

Abstract
The distribution of aqueous Pb(II) sorbed at the interface between Burkholderia cepacia biofilms and hematite (α-Fe2O3) or corundum (α-Al2O3) surfaces has been probed by using an application of the long-period x-ray standing wave technique. Attached bacteria and adsorbed organic matter may interfere with sorption processes on metal oxide surfaces by changing the characteristics of the electrical double layer at the solid–solution interface, blocking surface sites, or providing a variety of new sites for metal binding. In this work, Pb Lα fluorescence yield profiles for samples equilibrated with 10−7 to 10−3.8 M Pb(II) were measured and modeled to determine quantitatively the partitioning of Pb(II) at the biofilm–metal oxide interface. Our data show that the reactive sites on the metal oxide surfaces were not passivated by the formation of a monolayer biofilm. Instead, high-energy surface sites on the metal oxides form the dominant sink for Pb(II) at submicromolar concentrations, following the trend α-Fe2O3 (0001) > α-Al2O3 (11̄02) > α-Al2O3 (0001), despite the greater site density within the overlying biofilms. At [Pb] > 10−6 M, significant Pb uptake by the biofilms was observed.
The dissolved concentration of trace metals in terrestrial and marine environments is directly linked to sorption and precipitation reactions at mineral surfaces (1, 2). However, various types of natural organic matter form ubiquitous surface-coatings on minerals exposed to aqueous solutions (3–7). Among the most complex and dynamic types of common organic coatings are “biofilms”. Biofilms form when microbial organisms attach to a surface and produce a highly hydrated framework of extracellular polymers in which the microorganisms become embedded (8). These biofilms may have a sorptive capacity similar in magnitude to many reactive mineral substrates (e.g., ref. 9) and thus are potentially significant sinks for metals. The presence of organic coatings may also alter the reactivity of the underlying mineral surface through blocking of high-energy surface sites or modifying the electrical properties of the mineral–water interface (3, 10–14). In particular, other workers have noted that bacterial cells often preferentially attach to surface features such as scratches, pits, cleavage steps, and edges of mineral grains (e.g., refs. 6 and 15). Passivation of the mineral surfaces could result if functional groups present within bacterial surface polymers (or within the exopolysaccharide matrix) were directly bound by a ligand-exchange mechanism to these high-energy sites on the mineral surface (e.g., refs. 12 and 13). How these biofilm coatings may alter metal ion partitioning between mineral surfaces and the surrounding aqueous environment is largely unresolved and challenging to probe experimentally.
Experimental Approach.
Lead is one of the most ubiquitous heavy metal contaminants in soils and aquatic ecosystems (16), and is a well-known neurotoxin in children (17). Pb forms strong complexes with both organic matter and mineral surfaces, and thus Pb partitioning is particularly interesting to study in heterogeneous biofilm–mineral systems. For this study, we have chosen Burkholderia cepacia, a highly versatile Gram-negative chemoorganotrophic bacterium commonly found in soil environments, as a model biofilm-forming organism, because of its tendency to produce biofilms composed of nearly a monolayer of bacterial cells under nutrient-limited conditions (e.g., ref. 18 and Fig. 1B). This relatively simple type of biofilm provides a model system in which to investigate Pb(II) sorption at the biofilm–metal oxide interface without the complicated Pb distributions that may exist within a more dynamic biofilm with significant three-dimensional structure. The α-Al2O3 (0001) and (11̄02) and α-Fe2O3 (0001) substrates used in this work represent well-characterized model system analogs for naturally abundant iron and aluminum (hydr)oxide phases, which are among the most common materials involved in trace metal sorption in natural systems (19). From previous work on clean, uncoated surfaces, we expect the reactivity of the three substrates to follow the order α-Fe2O3 (0001) > α-Al2O3 (11̄02) > α-Al2O3 (0001). Studies under ambient conditions have shown that these oxides exhibit a significant difference in reactivity toward dissolved Pb(II). For example, Pb(II) sorption is relatively weak on the α-Al2O3 (0001) surface (20), and results in the formation of outer-sphere complexes at high [Pb]. In contrast, spectroscopic data show that Pb(II) uptake is stronger on α-Al2O3 (11̄02) (21) and α-Fe2O3 (0001) (J. Barga, personal communication), because of the formation of inner-sphere surface complexes. Therefore, we have used these substrates to assess whether these differences are preserved when the metal-oxide surfaces are coated with B. cepacia biofilms.
Figure 1.

(A) Schematic diagram of the x-ray standing wave (XSW) intensity above the surface as a function of x-ray incidence angle. The period and position of the standing wave antinodes is varied by scanning the incident angle through the region of total external reflection. (B) Epifluorescent image of DAPI-stained B. cepacia attached to a α-Al2O3 (0001) surface. The exopolysaccharide matrix surrounding the cells is not visible. (Scale bar = 10 μm.) (C) Example of a fluorescence yield (FY) profile derived from a two-component Pb(II) distribution. Fluorescence intensity at low angles is associated with atoms residing above the reflecting surface (i.e., biofilm), whereas signal localized at the critical angle is sensitive to atoms residing at the surface.
In this work we have applied the long-period x-ray standing wave (XSW) technique to determine quantitatively the partitioning of Pb(II) within the B. cepacia-coated α-Al2O3 and α-Fe2O3 single crystal substrates. Previous studies have shown the utility of the long-period XSW technique for determining the spatial distribution of heavy metal ions within a variety of organic thin films and model membranes formed on mirror surfaces (23–25). Long-period XSWs are generated from the interference between an incident and reflected x-ray beam at incident angles below the critical angle for total external reflection of the substrate (≈160 mdeg and 180 mdeg for α-Al2O3 and α-Fe2O3, at 14 keV) (Fig. 1A). The standing wave period varies from >1,000 Å at low angles to Dc at the critical angle of the substrate (Dc is ≈159 Å and 141 Å for α-Al2O3 and α-Fe2O3), where the first antinode is coincident with the surface plane. For a layer of fluorescing atoms at a fixed position z, the fluorescent intensity is proportional to the standing wave intensity at the atom layer position (26). For a diffuse or continuous metal distribution, which we assume for our experiments, the net fluorescence yield, Y(θ), is given by (24, 27, 28)
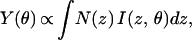 |
1 |
where N(z) is the distribution function for the metal atom and I(z, θ) is the standing wave intensity. For an atom layer localized near the reflecting substrate surface, the fluorescent yield profile will be sharply peaked near the critical angle. Strong fluorescent intensity at angles below the critical angle is indicative of atoms residing above the reflecting surface (Fig. 1C).
XSW experiments were conducted for each biofilm-coated crystal surface as a function of [Pb] (10−7 to 10−3.8 M) at 0.005 M ionic strength and pH 6. In situ XSW experiments at [Pb] from 10−5.2 to 10−3.2 M have also been conducted on clean α-Al2O3 surfaces in the absence of a biofilm (29). The x-ray reflectivity and Pb Lα fluorescence signal were collected as a function of incident angle at a fixed x-ray energy of 14 keV (above the Pb LIII absorption edge) (Fig. 2). Least-squares fits to the total normalized yield were carried out by using a two-component distribution function [N(z) in Eq. 1], to differentiate between Pb located at the crystal surface vs. Pb sorbed to the overlying biofilm. The partitioning data we have obtained demonstrate that the majority of sorbed Pb is sequestered at the metal oxide surface at submicromolar [Pb]. In particular, a small fraction of the metal oxide surface sites exhibits a considerably higher affinity toward Pb(II) than sites within the biofilm.
Figure 2.

Measured (dashed) and modeled (line) reflectivity [LogI1/Io] profiles and Pb Lα FY profiles (circles) with model fits (line) for the α-Al2O3 (0001), α-Al2O3 (11̄02) and α-Fe2O3 (0001) at 10−6 M (Upper) and 10−5 M (Lower) [Pb].
Materials and Methods
Sample Preparation.
Highly polished, 2-inch (5-cm) diameter (0001) and (11̄02) single-crystal α-Al2O3 substrates (Union Carbide) were obtained commercially. The α-Fe2O3 (0001) substrate was grown as a 1,400-Å thin film on α-Al2O3 (0001) by reactive oxygen multiple beam epitaxy methods at Pacific Northwest National Laboratories (30). Substrates were prepared as described by Eng et al. (31). Biofilms of B. cepacia (American Type Culture Collection no. 17616) were grown aerobically by using a minimal medium [200 μM CaCl2/150 μM MgSO4/90 μM (NH4)2SO4/150 μM KNO3,/10 μM NaHCO3/5 μM KH2PO4 and 500 μM sodium acetate as a carbon and energy source, pH 6] modified from Nelson et al. (18). Each crystal was inserted into a Teflon holder suspended vertically in a 125-ml Teflon flask equipped with ports for the introduction and removal of media and autoclaved before the introduction of 100 ml of an active culture of B. cepacia (OD600 ≈ 0.1). After a contact time of 12 h, the liquid was removed aseptically and exchanged with fresh medium. The medium was purged once every 24 h. After 6 days, the crystals were extracted, rinsed 3 times in 0.005 M NaNO3 solution (pH 6) to remove excess medium, and exposed to x-rays tuned to 14 keV for 5 min to render the cells metabolically inactive before equilibration with Pb(II). Metabolic effects were avoided to directly compare passive sorption processes within the biofilm vs. the mineral surface.
Each crystal was transferred to a CO2-free glove bag continuously flushed with N2, immersed in 100 ml of Pb(NO3)2 solution, and fixed at pH 6 and 0.005 M ionic strength, with NaNO3 as the background electrolyte. After equilibration for 2 h, each crystal was slowly withdrawn from solution under a jet of N2 to remove bulk water and inserted into a Teflon cell with a Kapton lid that was continuously purged with humidified N2. The cell was then mounted onto a grazing-incidence apparatus designed to control the x-ray incidence angle.
Data Collection.
XSW measurements were conducted on beamline VI-2 at the Stanford Synchrotron Radiation Laboratory. The x-rays were focused by using a Pt-coated mirror upstream of the monochromator, tuned to 14 keV by using a Si (111) double crystal monochromator, and collimated to 5 mm horizontal by 0.035–0.05 mm vertical. The incidence angle was scanned in 2.5 mdeg steps from 0 to 500 mdeg to collect the reflectivity [log (I1/Io)] by using Ar-filled ionization chambers. The fluorescence yield (FY) was collected simultaneously by using a 13-element Ge array detector (Canberra, Australia) coupled to digital x-ray processor (DXP) electronics (X-Ray Instrumentation Associates, Newark, CA) aligned perpendicular to the incident beam and inclined greater than 10° with respect to the sample to increase solid angle. The detector elements viewed the entire illuminated area on the sample throughout the measurements. The Pb FY profiles were generated by integrating the Pb Lα fluorescence peak in the emission spectrum, collected at each angle, followed by background subtraction and normalization relative to Io. The FY profiles were found to be reproducible [i.e., surface Pb/biofilm Pb (S/B) ratios differed by less than 5%] between samples, as a function of time (2–30 h) and following desorption/re-equilibration experiments.
Data Analysis.
The FY data were modeled by using a biofilm layer thickness of 10,000 Å (designated to represent a monolayer biofilm nominally 1 μm thick) and biofilm densities ranging from 0.3 to 0.9 g/cm3 as determined from the reflectivity and FY data. The Pb distribution function used in Eq. 1 was a simple two-box model in which the mineral surface component was designated as the 5-Å region adjacent to a reflecting surface and the biofilm component constituted the upper 9,995 Å of the overlayer. When the parameters derived from fitting the reflectivity data were used, the FY was calculated by using Eq. 1, assuming that the incident and reflected beams were fully coherent over the length scale of the biofilm. The maximum path-length difference for x-rays at the top and bottom of the 1-μm biofilm layer is ≈140 Å at 0.4°. Compared with our estimate of the longitudinal coherence length of the incident beam of ≈310 Å (based on the estimated energy resolution of 20 eV at 14 keV), we expect our assumption of coherent interference to be valid.
The intensity modulation of the XSW field along the direction of the surface normal (z) above the substrate surface as a function of the incidence angle (θ) is calculated from
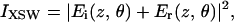 |
2 |
where Ei(z, θ) and Er(z, θ) are the complex amplitudes of the incident beam and reflected beams, respectively. The values of Ei(zo, θ) and Er(zo, θ) at any interface (zo) within a stratified model are calculated from the optical recursion formula (or equivalently by using a matrix method) (26, 32, 33). The E-field amplitudes at any position z (E(z, θ)) are then found by applying a phase shift from the interface which also accounts for attenuation within the layer (26, 32). The fluorescent yield calculated from Eq. 1 includes the change in illuminated sample area as a function of incident angle and attenuation of the fluorescent x-rays within the biofilm.
The most sensitive parameter in determining the shape of the FY profiles is the ratio of the surface-to-biofilm bound [Pb]. For example, similar FY profiles are calculated for biofilm thicknesses ranging from 1,000 Å to greater than 10,000 Å if the [Pb] concentration for the overlayer is changed proportionally. If the Pb(II) associated with the biofilm accumulated in a single discrete, two-dimensional layer (i.e., if the majority of biofilm Pb were bound to the top surface of the biofilm or if the layer thickness is <10% of the biofilm thickness), the FY profiles would exhibit significant fine structure below the critical angle, which was not observed. Therefore, we have no evidence for a vertical stratification of the biofilm-bound Pb. However, there may exist a random, heterogeneous three-dimensional Pb(II) distribution within the biofilm which results in FY profiles that can be successfully modeled by using an average [Pb].
A least-squares routine was used to fit the Pb Lα FY data between 0.04° and 0.4°. The [Pb] in the surface and biofilm layers was varied to simulate the FY profiles (reported as a surface Pb/biofilm Pb ratio, S/B). The S/B ratios extracted from the fitting had a precision of ± 10% within the sensitivity range of the measurement (S/B ratios 20 to 0.05). Outside of this range, there is no observed change in the calculated profiles, and we do not report fitting results for any spectra that fall into this category.
Microscopy.
Air-dried samples were examined by using a JEOL JSM-5600LV scanning electron microscope at 2-keV accelerating voltage to determine the cell density on each substrate. Samples stained with DAPI (4′,6-diamidino-2-phenylindole) nucleic acid stain were also examined by using a Nikon Eclipse E600 microscope with an epifluorescent attachment (Fig. 1B). Cell densities were similar for all three substrates and ranged from 1011.7 to 1011.9 cells per m2. Cells were typically closely packed, although numerous gaps of ≈5 μm2 were distributed across the crystal surfaces. A range of estimates of bacterial surface functional group site densities between 10−4.2 and 10−3.6 (mol of cell surface sites per m2 of oxide surface area) can be calculated by assuming an average bacterial coverage of 6.5 × 1011 cells per m2 of crystal surface area, cell weight of ≈2 × 10−13 g per cell, cell-surface area of ≈140 m2 per g cells (e.g., ref. 34), and cell-site density of 3.5 μmol of sites per m2 of cell surface obtained for the Gram-positive Bacillus subtilis (35) or 1.77 μmol/mg derived for the Gram-negative Shewanella putrefaciens (36), respectively.
Results and Discussion
There is a large variation in FY profiles among the different biofilm-coated substrates (Fig. 2). At 10−6 M Pb(II), the FY maximum is located at the critical angle for the α-Fe2O3 (0001) and α-Al2O3 (11̄02) surfaces, indicating that Pb is dominantly sorbed to the metal oxide surface. This is in sharp contrast to the much broader FY profile for the α-Al2O3 (0001) surface at 10−6 M Pb(II), which shows two maxima, indicating that a significant fraction of Pb(II) is sequestered within the overlying biofilm (Fig. 2 Upper). With a 10-fold increase in the equilibrium [Pb], there is a striking increase in the Pb FY at low incident angles for all three surfaces (Fig. 2 Lower, compared with Upper), which demonstrates that a larger fraction of the total adsorbed Pb is located within the biofilms (see Fig. 1C). At this higher [Pb], the FY profiles for both α-Al2O3 surfaces are strongly biofilm dominated, whereas the profile for the α-Fe2O3 now shows two maxima.
The FY spectra obtained for the entire set of substrates and experimental conditions were modeled to obtain a ratio of surface to biofilm bound Pb (S/B ratio) as a function of [Pb] (Fig. 3) (see Data Analysis). A similar trend is obtained for each of the substrates examined. The largest S/B ratios are observed at [Pb] ⩽ 10−6 M, whereas at greater [Pb] the S/B ratios rapidly decrease. The sharp decrease in S/B indicates that some fraction of metal oxide sites reach saturation and the biofilm becomes an important sink for Pb. At [Pb] > ≈10−4.5 M, S/B ratios for the α-Fe2O3 (0001) and α-Al2O3 (11̄02) surfaces no longer decrease. This change does not result from saturation of the biofilm sites because the total uptake continues to increase as determined by the total FY measurements. Therefore, Pb sorption to another lower affinity site on the mineral substrate must be occurring to balance the continued uptake of Pb within the biofilm.
Figure 3.

S/B ratios vs. [Pb] for α-Al2O3 (0001) (gray circles), α-Al2O3 (11̄02) (empty circles), and α-Fe2O3 (0001) (filled circles). Fit curves (lines) for the S/B ratios for each surface were derived by using site densities and Kapp values reported in Table 1.
Qualitative analysis of the Pb FY profiles is sufficient for understanding two of the most important phenomena regarding Pb(II) partitioning at the biofilm–metal oxide interfaces that we studied. First, the largest S/B ratios for each surface are observed from 10−7 M to 10−6 M Pb(II), which indicates that high-affinity metal oxide surface sites control the initial partitioning of Pb(II) between the biofilm and metal oxide surface. Second, at a given equilibrium [Pb], the S/B ratios follow the trend α-Fe2O3 (0001) > α-Al2O3 (11̄02) > α-Al2O3 (0001) (Fig. 3), which demonstrates that the differences in reactivity among these surfaces are preserved in the presence of the biofilm
The simplest method for more quantitatively describing the total Pb(II) uptake in our systems is to use a set of generalized mass-action equations, treating the bacterial surface reactions in an analogous manner to the mineral surface reactions (e.g. ref. 37), so that L is defined as a reactive site associated with either the metal oxide surface or biofilm,
 |
3 |
The equilibrium sorption density (Γ, mol/m2) for each type of site is described by a Langmuir-type isotherm, where n is total number of available sites, and Kapp is the apparent stability constant,
 |
4 |
This approach generalizes the specific stoichiometry and proton dependence of the sorption reaction, but is useful for making a relative comparison between the Pb(II) affinity constants for the metal oxide vs. biofilm sites under our experimental conditions.
Applying this generalized reaction scheme, we have defined a system in which the total Pb(II) uptake is the result of Pb complexation reactions at three discrete sites. The minimum number of reactions required to describe the changes in S/B ratios as a function of [Pb] involves two reactions at the mineral surface (M1 and M2 sites) and one reaction within the biofilm (B site) (Table 1). Therefore, the total surface-to-biofilm ratio is calculated as
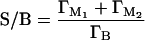 |
5 |
Two sites (one mineral site and one biofilm site) are not sufficient to model the full range of the uptake data on the α-Fe2O3 (0001) and α-Al2O3 (11̄02) surfaces because the plateau in the S/B ratios at [Pb] > 10−4.5 M is not due to a total saturation of all sites (the total FY continues to increase as [Pb] increases). Reactions at more than three sites would be severely under-constrained and therefore are not justified for the data, although there may be a large diversity of functional groups involved in Pb(II) uptake within the biofilm. Therefore, when the site densities in Table 1 were used, the Pb(II) partitioning data for each sample (S/B ratios vs. [Pb]; Fig. 3) were simulated by adjusting the Kapp values for the metal oxide and biofilm reactions, by using Eqs. 4 and 5. The site densities were scaled by fixing the M2 site density at 1 site per unit cell on the metal oxide surfaces, and the M1 site densities were fit with values approximately an order of magnitude smaller than the M2 site density. We applied the same estimated biofilm site density to all three surfaces, which is consistent with similar microscopic observations of the biofilm distribution on each sample.
Table 1.
Additive Langmuir model for Pb(II) uptake on biofilm-coated metal oxides
| Ligand | Substrate
|
|||||
|---|---|---|---|---|---|---|
| α-Al2O3 (0001)
|
α-Al2O3 (11̄02)
|
α-Fe2O3 (0001)
|
||||
| log (n) | log (Kapp) | log (n) | log (Kapp) | log (n) | log (Kapp) | |
| M1 (mineral) | −6.25 | 5.35 | −6.25 | 6.0 | −5.85 | 6.65* |
| M2 (mineral) | −5.05 | 2.7 | −5.15 | 3.55 | −5.1 | 3.5 |
| B (biofilm) | −4.5 | 3.5† | −4.5 | 3.5 | −4.5 | 3.5 |
Site densities (n; mol/m2) and affinity constants (Kapp; L/mol) for the mineral sites (M1 and M2) and biofilm site (B) invoked for Pb complexation. These parameters are used to derive a Langmuir isotherm for each site on each surface, using Eq. 4, to fit the trends in the S/B data, using Eq. 5. The Kapp are useful for a relative comparison of Pb(II) binding to the three reactive sites on the biofilm–α-Fe2O3 (0001) vs. –α-Al2O3 (11̄02) and –α-Al2O3 (0001) surfaces. The trends in the S/B data (Fig. 3) are sensitive to approximately ±0.1 log unit variations in the Kapp values for the given site densities.
A minimum value, because this site is saturated across the range of experimental conditions tested.
The FY profiles for the α-Al2O3 (0001) surface are beyond the sensitive range above 10−5.5 M Pb. Therefore, the estimated Kapp value for M2 site on the α-Al2O3 (0001) is a maximum value.
The greater S/B ratios on the α-Fe2O3 (0001) surface at lower [Pb] are due both to the larger affinity and higher site density of the M1 sites than on either alumina surface (Fig. 3) (Table 1). Comparison of the alumina surfaces shows that the M1 site densities are similar, but the greater affinity of the α-Al2O3 (11̄02) M1 sites makes this surface significantly more reactive than the α-Al2O3 (0001) at the smallest [Pb]. A similar trend in reactivity is observed for the M2 sites (Table 1).
The occurrence of two different types of reactive surface sites on the single-crystal surfaces is supported by spectroscopic information. For example, grazing-incidence x-ray absorption fine-structure (GI-XAFS) spectra for Pb(II) sorption to α-Al2O3 (0001) surfaces at low [Pb] in the absence of a biofilm (unpublished results) indicate that Pb binds in an inner-sphere mode to the α-Al2O3 (0001) surface at a different site than the outer-sphere complex observed by Bargar et al. (20) at higher [Pb]. We suggest that the high-affinity M1 sites are associated with surface defects (e.g., step edges associated with a terrace-and-step morphology; see ref. 38). Because the M1 site density corresponds to <1 site per 10 surface unit cells, the M1 distribution is too small to represent one of the regularly repeated terrace sites but does agree well with the defect density observed on similar α-Al2O3 substrates in ultrahigh vacuum photoemmision studies of water reaction (39). The larger number of high-affinity sites we have invoked on the α-Fe2O3 (0001) surface than on the alumina surfaces is indirectly supported by surface characterization of these substrates. Although the measured roughnesses of all of the substrates are on the order of 1–5 Å rms, the α-Fe2O3 thin films are more mosaic than the α-Al2O3 single crystals. The α-Fe2O3 thin films grow as columns with an in-plane domain size of ≈450 Å (30). Therefore, the occurrence of numerous domains bounded by step edges on the α-Fe2O3 crystal surface could be expected to provide more defect or high-energy sites than on the α-Al2O3 surfaces.
The greater reactivity of the α-Al2O3 (11̄02) surface M2 sites relative to the α-Al2O3 (0001) M2 sites may be because of the difference in the surface structure of the terminating plane. The fully hydroxylated α-Al2O3 (0001) surface is terminated by doubly coordinated hydroxyl groups (31), whereas singly, doubly, and triply coordinated oxygens are exposed at the hydroxylated α-Al2O3 (11̄02) surface (40). Similar differences in oxygen coordination have previously been invoked to predict differences in the acid-base character and reactivity toward heavy metals of the terminating planes (e.g., refs. 21 and 41).
Although the same biofilm site density is applied to all data sets in this study, the precise site density is poorly constrained. We used the smallest possible value for the site density within the biofilm given that we do not observe saturation ([B] = 10−4.5 mol of cell surface sites per m2 of crystal surface area) (Table 1), but the total biofilm site density on the bacterial surfaces may be significantly larger, given that our general estimates (see Microscopy) predict 10−4.2 to 10−3.6 mol of cell surface sites per m2 of crystal surface area. Moreover, a large number of additional biofilm sites are likely to be present within the exopolysaccharide matrix (e.g., ref. 18). Therefore, the Kapp value for Pb(II) sorption by the biofilm sites may be smaller than the value we have estimated.
Although a single reaction was sufficient to model Pb(II) uptake in the biofilm, the types of reactive sites present in the biofilm are potentially more heterogeneous than the sites present on the single-crystal oxide surfaces. Numerous types of amphoteric functional groups have been identified (e.g., refs. 42 and 43) and characterized (e.g., refs. 34 and 35) on bacterial surfaces. Therefore, the biofilm reaction used in the modeling procedure is likely an “effective reaction” accounting for Pb(II) associated with multiple functional groups in the biofilm and exopolysaccharide matrix.
Comparison of Pb(II) uptake on the two α-Al2O3 surfaces indicates that Pb coverage on the biofilm-coated metal oxides is similar to that observed in biofilm-free controls. X-ray photoelectron spectroscopy (XPS) data for uncoated crystals of α-Al2O3 (11̄02) and (0001) equilibrated with 10−4.3 M Pb(II) (pH 6) yield surface coverages of 1.5 and 0.5 μmol/m2, respectively, which is in good agreement with the mineral surface concentrations of 2.0 and 0.8 μmol/m2 predicted from the parameters reported in Table 1. Thus, we infer that the B. cepacia biofilms have not severely altered the intrinsic reactivity of the metal oxide surfaces, which is similar to the conclusions advocated by Zachara et al. (44) in their studies of mineral–humic acid mixtures. Our data indicate that at near-neutral pH and trace [Pb], Pb(II) sorption is dominated by complexation to high-affinity metal oxide surface sites, despite the excess of reactive sites within the overlying biofilms. In particular, the highly reactive mineral surface sites have not been “blocked” during biofilm formation, which has important implications for heavy metal transport in subsurface environments in which mineral surfaces may be only partially colonized by bacteria preferentially associated with surface defects.
This result validates efforts to predict the speciation of trace metals in natural systems by studying the reactivity of individual phases. Furthermore, the importance of high-energy mineral surface sites will be amplified in natural systems containing high surface-area grains or amorphous gels (particularly iron and manganese (hydr)oxides) (e.g., ref. 45). We would predict that biofilms will be more likely to form a significant sink for metal ions such as Pb(II) at high metal loadings, depending on the extent of biofilm formation (i.e., numbers of bacteria and amount of exopolysaccharides associated with the mineral surfaces in a given environment; Fig. 4). However, the interfacial properties of the biofilm–mineral interface, in particular the distribution of surface charge, remain challenging to probe or quantitatively describe (e.g., ref. 46).
Figure 4.

Total uptake (mol/m2) on biofilm-coated α-Al2O3 (0001) vs. α-Fe2O3 (0001) calculated by using the parameters in Table 1. At high [Pb], Pb is predominantly sorbed to the biofilms on both surfaces and the total Pb uptake will be sensitive to the amount of biofilm present. At low [Pb], Pb binding is dominated by high-affinity (M1) sites on the mineral surfaces. Enhanced uptake occurs on α-Fe2O3 (0001) even in the presence of a biofilm, demonstrating that sorption is mineralogically controlled at trace [Pb].
Although the results of the present study indicate that high-energy surface sites on corundum and hematite outcompete the majority of B. cepacia functional groups for Pb(II) on initial exposure to submicromolar [Pb], biofilm biomineralization processes will be critically important in heavy-metal sequestration over longer time periods. Localized metabolic activity may generate steep chemical gradients within the biofilm (47), potentially inducing precipitation with ligands such as CO and PO
and PO , or enhancing the mineral dissolution rates (e.g., ref. 6) and release of Al3+ (or Fe3+), which may ultimately lead to the formation of insoluble, mixed-metal phases such as plumbogummite [PbAl3(PO4)2(OH)5⋅(H2O)] in the case of Pb (e.g., ref. 22). Direct spectroscopic approaches, such as grazing-incidence x-ray absorption spectroscopy, coupled with the XSW approach described herein, promise to be invaluable tools for probing the long-term distribution and speciation of metal ions that accumulate at biofilm–mineral interfaces.
, or enhancing the mineral dissolution rates (e.g., ref. 6) and release of Al3+ (or Fe3+), which may ultimately lead to the formation of insoluble, mixed-metal phases such as plumbogummite [PbAl3(PO4)2(OH)5⋅(H2O)] in the case of Pb (e.g., ref. 22). Direct spectroscopic approaches, such as grazing-incidence x-ray absorption spectroscopy, coupled with the XSW approach described herein, promise to be invaluable tools for probing the long-term distribution and speciation of metal ions that accumulate at biofilm–mineral interfaces.
Acknowledgments
We thank two anonymous reviewers for constructive comments that helped improve this manuscript. We also thank Dr. Scott Chambers (Pacific Northwest National Laboratory, Richland, WA) for providing the molecular beam epitaxy (MBE)-grown thin-film α-Fe2O3 (0001) sample. This work was supported by National Science Foundation Grants NSF-EAR-9905755 and NSF-CHE-0089215, the Eugene Holman Stanford Graduate Fellowship (to A.S.T.), and a Corning Foundation Graduate Fellowship (to T.P.T.). We appreciate the assistance provided by the staff at the Stanford Synchrotron Radiation Laboratory. The Stanford Synchrotron Radiation Laboratory is funded by the Department of Energy (Offices of Basic Energy Sciences and Biological and Environmental Research) and by the National Institutes of Health.
Abbreviations
- XSW
x-ray standing wave
- S/B ratio
[(surface Pb)/(biofilm Pb)]
- FY
fluorescence yield
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Krauskopf K B. Geochim Cosmochim Acta. 1956;10:1–26. [Google Scholar]
- 2.Stumm W, Morgan J J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters. New York: Wiley; 1996. [Google Scholar]
- 3.Neihoff R A, Loeb G I. Limnol Oceanogr. 1972;17:7–16. [Google Scholar]
- 4.Hunter K A, Liss P S. Nature (London) 1979;282:873–825. [Google Scholar]
- 5.Mayer L M. Geochim Cosmochim Acta. 1994;58:1271–1284. [Google Scholar]
- 6.Barker W W, Welch S A, Chu S, Banfield J F. Am Mineral. 1998;83:1551–1563. [Google Scholar]
- 7.Bennet P C, Hiebert F K, Rogers J R. Hydrogeol J. 2000;8:47–62. [Google Scholar]
- 8.Characklis W G, Marshall K C. Biofilms. New York: Wiley; 1990. [Google Scholar]
- 9.Small T D, Warren L A, Roden E E, Ferris F G. Environ Sci Technol. 1999;33:4465–4470. [Google Scholar]
- 10.Tipping E, Cooke D. Geochim Cosmochim Acta. 1982;46:75–80. [Google Scholar]
- 11.Davis J A. Geochim Cosmochim Acta. 1982;48:679–691. [Google Scholar]
- 12.Gu B, Schmitt J, Chen Z, Liang L, McCarthy J F. Environ Sci Technol. 1994;28:38–46. doi: 10.1021/es00050a007. [DOI] [PubMed] [Google Scholar]
- 13.Mayer L M. Geochim Cosmochim Acta. 1999;63:207–215. [Google Scholar]
- 14.Floroiu M R, Davis A P, Torrents A. Environ Sci Technol. 2001;35:348–354. doi: 10.1021/es9913479. [DOI] [PubMed] [Google Scholar]
- 15.Edwards, K. J. & Rutenberg, A. D. Chem. Geol., in press.
- 16.Nriagu J O. The Biogeochemistry of Lead. New York: Wiley Interscience; 1986. [Google Scholar]
- 17.Agency for Toxic Substances and Disease Registry. The Nature and Extent of Lead Poisoning in Children in the United States: A Report to Congress. DHHS Doc. 99-2996. Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service; 1988. [Google Scholar]
- 18.Nelson Y M, Lion L W, Shuler M L, Ghiorse W C. Environ Sci Technol. 1996;30:2027–2035. [Google Scholar]
- 19.Brown G E, Jr, Henrich V E, Casey W H, Clark D L, Eggleston C, Felmy A, Goodman D W, Gratzel M, Maciel G, McCarthy M, et al. Chem Rev. 1999;99:77–174. doi: 10.1021/cr980011z. [DOI] [PubMed] [Google Scholar]
- 20.Bargar J R, Towle S N, Brown G E, Jr, Parks G A. Geochim Cosmochim Acta. 1996;60:3541–3547. [Google Scholar]
- 21.Bargar J R, Towle S N, Brown G E, Jr, Parks G A. J Colloid Interface Sci. 1997;185:473–492. doi: 10.1006/jcis.1996.4574. [DOI] [PubMed] [Google Scholar]
- 22.Morin G, Juillot F, Ildefonse P, Calas G, Samama J-C, Chevallier P, Brown G E., Jr Am Mineral. 2001;86:92–104. [Google Scholar]
- 23.Bedzyk M J, Bommarito G M, Caffrey M, Schildkraut J S. Science. 1988;241:1788–1791. doi: 10.1126/science.3175619. [DOI] [PubMed] [Google Scholar]
- 24.Bedzyk M J, Bommarito G M, Caffrey M, Penner T L. Science. 1990;248:51–56. doi: 10.1126/science.2321026. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Bedzyk M J, Penner T L, Caffrey M. Nature (London) 1991;543:377–380. doi: 10.1038/354377a0. [DOI] [PubMed] [Google Scholar]
- 26.de Boer D K G. Phys Rev B. 1991;44:498–511. doi: 10.1103/physrevb.44.498. [DOI] [PubMed] [Google Scholar]
- 27.Abruna H D, Bommarito G M, Acevedo D. Science. 1990;250:69–74. doi: 10.1126/science.250.4977.69. [DOI] [PubMed] [Google Scholar]
- 28.Bommarito G M, White J H, Abruna H D. J Phys Chem. 1990;94:8280–8288. [Google Scholar]
- 29.Trainor T P. Ph.D. Thesis. Stanford, CA: Stanford University; 2001. [Google Scholar]
- 30.Chambers S A. Surf Sci Rep. 2000;39:105–180. [Google Scholar]
- 31.Eng P J, Trainor T P, Brown G E, Jr, Waychunas G A, Newville M, Sutton S R, Rivers M L. Science. 2000;288:1029–1033. doi: 10.1126/science.288.5468.1029. [DOI] [PubMed] [Google Scholar]
- 32.Krol A, Sher C J, Kao Y H. Phys Rev B. 1988;38:8579–8592. doi: 10.1103/physrevb.38.8579. [DOI] [PubMed] [Google Scholar]
- 33.Parratt L G. Phys Rev. 1954;95:359–369. [Google Scholar]
- 34.Fein J B, Daughney C J, Yee N, Davis T A. Geochim Cosmochim Acta. 1997;61:3319–3328. [Google Scholar]
- 35.Cox J S, Smith S D, Warren L A, Ferris F G. Environ Sci Technol. 1999;33:4514–4521. [Google Scholar]
- 36.Sokolov I, Smith D S, Henderson G S, Gorby Y A, Ferris F G. Environ Sci Technol. 2001;35:341–347. doi: 10.1021/es001258s. [DOI] [PubMed] [Google Scholar]
- 37.Warren L A, Ferris F G. Environ Sci Technol. 1998;32:2331–2337. [Google Scholar]
- 38.Heffelfinger J R, Bench M W, Carter C B. Surf Sci. 1997;370:L168–L172. [Google Scholar]
- 39.Liu P, Kendelewicz T, Brown G E., Jr Surf Sci. 1998;412/413:315–332. [Google Scholar]
- 40.Trainor, T. P., Eng, P., Robinson, I. & Brown, G. E., Jr. Surf. Sci., in press.
- 41.Hiemstra T, Yong H, Van Riemsdijk W H. Langmuir. 1999;15:5942–5955. [Google Scholar]
- 42.Beveridge T J, Murray G E. J Bacteriol. 1980;148:876–887. doi: 10.1128/jb.141.2.876-887.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beveridge T J, Koval S F. Applied Environ Microbiol. 1981;42:325–335. doi: 10.1128/aem.42.2.325-335.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zachara J M, Resch C T, Smith S C. Geochim Cosmochim Acta. 1994;58:553–566. [Google Scholar]
- 45.Tessier A, Fortin D, Belzile N, DeVitre R R, Leppard G G. Geochim Cosmochim Acta. 1996;60:387–404. [Google Scholar]
- 46.Vermeer A W P, McCulloch J K, Van Riemsdijk W H, Koopal L K. Environ Sci Technol. 1998;33:3892–3897. [Google Scholar]
- 47.Costerton J W, Lewandowski Z, Caldwell D E, Korber D R, Lappin-Scott H M. Annu Rev Microbiol. 1995;49:711–745. doi: 10.1146/annurev.mi.49.100195.003431. [DOI] [PubMed] [Google Scholar]