

ABSTRACT
The quorum sensing (QS) system controls bacterial group behaviors in response to cell density. In vibrios, LuxR and AphA are two master QS regulators (MQSRs) controlling gene expression in response to high or low cell density. Other regulators involved in the regulation of these two MQSRs and QS pathways remain to be determined. Here, we performed bacterial one-hybrid (B1H)-assay-based screens of transcriptional factors (TFs) to identify TFs that can directly regulate the expression of luxR and aphA from a library of 285 TFs encoded by the fish pathogen Vibrio alginolyticus. A total of 7 TFs were identified to bind to the promoters of both luxR and aphA. Among these TFs, the novel LysR-type transcriptional regulator (LTTR) VqsA could activate LuxR and repress AphA transcription. Meanwhile, LuxR and AphA exerted feedback inhibition and activation of vqsA expression, respectively, indicating that VqsA coordinates QS and is also regulated by QS. In addition, VqsA inhibited its own expression by directly binding to its own promoter region. The VqsA-binding sites in the promoter regions of luxR and aphA as well as the binding sites of LuxR, AphA, and VqsA in the vqsA gene were uncovered by electrophoretic mobility shift assays (EMSAs) and DNase I footprinting analysis. Finally, VqsA was verified to play essential roles in QS-regulated phenotypes, i.e., type VI secretion system 2 (T6SS2)-dependent interbacterial competition, biofilm formation, exotoxin production, and in vivo virulence of V. alginolyticus. Collectively, our data showed that VqsA is an important QS regulator in V. alginolyticus.
IMPORTANCE Investigation of the mechanism of regulation of quorum sensing (QS) systems will facilitate an understanding of bacterial pathogenesis and the identification of effective QS interference (QSI) targets. Here, we systematically screened transcriptional factors (TFs) that modulate the expression of the master QS regulators (MQSRs) LuxR and AphA, and a novel LysR-type transcriptional regulator, VqsA, was identified. Our data illuminated the mechanisms mediating the interaction among LuxR, AphA, and VqsA as well as the effects of these regulators on the expression and output of QS. The impaired expression of virulence genes as a result of vqsA disruption demonstrated that VqsA is an important player in QS regulation and pathogenesis and may be the third MQSR involved in sensing environmental signals by vibrios to coordinate QS responses. This study will facilitate the development of strategies to interfere with QS and effectively control this pathogen that plagues the aquaculture industry.
KEYWORDS: Vibrio alginolyticus, quorum sensing, LuxR, AphA, VqsA, virulence, bacterial one-hybrid assay, MQSR
INTRODUCTION
Quorum sensing (QS) enables bacteria to collectively coordinate gene expression in response to cell density. LuxR and AphA are two master QS regulators (MQSRs) in the QS circuits of vibrios (1–3). AphA is involved in the regulation of gene expression in the low-cell-density (LCD) stage, while LuxR is involved in the high-cell-density (HCD) stage. The separate or combined activities of these two MQSRs regulate the expression of ∼600 genes in Vibrio harveyi, leading to a variety of cooperative behaviors, including bioluminescence, biofilm formation, motility, and virulence (4, 5). Reciprocal repression of these two MQSRs enables these regulators to be individually expressed at the low- or high-cell-density stage (3, 4). The mechanism of QS-based signaling in response to cell densities and autoinducer (AI) concentrations has been well established in V. harveyi and other vibrios (2).
MQSR production is strictly regulated at multiple levels to obtain concentration gradients for the temporal regulation of gene expression at different cell densities while avoiding the potentially high fitness cost of runaway expression and protein bursts under nonideal conditions (4, 6). Indeed, various HapR (a LuxR homolog) regulators are deployed in Vibrio cholerae to optimize the QS output at the transcriptional level (e.g., Fis, VqmA, HapR, AphA, VarS/VarA, and FliA/FlgM) (3, 7–11), the posttranscriptional level (e.g., small RNAs [sRNAs] associated with csrA and grr genes) (12, 13), and the posttranslational level (e.g., VarS/VarA) (10, 14), constituting a sophisticated regulation network. These regulators and other uncharacterized factors play essential roles in the interactions of QS pathways with environmental signals and thus are important targets for the development of nonantibiotic therapeutics and drugs, known as QS interference (QSI) therapies (15).
Vibrio alginolyticus is a Gram-negative bacterium and an opportunistic pathogen that causes intra- and extraintestinal diseases or wound infections in humans and infects a large number of sea animals (16). Known virulence factors of V. alginolyticus include extracellular proteases, e.g., Asp (16), MviN (17), ProA (18), and Pep (19). These virulence factors and other virulence-related genes are closely regulated by the LuxR-LuxO-centered QS regulatory system in V. alginolyticus (20). The QS signaling network of V. alginolyticus is quite similar to that of V. harveyi, including the three types of AIs that are produced and sensed by LuxS/PQ, LuxM/N, and CqsA/S (21–23). At low cell density, AphA binds to the promoter of luxR and 48 other genes and regulates the expression of these genes (24). At high cell density, LuxR can also act a global regulator, binding directly upstream of and regulating the expression of more than 100 genes in response to QS signals, as also seen in V. harveyi (reference 4 and our unpublished data). Autorepression of LuxR occurs via the binding of LuxR to the promoter at two sites, known as LuxR-binding site I (RBSI) and RBSII (6). In addition to LuxR and AphA, the promoter of luxR is also indirectly regulated by LuxT and directly regulated by AphB and the alternative sigma factor RpoE, which works in conjunction with core RNA polymerase in response to changes in temperature (6, 23, 25). However, whether there are other factors that can regulate QS in the bacterium remains largely unknown.
In this study, we employed a modified bacterial one-hybrid (B1H) system to systematically screen a library of 285 transcriptional factors (TFs) in V. alginolyticus to identify luxR and aphA regulators. A total of 7 TFs were identified to bind to and regulate the promoters of both luxR and aphA. Among these factors, the hypothetical protein V12G01_18672 (referred to here as VqsA [Vibrio quorum sensing activator]), which encodes a novel LysR-type transcriptional regulator (LTTR) and the deletion of which leads to the greatest reduction in exotoxin production, was chosen for further analysis. Electrophoretic mobility shift assays (EMSAs) and DNase I footprinting analysis confirmed that VqsA regulates luxR and aphA expression by binding directly to the promoter DNA of these genes. VqsA is important for the virulence of the bacterium and may be the third MQSR coordinating QS and virulence gene expression.
RESULTS
Systematic screening revealed multiple regulators binding to the promoters of luxR and aphA and regulating QS output.
We used a previously described strategy (26) to adapt the BacterioMatch II two-hybrid system (Stratagene, La Jolla, CA, USA) for identifying protein-protein interactions to a bacterial one-hybrid system (B1H) for the detection of specific protein-DNA interactions in vivo (Fig. 1A; see also Fig. S1 in the supplemental material). The B1H system consists of a selected bait promoter DNA cloned into the reporter plasmid pBXcmT carrying the selectable genes HIS3 and aadA. A specific transcription factor (TF) is expressed as a fusion with the alpha-subunit of RNA polymerase (RNAP) from the pTRG plasmid. This pair of recombinant vectors is cotransformed into the host strain (Fig. S1). If the TF protein interacts with the target DNA, the protein recruits and stabilizes the binding of RNAP at the promoter and thus activates the transcription of the HIS3 reporter gene in the presence of the compound 3-amino-1,2,4-triazole (3-AT). A second reporter gene, aadA, encoding a protein that confers streptomycin resistance (Strr), provides an additional form of selection to validate the DNA-protein interaction (Fig. 1A) (26). To search for potential TFs involved in the regulation of luxR and aphA expression in V. alginolyticus, the promoter regions of these genes (PluxR and PaphA) were individually cloned into the T/A cloning site upstream of HIS3-aadA in pBXcmT. The B1H system was first validated by the growth of the cotransformants harboring pTRG::LuxR and pBXcmT::PluxR plasmids with a known LuxR-PluxR interaction pair (6) on the plate with selective medium containing 3-AT and Str (positive control) (Fig. 1B). No growth of the cotransformants encoding LuxR and PluxR ΔRBSI/II with mutated LuxR-binding sites I and II (6) was observed on the selection plates (negative control). All these cotransformants grew on the LB plate in the absence of 3-AT and Str (Fig. 1B). To further validate the B1H system, AphA was observed to bind to the promoter regions of both aphA and luxR (Table S1), as previously documented (24).
FIG 1.

Screening of TFs bound to the promoter DNA of luxR and aphA in vitro. (A) Diagrams for the modified bacterial one-hybrid (B1H) assay. If the TF interacts with the target DNA, the TF recruits and stabilizes the binding of RNA polymerase at the weak promoter (shown as a turned arrow), thus activating the transcription of the HIS3 reporter gene, conferring 3-AT resistance, and the gene aadA, conferring Str resistance; this confirms the DNA-protein interaction. (B) B1H identification of TFs interacting with promoter DNA of aphA and luxR. Positive growth of cotransformants with pBXcmT harboring PluxR or PaphA and the pTRG derivatives expressing AsnC, 02325, SoxR, MetR, 01665, 06711, or 18672 on selective plates are shown. Cotransformants harboring pTRG::LuxR with pBXcmT::PluxR and pTRG::LuxR with pBXcmT::PluxR ΔRBSI/II were used as positive and negative controls (6), respectively. (C) EMSA analysis of protein-DNA interactions. Cy5-labeled PluxR or PaphA DNA was added to reaction mixtures with different concentrations of the AsnC, SoxR, MetR, 02323, 01665, and 06711 proteins and a 10-fold excess of nonspecific competitor [poly(dI:dC)]. The unrelated gyrB DNA was used as a negative control. B, bound DNA; F, unbound DNA. (D) Hide powder azure (HPA) digestion assays were performed to analyze extracellular Asp activity of the supernatants of 9-h cultures. Asp activity was normalized by dividing the total protease activity by the cell density of each strain. The results are presented as the mean ± SD (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001 based on analysis of variance (ANOVA) comparisons. (E) Western blot analysis was performed to analyze Asp yields in the WT and in the indicated deletion mutant strains using concentrated supernatant with Asp-specific antibodies. RNAP was used as the loading control for the supernatants that were obtained from the same number of cells. The numbers indicate the densitometric measurements.
The V. alginolyticus genome encodes ∼285 predicted TFs with putative DNA-binding domains. The TF genes were individually cloned into the pTRG plasmid and then screened using PluxR and PaphA as bait sequences in pBXcmT in the B1H system. The comprehensive PluxR and PaphA screens yielded 16 and 19 putative TFs, respectively, by interacting with the luxR and aphA promoters, respectively (Table S1). Many of the PluxR- and PaphA-associated TFs belong to the LysR-type transcriptional regulator (LTTR) family, the most common type of prokaryotic DNA-binding protein, suggesting the importance of LTTRs in regulating QS. Moreover, 7 TFs, i.e., AsnC, SoxR, MetR, 12G01_02325, 01665, 06711, and 18672, exhibited binding affinities toward both PluxR and PaphA and supported the growth of the cotransformants harboring the TF and the PluxR or PaphA pairs (Fig. 1B) (Table 1). The electrophoretic mobility shift assays (EMSAs) showed that all TFs bound efficiently to both PluxR and PaphA in a concentration-dependent manner, whereas the negative control (gyrB) remained unbound (Fig. 1C and 2C). These data confirmed the interaction between the promoters and the selected TFs.
TABLE 1.
Seven transcriptional regulators identified to bind to the promoters of both aphA and luxR
| Gene IDa | Proteinb | Molecular mass (kDa) | pI | Function | Location of HTHc | Binding withd: |
|
|---|---|---|---|---|---|---|---|
| PluxR | PaphA | ||||||
| V12G01_10813 | AsnC | 17.32 | 4.67 | Transcriptional regulator AsnC | N, C | +++ | ++++ |
| V12G01_02325 | HP | 42.19 | 4.66 | Transcriptional activator | N | ++++ | ++++ |
| V12G01_02620 | SoxR | 16.20 | 7.59 | SoxR protein | N | +++++ | ++++ |
| V12G01_16177 | MetR | 34.09 | 7.35 | LysR family MetR | N | +++ | ++++ |
| V12G01_01665 | HP | 15.47 | 7.57 | Hypothetical regulator | N | ++++ | +++ |
| V12G01_06711 | HP | 33.52 | 4.87 | LysR family protein | N | ++++ | +++ |
| V12G01_18672 | VqsA | 32.84 | 7.25 | LysR family protein | N | ++++ | ++++ |
ID, identification.
HP, hypothetical protein.
HTH, helix-turn-helix domain and the location of this domain in the protein (N- or C-terminal end).
The observed growth of the serial dilutions of cotransformants harboring both the indicated promoter DNA and the pTRG plasmid expressing the selected protein on LB agar plates containing 3-AT and Str. Plus signs indicate the level of growth of the indicator strains used for B1H assays.
FIG 2.

VqsA regulates luxR expression by directly binding to the luxR promoter. (A) Western blot analysis showing LuxR expression in WT and ΔluxR, ΔluxO, ΔvqsA, vqsA+, and ΔvqsA ΔluxO mutant cells grown in LBS for 9 h. RNAP was used as a loading control. The numbers indicate the densitometric measurements. (B) qRT-PCR analysis of luxR transcripts in the WT, ΔvqsA mutant, and vqsA+ mutant cells grown in LBS for 9 h; 16S rRNA was selected as a control. The results are displayed as the mean ± SD (n = 3). ***, P < 0.001 based on Student's t test. (C) EMSA examination of the binding of VqsA to PluxR. The amount of VqsA protein used was as indicated, and 20 ng of each Cy5-labeled probe was added to the EMSA reactions. The shifts were verified to be specific in experiments in which 25- to 50-fold excess of unlabeled specific DNA and nonspecific competitor DNA [poly(dI:dC)] were used. (D) Plot showing the binding affinity of VqsA for the luxR promoter. The densitometric intensities of bound DNA fragments were plotted against VqsA concentrations. The indicated dashed line represents the concentration of VqsA that caused half-maximal binding (Kd). (E) DNase I footprinting analysis of VqsA binding to a binding site in the luxR promoter (shown in the dashed box). Electropherograms show a DNase I digestion of the PluxR promoter probe after incubation with 0 or 3.6 μM VqsA. The corresponding nucleotide sequences that were protected by VqsA are indicated below, with the binding motif highlighted in blue. The direction of transcription (as depicted with an arrow), the location of the partial −10 box and −35 box (underlined), and the VqsA-binding site relative to the start codon ATG are shown. (F) EMSA investigation of the binding of VqsA to the specific VqsA-binding site (VBS) in the luxR promoter. The PluxR variant lacking the VBS sequence (PluxR ΔVBS) was also investigated.
We further generated the in-frame deletion mutants of the 7 selected TFs, and none of these mutants exhibited growth defects compared to the wild-type (WT) strain (Fig. S2). Hide powder azure (HPA) digest assays and Western blot analyses were performed to investigate the roles of these TFs in the production of alkaline serine protease (Asp), a QS-controlled exotoxin (16). Compared to the WT strain, the Asp yield was seen to be drastically reduced in the ΔluxR mutant and significantly increased in the ΔaphA mutant (Fig. 1D and E) (6, 24). The ΔsoxR, ΔmetR, and Δ06711 mutant strains showed markedly increased Asp production, while the ΔasnC, Δ02325, Δ01665, and Δ18672 mutants were significantly impaired in Asp production (Fig. 1D and E). Taken together, these data demonstrate that various novel positive and negative regulators bind directly to the promoters of MQSR-encoding genes and are involved in QS pathways.
VqsA regulates the expression of luxR by directly binding to a specific binding site.
Among the above-mentioned 7 selected TFs, we were intrigued by 12G01_18672 (here referred to as VqsA), as the disruption of this gene drastically impaired the ability of V. alginolyticus EPGS to produce the exotoxin (Fig. 1E). The vqsA gene encodes a 292-amino-acid (aa) protein annotated as an LTTR that is highly conserved in vibrios. We first confirmed that complementation with a plasmid carrying the vqsA gene restored LuxR expression in the ΔvqsA mutant to the WT level (Fig. 2A). In the QS cascade of vibrios, LuxO represses the expression of LuxR (2, 6). Compared to the ΔluxO (6) and ΔvqsA mutants, the ΔvqsA ΔluxO double-mutant strain showed significantly lower LuxR production than the ΔluxO mutant (Fig. 2A). These results contradict the idea that VqsA regulates LuxR expression via LuxO and suggest that VqsA might act on the LuxR regulatory pathway independent of LuxO and upstream QS cascades (Fig. 2A). The regulation by VqsA of the expression of luxR was further confirmed by reverse transcription-quantitative PCR (qRT-PCR) analysis, showing that the transcript levels of luxR in ΔvqsA mutant cells are significantly lower than those in vqsA+ mutant cells after 9 h of growth (Fig. 2B).
EMSAs showed that VqsA bound specifically to a DNA probe that included the promoter region of the luxR gene in a concentration-dependent manner in the presence of an excess amount (10-fold) of the nonspecific poly(dI:dC) competitor (Fig. 2C, lanes 1 to 8) and could be titrated out by the addition of the unlabeled luxR probe (Fig. 2C, lanes 9 and 10). The value of the dissociation constant (Kd) for the binding of VqsA to luxR was calculated to be 0.55 μM (Fig. 2D). Next, a dye-primer-based DNase I footprinting assay was performed on both strands of a DNA fragment encompassing the promoter of luxR. We compared the electropherograms with and without addition of the VqsA protein to determine the specific VqsA-binding site (VBS), 5′-TGAGTGTAATGGTGCATATGCACAAGGTCAA-3′ (88 bp upstream of ATG), which mapped to the −35 box and −10 box in the promoter region of luxR (Fig. 2E). The absence of the specific binding site abolished the binding of VqsA to the luxR promoter (Fig. 2F). Taken together, these data indicate that VqsA binds to the luxR promoter, positively regulating transcription and expression of the luxR gene.
VqsA directly represses aphA expression.
We next asked whether VqsA is a transcriptional regulator of aphA. The MQSR AphA governs the LCD QS gene expression and is highly expressed in the first 2 h of growth (24). Western blotting was performed with an AphA-specific antibody to test AphA expression in the WT, ΔaphA mutant, and ΔvqsA mutant strains and in the complement vqsA+ strain at 2 h in Luria-Bertani medium supplemented with 3% NaCl (LBS). A notable increase in aphA expression was observed in the ΔvqsA mutant strain, whereas aphA expression in the vqsA+ complement strain decreased to the WT level (Fig. 3A). qRT-PCR analysis also confirmed that the aphA transcript level was higher in the ΔvqsA mutant strain than in the WT strain and was significantly lower in the vqsA+ complement strain (Fig. 3B), indicating that VqsA represses the transcription of aphA.
FIG 3.

VqsA is involved in aphA expression by binding directly to the promoter of aphA. (A) Western blot analysis of AphA expression in WT, ΔvqsA mutant, and vqsA+ mutant cells cultured in LBS medium for 2 h. RNAP was used as a loading control. The numbers indicate the densitometric measurements. (B) qRT-PCR analysis of the aphA transcripts. The WT, ΔvqsA mutant, and vqsA+ mutant strains were cultured in LBS for 2 h, and mRNA transcripts were detected by qRT-PCR; 16S rRNA was selected as a control. The results are displayed as the mean ± standard deviation (SD) (n = 3), ***, P < 0.001 based on Student's t test. (C) EMSA analysis of the binding of VqsA to PaphA. The amount of VqsA protein used is indicated, and 20 ng of each Cy5-labeled probe was added to the EMSA reactions. The shifts were verified to be specific in experiments in which 25- to 50-fold excess of unlabeled specific DNA and nonspecific competitor DNA [poly(dI:dC)] were used. (D) Plot showing the affinity of the binding of VqsA to the aphA promoter. The densitometric intensities of bound DNA fragments were plotted against VqsA concentrations. (E) DNase I footprinting analysis of the binding of VqsA to a binding site in PaphA. Electropherograms show the PaphA promoter probe treated with DNase I after incubation with 0 or 2.0 μM VqsA. (F) EMSA performed with purified VqsA and the aphA promoter DNA (PaphA) or its variant lacking the specific VqsA-binding site (PaphA ΔVBS). Dashed boxes indicate the binding site revealed by DNase I footprinting assays.
EMSAs were then performed to investigate the binding of VqsA to the aphA promoter probe. The result showed that PaphA had affinity for the purified VqsA protein in a concentration-dependent manner in the presence of excess (10-fold) poly(dI:dC) competitor and could be titrated out by a higher concentration of unlabeled PaphA probe (Fig. 3C). The Kd value for the binding of VqsA to aphA was calculated to be 0.35 μM (Fig. 3D). In addition, the DNase I footprinting assay with both strands of the PaphA probe uncovered a specific VqsA-protected region, 5′-CACAAGTTAACACTATTGATTATTATGTTAGTTTT-3′ (223 bp upstream of ATG), in the aphA promoter region (Fig. 3E). The absence of the specific binding site abolished the capacity of VqsA to bind to the aphA promoter (Fig. 3F). Taken together, these data along with the results of B1H assays (Fig. 1B) demonstrated that VqsA binds to the aphA promoter, negatively regulating the transcription of the aphA gene.
LuxR, AphA, and VqsA bind to the promoter region of vqsA in vitro.
EMSAs and DNase I footprinting assays were carried out to test the binding of LuxR, AphA, and VqsA to the vqsA promoter region in vitro (Fig. 4). Both LuxR and AphA bound efficiently and specifically to a probe encompassing the entire PvqsA in a concentration-dependent manner in the presence of high concentrations (10-fold excess of the probe DNA) of a nonspecific poly(dI:dC) competitor and could be titrated out by the unlabeled DNA probe (Fig. 4A and C). The Kd values for the binding of LuxR and AphA to the vqsA promoter were calculated to be 1.5 μM and 1.74 μM, respectively. The specific binding sites of LuxR (RBS) and AphA (ABS) on PvqsA were further mapped with DNase I footprinting assays (Fig. 4B and D). These data suggested that vqsA could also be regulated via feedback by QS through the combined activities of LuxR and AphA.
FIG 4.

AphA, LuxR, and VqsA bind to PvqsA. (A, C, and E) EMSAs were performed with purified LuxR (A), AphA (C), and VqsA (E). The amounts of the proteins used are indicated, and 20 ng of each Cy5-labeled PvqsA probe was added to the EMSA reactions. The shifts were verified to be specific in experiments in which 25- to 50-fold excess of unlabeled specific DNA and nonspecific competitor DNA [poly(dI:dC)] were added. (B, D, and F) DNase I footprinting analysis of LuxR (B), AphA (D), or VqsA (F) binding to their distinct binding sites in PvqsA. Electropherograms of a DNase I digestion of 400 ng of the PvqsA promoter probe after incubation with 0 or 3.0 μM LuxR, 0 or 4.0 μM AphA, and 0 or 2.4 μM VqsA are shown. The nucleotide sequences that were protected by LuxR, AphA, or VqsA are as indicated below. The specific palindromic sequences are underlined. Dashed boxes indicate the binding site revealed by DNase I footprinting assays. (G) EMSA investigation of the binding of LuxR, AphA, or VqsA to their specific binding sites, RBS, ABS, and VBS, in the luxR promoter region. The WT and the variant PvqsA lacking the related binding sites were also investigated.
LTTRs usually exhibit autorepression (27). To investigate the autorepression of VqsA, an EMSA was first performed, and the results indicated that VqsA bound directly to the vqsA promoter at two sites (Fig. 4E) with a calculated Kd value of 0.73 μM. These results indicated that VqsA has a much higher binding affinity toward the vqsA promoter than LuxR and AphA and thus might exhibit strong autorepression. A DNase I footprinting assay on both strands of PvqsA confirmed that VqsA bound to the promoter at the 5′-TAACCTCATTTGATTGATAAACTCTGC-3′ (VBS1) site, −30 bp relative to the start codon (ATG), and the 5′-TTTATCAACTCTGGGTGTGCA-3′ (VBSII) site, immediately upstream of the start codon (Fig. 4F). Moreover, the perfect palindromic sequence GATAAA was identified in these two binding sites (Fig. 4F). When the respective LuxR, AphA, and VqsA-binding sites were deleted on PvqsA, the ability of these regulators to bind to the promoter was abolished (Fig. 4G, lanes 3, 7, and 11). A VqsA-specific-binding motif (TAAACTC) was generated with the MEME suite algorithms (http://meme-suite.org) (P < 10−12) by providing the sequences of the four binding sites in the promoter regions of luxR, aphA, and vqsA as input (Fig. 2E, 3E, and 4F). Taken together, these results showed that the vqsA promoter was bound to and could be regulated by LuxR, AphA, and VqsA.
Regulation of the activity of the vqsA promoter by LuxR, AphA, and VqsA.
We then tested the roles of VqsA, LuxR, and AphA in the expression of vqsA. As an investigation of the overall effect of these proteins on vqsA expression, cells cultured for 9 h and approaching the stationary-growth phase (24) were used for PvqsA-luxAB or qRT-PCR transcriptional assays. PvqsA activity was significantly enhanced in the cells with the deletion of the vqsA gene and was restored to the wild-type (WT) levels in the vqsA+ complement strain after 9 h of incubation (Fig. 5A), indicating negative regulation of vqsA expression by VqsA, a typical autoregulatory feature of LTTRs (27). In qRT-PCR assays, upon measuring the transcript levels of vqsA in the WT, ΔluxR mutant, and ΔaphA mutant strains and in the complemented luxR+ and aphA+ strains, the transcript level of vqsA was significantly higher, by 5-fold, in the ΔluxR mutant than that in the WT, but lower by ∼2-fold in the ΔaphA mutant; the levels of the vqsA transcripts in the luxR+ and aphA+ complemented strains were restored to WT levels (Fig. 5B). We also performed an epistatic test of the functions of LuxR and AphA in the transcriptional control of vqsA. The ΔluxR ΔaphA double mutant and the ΔluxR mutant strain overexpressing aphA showed levels of vqsA transcripts similar to those of the ΔluxR mutant strain, while the ΔaphA mutant strain overexpressing luxR exhibited a vqsA transcript level comparable to that of the luxR+ mutant strain in 9-h cultures (Fig. 5B), demonstrating that LuxR exerts dominant control over vqsA expression at the HCD stage. In addition, a 5′-RACE experiment was performed as previously described (28) using RNA extracted from the WT, ΔvqsA mutant, and ΔluxR mutant strains grown for 9 h in LBS. The deletion of vqsA or luxR resulted in a stronger sequencing signal than that observed for the WT after reverse transcriptional analysis (Fig. S3), which facilitated the identification of the transcriptional start site (TSS), a “G” at position −516 relative to the start codon ATG (Fig. 5C).
FIG 5.

The expression of vqsA is regulated by AphA, LuxR, and VqsA at the transcriptional level. (A) Transcriptional analysis of PvqsA-luxAB in the WT and ΔvqsA mutant strains cultured in LBS for 9 h. The results are shown as the mean ± SD (n = 3). ***, P < 0.001 based on Student's t test. (B) qRT-PCR analysis of the vqsA transcripts in ΔluxR, ΔaphA, luxR+, and aphA+ mutants and in the ΔluxR ΔaphA double mutant, as well as the related strains exhibiting epistatic expression (ΔluxR aphA mutant and ΔaphA luxR mutant) cultured in LBS for 9 h, and mRNA transcripts were detected by qRT-PCR; 16S rRNA was selected as a control. The results are displayed as the mean ± SD (n = 3); **, P < 0.01; ***, P < 0.001, based on Student's t test relative to the WT results. (C) The nucleotide sequence of PvqsA. The LuxR-binding site (RBS), AphA-binding site (ABS), VqsA-binding site I (VBSI), and VqsA-binding site II (VBSII) and the −10 box and −35 box are underlined. The PvqsA transcription start site as determined by 5′-RACE is labeled with an arrow, and the translational start codon (ATG) is in red. (D) The promoter activities of PvqsA and its variants fused to luxAB. Fluorescence levels were assayed for each of the strains. The data are shown as the mean ± standard error of the mean (SEM) (n = 3). **, P < 0.01; ***, P < 0.001, based on Student's t test relative to the WT PvqsA promoter.
We further characterized the roles of the binding sites of AphA (ABS), LuxR (RBS), and VqsA (VBSI and VBSII) in the PvqsA region on the expression of vqsA (Fig. 5C and D). As expected, PvqsA activity was significantly attenuated in the mutant of ABS, PvqsAMABS, indicating that this cis-acting element and AphA were essential for activation of the vqsA promoter. Moreover, the single nucleotide substitutions in the RBS PvqsAMRBS resulted in increasing the activity of the vqsA promoter to a level higher than that observed for WT PvqsA. Furthermore, the activity of the vqsA promoter with a mutated VBSI, PvqsA MVBSI, and/or VBSII (PvqsA MVBSII or PvqsA MVBSI/II) was higher than that of the WT promoter. Collectively, these data confirmed that vqsA expression was repressed by LuxR and VqsA but activated by AphA, supporting the idea that vqsA expression is subject to a sophisticated regulation network in which AphA, LuxR, and VqsA are participants.
Essential roles of VqsA in bacterial killing capacity, biofilm formation, exotoxin production, and in vivo virulence toward fish.
Previous investigations had demonstrated that QS regulates the bacterial killing via type VI secretion system 2 (T6SS2) in V. alginolyticus (29, 30). We tested whether VqsA is involved in bacterial killing capacity, a QS-controlled phenotype. We found that the WT strain killed Escherichia coli MC4100 in coculture and reduced the starting CFU of E. coli ∼100× when the strains were cocultured at 30°C for 4 h on LBS agar plates. Hcp2 is a structural component of T6SS2 that is essential for bacterial killing activity, and the ability to kill bacteria was abolished in the Δhcp2 mutant strain (30). Compared to the ΔluxR and Δhcp2 mutant strains, which are deficient in interbacterial competition activity (30), the ΔvqsA mutant also exhibited markedly decreased killing of E. coli, and the complement vqsA+ strain restored the bacterial killing capacities (Fig. 6A). Furthermore, biofilm formation was significantly decreased in the ΔvqsA mutant strain in comparison to the WT strain and the complement strain (Fig. 6B). Western blotting confirmed the essential role of vqsA in the positive regulation of exotoxin Asp production in V. alginolyticus (Fig. 1D, 1E, and 6C). Similarly, the transcription of MviN and Pep, two additional exotoxins that are essential for pathogenesis in V. alginolyticus (17, 18), was also observed to be positively regulated by VqsA (Fig. 6D). These data demonstrate that VqsA is involved in QS pathways to regulate a plethora of group behaviors.
FIG 6.

Essential roles of VqsA in interbacterial competition activity, biofilm formation, exotoxin production, and virulence. (A) CFU of E. coli before (t = 0) and after (t = 4) a 4-h coculture with the indicated V. alginolyticus predator strains or E. coli alone. The Δhcp2 mutant strain deficient in interbacterial competition capacity was used as a control. The data represent the results from three independent experiments. ***, P < 0.001 by an unpaired two-tailed t test. (B) Biofilm formation in glass tubes after 48 h was quantitatively assayed with crystal violet staining of the biofilm cells. For the colorimetric assay of the released crystal violet from the biofilm cells, the OD570 was measured and normalized with the OD600 of the cultures. **, P < 0.01 based on Student's t test relative to the WT strain. (C) Western blot analysis was performed to assay Asp yields in the ΔvqsA and vqsA+ mutant strains. RNAP was used as the loading control. (D) qRT-PCR analysis of the transcriptional levels of the pep and mviN genes. The WT, ΔvqsA mutant, and vqsA+ mutant strains were cultured in LBS for 9 h, and the mRNA transcripts were detected by qRT-PCR; 16S rRNA was selected as a control. ***, P < 0.001, Student's t test relative to the vqsA+ mutant strain. (E) Groups of 30 fish were infected with the indicated strains. The number of fish that survived was recorded for 4 days. The P values were calculated using a Kaplan-Meier survival analysis with a log rank test with Prism software (version 6.01).
To further evaluate the influence of VqsA on the virulence of V. alginolyticus, adult zebrafish (n = 30) were challenged via intramuscular (i.m.) injection with 5 × 106 CFU/fish of the WT, ΔvqsA mutant, or vqsA+ mutant strain, as previously described (23). The fish mortalities caused by the ΔvqsA mutant challenge were much lower than those observed with the WT and complementation strains. The survival rates of the fish challenged with the ΔvqsA mutant (∼45%) were significantly higher than those of fish challenged with the WT (∼17%) or vqsA+ mutant (∼16%) strains (Fig. 6E). The significant virulence attenuation resulting from vqsA abrogation suggested that VqsA is involved in the in vivo virulence of V. alginolyticus. Taken together, these data indicate that VqsA plays essential roles in coordinating QS and virulence expression in V. alginolyticus.
DISCUSSION
In vibrios, QS is essential for the regulation of virulence gene expression, and the identification of QS regulators will facilitate the development of QSI strategies. With this goal in mind, here, we performed a comprehensive screen of QS regulators and identified multiple novel QS regulators involved in the regulation of the MQSRs LuxR and/or AphA in the marine pathogen V. alginolyticus, uncovering the complicated regulation network of QS and pathogenesis in the bacterium. In addition, a large portion of the QS regulators, including the LTTR family proteins (Tables 1 and S1), contain ligand-binding domains, which suggest their roles in the sensing of various physiological or biochemical signals to modulate QS output. The LTTR protein MetR has been shown to be involved in QS regulation in response to methionine metabolism in V. harveyi (31). AphB, a low-pH- and oxygen-responsive LTTR protein (32, 33), regulates QS by directly binding to the luxR promoter and controlling LuxR expression at the transcriptional level in V. alginolyticus (25). Moreover, identification of the oxidative-stress-responsive SoxR (34) and the feast/famine regulatory protein AsnC (35) as being involved in QS regulation also suggested that various physiological signals might affect QS regulation.
As QS directly or indirectly regulates ∼600 (approximately 15%) genes in vibrios (4, 36, 37), it is reasonable to suggest that other unrelated regulatory pathways could be integrated into the preexisting QS regulatory circuits to modify gene expression in response to environmental cues. Our EMSA, DNase I footprinting assay, and transcriptional assay indicated that the LTTR family protein VqsA is involved in QS control (Fig. 1 and 6) via regulation of the expression of the MQSRs AphA and LuxR. We are greatly interested in the signals to which VqsA might respond to regulate QS. We modeled the structure of VqsA and found that the ligand-binding domain of VqsA is structurally related to AphB and CrgA. AphB has been reported to form a tetramer (38), while CrgA family proteins form octameric rings for DNA binding in response to specific stimuli to regulate gene expression (39). The molecular model indicated that the ligand-binding domain of VqsA also contains Cys residues Cys189 and Cys166 (Fig. S4), which might mediate intra-/intermolecular interactions in a manner similar to the thiol-switch Cys235 in AphB to sense oxidative stresses and ultimately regulate QS gene expression (38, 40).
Our transcriptional assays indicated that VqsA, similar to other LTTRs, acts as both an autorepressor and activator/repressor of target promoters (Fig. 2, 3, and 5) (27). Our analysis uncovered two specific binding sites (VBS1 and VBSII) upstream of the translational start site of PvqsA. In addition, the palindromic repeat GATAAA identified in these two flanking VqsA-binding sites of PvqsA might facilitate the formation of a hairpin structure to repress autotranscription (Fig. 4F). Although the expression of AphA was repressed by VqsA (Fig. 3A and B), our data did not disclose an additional VqsA-binding site in proximity to the binding site identified in the promoter region of aphA (Fig. 3E). We speculate that VqsA might interact with other proteins to collectively repress the expression of aphA. Unlike CrgA or other LTTRs that bind to a 50- to 60-bp stretch of DNA (27), our DNase I footprinting assays revealed a region of 21 to 34 bp protected by VqsA, including a previously unreported conserved 7-bp consensus sequence, TAAACTC, in both the activated and repressed promoter regions (Fig. 2E, 3E, and 4F). Moreover, VqsA appears to facilitate luxR transcription by binding to the template strand of the luxR promoter at nucleotides (nt) −40 to −9 relative to the transcription start site (Fig. 2) (6), a region usually essential for RNA polymerase binding and for the formation of an open promoter complex to initiate transcription. It is unusual to observe a factor binding to the −10 element to facilitate gene transcription in Gram-negative bacteria, although in mycobacteria, the transcription factors CarD and RbpA are essential for formation of the transcriptionally competent open complex by directly interacting with RNAP and the −10 region to activate transcription (41). Taking these analyses together, our data indicate that VqsA might adopt special DNA-binding mechanisms to regulate gene expression. It is important to note that all these binding analyses were performed in vitro. Chromatin immunoprecipitation sequencing (ChIP-seq) and structural analysis could further dissect the mechanisms via which VqsA regulates QS.
We were intrigued by the role of VqsA in the regulation of QS output. Our data indicated that similar to the MQSRs LuxR and AphA (6, 24), VqsA regulates the expression of LuxR and AphA (Fig. 2 and 3), and vqsA expression is also subject to tight regulation by LuxR, AphA, and VqsA (Fig. 4 and 5). Specifically, LuxR and VqsA repress vqsA expression, while AphA promotes the transcription of vqsA. Epistasis analysis of LuxR and AphA further revealed a dominant role for LuxR in the regulation of vqsA at HCD stages (Fig. 5B). All these analyses support a model in which VqsA is the third MQSR in V. alginolyticus (Fig. 7). As V. harveyi AphA and LuxR regulate the expression of a large number of genes (37), VqsA might also regulate the expression of various genes directly or indirectly via AphA or LuxR and may be associated with the QS regulatory network to control various virulence-associated phenotypes (Fig. 6 and 7). Future work should focus on (i) the individual and combined roles of these three MQSRs in controlling QS regulons in different vibrios, and (ii) the mutual and reciprocal control of the expression of these MQSRs to control QS in growing cells and in different growth stages, as well as under various environmental conditions. These studies shall enrich our knowledge of the architecture of QS systems and their regulation.
FIG 7.
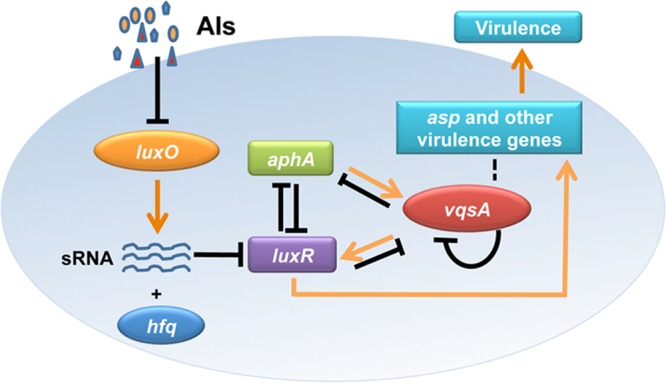
Schematic summarization of the regulatory roles of VqsA in QS circuits and virulence gene expression in V. alginolyticus. The pathways are illustrated with arrows (activation) or with bar-ended lines (repression) and are discussed in the text. The putative pathway indicating that VqsA might directly regulate the expression of some virulence-associated genes is depicted as dashed lines. sRNA, small RNA.
In summary, here, we identified that the LysR family regulator VqsA regulates QS by binding to the promoters of aphA and luxR to negatively and positively regulate the expression of these genes, respectively. The expression of vqsA is autorepressed and feedback controlled by AphA and LuxR. VqsA also regulates the expression of exotoxins and other QS-controlled virulence factors and plays essential roles in survival in zebrafish. The modified B1H system proved to be a powerful tool to uncover new regulators in QS signaling cascades and led to the discovery of VqsA as an important player in QS regulation and pathogenesis. VqsA may be the third master QS regulator (MQSR) in vibrios to coordinate QS-mediated responses (Fig. 7). The discovery of VqsA along with various as-yet-uncharacterized regulators involved in QS circuits provided a genetic framework to dissect the complex regulatory network of QS and thereby facilitate the development of QSI strategies to effectively control pathogens that plague the aquaculture industry.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids that were used in this study are listed in Table 2. The V. alginolyticus wild-type (WT) and derivative strains were grown at 30°C in Luria-Bertani (LB) medium supplemented with 3% NaCl (LBS). Escherichia coli strains were grown at 37°C in LB medium. When appropriate, the medium was supplemented with carbenicillin (Carb; 100 μg/ml), chloramphenicol (Cm; 25 μg/ml), kanamycin (Km; 100 μg/ml), or l-arabinose (0.04% [wt/vol]).
TABLE 2.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α λpir | Host for π-requiring plasmids | Lab collection |
| SM10 λpir | Host for π-requiring plasmids, conjugal donor | Lab collection |
| BL21(DE3) | Host strain for protein expression | Novagen |
| XL1-Blue MRF9 | B1H reporter; host strain for propagating pBXcmT and pTRG recombinants, Kmr | Agilent |
| MC4100 | Prey bacterium used for interbacterial competition assay | Lab collection |
| V. alginolyticus | ||
| EPGS | Wild type, isolated from aquiculture farm of the South China Sea (CCTCC AB 209306), Ampr | Lab collection |
| asp mutant | EPGS Δasp, Cmr | 16 |
| ΔluxR mutant | EPGS ΔluxR | 16 |
| ΔaphA mutant | EPGS ΔaphA | 24 |
| ΔluxO mutant | EPGS ΔluxO | 20 |
| luxR+ strain | ΔluxR mutant/pMMB-luxR, Cmr | 18 |
| aphA+ strain | ΔaphA mutant/pBAD33-aphA-flag-1, Cmr | 24 |
| ΔasnC mutant | EPGS ΔasnC | This study |
| Δ02325 mutant | EPGS Δ02325 | This study |
| ΔsoxR mutant | EPGS ΔsoxR | This study |
| ΔmetR mutant | EPGS ΔmetR | This study |
| Δ01665 mutant | EPGS Δ01665 | This study |
| Δ06711 mutant | EPGS Δ06711 | This study |
| ΔvqsA mutant | EPGS Δ18672 (vqsA) | This study |
| ΔvqsA ΔluxO mutant | EPGS ΔluxO Δ18672 | This study |
| ΔluxR ΔaphA mutant | EPGS ΔluxR ΔaphA | This study |
| BL21/pET28a-aphA | BL21/pET28a-aphA, Kmr | 6 |
| BL21/pET28a-luxR | BL21/pET28a-luxR, Kmr | 6 |
| XL1-Blue MRF9/pTRG::LuxR+pBXcmT::PluxR | XL1-Blue MRF9 carrying pTRG-LuxR+pBXcmT-PluxR | This study |
| XL1-Blue MRF9/pTRG::LuxR+pBXcmT::PluxR ΔRBSI/II | XL1-Blue MRF9 carrying pTRG-LuxR+pBXcmT-PluxR ΔRBSI/II | This study |
| XL1-Blue MRF9/pTRG::AsnC+pBXcmT::PluxR | XL1-Blue MRF9 carrying pTRG-AsnC+pBXcmT-PluxR | This study |
| XL1-Blue MRF9/pTRG::02325+pBXcmT::PluxR | XL1-Blue MRF9 carrying pTRG-02325+pBXcmT-PluxR | This study |
| XL1-Blue MRF9/pTRG::SoxR+pBXcmT::PluxR | XL1-Blue MRF9 carrying pTRG-SoxR+pBXcmT-PluxR | This study |
| XL1-Blue MRF9/pTRG::MetR+pBXcmT::PluxR | XL1-Blue MRF9 carrying pTRG-MetR+pBXcmT-PluxR | This study |
| XL1-Blue MRF9/pTRG::01665+pBXcmT::PluxR | XL1-Blue MRF9 carrying pTRG-01665+pBXcmT-PluxR | This study |
| XL1-Blue MRF9/pTRG::06711+pBXcmT::PluxR | XL1-Blue MRF9 carrying pTRG-06711+pBXcmT-PluxR | This study |
| XL1-Blue MRF9/pTRG::18672+pBXcmT::PluxR | XL1-Blue MRF9 carrying pTRG-18672+pBXcmT-PluxR | This study |
| XL1-Blue MRF9/pTRG::AsnC+pBXcmT::PaphA | XL1-Blue MRF9 carrying pTRG-AsnC+pBXcmT-PaphA | This study |
| XL1-Blue MRF9/pTRG::02325+pBXcmT::PaphA | XL1-Blue MRF9 carrying pTRG-02325+pBXcmT-PaphA | This study |
| XL1-Blue MRF9/pTRG::SoxR+pBXcmT::PaphA | XL1-Blue MRF9 carrying pTRG-SoxR+pBXcmT-PaphA | This study |
| XL1-Blue MRF9/pTRG::MetR+pBXcmT::PaphA | XL1-Blue MRF9 carrying pTRG-MetR+pBXcmT-PaphA | This study |
| XL1-Blue MRF9/pTRG::01665+pBXcmT::PaphA | XL1-Blue MRF9 carrying pTRG-01665+pBXcmT-PaphA | This study |
| XL1-Blue MRF9/pTRG::18672+pBXcmT::PaphA | XL1-Blue MRF9 carrying pTRG-18672+pBXcmT-PaphA | This study |
| vqsA+ strain | Δ18672 mutant carrying pBAD33-18672, Cmr | This study |
| ΔaphA luxR mutant | ΔaphA mutant carrying pMMB-luxR | This study |
| ΔluxR aphA mutant | ΔluxR mutant carrying pBAD33::aphA-flag-1 | This study |
| EPGS/pDM8-PluxR-luxAB | EPGS/pDM8-PluxR-luxAB, Cmr | This study |
| EPGS Δ18672/pDM8-PluxR-luxAB | Δ18672 mutant/pDM8-PluxR-luxAB, Cmr | This study |
| EPGS/pDM8-PaphA-luxAB | EPGS/pDM8-PaphA-luxAB, Cmr | This study |
| EPGS Δ18672/pDM8-PaphA-luxAB | Δ18672 mutant/pDM8-PaphA-luxAB, Cmr | This study |
| EPGS/pDM8-Pasp-luxAB | EPGS/pDM8-Pasp-luxAB, Cmr | This study |
| EPGS Δ18672/pDM8-Pasp-luxAB | Δ18672 mutant/pDM8-Pasp-luxAB, Cmr | This study |
| EPGS/pDM8-PvqsA-luxAB | EPGS/pDM8-PvqsA-luxAB, Cmr | This study |
| EPGS Δ18672/pDM8-PvqsA-luxAB | Δ18672 mutant/pDM8-PvqsA-luxAB, Cmr | This study |
| EPGS ΔluxR/pDM8-PvqsA-luxAB | ΔluxR mutant/pDM8-PvqsA-luxAB, Cmr | This study |
| EPGS ΔaphA/pDM8-PvqsA-luxAB | ΔaphA mutant/pDM8-PvqsA-luxAB, Cmr | This study |
| EPGS/pDM8-PvqsAMRBS-luxAB | EPGS/pDM8-PvqsAMRBS-luxAB, Cmr | This study |
| EPGS/pDM8-PvqsAMABS-luxAB | EPGS/pDM8- vqsAMABS-luxAB, Cmr | This study |
| EPGS/pDM8-PvqsAMVBSI-luxAB | EPGS/pDM8-PvqsAMVBSI-luxAB, Cmr | This study |
| EPGS/pDM8-PvqsAMVBSII-luxAB | EPGS/pDM8-PvqsAMVBSII-luxAB, Cmr | This study |
| EPGS/pDM8-PvqsAMVBSI/II-luxAB | EPGS/pDM8-PvqsAMVBSI/II-luxAB, Cmr | This study |
| BL21/pET28a-asnC | BL21/pET28a-asnC, Kmr | This study |
| BL21/pET28a-metR | BL21/pET28a-metR, Kmr | This study |
| BL21/pET28a-soxR | BL21/pET28a-soxR, Kmr | This study |
| BL21/pET28a-vqsA | BL21/pET28a-vqsA, Kmr | This study |
| Plasmids | ||
| pDM4 | Suicide vector, pir dependent, R6K, SacBR, Cmr | 47 |
| pBAD33-mob | pBAD33, mob, Cmr | 24 |
| pDM8 | pSup202 derivate containing promoterless lacZ, Cmr | 47 |
| pET28a-aphA | pET28a carrying aphA ORF, Kmr | 6 |
| pET28a-luxR | pET28a carrying luxR ORF, Kmr | 6 |
| pDM8-PluxR-luxAB | pDM8 with promoter region of luxR and luxAB, Cmr | 6 |
| pDM8-PaphA-luxAB | pDM8 with promoter region of aphA and luxAB, Cmr | 24 |
| pMMB-luxR | pMMB206 derivative containing luxRval from bp −782 to 1408, Cmr | 18 |
| pBAD33::aphA-flag-1 | pBAD33 derivative with PaphA-driven expression of AphA-Flag fusion protein, Cmr | 24 |
| pBXcmT-PaphA | pBXcmT with promoter region of aphA, Cmr | This study |
| pBXcmT-PluxR | pBXcmT with promoter region of luxR, Cmr | This study |
| pBXcmT-PluxR ΔRBSI/II | pBXcmT with luxR promoter deleted LuxR binding box I/II, Cmr | This study |
| pTRG-LuxR | pTRG with luxR ORF, Tetr | This study |
| pTRG-AsnC | pTRG with asnC ORF, Tetr | This study |
| pTRG-MetR | pTRG with metR ORF, Tetr | This study |
| pTRG-SoxR | pTRG with soxR ORF, Tetr | This study |
| pTRG-02325 | pTRG with 02325 ORF, Tetr | This study |
| pTRG-01665 | pTRG with 01665 ORF, Tetr | This study |
| pTRG-06711 | pTRG with 06711 ORF, Tetr | This study |
| pTRG-18672 | pTRG with 18672 ORF, Tetr | This study |
| pDM4-18672 | pDM4 with 18672 fragment deleted from nt 4 to 876, Cmr | This study |
| pDM4-02325 | pDM4 with 02325 fragment deleted from nt 4 to 1146, Cmr | This study |
| pDM4-asnC | pDM4 with asnC fragment deleted from nt 4 to 462, Cmr | This study |
| pDM4-metR | pDM4 with metR fragment deleted from nt 4 to 876, Cmr | This study |
| pDM4-soxR | pDM4 with soxR fragment deleted from nt 4 to 909, Cmr | This study |
| pDM4-06711 | pDM4 with 06711 fragment deleted from nt 4 to 876, Cmr | This study |
| pDM4-01665 | pDM4 with 18672 fragment deleted from nt 4 to 411, Cmr | This study |
| pBAD33-18672 | 18672-expressing plasmid driven by native promoter | This study |
| pET28a-18672 | pET28a carrying 18672 ORF, Kmr | This study |
| pET28a-asnC | pET28a carrying asnC ORF, Kmr | This study |
| pET28a-metR | pET28a carrying metR ORF, Kmr | This study |
| pET28a-soxR | pET28a carrying soxR ORF, Kmr | This study |
| pET28a-02325 | pET28a carrying 02325 ORF, Kmr | This study |
| pET28a-01665 | pET28a carrying 01665 ORF, Kmr | This study |
| pET28a-06711 | pET28a carrying 06711 ORF, Kmr | This study |
| pDM8-Pasp-luxAB | pDM8 with promoter region of asp and luxAB, Cmr | This study |
| pDM8-PvqsA-luxAB | pDM8 with promoter region of vqsA and luxAB, Cmr | This study |
| pDM8-PvqsAMRBS-luxAB | pDM8 with mutated LuxR box of vqsA promoter and luxAB, Cmr | This study |
| pDM8-PvqsAMABS-luxAB | pDM8 with mutated AphA box of vqsA promoter and luxAB, Cmr | This study |
| pDM8-PvqsAMVBSI-luxAB | pDM8 with mutated VqsA box I of vqsA promoter and luxAB, Cmr | This study |
| pDM8-PvqsAMVBSII-luxAB | pDM8 with mutated VqsA box II of vqsA promoter and luxAB, Cmr | This study |
| pDM8-PvqsAMVBSI/II-luxAB | pDM8 with mutated VqsA box I/II of vqsA promoter and luxAB, Cmr | This study |
Kmr, kanamycin resistance; Ampr, ampicillin resistance; Cmr, chloramphenicol resistance.
Construction of the deletion mutants, complement strains, and the protein expression strains.
In-frame deletion mutant strains and complement strains were constructed as previously described using specific primers (Table 3) and isothermal assembly (6, 42). Briefly, overlap PCR was used to generate DNA fragments for the in-frame deletion. These fragments were then cloned into the XbaI-digested R6K-based suicide vector pDM4 by isothermal assembly and transformed into E. coli DH5α λpir cells, as previously described (6). After being sequenced, the resulting plasmids were transformed into E. coli SM10 λpir and then mated into EPGS by conjugation. Double-crossover transformants were selected sequentially on LBS medium containing Carb and Cm and then on LBS with 12% (wt/vol) sucrose. Generation of the targeted mutants was confirmed by PCR using a primer pair targeting the upstream and downstream regions of the designed deletion region (Table 3) to sequence the region. To construct the complement strains, the genes spanning the predicted promoter region and open reading frame (ORF) were cloned into the modified pBAD33 plasmid harboring the mob gene (pBAD33-mob) and a FLAG tag downstream of the cloned gene (24). The derivative plasmid was conjugated into the related mutant strains.
TABLE 3.
Primers used in this study
| Primer name | Primer sequence (5′ to 3′) | Target |
|---|---|---|
| 18672-P1 | AGTACGCGTCACTAGTGGGGCCCTTCTAGAGAAGGTGATTTGGCAGTGCGATGTA | 18672 deletion mutant |
| 18672-P2 | CTTTATGGGGTGACGTACACACCCAGAGTTGATAAAG | |
| 18672-P3 | AACTCTGGGTGTGTACGTCACCCCATAAAGCGGCGACAATT | |
| 18672-P4 | TAACAATTTGTGGAATTCCCGGGAGAGCTTAAATGCAGTCAAGGGGTAGCGTCC | |
| 18672-OUT-F | AGGATCGCTTTCCCCTGCTCTGCCG | |
| 18672-OUT-R | AGAACCGCCGATGGAATGT | |
| 18672-IN-F | ATGGATAAATTAACAAGCATGAAGG | |
| 18672-IN-R | CGACCAAGGTACGGTTTTTAAGGGA | |
| 02325-P1 | AGTACGCGTCACTAGTGGGGCCCTTCTAGATCAGCCACGCATTGCGAATAGAACG | 02325 deletion mutant |
| 02325-P2 | TACACTCGCGTTCAAGTGCTGTGCTTCTCGTAATTGTCTT | |
| 02325-P3 | CGAGAAGCACAGCACTTGAACGCGAGTGTAAGAACCTGCCT | |
| 02325-P4 | TAACAATTTGTGGAATTCCCGGGAGAGCTGTATTCATCCTGATAACCCCTCTGT | |
| 02325-OUT-F | ATGCAAACCGTATTGAACCGAAAAG | |
| 02325-OUT-R | CGGCGAATGCGGTAAAAAGTGAAAG | |
| 02325-IN-F | ATGGAAATCGCACAGTCATTACAAC | |
| 02325-IN-R | TTACAACGTGTTGAGGACATCCGCG | |
| soxR-P1 | AGTACGCGTCACTAGTGGGGCCCTTCTAGATTCTTGGGTGAAGGCATTGTTGGTC | soxR deletion mutant |
| soxR-P2 | GAATAGAGATAATCGCCTTGACTCCCTGTGTATCAA | |
| soxR-P3 | CACAGGGAGTCAAGGCGATTATCTCTATTCTTTGTCGAA | |
| soxR-P4 | TAACAATTTGTGGAATTCCCGGGAGAGCTTAGCAAGCGAATAATGGAATAAACA | |
| soxR-OUT-F | AAGCAAACTCTTTCATACCCCACAT | |
| soxR-OUT-R | GATTTACTTGCCGAAGAACGAGACC | |
| soxR-IN-F | TTCTGGAGTGAAAGTATCTGCCCTA | |
| soxR-IN-R | CCGAGTGGTCTTTCCCTAATACATC | |
| asnC-P1 | AGTACGCGTCACTAGTGGGGCCCTTCTAGAGAGTGGTCACTGACGATGAGAACTT | asnC deletion mutant |
| asnC-P2 | ATTTACGTTTCGGTTACTATGATTTCCGTTAGGGATAGAG | |
| asnC-P3 | AACGGAAATCATAGTAACCGAAACGTAAATCCGT | |
| asnC-P4 | TAACAATTTGTGGAATTCCCGGGAGAGCTAAGAGAAAGTCGCGGTAGTT | |
| asnC-OUT-F | GTTGTTTTAGATCGATGATGCTGTC | |
| asnC-OUT-R | TATTTTGCGTCAGCTAGAATTCGGC | |
| asnC-IN-F | GCGCAATATTAAAAACACTGATGGC | |
| asnC-IN-R | GTTCTGCAACGATATCAATGTCTCC | |
| 06711-P1 | AGTACGCGTCACTAGTGGGGCCCTTCTAGATCTGCAATCCAATCAAAATGAACAA | 06711 deletion mutant |
| 06711-P2 | TCACTGATAAAATGTGATTTGACACAACCAATAAATGAATTC | |
| 06711-P3 | TGGTTGTGTCAAATCACATTTTATCAGTGAGCGTC | |
| 06711-P4 | TAACAATTTGTGGAATTCCCGGGAGAGCTTTCTAAGACCGCTTTTCCAAATGGG | |
| 06711-OUT-F | ACGCTCAGACTTCGCAAGGGTAA | |
| 06711-OUT-R | ACAACCGATAACATTCACGAAGCCG | |
| 06711-IN-F | GTATGTCGATTAAGGCATTAGAGAA | |
| 06711-IN-R | GCTCAATTTATCTGCTAACCACGTC | |
| 01665-P1 | AGTACGCGTCACTAGTGGGGCCCTTCTAGAACCTAAGCGAGATTGCAGCAGTTCA | 06115 deletion mutant |
| 01665-P2 | GCGAAATTATCGAGGATTTCGTACTCCAGTTCGAATTGCG | |
| 01665-P3 | ACTGGAGTACGAAATCCTCGATAATTTCGCCATCACTGCG | |
| 01665-P4 | TAACAATTTGTGGAATTCCCGGGAGAGCTAAGTGGAAGCTGGTATGGTTGCGAAG | |
| 01665-OUT-F | ATTTGTTCAAGCGTTCCATCGTGAG | |
| 01665-OUT-R | TACTTTCTTTATGTTGTTGTTCGGT | |
| 01665-IN-F | TGTCTTTTGCCCAACGCCTA | |
| 01665-IN-R | TCTTTATGGAGAGATAGGAAGGTCG | |
| metR-P1 | AGTACGCGTCACTAGTGGGGCCCTTCTAGAACCATTTGGTCATATCCGATGC | metR deletion mutant |
| metR-P2 | CATAACCCAAATCTTGAATTCCTTTCATCCTTTGTTGCTC | |
| metR-P3 | GGATGAAAGGAATTCAAGATTTGGGTTATGAAAGGCTTAGCG | |
| metR-P4 | TAACAATTTGTGGAATTCCCGGGAGAGCTGTCGATGACGAGCCTGCAAGGTTTA | |
| metR-OUT-F | GCGGCCAGCTCACTTCAAACGTATC | |
| metR-OUT-R | GCCGATCTTGCTGTCTTTAGCTATG | |
| metR-IN-F | AGCTAAAACATTTGCGGACACTCAC | |
| metR-IN-R | CGCGGTTGCGAAAAAAGCTTGTAAG | |
| 18672-Flag-F | CCATACCCGTTTTTTTGGGCTAGCGAATTCACAGGAGGAATTAACCATGAAATTAGATGACTTAAACCTG | 18672 expressed in pBAD33-mob |
| 18672-Flag-R1 | TCGTCGTCCTTGTAGTCTGAGCCACCGCCACCGTTGTTCACTTTTAGCTTTTGGAAG | |
| 18672-Flag-R2 | GGTCAGCATGGGTACCTTTCTCCTCTTTAATTACTTGTCGTCGTCGTCCTTGTAGTCTGA | |
| 18672-Flag2-F | AGAGGACACAATGGATAAATTAACAAGCATGAAGG | |
| pBAD33mob-C'Flag-R | CTTGCATGCCTGCAGGTCGATTACTTGTCGTCGTCGTCCTTGTAGTCTGA | |
| P18672-luxAB-F | ATTGCTGCAGGTCGACGGATCCGGGGAATTCAGCAACCGCGTACAGTTCACTCTT | Fluorescence assay |
| P18672-luxAB-R | GTCGACCTGCAGCCCAAGCTTATCGATTCGTACACACCCAGAGTTGATAAAG | |
| PaphA-luxAB-F | ATTGCTGCAGGTCGACGGATCCGGGGAATTAGCAGAGCTGCTATACCGCACGCGT | |
| PaphA-luxAB-R | GTCGACCTGCAGCCCAAGCTTATCGATTCGGCTACTTATACTTTCAGCTGCGCTC | |
| luxAB-pDM8-P4 | GTCGACCTGCAGCCCAAGCTTATCGATTCGTTACGAGTGGTATTTGACGATGTTG | |
| P18672-F | TGCCTGCAGGTCGACGATTCGGCATTCATTTTCAGTTCGGCGG | EMSA or DNase I footprinting |
| P18672-R | CATGGTCGGTGAAATATCAAAGTGG | |
| PluxR-F | AGTCGCACGCAAACGATCACC | |
| PluxR-R | ATCCATTTTCCTTGCCATTTG | |
| PaphA-F | ACTCCCGGGAGCAGAGCTGCTATACCGCACGCGT | |
| PaphA-R | ACTCCCGGGGCTACTTATACTTTCAGCTGCGCTC | |
| Pasp-F | TGCCTGCAGGTCGACGATAATCCATTCAGCATAGCAATCG | |
| Pasp-R | TTGCCCTATTCCTTATTTGAGC | |
| PluxRΔCBS-1 | CATCTGTAGAACAATTTATATACAG | PluxR ΔCBS |
| PluxRΔCBS-2 | TGATTATTTTAATAGTTGCTTAAAGCAGTGACATCTGTAGAACAATTTA | |
| PluxRΔCBS-3 | ATCCATTTTCCTTGCCATTTGAGTTGATATTGGGGGTAACCCCCTAATTGATTATTTTAATAGTTGCTT | |
| PaphAΔCBS-1 | AATTACCCTAAGTTTTTTATTCTTTTATTTTGGTAT | PaphA ΔCBS |
| PaphAΔCBS-2 | ATAAAAAACTTAGGGTAATTTTGTCCTACTATTC | |
| PaspΔCBS-1 | GATCATTCAATTAAAAATTATCTACAAAACCTTATAAAGC | Pasp ΔCBS |
| PaspΔCBS-2 | GATAATTTTTAATTGAATGATCTTTTGTACAAATTTG | |
| P18672ΔCBSI/II-1 | GAAATAACTAAAATTACAGTGGACGTATGATTAAGAA | P18672 ΔCBSI/II |
| P18672ΔCBSI/II-2 | CGTCCACTGTAATTTTAGTTATTTCATGGATAAATTAACAAGCATGAAGG | |
| P18672MRBS-1 | TCCGTATCAAGCATCGGAGCCAGAACCACAACGTGTGATT | P18672MRBS |
| P18672MRBS-2 | GCTTGATACGGATAGGTACCGCACCCCGCTTCGCTTTG | |
| P18672MABS-1 | GGTCGCGTACCCTATCAGCGGGGTGCTTGCAAGCGAAT | P18672MABS |
| P18672MABS-2 | GATAGGGTACGCGACCGGCAAAATTGTTCAATGGCTTCAACCA | |
| P18672MCBSI-1 | TACTCTGGGCGACCGACCCGTCTTGGCATTACAGTGGACGTATGATT | P18672MCBSI |
| P18672MCBSI-2 | CGGTCGCCCAGAGTATTTAGTTATTTCTTTATCAAC | |
| P18672MCBSII-1 | GTACACAAACTCTGGTCGCCCGAAATAACTAAAGCAGAGTT | P18672MCBSII |
| P18672MCBSII-2 | ACCAGAGTTTGTGTACATGGATAAATTAACAAGCATGA | |
| aphA-RT-F | GAAAGCAAGCCACCAACAGG | qRT-PCR |
| aphA-RT-R | CGACCAGCGTCTGTAATGGA | |
| luxR-RT-F | CCTACACGCGAAGACTTGGT | |
| luxR-RT-R | TCTTGGCTGACAAGCTCGAT | |
| 18672-RT-F | CCTTGAGCAACATGCCCCTA | |
| 18672-RT-R | ATCGGGTGTTGCTGTTTCCA | |
| asp-RT-F | TGCTCAGCGAAACGACTTCA | |
| asp-RT-R | TAGTACCAGCAACGCGGTTT | |
| 16sRNA-F | AAAGCACTTTCAGTCGTGAGGAA | |
| 16sRNA-R | TGCGCTTTACGCCCAGTAAT | |
| RACE-adapter | GCGCGAATTCCTGTAGAACGAAC | 5′-RACE |
| 18672-RACE-nested-1 | ACGGTTGATCAGCGTCTGG | |
| 18672-RACE-nested-2 | GATCAGCGTCTGGATCAGGTT | |
| 18672-RACE | TTACAGTTCCGGTTGGACGG | |
| RNA-Linker | AUAUGCGCGAAUUCCUGUAGAACGAACACUAGAAGAAA | |
| pBXcmT-F | CCTCGGACTGAACACGTTGGCT | BIH |
| pBXcmT-R | GTGTAAAGCCCGGGGTGCCTAA | |
| pTRG-LuxR-F | CGGGATCCATGGCAAGGAAAATGGATATGGACT | |
| pTRG-LuxR-R | CGGAATTCACAGCAGACAATAAAAAGCCCA | |
| pTRG-18672-F | CGGGATCCATGGATAAATTAACAAGCATGA | |
| pTRG-18672-R | CGGAATTCTTATGGATTCACATGAGAAAGA | |
| pTRG-02325-F | CGGGATCCATGGAAATCGCACAGTCATTAC | |
| pTRG-02325-R | CGGAATTCTTACAACGTGTTGAGGACATCC | |
| pTRG-06711-F | CGGGATCCATGAACTTAACTCAAGTAGAAG | |
| pTRG-06711-R | CGGAATTCTTAGAAGTCAAATAAATACTCG | |
| pTRG-AsnC-F | CGGGATCCATGAAATTAGATGACTTAAACCTG | |
| pTRG-AsnC-R | GGAATTCGTGAATGTTATAAGCGATCA | |
| pTRG-01665-F | CGGGATCCATGTCTTTTGCCCAACGCCTAGCCA | |
| pTRG-01665-R | CGGAATTCTCAAAGTGACTCGTTTTTAGATTCT | |
| pTRG-MetR-F | CGGGATCCATGATAGAGCTAAAACATTTGC | |
| pTRG-MetR-R | CGGAATTCTTAAGCCACCTTGATTCCGTCA | |
| pTRG-SoxR-F | CGGGATCCATGGAAATCAGCGTAGGAGAAG | |
| pTRG-SoxR-R | CGGAATTCCTACTCTTCTTCTAACGTTAAG | |
| pTRG-F | TGGCTGAACAACTGGAAGCT | |
| pTRG-R | ATTCGTCGCCCGCCATAA |
The pET28a derivative plasmids used for VqsA, AsnC, SoxR, MetR, 02325, 01665, 06711, AphA, and LuxR expression were generated as previously described (6). Briefly, the ORF sequences were amplified by PCR using the primers shown in Table 3. Each ORF sequence was inserted into pET28a with a His6 tag at the carboxyl terminal. Then, the recombinant plasmid was transformed into E. coli BL21(DE3). Successful generation of the plasmids was confirmed by PCR using primer pairs (Table 3) to sequence the recombinant region. The pDM8-PluxR-luxAB constructs used for the promoter activity assays of vqsA and aphA with pDM8 and luxAB were generated as previously described (24).
Cloning of V. alginolyticus transcription factors and regulatory sequences of the target genes for bacterial one-hybrid assays.
The genes of 285 probable transcription factors were amplified from V. alginolyticus EPGS by using gene-specific primers (a subset of the primers are shown in Table 3) and cloned into the pTRG plasmid. The promoters of the V. alginolyticus aphA and luxR genes were amplified using primers specific to these promoters (Table 3) and inserted into the reporter vector pBXcmT, as previously described (25, 26). The E. coli XL1-Blue MRF9 kanamycin-resistant (Kmr) strain was used for propagating all pBXcmT and pTRG recombinant plasmids. The cotransformants with pairs of pBXcmT/pTRG plasmids in the reporter strain were tested on LB medium containing 34 mg/ml Cm, 50 mg/ml Km, and 15 mg/ml tetracycline (Tet). Positive cotransformants were screened on plates containing 16 mg/ml streptomycin (Str) and 20 mM 3-amino-1,2,4-triazole (3-AT). The plates were incubated at 30°C for ∼3 to 4 days.
ECP activity assay and immunoblots of Asp, LuxR, and AphA.
Extracellular protease (ECP) activity was assayed by using hide powder azure (HPA) digestion, as previously described (43). In brief, the strains were grown in LBS at 30°C for 9 h. The cell density at 600 nm (OD600) was measured. The cultures were centrifuged at 8,000 × g, and the supernatants were filtered through 0.22-μm-pore-size filters. Then, 10 mg of HPA and 1 ml of phosphate-buffered saline (PBS [pH 7.2]) were added to 1 ml of the filtered supernatants. The mixture was incubated with shaking at 37°C for 2 h, followed by the addition of 10% trichloroacetic acid into the mixture to stop the reaction. Finally, the total protease activity was measured at 600 nm. ECP activity was normalized for each strain by dividing the total activity by the OD600.
For the Asp, AphA, and LuxR immunoblotting assays, bacterial cell pellets at low cell density (2 h) or high cell density (9 h) were suspended in 1 volume of PBS to normalize the culture densities based on OD600 values. The ECP from equal cell numbers was mixed with phenylmethylsulfonyl fluoride (PMSF) at a ratio of 1:100 on ice for 30 min, and the mixtures were ultrafiltered through a Millipore tube with a 5-kDa molecular weight cutoff. The concentrated ECP was mixed with the sample-loading buffer and processed as described previously (43). In addition, the samples were heated with 20% SDS-PAGE loading buffer. Then, 15 μl of each normalized sample was loaded onto a 12% denaturing polyacrylamide gel. The proteins were resolved using electrophoresis and then transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked in a 12% solution of skim milk powder, incubated with a 1:3,000 dilution of Asp-, LuxR- or AphA-specific rabbit antibodies, and then incubated with a 1:2,000 dilution of the horseradish peroxidase (HRP)-conjugated secondary antibody. RNAP was used for the loading controls, as previously described. Then, the blots were visualized with the enhanced chemiluminescence (ECL) reagent in a Western blotting kit provided by Thermo Fisher Scientific, Shanghai, China.
qRT-PCR assays.
Bacteria were cultured overnight and then diluted 1:100 in LBS. The cells were then grown at 30°C and harvested at the indicated time points. An RNA extraction kit (Tiangen, Beijing, China) was used to isolate the total RNA. In addition, the RNA samples were digested with DNase I to degrade DNA contaminants. One microgram of RNA was used to generate cDNA using random primers (TaKaRa, Dalian, China). In addition, the cDNA was amplified with specific primer pairs with FastStart universal SYBR green master (Tiangen) on a 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). The specific primers used are shown in Table 3. Three independent qRT-PCR experiments were performed (each in triplicate), and the transcript levels were normalized to 16S rRNA in each sample by the ΔΔCT method (44).
Bioluminescence assays.
Bioluminescence assays were performed at least three times, as previously described (24). Briefly, the strains were cultured overnight, diluted to 6 × 106 CFU/ml in LBS, and then incubated with shaking at 30°C. Every hour for 9 h, a 200-μl aliquot was sampled. After adding 1% decanal to the samples, the bioluminescence was measured on a Berthold Orion II bioluminescence reader (Berthold Detection Systems, Pforzheim, Germany), as previously described (45). Meanwhile, the bacterial growth was also monitored by measuring the optical density at 600 nm (OD600) (BioTek, Winooski, VT, USA).
Purification of VqsA and other proteins.
The VqsA open reading frame (ORF) sequence was amplified by PCR using the primers shown in Table 3. The ORF sequence was inserted into pET28a with a His6 tag at the carboxyl terminal. Then, the recombinant plasmid was transformed into E. coli BL21(DE3). The strain was grown to an OD600 of ∼0.8 to 1.0 at 25°C, and expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The cells were grown for ∼14 to 15 h to allow VqsA expression. Finally, the protein was purified using nickel affinity chromatography. The purification of overexpressed AsnC, SoxR, MetR, 02325, 01665, 06711, AphA, and LuxR was also conducted as described above.
Electrophoretic mobility shift and DNase I footprinting assays.
For the electrophoretic mobility shift assays (EMSAs), purified His6-tagged proteins were incubated with different Cy5-labeled DNA probes (Table 3) in 20 μl of loading buffer (10 mM NaCl, 0.1 mM dithiothreitol [DTT], 0.1 mM EDTA, 10 mM Tris [pH 7.4]). After the mixture was incubated at 25°C for 30 min, the samples were resolved by 6% polyacrylamide gel electrophoresis in 0.5× Tris-boric acid-EDTA (TBE) buffer on ice at 100 V for 120 min. Then, the gels were scanned using a Typhoon FLA 9500 imager (GE Healthcare, Uppsala, Sweden). Dye-primer-based DNase I footprinting assays were performed as previously described (6). In brief, the promoter regions of aphA, luxR, or vqsA were amplified by PCR with PFU DNA polymerase and specific primers labeled with 6-carboxyfluorescein (6-FAM) at the 5′ ends (Table 3). The probes with the FAM label were purified using a Wizard SV gel and PCR clean-up system (Promega, Beijing, China) and then quantified using a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific). In each test, 400 ng of the probe was incubated with different amounts of VqsA, AphA, or LuxR in a total volume of 40 μl. Subsequently, the mixtures were incubated for 30 min at 25°C. After incubation, 10 μl of a solution containing DNase I (0.015 units) and CaCl2 (100 nM) was added to the mixtures. The mixtures were then incubated for 60 s at 25°C. The reactions were stopped with 140 μl of DNase I stop solution (0.15% SDS 200 mM, 30 mM EDTA, and unbuffered sodium acetate). Next, the samples were extracted by the phenol-chloroform extraction method and precipitated with ethanol. After evaporation of the ethanol, the pellets were dissolved in 10 μl of Milli-Q water. Then, 0.1 μl of GeneScan-500 LIZ size standards (Applied Biosystems) and 7.9 μl of HiDi formamide (Applied Biosystems) were added to the 2 μl of the digested DNA. Finally, the mixtures were analyzed using a 3730 DNA Analyzer with a G5 dye set that was run on an altered default genotyping module that increased the injection time to 30 s and the injection voltage to 3 kV. GeneMapper 4.0 (Applied Biosystems) was used to analyze the results.
5′-RACE.
The 5′-rapid amplification of cDNA ends (RACE) experiments were performed as previously described (28). Six micrograms of total RNA was extracted using the TRIzol reagent (28) from WT, ΔvqsA mutant, and ΔluxR mutant cultures and subjected to dephosphorylation using tobacco acid pyrophosphatase (TAP) (Epicentre) for 60 min at 37°C. The total RNA was ligated with an RNA oligonucleotide linker by using T4 RNA ligase (New England BioLabs, Beverly, MA, USA), according to the manufacturer's instructions. cDNA was synthesized using avian myeloblastosis virus (AMV) reverse transcriptase (RT) (TaKaRa), according to the manufacturer's instructions with a specific primer, 18672-RACE. PCR amplification was performed using the nested primers RACE-adapter and either 18672-RACE-nested-1 or 18672-RACE-nested-2 (Table 3). Finally, the single band was extracted, subcloned, and sequenced.
Bacterial killing assay.
The interbacterial killing assays were performed three times, as previously described (30). Overnight cultures were used for the interbacterial killing assays. Cultures were first normalized to an OD600 of 0.5, and the cells were mixed in a predator-to-prey ratio of 4:1. Twenty-five microliters of these mixtures was spotted on LBS agar plates and then cocultured at 30°C for 4 h. The CFU counts of the strains in the mixtures spotted on LBS agar plates at t = 0 were determined by plating 10-fold serial dilutions on appropriate plates. After 4 h, the bacterial spots were collected from the LBS agar plates, and the CFU counts of the surviving predator and prey were determined. An empty pBAD33 or pRK415 plasmid that could replicate stably in the corresponding host was used to determine E. coli and V. alginolyticus resistance to chloramphenicol or tetracycline, respectively (30).
Biofilm formation assays.
The biofilm assay was performed as previously described (43). Overnight cultures of WT, ΔvqsA mutant, vqsA+ mutant, and other strains were diluted to an OD600 of 1.0. Fifty microliters of the cultures was inoculated into 5 ml of LBS in glass tubes and grown at 30°C without shaking for 48 h. After staining with 2% crystal violet for 20 min and washing, the biofilms were dissolved in 33% (vol/vol) acetic acid, and the absorbance of the solutions at 570 nm was measured. The experiments were performed at least three times (Fig. 6B).
Infection of fish.
WT and ΔvqsA mutant bacteria were incubated at 30°C overnight and then inoculated into fresh medium and grown for 9 h. All of the strains were harvested and then serially diluted with PBS. Zebrafish weighing approximately 0.25 g were infected with the strains at doses ranging from 104 to 107 CFU/fish via intramuscular injection, according to a previously described method (46). Thirty fish were infected with each dilution, and three parallel experiments were performed. Then, the fish were cultured for 72 h, and vibriosis-induced death was observed and confirmed by isolating V. alginolyticus strains from the dead fish. LD50 values were calculated according to a previously described method (46).
Ethics statement.
All animal experiments presented in this study were approved by the Animal Care Committee of the East China University of Science and Technology (grant 2006272). The Experimental Animal Care and Use Guidelines from the Ministry of Science and Technology of China (MOST-2011-02) were strictly followed.
Accession number(s).
The draft genome sequence of EPGS has been deposited in GenBank under the accession number GCA_001273715.1.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from National Natural Science Foundation of China (grants 31772891 to Q.W., 31772893 to Y.M., and 41376128 to Y.Z.), the Ministry of Agriculture of China (grant CARS-47-G17), the Shanghai Pujiang Program (grant 16PJD018), and the Science and Technology Commission of Shandong and Shanghai Municipality (grants 2017CXGC0103 and 17391902000). X.Z. was supported by NIH grant R01AI118943 and the National Natural Science Foundation of China (grant 31772741).
We thank Matthew K. Waldor from HHMI for his helpful comments and discussion about the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00444-18.
REFERENCES
- 1.Miller MB, Bassler BL. 2001. Quorum sensing in bacteria. Annu Rev Microbiol 55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- 2.Ng W-L, Bassler BL. 2009. Bacterial quorum-sensing network architectures. Annu Rev Genet 43:197–222. doi: 10.1146/annurev-genet-102108-134304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rutherford ST, van Kessel JC, Shao Y, Bassler BL. 2011. AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev 25:397–408. doi: 10.1101/gad.2015011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Kessel JC, Ulrich LE, Zhulin IB, Bassler BL. 2013. Analysis of activator and repressor functions reveals the requirements for transcriptional control by LuxR, the master regulator of quorum sensing in Vibrio harveyi. mBio 4:e00378-13. doi: 10.1128/mBio.00378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruwandeepika HAD, Bhowmick PP, Karunasagar I, Bossier P, Defoirdt T. 2012. Pathogenesis, virulence factors and virulence regulation of vibrios belonging to the Harveyi clade. Rev Aquacult 4:59–74. doi: 10.1111/j.1753-5131.2012.01061.x. [DOI] [Google Scholar]
- 6.Gu D, Guo M, Yang MJ, Zhang YX, Zhou XH, Wang QY. 2016. A σE-mediated temperature gauge controls a switch from LuxR-mediated virulence gene expression to thermal stress adaptation in Vibrio alginolyticus. PLoS Pathog 12:e1005645. doi: 10.1371/journal.ppat.1005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin W, Kovacikova G, Skorupski K. 2005. Requirements for Vibrio cholerae HapR binding and transcriptional repression at the hapR promoter are distinct from those at the aphA promoter. J Bacteriol 187:3013–3019. doi: 10.1128/JB.187.9.3013-3019.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z, Hsiao A, Joelsson A, Zhu J. 2006. The transcriptional regulator VqmA increases expression of the quorum-sensing activator HapR in Vibrio cholerae. J Bacteriol 188:2446–2453. doi: 10.1128/JB.188.7.2446-2453.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenz DH, Bassler BL. 2007. The small nucleoid protein Fis is involved in Vibrio cholerae quorum sensing. Mol Microbiol 63:859–871. doi: 10.1111/j.1365-2958.2006.05545.x. [DOI] [PubMed] [Google Scholar]
- 10.Tsou AM, Liu Z, Cai T, Zhu J. 2011. The VarS/VarA two-component system modulates the activity of the Vibrio cholerae quorum-sensing transcriptional regulator HapR. Microbiology 157:1620–1628. doi: 10.1099/mic.0.046235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Z, Miyashiro T, Tsou A, Hsiao A, Goulian M, Zhu J. 2008. Mucosal penetration primes Vibrio cholerae for host colonization by repressing quorum sensing. Proc Natl Acad Sci U S A 105:9769–9774. doi: 10.1073/pnas.0802241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. 2004. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Lenz DH, Miller MB, Zhu J, Kulkarni RV, Bassler BL. 2005. CsrA and three redundant small RNAs regulate quorum sensing in Vibrio cholerae. Mol Microbiol 58:1186–1202. doi: 10.1111/j.1365-2958.2005.04902.x. [DOI] [PubMed] [Google Scholar]
- 14.Lee KJ, Jung YC, Park SJ, Lee KH. 2018. Role of heat shock proteases in quorum-sensing-mediated regulation of biofilm formation by Vibrio species. mBio 9:e02086-17. doi: 10.1128/mBio.02086-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalia VC, Purohit HJ. 2011. Quenching the quorum sensing system: potential antibacterial drug targets. Crit Rev Microbiol 37:121–40. doi: 10.3109/1040841X.2010.532479. [DOI] [PubMed] [Google Scholar]
- 16.Rui HP, Liu Q, Wang QY, Ma Y, Liu H, Shi C, Zhang YX. 2009. Role of alkaline serine protease, asp, in Vibrio alginolyticus virulence and regulation of its expression by luxO-luxR regulatory system. J Microbiol Biotechnol 19:431–438. doi: 10.4014/jmb.0807.404. [DOI] [PubMed] [Google Scholar]
- 17.Cao XD, Wang QY, Liu Q, Liu H, He HH, Zhang YX. 2010. Vibrio alginolyticus MviN is a luxO-regulated protein and affects cytotoxicity towards epithelioma papulosum cyprini (EPC) cells. J Microbiol Biotechnol 20:271–280. doi: 10.1007/s00253-009-2377-x. [DOI] [PubMed] [Google Scholar]
- 18.Rui HP, Liu Q, Ma Y, Wang QY, Zhang YX. 2008. Roles of LuxR in regulating extracellular alkaline serine protease A, extracellular polysaccharide and mobility of Vibrio alginolyticus. FEMS Microbiol Lett 285:155–162. doi: 10.1111/j.1574-6968.2008.01185.x. [DOI] [PubMed] [Google Scholar]
- 19.Cao XD, Wang QY, Liu Q, Rui HP, Liu H, Zhang YX. 2011. Identification of a luxO-regulated extracellular protein Pep and its roles in motility in Vibrio alginolyticus. Microb Pathog 50:123–131. doi: 10.1016/j.micpath.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Wang QY, Liu Q, Ma Y, Rui HP, Zhang YX. 2007. LuxO controls extracellular protease, haemolytic activities and siderophore production in fish pathogen Vibrio alginolyticus. J Appl Microbiol 103:1525–1534. doi: 10.1111/j.1365-2672.2007.03380.x. [DOI] [PubMed] [Google Scholar]
- 21.Ye J, Ma Y, Liu Q, Zhao DL, Wang QY, Zhang YX. 2008. Regulation of Vibrio alginolyticus virulence by the LuxS quorum-sensing system. J Fish Dis 31:161–169. doi: 10.1111/j.1365-2761.2007.00882.x. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Fu K, Wang Y, Wu C, Li F, Shi L, Ge Y, Zhou L. 2017. Detection of diverse N-acyl-homoserine lactones in Vibrio alginolyticus and regulation of biofilm formation by N-(3-oxodecanoyl) homoserine lactone in vitro. Front Microbiol 16 8:1097. doi: 10.3389/fmicb.2017.01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Gu D, Cao XD, Liu Q, Wang QY, Zhang YX. 2012. Characterization of a new quorum sensing regulator luxT and its roles in the extracellular protease production, motility, and virulence in fish pathogen Vibrio alginolyticus. Arch Microbiol 194:439–452. doi: 10.1007/s00203-011-0774-x. [DOI] [PubMed] [Google Scholar]
- 24.Gu D, Liu H, Yang Z, Zhang YX, Wang QY. 2016. Chromatin immunoprecipitation sequencing technology reveals global regulatory roles of low-cell-density quorum-sensing regulator AphA in the pathogen Vibrio alginolyticus. J Bacteriol 198:2985–2999. doi: 10.1128/JB.00520-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao XT, Liu Y, Liu H, Yang Z, Liu Q, Zhang YX, Wang QY. 2017. Identification of the regulon of AphB and its essential roles in LuxR and exotoxin Asp expression in the pathogen Vibrio alginolyticus. J Bacteriol 199:e00252-17. doi: 10.1128/JB.00252-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo M, Feng H, Zhang J, Wang W, Wang Y, Li Y, Gao C, Chen H, Feng Y, He ZG. 2009. Dissecting transcription regulatory pathways through a new bacterial one-hybrid reporter system. Genome Res 19:1301–1308. doi: 10.1101/gr.086595.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maddocks SE, Oyston PC. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 28.Cao XD, Studer SV, Wassarman K, Zhang YX, Ruby EG, Miyashiro T. 2012. The novel sigma factor-like regulator RpoQ controls luminescence, chitinase activity, and motility in Vibrio fischeri. mBio 3:e00285-11. doi: 10.1128/mBio.00285-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salomon D, Klimko JA, Trudgian DC, Kinch LN, Grishin NV, Mirzaei H, Orth K. 2015. Type VI secretion system toxins horizontally shared between marine bacteria. PLoS Pathog 11:e1005128. doi: 10.1371/journal.ppat.1005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Z, Zhou XH, Ma Y, Zhou M, Waldor MK, Zhang YX, Wang QY. 2018. Serine/threonine kinase PpkA coordinates the interplay between T6SS2 activation and quorum sensing in the marine pathogen Vibrio alginolyticus. Environ Microbiol 20:903–919. doi: 10.1111/1462-2920.14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chatterjee J, Miyamoto CM, Zouzoulas A, Lang BF, Skouris N, Meighen EA. 2002. MetR and CRP bind to the Vibrio harveyi lux promoters and regulate luminescence. Mol Microbiol 46:101–111. doi: 10.1046/j.1365-2958.2002.03128.x. [DOI] [PubMed] [Google Scholar]
- 32.Rhee JE, Jeong HG, Lee JH, Choi SH. 2006. AphB influences acid tolerance of Vibrio vulnificus by activating expression of the positive regulator CadC. J Bacteriol 188:6490–6497. doi: 10.1128/JB.00533-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovacikova G, Lin W, Skorupski K. 2010. The LysR-type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. J Bacteriol 192:4181–4191. doi: 10.1128/JB.00193-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Demple B, Ding H, Jorgensen M. 2002. Escherichia coli SoxR protein: sensor/transducer of oxidative stress and nitric oxide. Methods Enzymol 348:355–364. doi: 10.1016/S0076-6879(02)48654-5. [DOI] [PubMed] [Google Scholar]
- 35.Yokoyama K, Ishijima SA, Clowney L, Koike H, Aramaki H, Tanaka C, Makino K, Suzuki M. 2006. Feast/famine regulatory proteins (FFRPs): Escherichia coli Lrp, AsnC and related archaeal transcription factors. FEMS Microbiol Rev 30:89–108. doi: 10.1111/j.1574-6976.2005.00005.x. [DOI] [PubMed] [Google Scholar]
- 36.van Kessel JC, Rutherford ST, Shao Y, Utria AF, Bassler BL. 2013. Individual and combined roles of the master regulators AphA and LuxR in control of the Vibrio harveyi quorum-sensing regulon. J Bacteriol 195:436–443. doi: 10.1128/JB.01998-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ball AS, Chaparian RR, van Kessel JC. 2017. Quorum sensing gene regulation by LuxR/HapR master regulators in vibrios. J Bacteriol 199:e00105-17. doi: 10.1128/JB.00105-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor JL, De Silva RS, Kovacikova G, Lin W, Taylor RK, Skorupski K, Kull FJ. 2012. The crystal structure of AphB, a virulence gene activator from Vibrio cholerae, reveals residues that influence its response to oxygen and pH. Mol Microbiol 83:457–470. doi: 10.1111/j.1365-2958.2011.07919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sainsbury S, Lane LA, Ren J, Gilbert RJ, Saunders NJ, Robinson CV, Stuart DI, Owens RJ. 2009. The structure of CrgA from Neisseria meningitidis reveals a new octameric assembly state for LysR transcriptional regulators. Nucleic Acids Res 37:4545–4558. doi: 10.1093/nar/gkp445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Z, Yang M, Peterfreund GL, Tsou AM, Selamoglu N, Daldal F, Zhong Z, Kan B, Zhu J. 2011. Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol-based switches of virulence regulator AphB. Proc Natl Acad Sci U S A 108:810–815. doi: 10.1073/pnas.1014640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hubin EA, Fay A, Xu C, Bean JM, Saecker RM, Glickman MS, Darst SA, Campbell EA. 2017. Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. Elife 6: e22520. doi: 10.7554/eLife.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibson DG, Young L, Chuang R, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 43.Sheng LL, Lv YZ, Liu Q, Wang QY, Zhang YX. 2013. Connecting type VI secretion, quorum sensing, and c-di-GMP production in fish pathogen Vibrio alginolyticus through phosphatase PppA. Vet Microbiol 162:652–662. doi: 10.1016/j.vetmic.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 44.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 45.Yin KY, Wang QY, Xiao JF, Zhang YX. 2017. Comparative proteomic analysis unravels a role for EsrB in the regulation of reactive oxygen species stress responses in Edwardsiella piscicida. FEMS Microbiol Lett 364:fnw269. doi: 10.1093/femsle/fnw269. [DOI] [PubMed] [Google Scholar]
- 46.Lv YZ, Xiao JF, Liu Q, Wu HZ, Zhang YX, Wang QY. 2012. Systematic mutation analysis of two-component signal transduction systems reveals EsrA-EsrB and PhoP-PhoQ as the major virulence regulators in Edwardsiella tarda. Vet Microbiol 157:190–199. doi: 10.1016/j.vetmic.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 47.Wang SY, Lauritz J, Jass J, Milton DL. 2002. A ToxR homolog from Vibrio anguillarum serotype O1 regulates its own production, bile resistance, and biofilm formation. J Bacteriol 184:1630–1639. doi: 10.1128/JB.184.6.1630-1639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.