

Key Points
We established a Nudt15 knockout mouse model with which to evaluate individualized thiopurine therapy.
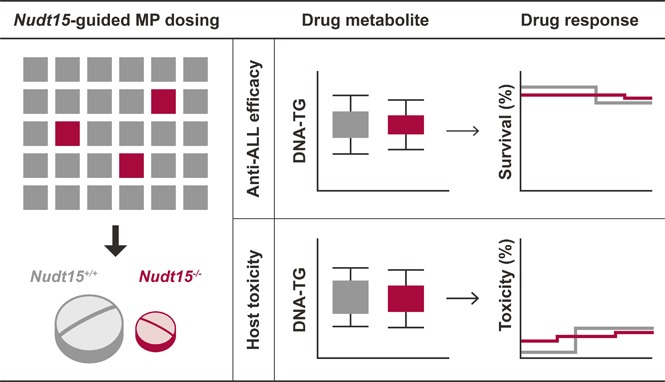
Preemptive NUDT15 genotype–guided thiopurine dosing can effectively prevent drug toxicity without compromising antileukemic efficacy.
Abstract
Thiopurines (eg, 6-mercaptopurine [MP]) are highly efficacious antileukemic agents, but they are also associated with dose-limiting toxicities. Recent studies by us and others have identified inherited NUDT15 deficiency as a novel genetic cause of thiopurine toxicity, and there is a strong rationale for NUDT15-guided dose individualization to preemptively mitigate adverse effects of these drugs. Using CRISPR-Cas9 genome editing, we established a Nudt15−/− mouse model to evaluate the effectiveness of this strategy in vivo. Across MP dosages, Nudt15−/− mice experienced severe leukopenia, rapid weight loss, earlier death resulting from toxicity, and more bone marrow hypocellularity compared with wild-type mice. Nudt15−/− mice also showed excessive accumulation of a thiopurine active metabolite (ie, DNA-incorporated thioguanine nucleotides [DNA-TG]) in an MP dose–dependent fashion, as a plausible cause of increased toxicity. MP dose reduction effectively normalized systemic exposure to DNA-TG in Nudt15−/− mice and largely eliminated Nudt15 deficiency–mediated toxicity. In 95 children with acute lymphoblastic leukemia, MP dose adjustment also directly led to alteration in DNA-TG levels, the effects of which were proportional to the degree of NUDT15 deficiency. Using leukemia-bearing mice with concordant Nudt15 genotype in leukemia and host, we also confirmed that therapeutic efficacy was preserved in Nudt15−/− mice receiving a reduced MP dose compared with Nudt15+/+ counterparts exposed to a standard dose. In conclusion, we demonstrated that NUDT15 genotype–guided MP dose individualization can preemptively mitigate toxicity without compromising therapeutic efficacy.
Visual Abstract
Introduction
As anticancer and immunosuppressive agents, thiopurines (eg, 6-mercaptopurine [MP], 6-thioguanine, and azathioprine) are highly efficacious in a variety of clinical settings.1-7 In adults and children with acute lymphoblastic leukemia (ALL), daily exposure to MP is the main component of maintenance therapy and essential for long-term cure of this disseminated cancer.3,5,8-11 However, thiopurines are associated with dose-limiting adverse effects, particularly in the hematopoietic tissues, with potential morbidity and even mortality.4,12-14 Therefore, monitoring of white blood cell count on a regular basis is clinically required to prevent excessive bone marrow suppression. Cytotoxic effects of thiopurines are thought to be mediated by their active metabolite thioguanosine triphosphate (TGTP), the incorporation of which into DNA (DNA-TG) triggers futile mismatch repair and eventually apoptosis.1,15-19
Genetic polymorphisms in genes involved in thiopurine metabolism (eg, TPMT) can strongly influence the risk of thiopurine toxicity.20-23 In fact, preemptive TPMT genotype–guided thiopurine dose individualization has been clinically implemented to mitigate drug toxicity and is a prototype of pharmacogenetics-driven precision medicine in cancers and autoimmune diseases.24-26 More recently, we and others identified NUDT15 as a novel genetic determinant of thiopurine metabolism and intolerance in patients with ALL27,28 or inflammatory bowel diseases,29 most prominently in Asians and Hispanics.27-29 As a nucleotide diphosphatase, NUDT15 is responsible for the inactivation of thiopurine metabolites by converting TGTP to TGMP and therefore negatively regulates the cytotoxic effects of this class of drugs.28,30 Therefore, patients with loss-of-function germ line variants in NUDT15 (eg, rs116855232) have increased MP activation and excessive DNA damage and require drastic thiopurine dose reduction to avoid myelosuppression (eg, ≤ 92% of standard MP dosage).27,28
Although there is growing evidence in support of incorporating NUDT15 into the pharmacogenetics-based thiopurine dosing algorithm, there is also a paucity of data to inform the precise level of dose reduction needed for patients with various NUDT15 genotypes to minimize toxicity without compromising therapeutic effects. Because NUDT15 is directly linked to the generation of thiopurine active metabolite DNA-TG, it is conceivable that MP dose could be rationally decreased in NUDT15-deficient patients to reduce their exposure to DNA-TG to a level that is comparable to that of wild-type patients receiving standard dose of MP, thus mitigating toxicity. However, the ability to test this hypothesis has been hindered by the lack of faithful laboratory model systems for preclinical evaluation. We established a Nudt15-knockout mouse model using CRISPR-Cas9–mediated genome editing, with which we systematically evaluated NUDT15 genotype–based thiopurine dose reduction and its effects on hematopoietic toxicity and antileukemic efficacy in vivo.
Methods
CRISPR-Cas9–mediated genome editing to generate the Nudt15−/− mouse
Single-guide RNAs (sgRNAs) were designed to target nucleotide position 1 to 150 bp of the coding sequence of mouse Nudt15 (exon 3; Figure 1A) using the CRISPR design tool (http://tools.genome-engineering.org).31 Five candidate sgRNAs were tested for targeting efficiency in NIH3T3 cells using the T7 Endonuclease I cleavage assay (New England Biolabs; supplemental Figure 1 [available on the Blood Web site]; oligos for each sgRNA listed in supplemental Table 1).32 sgRNA #3 was selected for Nudt15 targeting in mouse and was transcribed in vitro using the MEGAshortscript Transcription Kit (Thermo Fisher Scientific) with a T7 promoter. Purified sgRNA (50 ng/μL) and Cas9 protein (100 ng/μL; PNA Bio) were then coinjected into zygotes from FVB/N-strain mice, which were then transferred into the oviduct of pseudopregnant female mice from the same background. F0 mice were bred for germ line transmission, and Nudt15 genotype was confirmed by polymerase chain reaction and Sanger sequencing (primers listed in supplemental Table 1). Genomic loci with potential off-target edits by CRISPR-Cas9 were predicted using the Cas-OFFinder (http://www.rgenome.net/cas-offinder/) and individually ruled out by Sanger sequencing (supplemental Table 2).33
Figure 1.

Establishment of the Nudt15−/−mouse model using CRISPR-Cas9–mediated genome editing. (A) Mouse wild-type Nudt15 gene consists of 5 exons, and the start codon is located in exon 3 (top panel). Magnified region describes sgRNA target sequence in Nudt15 (red) and the protospacer adjacent motif (PAM) sequence (blue; bottom panel). (B) A single base insertion of c.78_79insT (red arrows) was created, resulting in frameshift and loss of functional Nudt15.
Animal experiments in this study were approved by the Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital.
MP metabolism and toxicity in mice
Mice between 8 and 10 weeks of age received MP therapy in vivo. MP was administered via intraperitoneal (IP) injection daily at 3 dosage levels: 1, 5, or 20 mg/kg per day (equivalent to 3, 15, or 60 mg/m2 per day in children, respectively).34 Body weight and signs of morbidity were monitored daily. Hematopoietic toxicity was evaluated by complete blood count weekly. Mice were euthanized when weight loss exceeded >20% (or they became moribund) and subjected to full necropsy to evaluate morphological changes induced by MP treatment. Organs were dissected and fixed in 10% neutral buffer formalin, and paraffin-embedded sections were analyzed by hematoxylin and eosin staining. To determine thiopurine metabolite DNA-TG accumulation, mice were treated with daily MP for 4 days IP (1, 5, 20, or 40 mg/kg per day) and euthanized on day 5. Peripheral blood and bone marrow were then harvested for DNA extraction. Intact DNA was digested into deoxynucleosides with enzyme cocktail (benzonase [Sigma], phosphodiesterase I [Sigma], and alkaline phosphatase [Sigma]) as previously described.35 DNA-TG levels in total pool of free deoxynucleosides were quantified by using a liquid chromatography–mass spectrometry assay that was modified from the previously published method.36 DNA-TG levels were normalized to DNA quantity.
Antileukemic efficacy of MP in mice
Nudt15+/+ or Nudt15−/− mice were crossed with Arf−/− mice (strain number, 01XB2; National Cancer Institute at Frederick) on an FVB/N background to generate transplantable leukemic cells, according to previous studies with slight modification.37 Briefly, bone marrow cells of donor mice with Nudt15+/+Arf−/− or Nudt15−/−Arf−/− genotype were transduced with retroviral particles containing MSCV-BCR-ABL1-GFP in RPMI 1640 medium supplemented with 5% fetal bovine serum, 4 mM of l-glutamine, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 55 μM of β-mercaptoethanol for 3 hours.37 Infected cells were cultured in virus-free media and became cytokine independent (transformed) between 7 and 10 days. Murine BCR-ABL1 leukemia cells with Nudt15+/+ or Nudt15−/− genotype were intravenously injected into the tail vein of genotype-matched recipients between 8 and 10 weeks of age (2000 leukemia cells per mouse; supplemental Figure 2). One day after transplantation, daily IP MP therapy (1 or 20 mg/kg per day) or vehicle was started. Leukemia burden was assessed twice per week by flow cytometric determination of GFP+ blast count in peripheral blood. Mice were euthanized when they became moribund (eg, excessive body weight loss, ruffled fur, hunched back, poor mobility, respiratory distress), and their bone marrow and spleen were evaluated for MP-induced toxicity and leukemia burden.
Leukocyte DNA-TG monitoring in children with ALL during MP dose adjustment
A total of 95 children with ALL from the Japanese Pediatric Leukemia Study Group ALL B-12 protocol (UMIN000009339) or Singaporean MaSpore 2003/2010 protocols38 were included in this study. The recommended MP dosage during the maintenance phase of treatment was 50 or 75 mg/m2 per day for standard/intermediate risk or high risk, respectively. The dosage was adjusted to a target white blood cell count of between 2.0 and 3.0 × 109 cells/L in the Japanese ALL B-12 protocol or 2.0 and 4.0 × 109 cells/L in MaSpore 2003/2010 standardized guidelines. Peripheral blood was collected at multiple time points during MP dose adjustments, and the average of the daily MP dosage over 14 days before sampling was used for additional analyses. A total of 153 samples were analyzed, with an average of 1.6 samples per patient (range, 1 to 5 time points for each patient). Of these, 22 samples from 16 individuals had been described previously.28,39 DNA was extracted from nucleated cells in peripheral blood and used for DNA-TG measurement using the same assay described in “MP metabolism and toxicity in mice,” and NUDT15 was genotyped by polymerase chain reaction and Sanger sequencing.28 This study was approved by the respective institutional review boards, and informed consent was obtained from the parents, guardians, and/or patients, as appropriate.
Statistical analyses
All statistical tests were 2 sided and were chosen as appropriate according to data distribution as indicated in figure legends. Toxicity phenotypes were compared between Nudt15−/− and wild-type mice using analysis of variance (weight loss and leukemia burden), Wilcoxon rank test (hematopoietic parameters), and Kaplan-Meier test (death resulting from toxicity and leukemia-free survival). Correlation between MP dosage and DNA-TG level was determined using Pearson’s correlation coefficient test. MP dose–dependent accumulation of DNA-TG was modeled following Michaelis-Menten kinetics. Longitudinal DNA-TG change in response to MP dose adjustments in patients was analyzed using a mixed effect model, assuming the Michaelis-Menten kinetics. R software (version 3.0; http://www.r-project.org/) was used for all analyses.
Additional experimental details are included in supplemental Methods.
Results
Mouse and human NUDT15 proteins share 89% homology at the amino acid level (supplemental Figure 3A) and showed comparable enzymatic activity for catalyzing the conversion of thiopurine metabolite TGTP to TGMP (supplemental Figure 3B). Therefore, we hypothesized that an Nudt15-knockout mouse would be a faithful model of MP-induced toxicity in NUDT15-deficient patients. Using CRISPR-Cas9–mediated genome editing, we established an Nudt15-deficient mouse strain on the FVB/N background in which frameshift mutation c.78_79insT was introduced in exon 3 of Nudt15 (Figure 1). We confirmed that this single-nucleotide insertion completely abolished the translation of mouse Nudt15 protein (supplemental Figure 4) and thus likely resulted in loss of Nudt15 functions. Nudt15−/− mice were viable and did not show any gross abnormality by histology across major organs compared with Nudt15+/+ mice (supplemental Figure 5).
To assess the effects of Nudt15 on thiopurine toxicities, Nudt15−/− and wild-type mice were first treated with daily MP at 20 mg/kg per day (equivalent to 60 mg/m2 per day in humans) and monitored for toxicity. Compared with wild-type animals, Nudt15−/− mice experienced more precipitous weight loss (P = 4.3 × 10−10; F = 40.5; Figure 2A) and earlier death resulting from toxicity (median survival, 12 vs 25 days in Nudt15−/− vs Nudt15+/+ mice, respectively; P = 5.3 × 10−8; Figure 2B). Hematopoietic toxicity developed within 7 days of initiating MP and was more severe in Nudt15−/− mice than wild-type counterparts (Figure 3A), as reflected by significantly lower peripheral neutrophil count (P = .006), hemoglobin (P = .016), and platelet count (P = .016). There were no differences in lymphocyte count by Nudt15 genotype (Figure 3A) or in hepatic toxicities (liver enzymes and total bilirubin; supplemental Figure 6). Similarly, severe bone marrow hypocellularity at time of death was confirmed by necropsy and was particularly pronounced in Nudt15−/− mice receiving MP (Figure 3B). MP treatment at this dosage level also caused damage to the mucosal epithelial cells of the digestive tract of Nudt15−/− mice (especially in the esophagus and small intestine), whereas this was not seen in Nudt15+/+ mice (Figure 3B). The esophageal mucosa in Nudt15−/− mice exhibited dysplasia, hyperplasia, and hyperkeratosis, with large areas of ulceration. In the small intestine, there was an increase in apoptotic crypt epithelial cells. Collectively, these results provided unequivocal evidence that directly linked Nudt15 deficiency to MP toxicity in the hematopoietic tissues in vivo.
Figure 2.

Nudt15 deficiency caused accelerated weight loss and early death resulting from toxicity during thiopurine therapy in mice. Nudt15−/− or wild-type mice received MP therapy (20, 5, or 1 mg/kg per day via IP injection) and monitored daily for weight loss (A) and survival (B). Across all dosage levels (red, brown, and green for 20, 5, and 1 mg/kg per day, respectively), weight loss was significantly more severe and precipitous in Nudt15−/− mice compared with wild-type mice (indicated by dashed vs solid lines, respectively), leading to significantly earlier death resulting from toxicity in the former. In panel A, each line represents the mean percent weight loss relative to baseline, and the shade of the same color indicates standard deviation within the respective group.
Figure 3.

Thiopurine-induced hematopoietic and gastrointestinal toxicity in Nudt15−/−and wild-type mice. (A) Hematological parameters were measured after 7 days of MP therapy (20, 5, or 1 mg/kg per day, shown in red, brown, and green, respectively) by complete peripheral blood count in Nudt15−/− and wild-type animals (crosses and dots, respectively). (B) Thiopurine-induced damage to the bone marrow, esophagus, and small intestine was examined at the time of death resulting from toxicity. Wild-type mice receiving 1 mg/kg per day showed no sign of morbidity from thiopurine treatment and were euthanized on day 26. Bone marrow cellularity was normal in wild-type mice receiving 1 mg/kg per day, whereas in all other conditions, loss of bone marrow was evident by increased empty spaces (upper panel). In the esophagus, an area of severe mucosal ulceration is shown in the Nudt15−/− mice receiving 20 mg/kg per day; the starred area indicates a mat of bacteria overlying the ulcer (middle panel). Black arrowheads point to apoptotic crypt epithelial cells in the small intestine (lower panel). Hematoxylin and eosin staining; scale bars: upper panel, 100 µm; middle panel, 250 µm; lower panel, 50 µm. *P < .05, **P < .01, as estimated using Wilcoxon rank test.
To model NUDT15 genotype–guided MP dose individualization, we next evaluated toxicity in Nudt15−/− vs Nudt15+/+ mice exposed to progressively decreasing MP dosages. In wild-type mice, lowering MP dosage from 20 to 5 mg/kg per day significantly delayed weight loss, improved survival, and alleviated hematopoietic toxicity; these adverse effects were largely eliminated when dosage was further reduced to 1 mg/kg per day (Figures 2 and 3A). In Nudt15−/− mice, toxicity was also mitigated with decreasing MP dosage (slower weight loss [Figure 2A], prolonged survival [Figure 2B], and less myelosuppression [Figure 3A]), but it remained consistently more severe than that in wild-type mice treated at the same dosages. Importantly, dose reduction from 20 to 1 mg/kg per day in Nudt15−/− mice resulted in weight loss and survival patterns that were indistinguishable from those in wild-type mice treated at 20 mg/kg per day (Figure 2). Similarly, the peripheral blood parameters were comparable between Nudt15−/− mice receiving this reduced MP dosage and wild-type mice treated at the full MP dosage (Figure 3A), although the improvement was most significant in neutrophil and platelet counts and less so in hemoglobin. Furthermore, loss of bone marrow and damage to the digestive tract were substantially mitigated or largely avoided by MP dose reduction in Nudt15-deficient mice (Figure 3B). Taken together, these data indicated that Nudt15 deficiency–mediated MP toxicity can be effectively eliminated by preemptive MP dose reduction.
To establish the pharmacological basis of NUDT15 genotype–guided MP dose adjustments, we also characterized thiopurine metabolism in Nudt15−/− and Nudt15+/+ mice. After 4 days of MP therapy, DNA-TG was readily detectable in peripheral leukocytes and was positively correlated with MP dosage regardless of Nudt15 genotype (supplemental Figure 7). Although also MP dosage dependent, DNA-TG accumulation was significantly higher in bone marrow cells and reached saturation at high dosages (Figure 4A). Across dosage levels and tissue types, DNA-TG levels were drastically higher in Nudt15−/− mice than in wild-type mice. Assuming the Michaelis-Menten kinetics model, we estimated that the efficiency by which MP was metabolized to DNA-TG in bone marrow tissues was 1.8-fold higher in Nudt15−/− mice than in wild-type mice (Vmax/Km of 10.1 ± 2.1 and 5.6 ± 1.5, respectively). In fact, MP dose adjustment to 1 mg/kg per day in Nudt15−/− mice reduced DNA-TG level to 243.7 ± 121.8 fmol/μg of DNA, comparable to the level in Nudt15+/+ mice receiving the full MP dosage of 20 mg/kg per day (299.1 ± 63.9 fmol/μg of DNA), which supported the reduced toxicity in Nudt15-deficient mice with this degree of dose decrease. Therefore, our results indicated that rational MP dose reduction in Nudt15-deficient mice normalized their exposure to thiopurine active metabolite DNA-TG and thus prevented MP toxicity.
Figure 4.

Effects of MP dose adjustments on active metabolite DNA-TG accumulation in vivo. (A) Mice received daily MP injections at 4 different doe levels, and DNA-TG was measured in bone marrow mononucleated cells on day 5. At each dosage level, 8 mice of each genotype were included. (B) In 95 children with ALL, DNA-TG was monitored longitudinally during MP dosage titration. In both mice and patients, trend lines were established to describe the relationship of MP dosage with DNA-TG, using Michaelis-Menten kinetics model. Shades indicate 95% confidence intervals of the model.
To validate these findings in patients, we also characterized DNA-TG change in peripheral leukocytes in 95 children with ALL during MP dose adjustments. On an individual-patient basis, MP dosage fluctuated substantially to maintain the proper degree of myelosuppression during ALL therapy. As in mice, there was also a significant positive correlation of DNA-TG in peripheral blood leukocytes with MP dosage (P = 3.4 × 10−8; Figure 4B). Following Michaelis-Menten kinetics, in NUDT15 wild-type patients, DNA-TG level plateaued at 543.1 fmol/μg of DNA (range, 436.3-676.1) as MP dosage increased, whereas the maximal DNA-TG accumulation was estimated as 830.0 fmol/μg of DNA (range, 576.1-1195.7) and 1268.3 fmol/μg of DNA (range, 625.4-2572.0) in NUDT15+/− and NUDT15−/− patients, respectively. In vivo MP metabolism efficiency (Vmax/Km ratio) increased significantly with increasing number of the copy of defective alleles in NUDT15 (0.97 ± 0.19, 1.3 ± 0.17, and 2.2 ± 0.81 for NUDT15+/+, NUDT15+/−, and NUDT15−/− patients, respectively; P = 9.1 × 10−5). Therefore, as we demonstrated in mouse models, MP dosage could be proportionally reduced in NUDT15-deficient patients to achieve the same level of DNA-TG exposure as that in wild-type patients receiving the full MP dosage thus mitigating NUDT15 deficiency–mediated toxicity.
Next, we sought to evaluate the effect of MP dose reduction on antileukemic efficacy of thiopurine treatment in vivo. Murine Arf−/−, BCR-ABL1, GFP leukemic cells with Nudt15−/− genotype were significantly more sensitive to MP with concomitant increase in intracellular DNA-TG accumulation across all MP concentrations tested, compared with Arf−/− BCR-ABL1 leukemic cells with wild-type Nudt15 (supplemental Figure 8). These murine ALL cells were implanted in Nudt15 genotype–matched recipient mice to establish leukemia-bearing mouse models (ie, concordant Nudt15 genotype in host and leukemia; supplemental Figure 2). Because MP dose reduction from 20 to 1 mg/kg per day effectively mitigated host toxicity, we sought to determine to what extent this dose reduction would affect leukemia control in Nudt15−/− mice. Without MP treatment, Nudt15+/+ or Nudt15−/− mice showed indistinguishable patterns of leukemia progression (blast percentage in peripheral blood shown in Figure 5A; leukemia-free survival rates shown in Figure 5B; median survival, 14 days for both Nudt15+/+ and Nudt15−/− mice). In wild-type mice with wild-type leukemia, daily MP treatment at 1 mg/kg per day only slightly delayed disease progression (Figure 5A), with a modest increase in survival compared with nontreated mice (Figure 5B). By contrast, Nudt15−/− mice treated at this low MP dosage were completely leukemia free for the duration of the experiment, mirroring wild-type mice receiving the full MP dosage of 20 mg/kg per day (Figure 5). Together, these results compellingly indicate that MP dose reduction mitigates Nudt15 deficiency–mediated toxicity but without negative impact on therapeutic efficacy.
Figure 5.

MP dose reduction and antileukemic efficacy in Nudt15−/−mice. Nudt15−/− mice were used to derive murine leukemia (Arf−/−, BCR-ABL1, GFP) with Nudt15 deficiency. Leukemia-bearing mice (with concordant Nudt15 genotype between host and leukemia) received IP MP therapy (20 or 1 mg/kg per day; red or green, respectively) or vehicle (blue). Leukemia progression was monitored by quantifying the percentage of GFP+ cells (leukemia blast) in peripheral blood. In panel A, each line represents the mean blast percentage, and the shade of the same color indicates standard deviation within the respective group. In panel B, vertical ticks indicate death resulting from toxicity.
Discussion
Only 3 years ago, NUDT15 was identified by agnostic genome-wide studies as a major genetic risk locus for thiopurine toxicity in patients with inflammatory bowel disease29 or ALL.27 In fact, patients homozygous for the defective NUDT15 alleles are at such an elevated risk of severe toxicity that drastic dose reduction is inevitable,27,40-43 pointing to direct clinical utility of preemptive genetic testing. The rise of NUDT15 from a nucleotide hydrolase with relatively unknown endogenous functions to a new pharmacogenetic gene with actionable effects on the toxicity of an important class of drugs exemplifies the potential of pharmacogenomic research and its immediate impact on precision medicine. In this study, we developed a Nudt15−/− mouse model to preclinically characterize the pharmacological basis for genotype-guided thiopurine dose reduction to mitigate toxicity. Using this model, we demonstrated that preemptive thiopurine dose adjustment effectively prevented NUDT15-related toxicity in vivo by normalizing systemic exposure to cytotoxic thiopurine metabolite DNA-TG, thus establishing the principle by which NUDT15-guided thiopurine dose individualization can be applied in patients.
In our mouse model, a 95% MP dose reduction (from 20 to 1 mg/kg per day [5% of standard dose]) seemed to be necessary to normalize DNA-TG exposure and eliminate toxicity in Nudt15−/− mice (Figures 2-4), largely in line with our prior observation that patients with complete loss of NUDT15 tolerated only 8.3% of standard MP dosages. It is particularly encouraging that MP dose reduction in Nudt15−/− mice safely preserved leukemia-free survival using our preclinical models (Figure 5), plausibly because of increased sensitivity to MP (cell-autonomous effects) and also greater systemic exposure to active MP metabolites (host effects). With the assumption that 1 mg/kg per day of MP in mice is approximately equivalent to 3 mg/m2 per day of MP in a child, this dosage is also comparable to that tolerated empirically by NUDT15-deficient patients when MP dosage was clinically titrated according to leukocyte count. However, it should be noted that MP was administered IP in our mouse model, whereas patients receive chronic MP orally, a difference with potential effects on MP distribution. Also, MP is often used in a multidrug context (eg, with methotrexate in children with ALL), and thus, future mouse studies are warranted to evaluate the effects of route of administration and comedications on thiopurine dose reduction in those with NUDT15 deficiency before dose reduction is applied in these groups of patients. Combining evidence from our preclinical models and retrospective studies, it is reasonable to postulate that a dose reduction of ∼90% (ie, ∼10 mg/m2 per day of MP) might be appropriate to avoid excessive toxicity without negative effects on leukemia-free survival. That said, the impact of genotype-guided MP dosing on treatment outcome in patients with ALL might be more complicated, given the plethora of factors contributing to leukemia cell sensitivity to thiopurines.44-47
Although myelosuppression is the main adverse effect of thiopurines, this class of drugs can also cause hepatic and gastrointestinal toxicities.48,49 MP liver toxicities are influenced by TPMT, as clearly documented in mice,50 likely attributed to methylated thiopurine metabolites.51 In contrast, we did not observe any difference in hepatic damage by Nudt15 genotype during thiopurine therapy, implying that Nudt15 is not involved in anabolism of methylated thioguanine nucleotides.39 Interestingly, thiopurine-induced damage to the digestive tract was readily detected in Nudt15−/− mice, especially at high doses (Figure 3B). Largely restricted to intestinal segments, lesions were related to death of proliferative crypt cells, suggesting excessive cytotoxic effects of thiopurines as the underlying causes (similar to thiopurine hematopoietic toxicity). Therefore, preemptive dose reduction in NUDT15-deficient patients may also alleviate gastrointestinal adverese effects of thiopurines.
Nudt15−/− mice did not seem to have any developmental defects, and we did not observe any gross functional consequences associated with the loss of this gene other than thiopurine toxicity. Although NUDT15 has been linked to purine (especially guanosine nucleotide) metabolism in vitro,52,53 defective NUDT15 alleles are not known to cause health conditions in human. Because there are 22 NUDT genes with various nucleotide hydrolase activities,54 it is possible that NUDT15 plays a nonexclusive function in nucleotide biosynthesis, which can be easily compensated for by other NUDT genes. A comprehensive metabolomic profiling of Nudt15−/− vs wild-type mice might identify endogenous metabolites regulated by Nudt15 and shed light on the physiological functions of this gene.
In conclusion, using a novel Nudt15-knockout mouse model, we established the principle of Nudt15 genotype–guided thiopurine dose individualization, which may inform clinical implementation of this pharmacogenetic precision medicine strategy in the large number of patients receiving this class of essential drugs.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and parents who participated in the clinical trials.
This work was supported by the National Institutes of Health, National Institute of General Medical Sciences (GM118578 and GM115279), the National Institutes of Health, National Cancer Institute (CA021765), American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital, V Foundation for Cancer Research (T2015-006), National Medical Research Council, Singapore (NMRC/CSA/0053/2013), Singapore Totalisator Board, Children’s Cancer Foundation, VIVA Foundation for Children with Cancer, and National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative. The JPLSG ALL-B12 study is supported by the Japan Agency for Medical Research and Development (grant 17ck0106334). T. Moriyama is supported by the Garwood Fellowship at St. Jude Children’s Research Hospital. M.S. and U.H. are supported by the Robert Bosch Stiftung Stuttgart.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.J.Y. designed the study; R.N., T. Moriyama, C.C.S., T.-N.L., and L.L. performed the experiments; L.J.J. contributed to necropsy analysis; R.N., T. Moriyama, W.Y., and J.J.Y. performed and analyzed statistical studies; K.K., H.T., U.H., M.S., M.T., T. Morio, A.M., S.K., N.J., K.R.R., M.K., K.K., A.E.-J.Y., and H.H. contributed to collections of human samples and clinical data; R.N., T. Moriyama, L.J.J., and J.J.Y. contributed to the writing of the manuscript; and all authors critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jun J. Yang, Hematological Malignancies Program, Comprehensive Cancer Center, Department of Pharmaceutical Sciences, Department of Oncology, St. Jude Children’s Research Hospital, MS313, 262 Danny Thomas Place, Memphis, TN 38105-3678; e-mail: jun.yang@stjude.org.
References
- 1.Karran P, Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat Rev Cancer. 2008;8(1):24-36. [DOI] [PubMed] [Google Scholar]
- 2.Elion GB. The purine path to chemotherapy. Science. 1989;244(4900):41-47. [DOI] [PubMed] [Google Scholar]
- 3.Koren G, Ferrazini G, Sulh H, et al. Systemic exposure to mercaptopurine as a prognostic factor in acute lymphocytic leukemia in children. N Engl J Med. 1990;323(1):17-21. [DOI] [PubMed] [Google Scholar]
- 4.Goldberg R, Irving PM. Toxicity and response to thiopurines in patients with inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2015;9(7):891-900. [DOI] [PubMed] [Google Scholar]
- 5.Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood. 1999;93(9):2817-2823. [PubMed] [Google Scholar]
- 6.Reinisch W, Angelberger S, Petritsch W, et al. ; International AZT-2 Study Group. Azathioprine versus mesalazine for prevention of postoperative clinical recurrence in patients with Crohn’s disease with endoscopic recurrence: efficacy and safety results of a randomised, double-blind, double-dummy, multicentre trial. Gut. 2010;59(6):752-759. [DOI] [PubMed] [Google Scholar]
- 7.Maltzman JS, Koretzky GA. Azathioprine: old drug, new actions. J Clin Invest. 2003;111(8):1122-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vora A, Goulden N, Wade R, et al. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2013;14(3):199-209. [DOI] [PubMed] [Google Scholar]
- 9.Escherich G, Richards S, Stork LC, Vora AJ; Childhood Acute Lymphoblastic Leukaemia Collaborative Group (CALLCG). Meta-analysis of randomised trials comparing thiopurines in childhood acute lymphoblastic leukaemia. Leukemia. 2011;25(6):953-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vora A, Mitchell CD, Lennard L, et al. ; National Cancer Research Network Childhood Leukaemia Working Party. Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trial. Lancet. 2006;368(9544):1339-1348. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen SN, Grell K, Nersting J, et al. DNA-thioguanine nucleotide concentration and relapse-free survival during maintenance therapy of childhood acute lymphoblastic leukaemia (NOPHO ALL2008): a prospective substudy of a phase 3 trial. Lancet Oncol. 2017;18(4):515-524. [DOI] [PubMed] [Google Scholar]
- 12.Connell WR, Kamm MA, Ritchie JK, Lennard-Jones JE. Bone marrow toxicity caused by azathioprine in inflammatory bowel disease: 27 years of experience. Gut. 1993;34(8):1081-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmiegelow K, Nielsen SN, Frandsen TL, Nersting J. Mercaptopurine/methotrexate maintenance therapy of childhood acute lymphoblastic leukemia: clinical facts and fiction. J Pediatr Hematol Oncol. 2014;36(7):503-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lennard L, Cartwright CS, Wade R, Vora A. Thiopurine dose intensity and treatment outcome in childhood lymphoblastic leukaemia: the influence of thiopurine methyltransferase pharmacogenetics. Br J Haematol. 2015;169(2):228-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fotoohi AK, Coulthard SA, Albertioni F. Thiopurines: factors influencing toxicity and response. Biochem Pharmacol. 2010;79(9):1211-1220. [DOI] [PubMed] [Google Scholar]
- 16.Hedeland RL, Hvidt K, Nersting J, et al. DNA incorporation of 6-thioguanine nucleotides during maintenance therapy of childhood acute lymphoblastic leukaemia and non-Hodgkin lymphoma. Cancer Chemother Pharmacol. 2010;66(3):485-491. [DOI] [PubMed] [Google Scholar]
- 17.Ebbesen MS, Nersting J, Jacobsen JH, et al. Incorporation of 6-thioguanine nucleotides into DNA during maintenance therapy of childhood acute lymphoblastic leukemia-the influence of thiopurine methyltransferase genotypes. J Clin Pharmacol. 2013;53(6):670-674. [DOI] [PubMed] [Google Scholar]
- 18.Diouf B, Cheng Q, Krynetskaia NF, et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17(10):1298-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krynetskaia NF, Brenner TL, Krynetski EY, et al. Msh2 deficiency attenuates but does not abolish thiopurine hematopoietic toxicity in msh2-/- mice. Mol Pharmacol. 2003;64(2):456-465. [DOI] [PubMed] [Google Scholar]
- 20.Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91(23):2001-2008. [DOI] [PubMed] [Google Scholar]
- 21.Lennard L, Lilleyman JS, Van Loon J, Weinshilboum RM. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336(8709):225-229. [DOI] [PubMed] [Google Scholar]
- 22.Gardiner SJ, Gearry RB, Begg EJ, Zhang M, Barclay ML. Thiopurine dose in intermediate and normal metabolizers of thiopurine methyltransferase may differ three-fold. Clin Gastroenterol Hepatol. 2008;6(6):654-660, quiz 604. [DOI] [PubMed] [Google Scholar]
- 23.Schwab M, Schäffeler E, Marx C, et al. Azathioprine therapy and adverse drug reactions in patients with inflammatory bowel disease: impact of thiopurine S-methyltransferase polymorphism. Pharmacogenetics. 2002;12(6):429-436. [DOI] [PubMed] [Google Scholar]
- 24.Relling MV, Pui CH, Cheng C, Evans WE. Thiopurine methyltransferase in acute lymphoblastic leukemia. Blood. 2006;107(2):843-844. [DOI] [PubMed] [Google Scholar]
- 25.Relling MV, Altman RB, Goetz MP, Evans WE. Clinical implementation of pharmacogenomics: overcoming genetic exceptionalism. Lancet Oncol. 2010;11(6):507-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Relling MV, Gardner EE, Sandborn WJ, et al. ; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011;89(3):387-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang JJ, Landier W, Yang W, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol. 2015;33(11):1235-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moriyama T, Nishii R, Perez-Andreu V, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet. 2016;48(4):367-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang SK, Hong M, Baek J, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet. 2014;46(9):1017-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valerie NC, Hagenkort A, Page BD, et al. NUDT15 hydrolyzes 6-thio-deoxyGTP to mediate the anticancer efficacy of 6-thioguanine. Cancer Res. 2016;76(18):5501-5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247-256. [DOI] [PubMed] [Google Scholar]
- 33.Pelletier S, Gingras S, Green DR. Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity. 2015;42(1):18-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659-661. [DOI] [PubMed] [Google Scholar]
- 35.Quinlivan EP, Gregory JF III. DNA digestion to deoxyribonucleoside: a simplified one-step procedure. Anal Biochem. 2008;373(2):383-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobsen JH, Schmiegelow K, Nersting J. Liquid chromatography-tandem mass spectrometry quantification of 6-thioguanine in DNA using endogenous guanine as internal standard. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;881-882:115-118. [DOI] [PubMed] [Google Scholar]
- 37.Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2006;103(17):6688-6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeoh AE, Ariffin H, Chai EL, et al. Minimal residual disease-guided treatment deintensification for children with acute lymphoblastic leukemia: results from the Malaysia-Singapore acute lymphoblastic leukemia 2003 study. J Clin Oncol. 2012;30(19):2384-2392. [DOI] [PubMed] [Google Scholar]
- 39.Moriyama T, Nishii R, Lin TN, et al. The effects of inherited NUDT15 polymorphisms on thiopurine active metabolites in Japanese children with acute lymphoblastic leukemia. Pharmacogenet Genomics. 2017;27(6):236-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ailing Z, Jing Y, Jingli L, Yun X, Xiaojian Z. Further evidence that a variant of the gene NUDT15 may be an important predictor of azathioprine-induced toxicity in Chinese subjects: a case report. J Clin Pharm Ther. 2016;41(5):572-574. [DOI] [PubMed] [Google Scholar]
- 41.Asada A, Nishida A, Shioya M, et al. NUDT15 R139C-related thiopurine leukocytopenia is mediated by 6-thioguanine nucleotide-independent mechanism in Japanese patients with inflammatory bowel disease. J Gastroenterol. 2016;51(1):22-29. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki H, Fukushima H, Suzuki R, et al. Genotyping NUDT15 can predict the dose reduction of 6-MP for children with acute lymphoblastic leukemia especially at a preschool age. J Hum Genet. 2016;61(9):797-801. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka Y, Kato M, Hasegawa D, et al. Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol. 2015;171(1):109-115. [DOI] [PubMed] [Google Scholar]
- 44.Zhang R, Haag JD, Gould MN. Reduction in the frequency of activated ras oncogenes in rat mammary carcinomas with increasing N-methyl-N-nitrosourea doses or increasing prolactin levels. Cancer Res. 1990;50(14):4286-4290. [PubMed] [Google Scholar]
- 45.Tzoneva G, Perez-Garcia A, Carpenter Z, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19(3):368-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyer JA, Wang J, Hogan LE, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45(3):290-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li B, Li H, Bai Y, et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med. 2015;21(6):563-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ben Salah L, Belkhiria el Haj Amor M, Chbili C, et al. Analysis of thiopurine S-methyltransferase phenotype-genotype in a Tunisian population with Crohn’s disease. Eur J Drug Metab Pharmacokinet. 2013;38(4):241-244. [DOI] [PubMed] [Google Scholar]
- 49.Liu YP, Xu HQ, Li M, et al. Association between thiopurine S-methyltransferase polymorphisms and azathioprine-induced adverse drug reactions in patients with autoimmune diseases: a meta-analysis. PLoS One. 2015;10(12):e0144234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartford C, Vasquez E, Schwab M, et al. Differential effects of targeted disruption of thiopurine methyltransferase on mercaptopurine and thioguanine pharmacodynamics. Cancer Res. 2007;67(10):4965-4972. [DOI] [PubMed] [Google Scholar]
- 51.Evans WE, Hon YY, Bomgaars L, et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J Clin Oncol. 2001;19(8):2293-2301. [DOI] [PubMed] [Google Scholar]
- 52.Takagi Y, Setoyama D, Ito R, Kamiya H, Yamagata Y, Sekiguchi M. Human MTH3 (NUDT18) protein hydrolyzes oxidized forms of guanosine and deoxyguanosine diphosphates: comparison with MTH1 and MTH2. J Biol Chem. 2012;287(25):21541-21549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter M, Jemth AS, Hagenkort A, et al. Crystal structure, biochemical and cellular activities demonstrate separate functions of MTH1 and MTH2. Nat Commun. 2015;6:7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McLennan AG. The Nudix hydrolase superfamily. Cell Mol Life Sci. 2006;63(2):123-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.