in hepatic macrophages. The reductions of these markers of injury in our study targeting myeloid cells are similar to our previous study investigating Nrf2 overactivation in hepatocytes(10) and that seen with hepatocyte-specific Nrf2 induction in cold preservation IRI performed by Ke et al.(11) We are able to achieve comparable effects by manipulating these nonparenchymal cells, which is a significantly smaller cell population than that of the parenchymal cells. This suggests and re-affirms that myeloid cells are a significant source of ROS, causing oxidative damage to hepatocytes in the setting of IRI.
However, the details regarding the mechanism of protection in this setting remain uncertain. We do see a reduction in mRNA abundance of proinflammatory cytokines in the liver as a result of Nrf2 overactivation in myeloid cells during IRI. It is unclear whether this is due to a reduction of proinflammatory cytokine release or an increase in anti-inflammatory cytokine release from the myeloid cells. This may also be the reason why we observed a reduction in monocytic and neutrophilic infiltration after 6 hours of reperfusion. As resident liver macrophages, KCs have long been thought of as one of the main generators of ROS during the initial period of hepatic IRI.(30) It is likely that upregulation of the Nrf2 pathway decreases the ability of myeloid cells, such as KCs, to release ROS in the setting of IRI, due to their more efficient detoxification. This may lead to decreased injury and subsequent suppression of the inflammatory cascade. This is evident by the overall decreased tissue oxidative stress as seen in our study. However, at the same time, it is also known that ROS can function as small molecule messengers leading to further signaling cascades, such as inhibition of cytokine production via the NF-κB pathway.(31) Thus, it remains possible that up-regulation of the Nrf2-ARE pathway may in itself decrease myeloid cell-induced inflammatory potential.
Though Nrf2-ARE is well-known for its antioxidative and cell protective effects, there have also been studies suggesting potential deleterious effects of Nrf2 overinduction. This is of particular importance in the clinical setting where we envision the use of an Nrf2 activator to ameliorate IR injury. In a murine model of loss of autophagy via autophagy-related 5 knockout model system, activation of Nrf2 leads to increased inflammation and fibrosis.(32) Furthermore, in an experimental murine model of liver regeneration after partial hepatectomy, hepatocyte-specific Nrf2 overactivation leads to increased apoptosis and decreased hepatic regeneration.(13) In living donor liver transplantation or partial hepatectomy, where IRI can be a relevant entity, the liver’s regenerative ability would be an important factor for the patient’s postoperative recovery. Overactivation of Nrf2 in hepatocytes that could potentially impede hepatic regeneration would be suboptimal. In addition, transplant recipients would be expected to receive lifelong immunosuppressive therapy in order to maintain tolerance to their newly received allografts. However, 1 consequence of immunosuppressive therapy is increased risk of de novo malignancy. Previous studies have shown that Nrf2 activation in premalignant cells can lead to prolonged cancer cell survival,(33,34) and tumors exhibit increased chemotherapy resistance.(35–37) Thus, longterm systemic Nrf2 activation in an immunosuppressed state could lead to development of chemotherapy-resistant neoplasm and an unfavorable outcome. These potential risks have led some to question the benefits of Nrf2 inducers in the treatment of disease states. However, there appears to be a level of Nrf2 activation below which the benefits of antioxidant protection exist and above which the risk of disease states increase.(38) With these limitations in mind, a short-term, celltargeted treatment with Nrf2 inducers may be sufficient to decrease hepatocellular damage and increase cellular recovery after hepatic IRI.
In conclusion, we used a novel transgenic system to up-regulate the Nrf2-ARE pathway in a cell-specific manner to study the effect of antioxidant defense during hepatic IRI. We demonstrated that when the Nrf2-ARE pathway is over activated in myeloid-derived cells, the resulting IRI is reduced. This is substantiated with decreased tissue oxidative stress, decreased hepatocellular damage, decreased inflammatory cell infiltrate, as well as decreased necrosis and apoptosis. This study further illustrates the critical role of the Nrf2-ARE pathway in KCs, neutrophils, and monocytes in the setting of hepatic IRI. Because of these results and the potentially damaging effects of hepatocyte-specific Nrf2 induction, the myeloid cells may be a more favorable target for Nrf2-ARE induction to protect the liver from hepatic IRI. Additional studies are needed to delineate which group of myeloid cells is responsible for this Nrf2-induced protection and what are the intercellular mechanisms involved.
Supplementary Material
Acknowledgments
David P. Foley was supported by the Clinical and Translational Science Award program through the National Institutes of Health Center for Advancing Translational Sciences (UL1TR000427). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. David P. Foley was also supported by the American Society of Transplant Surgeons-Pfizer Mid-Level Faculty Award. Lung-Yi Lee was supported by the National Institutes of Health (T32 CA090217). Sabine Werner was supported by the Swiss National Science Foundation (310030_132884).
Abbreviations
- Alb
albumin
- ALT
alanine aminotransferase
- ARE
antioxidant response element
- BM
bone marrow
- bp
base pair
- caNrf2
constitutively active nuclear erythroid 2 p45-related factor 2
- CCL2
chemokine (C-C motif) ligand 2
- cDNA
complementary DNA
- Cre
cyclization recombination
- CXCL10
chemokine (C-X-C motif) ligand 10
- DMEM
Dulbecco’s modified Eagle’s medium
- DNaseI
deoxyribonuclease I
- FOV
field of view
- GCLM
glutamate-cysteine ligase, modifier subunit
- GSTA2
glutathione Stransferase alpha 2
- GSTA4
glutathione S-transferase alpha 4
- GSTM1
glutathione S-transferase mu 1
- GSTM2
glutathione S-transferase mu 2
- H & E
hematoxylin-eosin
- ICAM1
intercellular adhesion molecule 1
- IHC
immunohistochemistry
- IL
interleukin
- IR
ischemia/reperfusion
- IRI
ischemia/reperfusion injury
- KC
Kupffer cell
- KEAP1
kelch-like ECH-associated protein 1
- LY6-G
lymphocyte antigen 6 complex locus G
- LysMcre
lysozyme M cre recombinase
- Lyz
lysozyme
- Lyz2
lysozyme 2
- MACS
magnetic cell sorting
- mRNA
messenger RNA
- Neh2
Nrf2-ECH homology
- NQO1
nicotinamide adenine dinucleotide phosphate dehydrogenase, reduced form, quinone 1
- Nrf2
nuclear erythroid 2 p45-related factor 2
- NPC
nonparenchymal cell
- PCR
polymerase chain reaction
- PRDX1
peroxiredoxin 1
- ROS
reactive oxygen species
- RT-PCR
real-time polymerase chain reaction
- SEM
standard error of the mean
- TNF-α
tumor necrosis factor α
- TXNRD1
thioredoxin reductase 1
Footnotes
Additional supporting information may be found in the online version of this article.
View this article online at wileyonlinelibrary.com.
Potential conflict of interest: Nothing to report.
References
- 1.Ploeg RJ, D’Alessandro AM, Knechtle SJ, Stegall MD, Pirsch JD, Hoffmann RM, et al. Risk factors for primary dysfunction after liver transplantation–a multivariate analysis. Transplantation. 1993;55:807–813. doi: 10.1097/00007890-199304000-00024. [DOI] [PubMed] [Google Scholar]
- 2.Clavien PA, Harvey PR, Strasberg SM. Preservation and reperfusion injuries in liver allografts. An overview and synthesis of current studies. Transplantation. 1992;53:957–978. doi: 10.1097/00007890-199205000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. 2011;11:1563–1569. doi: 10.1111/j.1600-6143.2011.03579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson CD, Belous A, Pierce J, Nicoud IB, Knox C, Wakata A, et al. Mitochondrial calcium uptake regulates cold preservation-induced Bax translocation and early reperfusion apoptosis. Am J Transplant. 2004;4:352–362. doi: 10.1111/j.1600-6143.2004.00357.x. [DOI] [PubMed] [Google Scholar]
- 5.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 8.Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12,029–12,038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 9.Kudoh K, Uchinami H, Yoshioka M, Seki E, Yamamoto Y. Nrf2 activation protects the liver from ischemia/reperfusion injury in mice. Ann Surg. 2014;260:118–127. doi: 10.1097/SLA.0000000000000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee LY, Harberg C, Matkowskyj KA, Cook S, Roenneburg D, Werner S, et al. Overactivation of the nuclear factor (erythroid-derived 2)-like 2-antioxidant response element pathway in hepatocytes decreases hepatic ischemia/reperfusion injury in mice. Liver Transpl. 2016;22:91–102. doi: 10.1002/lt.24303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ke B, Shen XD, Zhang Y, Ji H, Gao F, Yue S, et al. KEAP1-Nrf2 complex in ischemia-induced hepatocellular damage of mouse liver transplants. J Hepatol. 2013;59:1200–1207. doi: 10.1016/j.jhep.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schäfer M, Farwanah H, Willrodt AH, Huebner AJ, Sandhoff K, Roop D, et al. Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol Med. 2012;4:364–379. doi: 10.1002/emmm.201200219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Köhler UA, Kurinna S, Schwitter D, Marti A, Schäfer M, Hellerbrand C, et al. Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology. 2014;60:670–678. doi: 10.1002/hep.26964. [DOI] [PubMed] [Google Scholar]
- 14.Lee LY, Köhler UA, Zhang L, Roenneburg D, Werner S, Johnson JA, Foley DP, et al. Activation of the Nrf2-ARE pathway in hepatocytes protects against steatosis in nutritionally induced non-alcoholic steatohepatitis in mice. Toxicol Sci. 2014;142:361–374. doi: 10.1093/toxsci/kfu184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasband WS. ImageJ. 1997–2012 http://imagej.nih.gov/ij/. Accessed May 2, 2015.
- 16.Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation. 1993;55:1265–1272. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YK, Yeager RL, Tanaka Y, Klaassen CD. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol Appl Pharmacol. 2010;245:326–334. doi: 10.1016/j.taap.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB, et al. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol Sci. 2008;104:218–227. doi: 10.1093/toxsci/kfn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reisman SA, Buckley DB, Tanaka Y, Klaassen CD. CDDO-Im protects from acetaminophen hepatotoxicity through induction of Nrf2-dependent genes. Toxicol Appl Pharmacol. 2009;236:109–114. doi: 10.1016/j.taap.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reisman SA, Aleksunes LM, Klaassen CD. Oleanolic acid activates Nrf2 and protects from acetaminophen hepatotoxicity via Nrf2-dependent and Nrf2-independent processes. Biochem Pharmacol. 2009;77:1273–1282. doi: 10.1016/j.bcp.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weerachayaphorn J, Cai SY, Soroka CJ, Boyer JL. Nuclear factor erythroid 2-related factor 2 is a positive regulator of human bile salt export pump expression. Hepatology. 2009;50:1588–1596. doi: 10.1002/hep.23151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M, Grigoryev DN, Crow MT, Haas M, Yamamoto M, Reddy SP, Rabb H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009;76:277–285. doi: 10.1038/ki.2009.157. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Sano M, Shinmura K, Tamaki K, Katsumata Y, Matsuhashi T, et al. 4-hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. J Mol Cell Cardiol. 2010;49:576–586. doi: 10.1016/j.yjmcc.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 25.Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Doré S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10,321–10,335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang J, Yue S, Ke B, Zhu J, Shen XD, Zhai Y, et al. Nuclear factor erythroid 2-related factor 2 regulates toll-like receptor 4 innate responses in mouse liver ischemia-reperfusion injury through Akt-forkhead box protein O1 signaling network. Transplantation. 2014;98:721–728. doi: 10.1097/TP.0000000000000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hume DA. Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol. 2011;89:525–538. doi: 10.1189/jlb.0810472. [DOI] [PubMed] [Google Scholar]
- 29.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolksac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol. 1991;260(Pt 1):G355–G362. doi: 10.1152/ajpgi.1991.260.3.G355. [DOI] [PubMed] [Google Scholar]
- 31.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP, et al. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61:617–625. doi: 10.1016/j.jhep.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in Nrf2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13,568–13,573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rolfs F, Huber M, Kuehne A, Kramer S, Haertel E, Muzumdar S, et al. Nrf2 activation promotes keratinocyte survival during early skin carcinogenesis via metabolic alterations. Cancer Res. 2015;75:4817–4829. doi: 10.1158/0008-5472.CAN-15-0614. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci. 2013;34:340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 36.Slocum SL, Kensler TW. Nrf2: control of sensitivity to carcinogens. Arch Toxicol. 2011;85:273–284. doi: 10.1007/s00204-011-0675-4. [DOI] [PubMed] [Google Scholar]
- 37.Nair S, Doh ST, Chan JY, Kong AN, Cai L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br J Cancer. 2008;99:2070–2082. doi: 10.1038/sj.bjc.6604703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–99. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.