

ABSTRACT
Type II thioesterases typically function as editing enzymes, removing acyl groups that have been misconjugated to acyl carrier proteins during polyketide secondary metabolite biosynthesis as a consequence of biosynthetic errors. Streptomyces chartreusis NRRL 3882 produces the pyrrole polyether ionophoric antibiotic, and we have identified the presence of a putative type II thioesterase-like sequence, calG, within the biosynthetic gene cluster involved in the antibiotic's synthesis. However, targeted gene mutagenesis experiments in which calG was inactivated in the organism did not lead to a decrease in calcimycin production but rather reduced the strain's production of its biosynthetic precursor, cezomycin. Results from in vitro activity assays of purified, recombinant CalG protein indicated that it was involved in the hydrolysis of cezomycin coenzyme A (cezomycin-CoA), as well as other acyl CoAs, but was not active toward 3-S-N-acetylcysteamine (SNAC; the mimic of the polyketide chain-releasing precursor). Further investigation of the enzyme's activity showed that it possessed a cezomycin-CoA hydrolysis Km of 0.67 mM and a kcat of 17.77 min−1 and was significantly inhibited by the presence of Mn2+ and Fe2+ divalent cations. Interestingly, when S. chartreusis NRRL 3882 was cultured in the presence of inorganic nitrite, NaNO2, it was observed that the production of calcimycin rather than cezomycin was promoted. Also, supplementation of S. chartreusis NRRL 3882 growth medium with the divalent cations Ca2+, Mg2+, Mn2+, and Fe2+ had a similar effect. Taken together, these observations suggest that CalG is not responsible for megasynthase polyketide precursor chain release during the synthesis of calcimycin or for retaining the catalytic efficiency of the megasynthase enzyme complex as is supposed to be the function for type II thioesterases. Rather, our results suggest that CalG is a dedicated thioesterase that prevents the accumulation of cezomycin-CoA when intracellular nitrogen is limited, an apparently new and previously unreported function of type II thioesterases.
IMPORTANCE Type II thioesterases (TEIIs) are generally regarded as being responsible for removing aberrant acyl groups that block polyketide production, thereby maintaining the efficiency of the megasynthase involved in this class of secondary metabolites' biosynthesis. Specifically, this class of enzyme is believed to be involved in editing misprimed precursors, controlling initial units, providing key intermediates, and releasing final synthetic products in the biosynthesis of this class of secondary metabolites. Our results indicate that the putative TEII CalG present in the calcimycin (A23187)-producing organism Streptomyces chartreusis NRRL 3882 is not important either for the retention of catalytic efficiency of, or for the release of the product compound from, the megasynthase involved in calcimycin biosynthesis. Rather, the enzyme is involved in regulating/controlling the pool size of the calcimycin biosynthetic precursor, cezomycin, by hydrolysis of its CoA derivative. This novel function of CalG suggests a possible additional activity for enzymes belonging to the TEII protein family and promotes better understanding of the overall biosynthetic mechanisms involved in the production of this class of secondary metabolites.
KEYWORDS: calcimycin biosynthesis, type II thioesterase, CoA derivatives, divalent cations
INTRODUCTION
Type II thioesterases (TEIIs) are encoded by either polyketide synthase (PKS) or nonribosomal peptide synthetase (NRPS) gene clusters and are discrete 25- to 29-kDa proteins that hydrolyze the thioester bonds between the 4′-phosphopantetheine arm of acyl carrier proteins (ACP) or peptidyl carrier proteins (PCP) and various residues during polyketide biosynthesis (1–3). In most polyketide cases, these enzymes are unessential for the synthesis of the end product but appear to play an important role in maintaining the efficiency of the “enzymatic assembly line” leading to product formation. Particularly, they are involved in removing nonreactive acyl residues that block the PKS/NRPS megasynthase (4, 5), maintaining the efficiency of the enzyme complex by editing misprimed precursors (6–8), removing aberrant intermediates (2), controlling starter units (3), providing key intermediates (9), or releasing final products (10–14). Consequently, it has been observed that deletion of TEII-encoding genes from gene clusters involved in polyketide biosynthesis usually leads to a decrease in the final product yield of the pathway concerned (6–8) rather than an absence thereof. Overexpression of TEII protein, on the other hand, has been observed to increase yield (9, 15). In a few limited cases, some TEIIs can function differently, and one of these is PikAV, involved in picromycin biosynthesis. In this case, it has been observed that the presence of PikAV protein has a negative effect on the polyketide's biosynthesis and that deletion of the pikAV gene had no effect on picromycin product yield (2, 16).
Calcimycin (A23187), produced by Streptomyces chartreusis NRRL 3882, is one of a few naturally occurring polyketide ionophores that specifically chelate divalent cations such as calcium and magnesium (17). Due to its ionophoric property, it is active against some Gram-positive bacteria and fungi and has been observed to inhibit ATPase activity in mammalian cells (17). It can also induce apoptosis in mammalian cells via activation of intracellular signaling (18, 19) and is widely used as a biochemical tool both for pharmacological and in vitro toxicological studies (5, 18, 20). In S. chartreusis NRRL 3882, N-demethyl calcimycin and cezomycin (21), two other naturally occurring pyrrole polyether ionophores, are also present. All three compounds share a great deal of structural similarity, possessing an α-ketopyrrole, substituted benzoxazole, and spiroketal ring that differ only in the structure of their side groups (21). X-ray crystallographic studies have shown that calcimycin's benzoxazole carboxylate oxygen, ketopyrrole oxygen, and benzoxazole (ring) nitrogen atom are crucial for calcium ion binding, while the hydrogen bond between the pyrrole NH proton and benzoxazole carboxylate oxygen may be involved in stabilizing the calcimycin-calcium ion conjugate structure (22, 23). Efforts are continuing to produce modifications of the benzoxazole moiety of calcimycin in attempts to better understand cation binding properties of the compound (24–27).
Our previous work in sequencing the gene cluster involved in calcimycin biosynthesis led us to propose the compound's biosynthetic pathway (21), in which the proteins CalN1 to CalN3 and CalB1 to CalB4 are responsible for the biosynthesis of its α-ketopyrrole moiety and 3-hydroxyanthranilic acid precursors, respectively. Also, we proposed that the calA1 to calA5 genes encode a polyketide synthase involved in the synthesis of the polyketide chain. Our previous work failed to identify any region that might have encoded a thioesterase, a region that is often present in this type of biosynthetic gene cluster and is normally located at the C terminus of the final extension module of a type I PKS protein complex.
The work and results reported here allow us to advance our previous findings by proposing a role in calcimycin biosynthesis for the product of calG, a gene present in the calcimycin biosynthesis gene cluster from S. chartreusis NRRL (21), which encodes a putative TEII protein. To explore and characterize the physicochemical and biochemical properties of the CalG TEII enzyme, we used a combination of genetic and biochemical approaches, and our results strongly suggest that CalG is responsible neither for the release of the polyketide precursor nor for maintaining the catalytic efficiency of the PKS megasynthase. Rather, it actually recycles or regulates the pool size of a calcimycin precursor, cezomycin coenzyme A (cezomycin-CoA), under growth conditions where nitrogen supply is limited.
RESULTS
CalG is involved in cezomycin production.
From its DNA sequence, the calG protein was estimated to be formed of 251 amino acids and shares 52% sequence identity with TylO (accession number AAS79448) from Streptomyces bikiniensis, 51% with FscTE (accession number AAQ82559) from Streptomyces sp. strain FR-008 (1, 28), 23% with TycF (accession number BAH43770) from Brevibacillus brevis, and 33% with GrsT (accession number AEI41826) from Paenibacillus mucilaginosus (see Fig. S3 in the supplemental material). TylO, involved in tylosin biosynthesis and the closest match to calG, represents an example of a TEII encoded by a type I PKS gene cluster that was identified as being responsible for hydrolysis of acyl-N-acetylcysteamine (acyl-NAC) as well as removing misprimed residues bound to ACP (6).
calG function was analyzed by creating an apramycin-resistant (Aprr) derivative strain of S. chartreusis NRRL 3882, named GLX16, in which almost the entire putative TEII gene had been replaced with the apramycin resistance gene (see Fig. S4A and B in the supplemental material). This strain and the wild-type (WT) strain, for comparison, were grown in soya flour mannitol (SFM) liquid medium and culture supernatants and subjected to high-pressure liquid chromatography (HPLC) analysis. Results showed that the mutant accumulated calcimycin alone whereas the wild-type strain accumulated both cezomycin and calcimycin (Fig. 1; see also Table S2 in the supplemental material). Interestingly, the wild-type strain accumulated less calcimycin than cezomycin. (Fig. 1 and Table S2). To confirm if deletion of calG was responsible for the loss of cezomycin production, GLX16 was complemented with a functional copy of calG to produce strain GLX17 (Fig. 1). When tested in a fashion similar to that described above for GLX16, it was observed that functional complementation of the defective calG gene restored the ability to produce cezomycin (Fig. 1) and that the total yield for the two ionophores (cezomycin and calcimycin) in the WT and GLX17 were approximately equal (Table S2). These results together suggest that calG is unlikely to be involved in the release of the polyketide precursor from the megasynthase enzyme complex or in maintaining its efficiency (since the yield for calcimycin was not affected in either case). Rather, the results suggest that CalG is involved only in the generation of cezomycin, the precursor of calcimycin.
FIG 1.

The calG disruption mutant does not accumulate cezomycin. HPLC analysis of calcimycin production in S. chartreusis NRRL 3882, GLX16, and GLX17 (ΔcalG/calG). The identity of the substances in the HPLC peaks was confirmed via LC/MS analysis.
CalG hydrolyzes cezomycin-CoA but is not involved in the release of calcimycin from the polyketide megasynthase.
The in vitro activity of His-tagged CalG was assessed by constructing a recombinant strain in which the calG gene was placed under the control of a T7 promoter, expressed at high level after induction with IPTG. Figure 2A revealed a typical SDS-PAGE of the purified protein.
FIG 2.

Characterization of cezomycin-CoA CalG thioesterase hydrolytic activity. (A) SDS-PAGE analysis of purified CalG protein. (B) HPLC analysis of the CalG hydrolysis reaction products. (C) High-resolution mass spectrometry analysis of cezomycin-CoA and CalG reaction product as well as CoA and cezomycin. The mass fragment peak at m/z of 94 is characteristic of the pyrrole moiety, and that at m/z of 160 is characteristic of benzoxazole. Mass fragments of CoA are marked.
We had previously observed that the disruption of the calB1 gene in S. chartreusis NRRL 3882 resulted in the accumulation of compound 3 (29), and we had therefore tentatively assigned it as the megasynthase release product (29). S-N-acetylcysteamine thioesters are close structural mimics of the phosphopantetheine moiety of acyl carrier proteins (ACP) and are typically used as the substrates in thioesterase activity assays (1, 13, 30). So, in order to test the possible involvement of CalG in the release of the polyketide chain during calcimycin biosynthesis (21), we synthesized compound 3-S-N-acetylcysteamine (SNAC) adduct to simulate the acyl-ACP substrate and tested it in CalG activity assays (see Table S1 and Fig. S1 in the supplemental material). We observed that even after prolonged incubation with CalG, no hydrolysis of compound 3-SNAC was detected (see Fig. S5 in the supplemental material). In the same context, cezomycin-SNAC, N-demethyl-calcimycin-SNAC, and calcimycin-SNAC were also synthesized for testing as possible substrates of CalG, and no hydrolysis of these compounds was observed either (Fig. S5).
Cezomycin is the modification precursor of calcimycin (31), which is converted to a CoA adduct by an acyl-CoA ligase (CalC) for further modification/processing (31) to calcimycin. Consequently, one possibility was that cezomycin-CoA could have been the canonical substrate of CalG. To test this hypothesis, we performed an experiment in which we incubated cezomycin-CoA (31) with CalG while monitoring the reaction via HPLC/mass spectrometry (MS), and the results are shown in Fig. 2B and C, where it can be observed that after 1 h of incubation all the cezomycin-CoA originally present had been completely hydrolyzed. Consequently, it can be inferred that cezomycin-CoA is indeed the substrate of CalG.
Characterization of CalG kinetic parameters with respect to cezomycin-CoA involved using an established photometric assay based on the reaction of free thiols with 5,5-dithiobis (2-nitrobenzoic acid) (DTNB) (1, 30, 32). Cezomycin-CoA and five other CoA adducts, acetyl-CoA, benzoyl-CoA, butyryl-CoA, butyryl-CoA, and palmitoyl-CoA, were tested in these experiments as possible substrates of CalG. Figures S6 and S7 in the supplemental material as well as Table 1 illustrate the results, which all indicate that CalG hydrolyzed all of the potential substrates tested. In fact, acetyl-CoA and butyryl-CoA were both hydrolyzed by CalG at a higher rate than cezomycin-CoA, while the hydrolysis rates of CalG for benzoyl-CoA and long-chain palmitoyl-CoA were lower. To further test the substrate specificity of the CalG protein for acyl-CoA esters and for the existence of other possible CoA thioester intermediates in calcimycin biosynthesis, compound 3-CoA, N-demethyl-calcimycin-CoA, and calcimycin-CoA were also synthesized (31) (see Table S1 and Fig. S2 in the supplemental material). See Table S3 for results that demonstrate that CalG appeared to be capable of slowly hydrolyzing all the compounds.
TABLE 1.
Kinetic parameters of CalG
| Substrate | Km (μM) | kcat (min−1) | kcat/Km (M−1 s−1) | Relative kcat/Km |
|---|---|---|---|---|
| Cezomycin-CoA | 670 ± 61 | 17.77 ± 0.55 | 442 | 1 |
| Acetyl-CoA | 200 ± 21 | 1.26 ± 0.03 | 105 | 0.23 |
| Benzoyl-CoA | 216 ± 15 | 0.92 ± 0.016 | 71 | 0.16 |
| Butyryl-CoA | 190 ± 22 | 1.26 ± 0.03 | 110 | 0.24 |
| Malonyl-CoA | 188 ± 12 | 1.02 ± 0.014 | 90 | 0.2 |
| Palmitoyl-CoA | 141 ± 15 | 0.053 ± 0.001 | 6.2 | 0.01 |
These results confirm that CalG was acting as a thioesterase and was most likely involved in regulating the relative pool sizes of cezomycin and cezomycin-CoA.
Effects of divalent cations on calcimycin/cezomycin production and CalG hydrolytic activity. (i) Divalent cations can reduce the production of cezomycin.
Both calcimycin and cezomycin have the ability to bind divalent cations, but the binding affinity of cezomycin is 10-fold weaker than that of calcimycin (33), which might imply diverse physiological roles for the two compounds in different/changing ionic environments. To test this hypothesis, S. chartreusis NRRL 3882 was grown in SFM liquid medium containing a variety of divalent cations (1 to 10 mM MnCl2, CaCl2, or MgCl2 or 0.5 to 1 mM ZnCl2 or FeCl2), and the amount of calcimycin and cezomycin present was monitored using HPLC. From the results, it can be observed that when growth medium was supplemented with Ca2+, Mg2+, Mn2+, and Fe2+ ions, the cezomycin level decreased and consequently the relative ratio of calcimycin to cezomycin increased (Fig. 3A and B; see also Table S4 in the supplemental material). However, the total amount of cezomycin and calcimycin remained almost the same (Table S4). In fact, when either 10 mM Mn2+ or 1 mM Fe2+ was present in growth medium, the production/presence of cezomycin was almost completely suppressed (Fig. 3A and B and Table S4).
FIG 3.

Divalent cation effects on calcimycin and cezomycin production by, and concentration ratio in, S. chartreusis NRRL 3882 as well as on CalG hydrolytic activity. (A) S. chartreusis NRRL 3882 treatment with 0.5 mM and 1 mM FeCl2. (B) Ratios of calcimycin to cezomycin after treatment with different divalent cations (1 to 10 mM CaCl2, MgCl2, and MnCl2 or 0.5 to 1 mM FeCl2 and ZnCl2). (C) Initial rates (measured from 0 to 15 min) of CalG hydrolysis in the presence of CaCl2, MgCl2, and MnCl2 at 10 mM and in the presence of FeCl2 and ZnCl2 at 1 mM.
(ii) Divalent cations can influence the transcription of calG.
The influence of the supplementation of growth medium with various cations on the expression level of calG was also investigated, and results from these experiments are shown in Fig. S9 in the supplemental material. From the data, it can be observed that almost all the cations tested in this way led to an increase in the relative copy number of calG transcripts observed to be present, which correlates well with results from experiments involving the effect of divalent cations on calcimycin/cezomycin biosynthesis. The only exception was in the case of Zn2+, which had no observable effect on the relative copy number of calG transcripts present at the concentration tested (Fig. S9). This also correlated with the observation that this ion had no effect on calcimycin/cezomycin ratio.
(iii) Divalent cations can reduce CalG activity.
The hydrolytic activity of CalG for cezomycin-CoA and acetyl-CoA was measured in the presence of MnCl2, CaCl2, MgCl2, ZnCl2, and FeCl2 at various concentrations to investigate their effects on CalG enzyme activity. Results are shown in Fig. 3C, as well as in Fig. S10 in the supplemental material, and to summarize, it can be stated that CalG hydrolytic activity was diminished by Mg2+ and Ca2+ ions at 10 mM while Mn2+ and Fe2+ at 10 mM and 1 mM completely abolished its hydrolytic activity (Fig. 3C and S10). Zn2+ at the concentration (1 mM) tested had no obvious effect on CalG hydrolytic activity (Fig. 3C and S10).
These results indicate that the divalent cationic environment of S. chartreusis NRRL 3882 influences its production of calcimycin and cezomycin, which correlates with the effect of divalent cations on CalG enzymatic activity.
Nitrogen influences the conversion of cezomycin to calcimycin by S. chartreusis NRRL 3882.
Recently, the production of nitrous acid has been reported as being responsible for the formation of the diazo group important for the biosynthesis of the ortho-diazoquinone secondary metabolite, cremeomycin, in Streptomyces cremeus (34). In that organism, the genes involved were identified as creE and creD.
After comparison, it was observed that the calU3 and calF (21) genes of S. chartreusis NRRL 3882 showed a high degree of sequence similarity to creE (68%) and creD (60%), suggesting that the nitrogen of the C-3 amino group of calcimycin might originate from nitrous acid, by way of l-asparagic acid (31) and that under conditions of nitrogen limitation the accumulation of the CoA-activated calcimycin precursor might occur, and the precursor might then be hydrolyzed back to cezomycin by CalG rather than be metabolized on to calcimycin. To test this hypothesis, we cultured S. chartreusis NRRL 3882 in LB liquid medium supplemented with inorganic nitrogen,1 mM NaNO2. This level of supplementation was chosen because we had observed that 1-liter cultures of S. chartreusis NRRL 3882 accumulated approximately 5 mg (about 0.01 mmol) calcimycin. Given that the stoichiometry of the reaction of formation of the calcimycin C-3 amino group was likely to be 1:1, supplementing cultures with 1 mM NaNO2 meant that its amount was in significant excess to that required for the production of the calcimycin observed. Cultures that had been supplemented with NaNO2 showed an increase in the level of calcimycin present and in the calcimycin-to-cezomycin ratio compared to an unsupplemented control (Fig. 4A). Data also indicated that without nitrite supplementation the relative amounts of cezomycin and calcimycin present were about 0.037 mM and 0.009 mM, respectively, while with NaNO2 supplementation the level of calcimycin was boosted to 0.042 mM and that of cezomycin reduced to 0.007 mM (Fig. 4B).
FIG 4.

Effects of NaNO2 on calcimycin and cezomycin production by S. chartreusis NRRL 3882. (A) Calcimycin and cezomycin presence in cultures of S. chartreusis NRRL 3882 with and without 1 mM NaNO2 supplementation assayed by HPLC with a control standard of calcimycin and cezomycin for comparison; (B) quantification of calcimycin and cezomycin in the cultures described for panel A.
DISCUSSION
TEIIs were believed to be responsible for the removal of misprimed acyl groups from acyl carrier proteins restoring PKS or NRPS functionality to maximize polyketide production (3, 6–8, 16). This hypothesis is supported by the observation that their absence often leads to a drastic decrease in polyketide yield (6–8, 35).
Our experiments showed that the TEII-like CalG protein of S. chartreusis NRRL 3882 was neither responsible for the removal of misprimed acyls nor acting as a polyketide chain-releasing enzyme. This was because S. chartreusis NRRL 3882's level of calcimycin production did not decrease with inactivation of calG. Rather, it was the strain's production of cezomycin, a calcimycin precursor, that was terminated. This observation and this conclusion are at odds with the previous prediction of this enzyme class's functionality (21), but the apparent paradox may be explained if other PKS gene clusters exist elsewhere in the S. chartreusis NRRL 3882 genome, possessing TEIIs that might compensate for CalG's lack of “classical” activity. In that context, it is known that genomes of streptomycetes can possess many polyketide biosynthetic gene clusters (36–38), although at the current time the situation with respect to S. chartreusis NRRL 3882 in this context is yet to be defined.
Although structurally similar, cezomycin and calcimycin show different properties with respect to their binding affinities for different metal cations, interactions with proteins, and passage through the cell membrane. In particular, the binding affinity of cezomycin for calcium is 10-fold lower than that of calcimycin (33). In vitro, we found that CalG catalyzed the hydrolysis of cezomycin-CoA, indicating that the enzyme might be involved in in recycling “overproduced acyls” to the non-thioester-related precursor (Fig. 5). The relatively high Km of CalG and its high turnover rate for cezomycin-CoA (Table 1) suggest that its hydrolysis activity may commence under conditions where the intracellular cezomycin-CoA concentration has risen. Our results also suggest that if a suitable nitrogen source is available to the cell, then the accumulating cezomycin-CoA is metabolized onto calcimycin (results from our experiments, which involved culturing WT S. chartreusis NRRL 3882 in the presence of additional NaNO2, are shown in Fig. 4).
FIG 5.
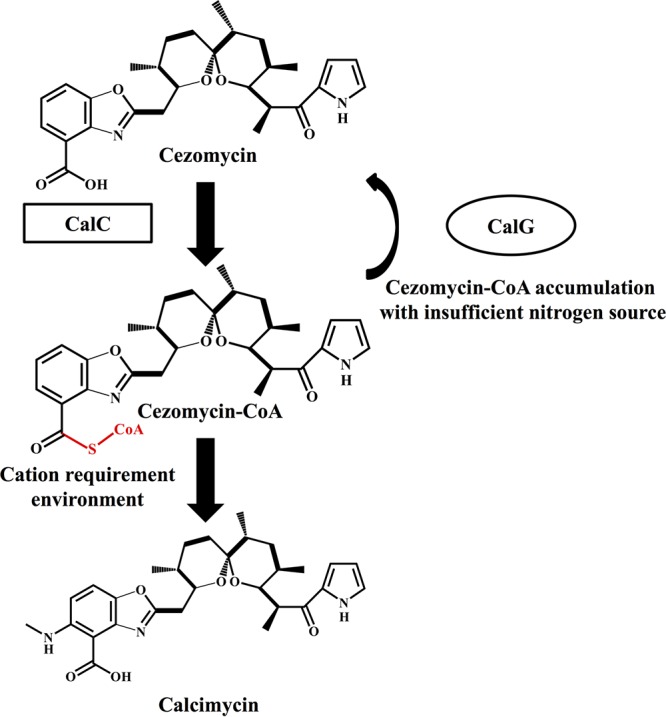
Proposed model for the function of CalG in the calcimycin biosynthetic pathway. Cezomycin was activated to form cezomycin-CoA and then modified to generate calcimycin. The hydrolysis of cezomycin-CoA by CalG leads to the accumulation of cezomycin under conditions of low levels of available nitrogen.
To maintain metal ion homeostasis, bacteria critically depend on membrane integrity and controlled ion translocation (39), and terrestrial Streptomyces species can undermine the cytoplasmic membrane function of their competing bacteria as a diffusion barrier for metal cations by using ionophores, like calcimycin and cezomycin (39). In fact, divalent cation ionophores appear to be particularly effective against competing microorganisms in soil environments rich in calcium but low in iron and manganese ions (40). Consequently, the production of calcimycin is a possible enhancer of host competition against other environmental microbes, and some form of regulation of calcimycin biosynthesis (and perhaps other ionophore secondary metabolites) by cations is not only imaginable but also probably desirable. A mechanism involving the CalG protein in that context is perfectly possible given the results of our experiments. We have demonstrated that increasing concentrations of Ca2+, Mg2+, and particularly Mn2+ or Fe2+ reduce the level of cezomycin produced by S. chartreusis NRRL 3882 without changing the total amount of the two compounds, cezomycin and calcimycin, present (see Table S4 in the supplemental material). Specifically, the cezomycin-CoA hydrolytic activity of CalG was reduced when concentrations of those cations were raised. Equally, when CalG was absent or its gene was mutated to an inactive form, cezomycin production was ceased but calcimycin continued to be produced. Interestingly, we observed that certain divalent cations elevated the transcriptional level of calG but not CalG protein activity for converting cezomycin-CoA to cezomycin (Fig. 3C and S9 and S10). It could be that the inhibition or absence of CalG activity at low cation concentration is what promotes the production of calcimycin by the bacterial host, thereby enhancing its cation scavenging ability and consequently survival chance. However, the detailed molecular mechanism involved is as yet unclear.
In summary, a new enzyme, CalG, involved in the biosynthesis of calcimycin has been discovered, characterized, and defined as a TEII type protein. Interestingly, this TEII appears to be different in function from those already reported. Rather than acting as a mechanism for enhancing polyketide synthesis by elimination of pathway errors, etc., the new protein appears to function in a specific way that regulates the flow of, and pool size related to, specific precursor molecules important in calcimycin formation.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Bacterial strains and plasmids used in the study are listed in Table 2. Escherichia coli plasmid isolation, gene cloning, and other routine molecular biological procedures were performed as described by Sambrook and Russell (41). S. chartreusis NRRL 3882 genomic DNA was isolated according to the protocol of Kieser et al. (42).
TABLE 2.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Streptomyces chartreusis | ||
| NRRL 3882 | Calcimycin production, wild type | NRRL |
| GLX16 (ΔcalG) | calG deletion mutant, no cezomycin production | This work |
| GLX17 (ΔcalG/calG) | ΔcalG complementation strain, restores cezomycin production | This work |
| Escherichia coli strains | ||
| DH10B | recA lacZΔM15 | Invitrogen |
| ET12567(pUZ8002) | Cml, Kan, dam dcm hsdS Tra+ Cml | 41 |
| BW25113/pIJ790 | RepA101(ts), araBp-gam-bet-exo, AraC, RepA101(ts) Cml | 43 |
| BL21(DE3)/plysS | F− dcm ompT hsdS (rB− mB−) gal λ(DE3) [pLysS Cml] | Stratagene |
| GLX-ET17 | ET12567(pUZ8002) containing pJTU3783 for calG complementation | This work |
| Plasmids | ||
| p6F5 | Cml | 21 |
| pJTU2170 | Integrative vector for gene complementation, aac(3)IV from pIB139 was replaced by bla and neo cassette | 44 |
| pET28a(+) | Plasmid for gene expression | Novagen |
| pJTU3661 | pET28a(+)-derived plasmid for calG expression | This work |
| pJTU3772 | p6F5-derived plasmid carrying an apramycin resistance gene and a defective calG | This work |
| pJTU3783 | pJTU2170-derived plasmid carrying calG for expression in Streptomyces | This work |
Cml, chloramphenicol resistance; Kan, kanamycin resistance; aac(3)IV, apramycin resistance.
E. coli strains were maintained and grown in or on liquid or solid Luria-Bertani medium (LB). Small-scale growth of S. chartreusis NRRL 3882 and its derivative strains was by culture in tryptic soy yeast extract (TSBY) liquid medium, containing 3% tryptone soy broth, 10.3% sucrose, and 0.5% yeast extract (for extraction of chromosomal DNA), or on soya flour mannitol (SFM) agar containing 2% mannitol, 2% soybean powder, and 2% agar [pH 7.2] (for sporulation and conjugation). Large-scale growth of S. chartreusis NRRL 3882 and its derivative mutant strains was performed in SFM medium without agar. Media were supplemented when necessary with 50 μg liter−1 apramycin.
Inactivation and complementation of the calG gene.
The calG gene in S. chartreusis NRRL 3882 was replaced by the apr resistance gene, using Redirect Technology (43) as described in the product literature. Briefly, the apr resistance gene from pIJ773 (43) (Table 2) was amplified using KOD-plus DNA polymerase (Toyobo Biotech Co. Ltd.) and calG gene-specific primers (Table 3). The resulting amplification products were introduced into E. coli BW25113/pIJ790 harboring p6F5 (21) to generate plasmid pJTU3772 (ΔcalG). This plasmid was then introduced into S. chartreusis NRRL 3882 by conjugation with E. coli ET12567/pUZ8002, and double-crossover Aprr mutants were isolated by selection on SFM medium containing apramycin. The identity of the mutants was confirmed by PCR and double-stranded sequencing of amplification products. The strain in which calG was deleted (ΔcalG mutant) was designated GLX16.
TABLE 3.
Primers used in this study
| Primer | Sequence (5′–3′)a | Use |
|---|---|---|
| G-F1 | CAGTACCCGGGGCGCCAGGACCGGCACCACGAGCCCATGATTCCGGGGATCCGTCGACC | Replacement of calG by Redirect Technology |
| G-F2 | ATGGCTCGCCTTGGGGTCGTCGTCTCCGGTCAGCGCGGTTGTAGGCTGGAGCTGCTTC | |
| G-F3 | ATGACCGCCACCGATCCGTG | PCR analysis of GLX16 (ΔcalG) |
| G-F4 | TCAGCGTGGCGCGACGGCGG | |
| G-F5 | CCGGAATTCTTGTGCGACCCGGCCGACTA | PCR amplification of calG for complementation |
| G-F6 | GGAATTCCATATGACCGCCACCGATCCGTG | |
| 28aG-F7 | GAATTCCATATGACCGCCACCGATCCGTGGATACG | Amplification of calG for expression |
| 28aG-F8 | CTTCCTCGAGTCAGCGTGGCGCGACGGCGGACTC |
Boldface indicates the 20-nt (G-F1) and 19-nt (G-F2) sequences for amplification of the apramycin resistance gene.
Gene complementation of the S. chartreusis ΔcalG mutant strain was achieved by constructing and introducing a full-length calG DNA-possessing plasmid pJTU3783 (Table 2) into the strain. Briefly, the procedure for construction of the complemented ΔcalG mutant strain was as follows. The calG gene from purified genomic DNA of S. chartreusis NRRL 3882 was PCR amplified using the gene-specific primers shown in Table 3. After size and sequence confirmation, PCR products were ligated into vector plasmid pJTU2170 (44) to generate the complementation plasmid, pJTU3783, which was stabilized in E. coli ET12567 to create strain GLX-ET17 (Table 2). This plasmid was then introduced into S. chartreusis calG-deleted strain GLX16 (ΔcalG) (Table 2) via conjugation with E. coli GLX-ET17 to produce GLX17 (ΔcalG/calG). calG gene expression in the complementation plasmid was under the control of an ermE promoter (42) and constitutive. Complemented conjugation products were selected by virtue of their kanamycin resistance (growth on LB supplemented with 50 μg liter−1 kanamycin), and their identities were confirmed by extracting, PCR amplifying, and sequencing the full-length plasmid-borne gene.
LC/MS analysis of molecules of interest from fermentation culture.
For liquid chromatography-mass spectrometry (LC/MS) analysis, S. chartreusis NRRL 3882 and its ΔcalG mutant strain were precultured in 10 ml of TSBY medium in a 50-ml conical flask at 30°C with gentle shaking at 220 rpm for 48 h. Subsequently, 5 ml of the resultant culture was aseptically removed and inoculated into a 500-ml baffled conical flask containing 100 ml liquid SFM medium (pH 7.3). Cultivation was continued at 30°C with shaking at 220 rpm for 9 days. For the analysis of the effects that divalent ions might have on calcimycin or cezomycin production, 1 to 10 mM MnCl2, CaCl2, or MgCl2 or 0.5 to 1 mM ZnCl2 or FeCl2, respectively, was included in the medium. The effect of NaNO2 was also investigated by its addition in SFM medium at 1 mM. All experiments were performed in triplicate.
After 9 days of fermentation, the culture medium was centrifuged at 6,000 × g for 30 min to remove cells and cellular debris, and the supernatant was used to assay for calcimycin/cezomycin after extraction with 1.5 volumes of ethyl acetate. Ethyl acetate extracts were dried under vacuum in a rotary evaporator and then redissolved in 0.5 ml methanol and assayed using an Agilent 1100 series LC/MSD Trap system (Agilent Technologies, Tokyo, Japan) fitted with an Agilent Zorbax SB-C18 (4.6- by 150-mm) column (Agilent Technologies). Sample separation was performed using gradient mixtures of solution A (0.1% formic acid in water) and solution B (0.1% formic acid in methanol) as follows: 75% to 85% of solution B for 8 min, followed by 85% to 95% for 14 min, 95% to 100% for 7 min, and finally 100% of solution B for 6 min at a flow rate of 0.4 ml min−1. Eluate was monitored by UV adsorption at a wavelength of 280 nm (λ280 nm), and a calcimycin standard was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) for comparison. Eluate producing a peak at λ280 nm was collected and subjected to high-resolution mass spectrometry using an Agilent 6530 series accurate-mass quadrupole time of flight (Q-TOF) LC/MS (Agilent Technologies) to reconfirm product (calcimycin and cezomycin) identity by Mr.
Concentrations of cezomycin and calcimycin present in S. chartreusis NRRL 3882, GLX16 (ΔcalG mutant), and GLX17 (ΔcalG/calG mutant) culture supernatants were determined by calculation from HPLC fraction peak areas using cezomycin (31) and calcimycin (obtained as described previously) as reference samples.
Cloning, expression, and purification of CalG-His protein.
The calG gene was cloned in frame with the poly-histidine codons in the expression vector pET28a(+) (Novagen, Merck KGaA) according to the manufacturer's instructions. Initially, the calG gene was amplified by PCR using purified genomic DNA extracted from S. chartreusis NRRL 3882 and primers 28aG-F7 and 28aG-F8 (Table 3) and after product identity confirmation by agarose gel electrophoresis and bidirectional sequencing ligated into pET28a(+), which had been linearized by double digestion with NdeI and XhoI. The ligation product, pJTU3783, was stabilized by introduction into CaCl2-treated E. coli DH10B (41). Production of cloned poly-His-tagged calG protein followed the protocol given by Novagen and was carried out by growth of the calG-transformed E. coli strain in 1 liter of LB medium at 37°C containing 50 μg liter−1 kanamycin and 25 μg liter−1 chloramphenicol while shaking at 250 rpm to A600 nm of 0.6. Protein expression was then induced by addition of 1 ml of 0.4 mM isopropyl-d-thiogalactopyranoside (IPTG) solution, and the culture was allowed to incubate for a further 24 h at 16°C. Cells were then harvested by centrifugation and stored frozen at −80°C for subsequent protein extraction and purification.
Recombinant His-tagged CalG protein was recovered from 4 g of the frozen cells following the GE Healthcare product instructions. Briefly, cells were thawed on ice, resuspended in 50 ml of buffer A (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5% glycerol, 40 mM imidazole), and lysed by sonication. Cell debris was removed by centrifugation at 20,000 × g for 40 min at 4°C, and the resulting supernatant was loaded onto a nickel-nitrilotriacetic acid (Ni-NTA) resin, HisTrap HP 1-ml column (GE Healthcare Life Sciences, Little Chalfont, UK) preequilibrated with buffer A. The column was then washed with 5 bed volumes of buffer C (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5% glycerol, and 80 mM imidazole) to facilitate His-tagged protein binding, followed by 2.5 bed volumes of elution buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5% glycerol, and 500 mM imidazole). The total eluate following the elution buffer addition (2.5 ml) was collected and dialyzed against buffer TK (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 5% glycerol) overnight. Protein purity was checked by SDS-PAGE: 10 μl of purified CalG protein was loaded onto a 15% polyacrylamide gel along with a size marker (Tiangen, Shanghai, China) for comparison, and samples were electrophoresed at a constant 25 V for 1 h or until the loading dye had reached the bottom of the gel. Protein was stored frozen at −80°C until required.
Production and purification of cezomycin, N-demethyl-calcimycin, and calcimycin and related compounds.
Production and purification of cezomycin, N-demethyl-calcimycin, and calcimycin was achieved by first culturing 6 liters of S. chartreusis NRRL 3882 in 15 1-liter baffled conical flasks each containing 400 ml SFM medium at 30°C for 9 days with gentle shaking at 220 rpm. After this time, cells and cellular debris were removed by centrifugation at 6,000 × g for 30 min in 500-ml polypropylene centrifuge bottles in an Eppendorf 5810 R centrifuge (Eppendorf AG, Hamburg, Germany); the resulting supernatant was pooled and divided into 500-ml volumes, each of which was carefully transferred to a 2-liter separating funnel and thoroughly mixed with 750 ml of dried ethyl acetate. The mixture was allowed to settle for 10 min to permit phase separation, and the upper ethyl acetate layer was collected, placed in a rotary evaporator, and dried in vacuo for 1 h. The total dried product from 6 liters of fermentation broth was then redissolved in 5 ml of 90% (vol/vol) aqueous methanol, and the whole volume was layered onto a reverse-phase silica gel (AAG 12S50; YMC Co. Ltd.) column and eluted with aqueous methanol in gradients from 70% to 100% and a flow rate of 0.5 ml min−1. Ten-milliliter fractions were collected from the column, and 20 μl from each fraction was analyzed using HPLC/MS as described above. The identity of compounds in eluates was confirmed by comparison of HPLC retention time, UV spectrometry, and mass spectrometry results with those obtained from standards (see above) (6). Normally, fractions containing the molecule of interest (cezomycin, N-demethyl-calcimycin, or calcimycin) totaled 20 ml, i.e., the amount from two collections; these were pooled and concentrated in a rotary evaporator under vacuum for 1 h, and the dried product was stored at −80°C until required for use. Typically, the approximate yields obtained from 6 liters of culture were 60 mg cezomycin, 5 mg N-demethyl-calcimycin, and 15 mg calcimycin.
Production and purification of compound 3 followed the same general procedure as that described above with the exception that strain GLX1 (ΔcalB1) (29) was cultured in a total volume of 3 liters medium. Typically, 45 mg of compound 3 was obtained from 3 liters of liquid culture.
The synthesis of the S-N-acetylcysteamine (SNAC) derivatives of compound 3, cezomycin, N-dimethyl-calcimycin, and calcimycin was conducted using the method of Zhou et al. (28), and all reagents involved were purchased from Sigma-Aldrich (Merck KGaA). Briefly, 5 mg compound 3 (0.014 mmol), cezomycin (0.01 mmol), N-demethyl-calcimycin (0.01 mmol), or calcimycin (0.01 mmol) was mixed with 200 mg (1.26 mmol) EDC-HCl [1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride], 25 mg (0.2 mmol) DMAP (4-dimethylaminopyridine), and 50 mg (0.42 mmol) SNAC (S-N-acetylcysteamine) in a 50-ml conical flask, and 10 ml dry CH2Cl2 was added. The mixtures were gently agitated for 3 h at room temperature, after which the precipitate was removed by centrifugation at 15,000 × g for 10 min and the supernatant containing SNAC adduct derivatives was subjected to reversed-phase silica gel (AAG 12S50; YMC Co. Ltd.) column chromatography by elution with aqueous methanol in gradients from 70% to 100% and a flow rate of 0.5 ml min−1. Ten-milliliter fractions were collected, and 20 μl of each fraction was analyzed as described previously; those found to contain the SNAC derivatives (a total of around 10 ml for each product [a single fraction]) were concentrated under vacuum for 1 h in a rotary evaporator, and dried products were subsequently redissolved in 2 ml of 90% (vol/vol) aqueous methanol. Product molecular identities were confirmed by Q-TOF LC-MS (Agilent 6530 series accurate-mass Q-TOF LC/MS; Agilent Technologies) by virtue of “signature” ion peaks (m/z) at 479.2574 (compound 3-SNAC), 596.2789 (cezomycin-SNAC), 611.2898 (N-demethyl-calcimycin-SNAC), and 625.3054 (calcimycin-SNAC) (Table S1 and Fig. S1).
CalG protein activity assays.
CalG protein activity assay buffer (total volume, 100 μl) was composed of 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5% glycerol, and 0.2 mM cezomycin-CoA or compound 3, cezomycin, N-demethyl-calcimycin, calcimycin, or their SNAC derivatives in a sterile Eppendorf tube. Reactions were initiated by addition of 0.2 μM purified CalG-His or boiled (denatured) protein (as a negative control) to the reaction mixture and were performed in triplicate at 30°C. Reaction termination after 1 h of incubation was via enzyme denaturation by addition of 100 μl of methanol, after which precipitated protein was removed by centrifugation at 15,000 × g. The supernatant was then removed and subjected to Q-TOF LC/MS analysis as described previously.
Hydrolysis of CoA adducts releases free thiol groups, which can react with the reagent 5,5-dithiobis (2-nitrobenzoic acid) (DTNB) to produce a yellow product, 5-thio-2-nitrobenzoate, whose formation can be monitored spectrophotometrically in solution at λ412 nm. We took advantage of this to measure CalG initial CoA adduct hydrolysis rates in this study. Reactions were performed in a total volume of 100 μl containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5% glycerol, 2 μM CalG, 0.2 mM DTNB, and 0.1 to 3 mM the acyl-CoA compound in the cuvette of a PerkinElmer Lambda 650 spectrophotometer at 30°C over a period of 2 h at 1-min intervals. The effects of various divalent cations, Mn2+, Ca2+, Mg2+, Zn2+, and Fe2+, on CalG hydrolysis of cezomycin-CoA or acetyl-CoA were also investigated using the same approach at various ion concentrations, between 0.5 to 10 mM depending on the ion involved.
Prism 5 software (Graph-Pad Software, Inc.) was used to calculate the resulting enzyme kinetic parameters.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Tang Berkeley Scholarship, the Ministry of Science and Technology (973 program, 2015CB554203), and the National Science Foundation of China (31470830, 21661140002, 91753123).
Footnotes
For a companion article on this topic, see https://doi.org/10.1128/AEM.00586-18.
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00587-18.
REFERENCES
- 1.Heathcote ML, Staunton J, Leadlay PF. 2001. Role of type II thioesterases: evidence for removal of short acyl chains produced by aberrant decarboxylation of chain extender units. Chem Biol 8:207–220. [DOI] [PubMed] [Google Scholar]
- 2.Kim BS, Cropp TA, Beck BJ, Sherman DH, Reynolds KA. 2002. Biochemical evidence for an editing role of thioesterase II in the biosynthesis of the polyketide pikromycin. J Biol Chem 277:48028–48034. doi: 10.1074/jbc.M207770200. [DOI] [PubMed] [Google Scholar]
- 3.Schwarzer D, Mootz HD, Linne U, Marahiel MA. 2002. Regeneration of misprimed nonribosomal peptide synthetases by type II thioesterases. Proc Natl Acad Sci U S A 99:14083–14088. doi: 10.1073/pnas.212382199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David L, Kergomard A. 1982. Production by controlled biosynthesis of a novel ionophore antibiotic, cezomycin (demethylamino A23187). J Antibiot (Tokyo) 35:1409–1411. doi: 10.7164/antibiotics.35.1409. [DOI] [PubMed] [Google Scholar]
- 5.Boot JH, van Hilten J. 1996. The use of the divalent calcium-ionophore A23187 as a biochemical tool in pharmacological and in vitro toxicological studies. Cell Struct Function 21:97–99. doi: 10.1247/csf.21.97. [DOI] [PubMed] [Google Scholar]
- 6.Butler AR, Bate N, Cundliffe E. 1999. Impact of thioesterase activity on tylosin biosynthesis in Streptomyces fradiae. Chem Biol 6:287–292. [DOI] [PubMed] [Google Scholar]
- 7.Doi-Katayama Y, Joon YJ, Choi CY, Yu TW, Floss HG, Hutchinson CR. 2000. Thioesterases and the premature termination of polyketide chain elongation in rifamycin B biosynthesis by Amycolatopsis mediterranei S699. J Antibiot (Tokyo) 53:484–495. doi: 10.7164/antibiotics.53.484. [DOI] [PubMed] [Google Scholar]
- 8.Schneider A, Marahiel MA. 1998. Genetic evidence for a role of thioesterase domains, integrated in or associated with peptide synthetases, in non-ribosomal peptide biosynthesis in Bacillus subtilis. Arch Microbiol 169:404–410. doi: 10.1007/s002030050590. [DOI] [PubMed] [Google Scholar]
- 9.Pfeifer B, Hu Z, Licari P, Khosla C. 2002. Process and metabolic strategies for improved production of Escherichia coli-derived 6-deoxyerythronolide B. Appl Environ Microbiol 68:3287–3292. doi: 10.1128/AEM.68.7.3287-3292.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu T, Cane DE, Deng Z. 2009. The enzymology of polyether biosynthesis. Methods Enzymol 459:187–214. doi: 10.1016/S0076-6879(09)04609-6. [DOI] [PubMed] [Google Scholar]
- 11.Liu T, You D, Valenzano C, Sun Y, Li J, Yu Q, Zhou X, Cane DE, Deng Z. 2006. Identification of NanE as the thioesterase for polyether chain release in nanchangmycin biosynthesis. Chem Biol 13:945–955. doi: 10.1016/j.chembiol.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Liu T, Lin X, Zhou X, Deng Z, Cane DE. 2008. Mechanism of thioesterase-catalyzed chain release in the biosynthesis of the polyether antibiotic nanchangmycin. Chem Biol 15:449–458. doi: 10.1016/j.chembiol.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey BM, Hong H, Jones MA, Hughes-Thomas ZA, Goss RM, Heathcote ML, Bolanos-Garcia VM, Kroutil W, Staunton J, Leadlay PF, Spencer JB. 2006. Evidence that a novel thioesterase is responsible for polyketide chain release during biosynthesis of the polyether ionophore monensin. ChemBioChem 7:1435–1442. doi: 10.1002/cbic.200500474. [DOI] [PubMed] [Google Scholar]
- 14.Harvey BM, Mironenko T, Sun Y, Hong H, Deng Z, Leadlay PF, Weissman KJ, Haydock SF. 2007. Insights into polyether biosynthesis from analysis of the nigericin biosynthetic gene cluster in Streptomyces sp. DSM4137. Chem Biol 14:703–714. doi: 10.1016/j.chembiol.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 15.Tang L, Fu H, Betlach MC, McDaniel R. 1999. Elucidating the mechanism of chain termination switching in the picromycin/methymycin polyketide synthase. Chem Biol 6:553–558. doi: 10.1016/S1074-5521(99)80087-8. [DOI] [PubMed] [Google Scholar]
- 16.Chen S, Roberts JB, Xue Y, Sherman DH, Reynolds KA. 2001. The Streptomyces venezuelae pikAV gene contains a transcription unit essential for expression of enzymes involved in glycosylation of narbonolide and 10-deoxymethynolide. Gene 263:255–264. doi: 10.1016/S0378-1119(00)00560-6. [DOI] [PubMed] [Google Scholar]
- 17.Pressman BC. 1976. Biological applications of ionophores. Annu Rev Biochem 45:501–530. doi: 10.1146/annurev.bi.45.070176.002441. [DOI] [PubMed] [Google Scholar]
- 18.Kozian D, Proulle V, Nitsche A, Galitzine M, Martinez M-C, Schumann B, Meyer D, Herrmann M, Freyssinet J-M, Kerbiriou-Nabias D. 2005. Identification of genes involved in Ca2+ ionophore A23187-mediated apoptosis and demonstration of a high susceptibility for transcriptional repression of cell cycle genes in B lymphoblasts from a patient with Scott syndrome. BMC Genomics 6:146. doi: 10.1186/1471-2164-6-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reed PW, Lardy HA. 1972. A23187: a divalent cation ionophore. J Biol Chem 247:6970–6977. [PubMed] [Google Scholar]
- 20.Kajitani N, Kobuchi H, Fujita H, Yano H, Fujiwara T, Yasuda T, Utsumi K. 2007. Mechanism of A23187-induced apoptosis in hl-60 cells: dependency on mitochondrial permeability transition but not on NADPH oxidase. Biosci Biotechnol Biochem 71:2701–2711. doi: 10.1271/bbb.70304. [DOI] [PubMed] [Google Scholar]
- 21.Wu Q, Liang J, Lin S, Zhou X, Bai L, Deng Z, Wang Z. 2011. Characterization of the biosynthesis gene cluster for the pyrrole polyether antibiotic calcimycin (A23187) in Streptomyces chartreusis NRRL. 3882. Antimicrob Agents Chemother 55:974–982. doi: 10.1128/AAC.01130-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaney MO, Debono JNM. 1976. The structure of the calcium complex of A23187, a divalent cation ionophore antibiotic. J Antibiot (Tokyo) 29:424–427. doi: 10.7164/antibiotics.29.424. [DOI] [PubMed] [Google Scholar]
- 23.Smith GD, WLD. 1976. Crystal and molecular structure of the calcium ion complex of A23187. J Am Chem Soc 98:1578–1580. doi: 10.1021/ja00422a050. [DOI] [Google Scholar]
- 24.Prudhomme M, Guyot DGJ, Jeminet G. 1984. Semisynthesis of A23187 (calcimycin) analogs. II. Introduction of a methyl group on the benzoxazole ring. J Antibiot (Tokyo) 37:627–634. [DOI] [PubMed] [Google Scholar]
- 25.Prudhomme M, Jeminet DGG. 1986. Semi-synthesis of A23187 (calcimycin) analogs. III. Modification of benzoxazole ring substituents, ionophorous properties in an organic phase. J Antibiot (Tokyo) 39:922–933. [DOI] [PubMed] [Google Scholar]
- 26.Erdahl WL, Chapman CJ, Wang E, Taylor RW, Pfeiffer DR. 1996. Ionophore 4-BrA23187 transports Zn2+ and Mn2+ with high selectivity over Ca2+. Biochemistry 35:13817–13825. doi: 10.1021/bi961391q. [DOI] [PubMed] [Google Scholar]
- 27.Vila S, Guyot ICJ, Jeminet G, Pointud Y. 2003. Molecular design of calcimycin(A23187) evidenced by the complexing behaviour of its 4-bromo and 19-demethyl analogues. New J Chem 27:1246–1250. doi: 10.1039/b300338h. [DOI] [Google Scholar]
- 28.Zhou Y, Meng Q, You D, Li J, Chen S, Ding D, Zhou X, Zhou H, Bai L, Deng Z. 2008. Selective removal of aberrant extender units by a type II thioesterase for efficient FR-008/candicidin biosynthesis in Streptomyces sp. strain FR-008. Appl Environ Microbiol 74:7235–7242. doi: 10.1128/AEM.01012-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gou L, Wu Q, Lin S, Li X, Liang J, Zhou X, An D, Deng Z, Wang Z. 2013. Mutasynthesis of pyrrole spiroketal compound using calcimycin 3-hydroxy anthranilic acid biosynthetic mutant. Appl Microbiol Biotechnol 97:8183–8191. doi: 10.1007/s00253-013-4882-1. [DOI] [PubMed] [Google Scholar]
- 30.Gokhale RS, Hunziker D, Cane DE, Khosla C. 1999. Mechanism and specificity of the terminal thioesterase domain from the erythromycin polyketide synthase. Chem Biol 6:117–125. doi: 10.1016/S1074-5521(99)80008-8. [DOI] [PubMed] [Google Scholar]
- 31.Wu H, Liang J, Wang J, Liang W-J, Gou L, Wu Q, Zhou X, Bruce IJ, Deng Z, Wang Z.. 2018. Cezomycin is activated by CalC to its ester form for further biosynthesis steps in the production of calcimycin in Streptomyces chartreusis NRRL 3882. Appl Environ Microbiol 84:e00586-18. doi: 10.1128/AEM.00586-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naggert J, Williams B, Cashman DP, Smith S. 1987. Cloning and sequencing of the medium-chain S-acyl fatty acid synthetase thioester hydrolase cDNA from rat mammary gland. Biochem J 243:597. doi: 10.1042/bj2430597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Albrecht-Gary A-M, Blanc S, David L, Jeminets G. 1994. Closely related ionophores cezomycin and calcimycin (A 23187): cooperative formation of the transporting species. Inorg Chem 33:518–524. doi: 10.1021/ic00081a020. [DOI] [Google Scholar]
- 34.Sugai Y, Katsuyama Y, Ohnishi Y. 2016. A nitrous acid biosynthetic pathway for diazo group formation in bacteria. Nat Chem Biol 12:73–75. doi: 10.1038/nchembio.1991. [DOI] [PubMed] [Google Scholar]
- 35.Geoffroy VA, Fetherston JD, Perry RD. 2000. Yersinia pestis YbtU and YbtT are involved in synthesis of the siderophore yersiniabactin but have different effects on regulation. Infect Immun 68:4452–4461. doi: 10.1128/IAI.68.8.4452-4461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF. 2007. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25:447–453. doi: 10.1038/nbt1297. [DOI] [PubMed] [Google Scholar]
- 37.Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M. 2001. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci U S A 98:12215–12220. doi: 10.1073/pnas.211433198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y, Zhou X, Liu J, Bao K, Zhang G, Tu G, Kieser T, Deng Z. 2002. ‘Streptomyces nanchangensis’, a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin, contains multiple polyketide gene clusters. Microbiology 148:361–371. doi: 10.1099/00221287-148-2-361. [DOI] [PubMed] [Google Scholar]
- 39.Klein JS, Lewinson O. 2011. Bacterial ATP-driven transporters of transition metals: physiological roles, mechanisms of action, and roles in bacterial virulence. Metallomics 3:1098–1108. doi: 10.1039/c1mt00073j. [DOI] [PubMed] [Google Scholar]
- 40.Raatschen N, Wenzel M, Ole Leichert LI, Düchting P, Krämer U, Bandow JE. 2013. Extracting iron and manganese from bacteria with ionophores—a mechanism against competitors characterized by increased potency in environments low in micronutrients. Proteomics 13:1358–1370. doi: 10.1002/pmic.201200556. [DOI] [PubMed] [Google Scholar]
- 41.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 42.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical streptomyces genetics. John Innes Foundation, Colney, Norwich, England. [Google Scholar]
- 43.Gust B, Kieser T, Chater K. 2002. PCR-targeting system in Streptomyces coelicolor A3(2). John Innes Foundation, Colney, Norwich, England. [Google Scholar]
- 44.Huang T, Wang Y, Yin J, Du Y, Tao M, Xu J, Chen W, Lin S, Deng Z. 2011. Identification and characterization of the pyridomycin biosynthetic gene cluster of Streptomyces pyridomyceticus NRRL B-2517. J Biol Chem 286:20648–20657. doi: 10.1074/jbc.M110.180000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.