

Abstract
The myeloperoxidase system of neutrophils uses hydrogen peroxide and chloride to generate hypochlorous acid, a potent bactericidal oxidant in vitro. In a mouse model of polymicrobial sepsis, we observed that mice deficient in myeloperoxidase were more likely than wild-type mice to die from infection. Mass spectrometric analysis of peritoneal inflammatory fluid from septic wild-type mice detected elevated concentrations of 3-chlorotyrosine, a characteristic end product of the myeloperoxidase system. Levels of 3-chlorotyrosine did not rise in the septic myeloperoxidase-deficient mice. Thus, myeloperoxidase seems to protect against sepsis in vivo by producing halogenating species. Surprisingly, levels of 3-bromotyrosine also were elevated in peritoneal fluid from septic wild-type mice and were markedly reduced in peritoneal fluid from septic myeloperoxidase-deficient mice. Furthermore, physiologic concentrations of bromide modulated the bactericidal effects of myeloperoxidase in vitro. It seems, therefore, that myeloperoxidase can use bromide as well as chloride to produce oxidants in vivo, even though the extracellular concentration of bromide is at least 1,000-fold lower than that of chloride. Thus, myeloperoxidase plays an important role in host defense against bacterial pathogens, and bromide might be a previously unsuspected component of this system.
Oxidants are thought to be key components of the neutrophil host defense system (1). Upon contact with a pathogen, neutrophils produce a respiratory burst characterized by intense uptake of oxygen. The resulting superoxide dismutates into hydrogen peroxide (H2O2) (2). The toxicity of H2O2 is greatly enhanced by the heme enzyme myeloperoxidase, which uses H2O2 to convert chloride (Cl−) into hypochlorous acid (HOCl) (3–8).
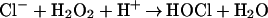 |
Remarkably, myeloperoxidase is the only mammalian enzyme known to oxidize Cl− to HOCl at plasma concentrations of halide (3–6).
Chloride is considered the major halide used by myeloperoxidase. Bromide (Br−) has attracted little attention because its extracellular concentration is at least 1,000-fold lower than that of Cl− (plasma [Cl−] 100 mM, plasma [Br−] 20–100 μM) (9). However, brominating intermediates such as HOBr are also potent antimicrobial oxidants in vitro (10, 11).
We recently demonstrated that myeloperoxidase can both chlorinate and brominate nucleobases at plasma levels of halide (12). In the reaction pathway, myeloperoxidase initially produces HOCl, which reacts with Br− to generate brominating intermediates (12). It has yet to be established whether this brominating pathway is physiologically relevant.
It also is not known whether the myeloperoxidase system is cytotoxic to bacteria in vivo, although myeloperoxidase-deficient mice are susceptible to fungal infection (13, 14). In the current study, we investigate the enzyme's role in host defense against invading bacteria by using a clinically relevant model of sepsis (15–18) and gas chromatography/mass spectrometry (GC/MS). Our results provide strong evidence that myeloperoxidase generates antimicrobial brominating and chlorinating intermediates in vivo.
Experimental Procedures
Materials.
All materials were purchased from Sigma or Fisher unless otherwise indicated. Isotope-labeled amino acids were from Cambridge Isotope Laboratories (Cambridge, MA). Rabbit polyclonal antibody was raised against a peptide present in mouse myeloperoxidase (NTLPKLNLTSWKET) (14).
Animals.
The Animal Studies Committee of Washington University School of Medicine approved all animal studies. Myeloperoxidase-deficient mice were generated in a 129/SvJ background; they were backcrossed at least four generations into the C57BL/6J background before use (14). C57BL/6J and 129/SvJ mice were from The Jackson Laboratory. Mice were maintained under pathogen-free conditions on a 12-h light-dark schedule and allowed ad libitum access to rodent diet 5001 (Harlan-Teklad, Madison, WI).
General Procedures.
Myeloperoxidase was purified from HL60 cells (19). Enzyme was apparently pure as assessed by its absorption spectrum (A430/A280 ratio > 0.8), SDS/PAGE, and peroxidase activity gel electrophoresis (20). Porcine eosinophil peroxidase (A415/A280 > 0.9) was from ExOxEmis (Little Rock, AR). Chloride-free NaOCl and HOBr were prepared as described (12, 21, 22). Peritoneal white blood cells were elicited with i.p. injection of 4% thioglycollate and harvested 24 h after injection (23). SDS/PAGE was performed by using 10% Bis-Tris NuPAGE gels (Invitrogen). The separated proteins were transferred onto a nitrocellulose membrane, which was blocked overnight at 4°C with 5% milk and 0.05% Tween 20 in PBS (10 mM phosphate/138 mM NaCl/2.7 mM KCl, pH 7.4). The blot was then incubated (1 h) with rabbit anti-mouse myeloperoxidase antibody (1:1,000) followed by goat anti-rabbit Ig antibody (1:10,000; Santa Cruz Biotechnology) conjugated with horseradish peroxidase. West Pico chemiluminescent substrate (Pierce) was added, and the membrane was exposed to autoradiography film and developed.
Reaction Conditions.
Reactions were carried out for 60 min at 37°C in buffer A (100 mM NaCl/50 mM sodium phosphate/100 μM diethylenetriaminepentaacetic acid) supplemented with 1 mM N-acetyl-L-tyrosine and either 50 μM hypohalous acid or 3 nM peroxidase and 50 μM H2O2. Buffer A was passed over a Chelex resin column before use to remove redox active transition metal ions. Reactions were initiated by the addition of oxidant and terminated with 0.1 mM methionine.
Reverse-Phase HPLC Analyses of Halogenated Amino Acids.
Analyses were performed by using a flow rate of 1 ml/min and an Ultrasphere ODS reverse-phase column (4.6 mm × 25 cm, 5 μm; Beckman Instruments) coupled to a Waters 484 tunable absorbance detector with monitoring at 275 nm. Solvent A was 0.1% trifluoroacetic acid (pH 2.5), and solvent B was 0.1% trifluoroacetic acid in methanol (pH 2.5). The gradient profile was isocratic elution at 66:34 (vol/vol) A/B over 2.1 min, linear increase of B from 34% to 40% over 30 min, isocratic elution at 60:40 A/B for 3 min, increase of B from 40% to 100% over 2 min, and isocratic elution at 100% B for 3 min. Products were quantified using integrated peak areas by comparison to standard curves of authentic N-Ac-chlorotyrosine and N-Ac-bromotyrosine.
Sepsis Model.
Cecal ligation and puncture of age-matched (8–16 weeks) and sex-matched mice were performed as described (16). The peritoneum was lavaged 24 h after surgery with 5 ml of PBS or with PBS supplemented with 50 μM butylated hydroxytoluene/100 μM diethylenetriaminepentaacetic acid/1 mM sodium azide/10 mM aminotriazole. Lavage fluid was stored at −80°C until analysis. Control experiments demonstrated that inclusion of antioxidants had no effect on the amount of products detected. For survival studies, animals were given i.p. injections of metrinodizole (35 mg/kg) and ceftriazone (50 mg/kg) ≈1 h after surgery. Antibiotic injections were repeated once 24 h after surgery. Mice were allowed ad libitum access to water and food.
Cell Analysis.
Cells were pelleted onto slides by centrifugation and stained with a modified Wright's stain (Dimscio & Associates, Manchester, MO). Cell counts were performed by the Department of Comparative Medicine (Washington University School of Medicine).
Peroxidase Activity Assay.
Peritoneal lavage fluid was centrifuged at 4°C for 10 min at 16,000 × g. Cetyltrimethylammonium bromide buffer (0.3%, 500 μl) containing 25% glycerol and 35 mM β-alanine (pH 4.5) was added to the pellet, and the mixture was sonicated for 10 min. After a second 10-min centrifugation at 16,000 × g, soluble proteins in the supernatant were subjected to native PAGE and peroxidase activity staining (20).
GC/MS Analysis.
Isotope-labeled internal standards (1 pmol of L-3-chloro[13C6]tyrosine, 500 fmol of L-3-bromo[13C6]tyrosine, and 10 nmol of L-[13C9,15N]tyrosine) were added to 500 μl of peritoneal fluid and prepared for GC/MS analysis as described (24, 25). Briefly, amino acids were isolated by using solid-phase extraction with a C18 column followed by a Supelco Chrom P column (24, 25). Samples were dried under anhydrous N2 and stored at −20°C until analysis.
After derivatization with ethyl heptafluorobutyrate and N-methyl-N-(t-butyldimethylsilyl)-trifluoroacetamide + 1% trimethylchlorosilane (MtBSTFA), the samples were dried under anhydrous N2 and resuspended in 50 μl of undecane/MtBSTFA (3:1 vol/vol). A 1-μl sample was analyzed by selected ion monitoring by using a Hewlett–Packard HP 6890 gas chromatograph coupled to an HP 5973 mass detector in the electron capture negative chemical ionization mode. The injector, transfer line, and source temperatures were initially set at 183°C, 300°C, and 250°C, respectively. The injector and transfer line temperatures were set to increase incrementally with the temperature gradient (180°C to 300°C at 40°C/min). 3-Chlorotyrosine and 3-bromotyrosine were quantified by using the respective ions at m/z 489 ([M–halide–t-butyl-dimethylsilyl]−) and their isotopically ring-labeled [13C6]-internal standards (m/z 495). Potential artifact formation during sample work-up was monitored as the appearance of ions at m/z 499 derived from L-[13C9,15N]tyrosine (24, 25).
Liquid Chromatography–Electrospray Ionization-MS.
N-acetyl amino acids were separated by reverse-phase HPLC and analyzed with a Finnigan LCQ (26). Full mass scanning (m/z 100–350) and tandem MS analyses were performed in the negative ionization mode.
Bacterial Killing.
The bactericidal activity of myeloperoxidase was quantified as described (27). Briefly, Klebsiella pneumoniae were cultured in Luria–Bertani broth to midlog growth phase, washed with PBS, and used at 109 bacteria/ml. Bacteria were incubated with 5 nM enzyme, 5 nM H2O2, and 100 mM NaCl in PBS (pH 5.5) for 15 min at 37°C. Serial dilutions were immediately spread on agar plates, and the colony-forming units were determined after overnight incubation at 37°C.
Statistical Analysis.
The survival study was evaluated by using a Fisher's exact P test. All other data were analyzed with a Student's t test. Significance was accepted at P < 0.05.
Results
Myeloperoxidase Deficiency Increases Mortality in the Cecal Ligation and Puncture (CLP) Model of Sepsis.
To evaluate the potential role of the myeloperoxidase system in host defense, we used myeloperoxidase-deficient mice and a clinically relevant and widely used model of intraabdominal infection and sepsis. In the CLP model, the blind-ended cecum is ligated and punctured (16), releasing intestinal microflora into the abdominal cavity. Previous studies have demonstrated that 35–45% of wild-type mice survive this procedure for longer than 7 days (16, 18). When we subjected myeloperoxidase-deficient mice to CLP, none of the animals were alive 5 days after surgery (Fig. 1). In contrast, 63% of the wild-type littermate control mice were alive after 5 days, and 38% were alive after 1 week (Fig. 1; P = 0.02).
Figure 1.

Myeloperoxidase deficiency impairs survival in a CLP model of sepsis. Mortality was monitored in myeloperoxidase-deficient (MPO−/−) mice and littermate wild-type (WT) controls.
Neutrophils predominated in the cell populations elicited from both the wild-type mice (n = 7; 77 ± 3% neutrophils, 23 ± 3% macrophages, 0.3 ± 0.2% eosinophils) and the myeloperoxidase-deficient mice (n = 11; 77 ± 4% neutrophils, 23 ± 3% macrophages, 0.9 ± 0.5% eosinophils). Red blood cells, cellular debris, and microorganisms were also apparent in the peritoneal fluid after the mice were subjected to CLP. Importantly, the cellular response to sepsis of the myeloperoxidase-deficient animals was comparable to that of the wild-type animals.
Levels of Free 3-Chlorotyrosine and 3-Bromotyrosine Rise Markedly During Sepsis.
To determine whether products of the myeloperoxidase system accumulate during sepsis, we focused on 3-chlorotyrosine, a product of cytotoxic HOCl (28). To explore the physiological significance of bromination by myeloperoxidase, we also quantified levels of 3-bromotyrosine. To recover free 3-chlorotyrosine and 3-bromotyrosine in the cellular and extracellular components of the lavage fluid, wild-type mice were subjected to CLP. After 24 h, inflammatory fluid was harvested from the peritoneum by lavage. The lavage fluid, which contained white blood cells, bacteria, and inflammatory exudate, was frozen. To recover free amino acids, we centrifuged thawed fluid to pellet cellular debris and harvested the supernatant.
Amino acids were derivatized with ethyl heptafluorobutyrate and MtBSTFA and analyzed by GC/MS in the electron capture-negative chemical ionization mode. To prevent the generation of halogenated tyrosines during sample preparation (24), we used a highly sensitive (<100 amol) and specific GC/MS method that avoids acidic conditions (24, 25).
The procedure detected compounds that exhibited major ions and retention times identical to those of authentic 3-chlorotyrosine and 3-bromotyrosine (Fig. 2a). Selected ion monitoring showed that the ions derived from the amino acids coeluted with those derived from 13C-labeled internal standards (Fig. 2 a and b). The identity of each compound was confirmed by comparison with authentic standards, using MtBSTFA derivatives of each oxidized amino acid. To monitor any artifactual generation of halogenated amino acids, we included L-[13C9,15N]tyrosine in the samples and looked for L-3-chloro[13C9,15N]tyrosine or L-3-bromo[13C9,15N]tyrosine (Fig. 2c). Artifact formation was negligible when either biological material or pure tyrosine was analyzed with this method.
Figure 2.

Electron capture-negative chemical ionization GC/MS analysis of the ethyl heptafluorobutyrate, MtBSTFA derivatives of 3-bromotyrosine and 3-chlorotyrosine in peritoneal inflammatory fluid of a wild-type mouse subjected to CLP. Note the simultaneous monitoring of endogenous (m/z 489; a), isotope-labeled (m/z 495; b), and artifactual (m/z 499; c) 3-chlorotyrosine (3-Cl-Tyr) and 3-bromotyrosine (3-Br-Tyr).
Peritoneal fluid from the sham-operated animals contained only low levels of 3-chlorotyrosine and 3-bromotyrosine (Fig. 3). Fluid from the CLP animals contained 16-fold higher levels of 3-chlorotyrosine (P = 0.03) and 6-fold higher levels of 3-bromotyrosine (P = 0.03). These observations indicate that generation of halogenating intermediates increases dramatically during acute inflammation induced by sepsis.
Figure 3.

Isotope dilution GC/MS quantification of 3-chlorotyrosine (3-Cl-Tyr; a) and 3-bromotyrosine (3-Br-Tyr; b) in the peritoneal inflammatory fluid of sham-operated and CLP-subjected mice. Oxidation products were monitored in myeloperoxidase-deficient (MPO−/−) mice and in wild-type (WT) mice in the 129/SvJ and C57BL/6J background.
Myeloperoxidase Generates 3-Chlorotyrosine and 3-Bromotyrosine During Sepsis.
To determine whether a pathway involving myeloperoxidase is responsible for the elevated levels of halogenated L-tyrosines seen in inflammatory exudates, we quantified levels of the amino acids in septic myeloperoxidase-deficient mice. Whereas lavage fluid levels of free 3-chlorotyrosine and 3-bromotyrosine rose when the wild-type mice became septic, 3-chlorotyrosine levels barely increased after the myeloperoxidase-deficient animals were subjected to CLP (P = 0.0004). The genetically altered mice also produced 59% less 3-bromotyrosine after CLP than the wild-type mice (P = 0.009).
The myeloperoxidase-deficient animals used for these experiments had been backcrossed at least four generations into the C57BL/6J background. To ensure that variability in the animals' genetic background had no bearing on our results, we performed CLP on two groups of wild-type mice that represented the two backgrounds of the myeloperoxidase-deficient mice. The C57BL/6J (n = 11) and 129/SvJ (n = 9) wild-type animals displayed similar patterns of tyrosine chlorination and bromination.
We were unable to detect myeloperoxidase when we immunoblotted peritoneal cells (predominantly macrophages) isolated from the sham-operated animals. Peritoneal cells from wild-type mice subjected to CLP had markedly higher levels of immunoreactive myeloperoxidase, as would be expected for an acute neutrophilic inflammatory response (data not shown). In contrast, peritoneal cells isolated from the septic myeloperoxidase-deficient mice contained no immunoreactive protein. These results indicate that the number of myeloperoxidase-containing cells increases markedly in the normal mouse peritoneum during sepsis and that myeloperoxidase-deficient mice lack immunoreactive enzyme.
The decreased ability of myeloperoxidase-deficient animals to generate 3-bromotyrosine suggested that myeloperoxidase produces brominating intermediates in vivo. Alternatively, the genetic manipulation might have deleted eosinophil peroxidase as well as myeloperoxidase. In humans, the gene for this brominating enzyme (11) lies near the myeloperoxidase gene, on chromosome 17 (29). To exclude the possibility of a double deletion, we measured peroxidase activity (20) in inflammatory cells isolated from wild-type and myeloperoxidase-deficient mice subjected to CLP. The wild-type cells generated a single major band of material with peroxidase activity that comigrated with human myeloperoxidase (data not shown). This material was undetectable in cells isolated from myeloperoxidase-deficient CLP-treated mice. Thus, the peroxidase in the inflammatory cells that appears in the peritoneum after CLP is myeloperoxidase rather than eosinophil peroxidase.
In contrast, extracts of white blood cells isolated from the peritoneal cavity of wild-type mice injected with thioglycollate produced two bands of peroxidase activity on nondenaturing PAGE (data not shown). The rapidly and slowly migrating bands comigrated with human eosinophil peroxidase and human myeloperoxidase, respectively. Cell extracts from the myeloperoxidase-deficient mice produced only the rapidly migrating band that corresponded to eosinophil peroxidase. These results suggest that both the wild-type mice and the genetically altered mice were able to make eosinophil peroxidase. Collectively, these observations strongly suggest that myeloperoxidase was the enzyme that generated 3-chlorotyrosine and 3-bromotyrosine in septic wild-type mice subjected to CLP.
Physiological Concentrations of Br− Modulate the Bactericidal Activity of Myeloperoxidase in Vitro.
Having shown that mice without myeloperoxidase are more vulnerable to sepsis than wild-type mice and also generate much lower levels of halogenated tyrosines, we looked directly at the effects of halides and myeloperoxidase on a bacterial pathogen, K. pneumoniae. The complete myeloperoxidase-H2O2-Cl− system killed 47% of the bacteria in the incubation mixture. Adding a low concentration (1 μM) of Br− markedly increased bacteria killing (72% dead); higher concentrations (10 μM) of Br− were completely inhibitory. Therefore, physiologically plausible variations in [Br−] markedly affect the ability of myeloperoxidase to kill K. pneumoniae in vitro.
Myeloperoxidase Brominates Tyrosine in Vitro.
We explored possible mechanisms of this cytotoxic effect of bromide by determining whether the transhalogen pathway that brominates nucleosides and nucleobases (12) can also halogenate tyrosine, which was brominated in our CLP experiments. For these experiments, we used N-acetyl-L-tyrosine rather than L-tyrosine itself to avoid chloramine formation and to prevent the conversion of L-tyrosine to p-hydroxyphenylacetaldehyde (22).
After we exposed N-acetyl-L-tyrosine to enzyme, H2O2, and plasma concentrations of halide (100 mM Cl− and 10 μM Br−), reverse-phase HPLC detected early and late eluting products that, respectively, comigrated with authentic N-acetyl-L-3-chlorotyrosine and N-acetyl-L-3-bromotyrosine (Fig. 4). Negative-ion electrospray ionization tandem mass spectrometry confirmed the identities of the halogenated amino acids (data not shown).
Figure 4.

Reverse-phase HPLC analysis of N-acetyl-L-tyrosine (N-Ac-tyrosine) exposed to the myeloperoxidase-H2O2-Cl−-Br− system. Reactions proceeded for 60 min at 37°C in Chelex-treated buffer A (100 mM NaCl/50 mM sodium phosphate/100 μM diethylenetriaminepentaacetic acid, pH 4.5) supplemented with 3 nM myeloperoxidase, 1 mM N-Ac-tyrosine, 10 μM NaBr, and 50 μM H2O2. The reactions were initiated with H2O2 and terminated with 0.1 mM methionine. MPO, myeloperoxidase; Ac, acetyl.
Both chlorination and bromination of N-acetyl-L-tyrosine were optimal under acidic conditions, but significant levels of the halogenated amino acids were also generated at neutral pH (Fig. 5a). Under acidic (pH 5.9) and neutral conditions, bromination by myeloperoxidase required both enzyme and H2O2; it was inhibited by catalase (a peroxide scavenger), sodium azide (a heme poison), and taurine (a scavenger of hypohalous acids).
Figure 5.

Reaction requirements for the generation of N-acetyl-L-3-bromotyrosine by phagocyte peroxidases and hypohalous acids at neutral pH. Reactions were carried out in buffer A (Fig. 4) supplemented with 10 μM Br−. (a) Effect of pH on the generation of N-acetyl-bromotyrosine by myeloperoxidase. (b–d) Effect of taurine (200 μM) on the generation of N-acetyl-bromotyrosine by HOCl, HOBr, myeloperoxidase, or eosinophil peroxidase at pH 7. Amino acids were quantified by reverse-phase HPLC. Results are representative of those found in three independent experiments. MPO, myeloperoxidase; EPO, eosinophil peroxidase; Ac, acetyl.
Myeloperoxidase Brominates N-Acetyl-L-Tyrosine at Physiologically Plausible Concentrations of Halide Ion.
We next determined whether myeloperoxidase prefers to use bromide or chloride when it oxidizes tyrosine under physiological conditions. In the presence of 100 mM Cl− alone, N-acetyl-L-3-chlorotyrosine was the principal product when either reagent HOCl or the myeloperoxidase-H2O2 system oxidized N-acetyl-L-tyrosine under acidic (Fig. 6 a and b) or neutral conditions (data not shown). Adding μM levels of Br− (in the presence of 100 mM Cl−) to either oxidation system generated N-acetyl-L-3-bromotyrosine and caused a corresponding decrease in N-acetyl-L-3-chlorotyrosine production (Figs. 5a and 6 a and b). The relative yields of N-acetyl-L-3-bromotyrosine and N-acetyl-L-3-chlorotyrosine depended on the pH and [Br−] in the reaction mixture (Figs. 5a and 6 a and b).
Figure 6.

Reaction requirements for the generation of N-acetyl-L-3-chlorotyrosine and N-acetyl-L-3-bromotyrosine by phagocyte peroxidases and hypohalous acids under acidic conditions. Effect of [Br−] on the generation of N-acetyl-chlorotyrosine and N-acetyl-bromotyrosine by: HOCl (a) or myeloperoxidase (b). Effect of taurine on the generation of N-acetyl-chlorotyrosine and N-acetyl-bromotyrosine by hypohalous acid (HOCl, HOBr) (c) or phagocyte peroxidases (MPO, EPO) (d). Reactions for a and b were carried out as described in the legend to Fig. 4. Reactions for c and d were performed as described in the legend to Fig. 4, except the pH was 5.9. Amino acids were quantified by reverse-phase HPLC. Results are representative of those found in three independent experiments. MPO, myeloperoxidase; EPO, eosinophil peroxidase; Ac, acetyl.
Under mildly acidic conditions (pH 5.9), taurine—a scavenger of HOCl—inhibited the formation of N-acetyl-L-3-bromotyrosine by the HOCl-Cl−-Br− system (Fig. 6c). In striking contrast, it stimulated N-acetyl-L-3-bromotyrosine formation by hypobromous acid (HOBr), suggesting that bromamines are potent brominating agents. Taurine also inhibited L-tyrosine bromination by the myeloperoxidase-H2O2-Cl−-Br− system (Fig. 6d). However, it failed to affect production of N-acetyl-L-3-bromotyrosine by the eosinophil peroxidase-H2O2-Cl−-Br− system (Fig. 6d). These observations strongly imply that myeloperoxidase first oxidizes Cl− to HOCl and that HOCl then reacts with Br− to generate reactive brominating intermediates.
Under neutral conditions, taurine completely inhibited the formation of N-acetyl-L-3-bromotyrosine by the HOCl-Cl−-Br− system but only partially inhibited the myeloperoxidase-H2O2-Cl−-Br− system (Fig. 5 c and d). In contrast, it stimulated N-acetyl-L-3-bromotyrosine formation by HOBr and had little affect on the eosinophil peroxidase-H2O2-Cl−-Br− system (Fig. 5 c and d). These results suggest that myeloperoxidase oxidizes Br− by two different pathways. In the major pathway, the enzyme initially generates HOCl, which then oxidizes Br−. In the minor pathway myeloperoxidase directly oxidizes Br−.
Discussion
More than 30 years ago, Klebanoff proposed that halogenating intermediates generated by myeloperoxidase are of major importance in killing bacteria (30). However, the enzyme's role in host defense against invading pathogens has remained unclear. Our experiments reveal that myeloperoxidase-deficient mice are more likely than wild-type mice to die after CLP, a clinically relevant model of sepsis that releases bacteria into the peritoneum. This observation supports the hypothesis that myeloperoxidase generates bacterial cytotoxins in vivo. Moreover, the enzyme was able to use halides to kill K. pneumoniae in vitro. We have since discovered that myeloperoxidase-deficient mice are more likely than wild-type mice to die after K. pneumoniae is injected into the peritoneum (unpublished results). Thus, myeloperoxidase seems critical to antibacterial defense mechanisms in mice.
Further evidence that myeloperoxidase contributes to antibacterial defense in vivo came from our observation that levels of 3-chlorotyrosine, one of the enzyme's characteristic products, rise markedly in peritoneal lavage fluid after wild-type mice are subjected to CLP. Levels of 3-bromotyrosine were also increased. Septic mice that were deficient in myeloperoxidase failed to generate 3-chlorotyrosine at levels above those observed in sham-operated animals, indicating that myeloperoxidase is the major source of chlorinating oxidants in this sepsis model. Levels of 3-bromotyrosine were also markedly lower in the septic myeloperoxidase-deficient animals, although they were slightly higher than in nonseptic wild-type mice, presumably because of eosinophil peroxidase activity. These observations indicate that myeloperoxidase generates chlorinating and brominating oxidants; the production of the latter was previously ascribed solely to eosinophil peroxidase (11, 31, 32). Thus, both myeloperoxidase-dependent chlorination and myeloperoxidase-dependent bromination may represent physiologically relevant pathways for bacterial killing. Hypothiocyanite and other oxidants derived from thiocyanate may also play a role because this pseudohalide is present at high concentrations in extracellular fluids and is readily oxidized by peroxidases (33). In contrast, other reactive intermediates generated by myeloperoxidase, such as amino acid-derived aldehydes, are likely to be less important because they are only weakly bactericidal in vitro (J.P.G., unpublished observation).
The halogenated intermediates produced by myeloperoxidase include HOCl (3, 4), a potent cytotoxic oxidant that converts tyrosine to 3-chlorotyrosine (28). Our observations indicate that myeloperoxidase will also brominate tyrosine in vitro at plasma concentrations of halide ions. Remarkably, the bromination pathway operates when Cl− concentrations are 1,000-fold to 10,000-fold higher than Br− concentrations. Therefore, it may be physiologically relevant.
N-Acetyl-bromotyrosine production by myeloperoxidase occurred at neutral pH but was optimal under acidic conditions. In vivo, therefore, myeloperoxidase might halogenate tyrosine extracellularly at neutral pH and also in the phagolysosome (or hypoxic inflamed tissue) under acidic conditions. At acidic pH, taurine almost completely inhibited bromination, suggesting that HOCl is an intermediate in the pathway. In contrast, taurine only partly inhibited N-acetyl-L-bromotyrosine production by myeloperoxidase at neutral pH, suggesting the existence of a bromination pathway not involving HOCl. One possibility for this second pathway is that myeloperoxidase might directly oxidize Br− to HOBr, as does eosinophil peroxidase (11, 20). At neutral pH and 10 μM Br−, myeloperoxidase produced similar concentrations of N-acetyl-L-bromotyrosine and N-acetyl-L-chlorotyrosine, suggesting that the enzyme could produce halogenating intermediates in the extracellular environment. In contrast, Wu et al. (34) found that neutrophils generated protein-bound 3-chlorotyrosine much more effectively than 3-bromotyrosine. One possible explanation for the discrepancy is that Wu et al. studied halogenation of proteins, whereas we focused on halogenation of free amino acids.
These observations indicate that mice lacking functional myeloperoxidase are more likely to die from polymicrobial sepsis, that in vivo levels of free 3-chlorotyrosine and 3-bromotyrosine rise during sepsis, and that production of these halogenated amino acids is markedly reduced when myeloperoxidase is absent. Thus, myeloperoxidase can produce a variety of chlorinating and brominating intermediates that seem capable of defending mice against CLP-induced sepsis. In the enzyme's absence, end products of these intermediates fail to accumulate in peritoneal fluid, and mice become more vulnerable to infection.
Acknowledgments
Mass spectrometry experiments were performed at the Washington University School of Medicine Mass Spectrometry Resource. This work was supported by grants from the National Institutes of Health (AG19309, AG15013, DK56341, and RR00954) and the Washington University-Pharmacia-Monsanto-Searle Research Program. J.P.G. was supported by the Cardiovascular Training Grant at Washington University School of Medicine and by the Glenn/American Federation of Aging Research. G.M.R. was supported by the Howard Hughes Medical Institute as a Washington University Prefreshmen Summer Scholar in Biology and Biomedical Research.
Abbreviations
- HOCl
hypochlorous acid
- HOBr
hypobromous acid
- MtBSTFA
N-methyl-N-(t-butyldimethylsilyl)-trifluoroacetamide + 1% trimethylchlorosilane
- CLP
cecal ligation and puncture
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Klebanoff S. Science. 1970;169:1095–1097. doi: 10.1126/science.169.3950.1095. [DOI] [PubMed] [Google Scholar]
- 2.Babior B. Am J Med. 2000;109:33–44. doi: 10.1016/s0002-9343(00)00481-2. [DOI] [PubMed] [Google Scholar]
- 3.Harrison J E, Schultz J. J Biol Chem. 1976;251:1371–1374. [PubMed] [Google Scholar]
- 4.Klebanoff S. Proc Assoc Am Phys. 1999;111:383–389. doi: 10.1111/paa.1999.111.5.383. [DOI] [PubMed] [Google Scholar]
- 5.Hurst J K, Barrette W C., Jr CRC Crit Rev Biochem Mol Biol. 1989;24:271–328. doi: 10.3109/10409238909082555. [DOI] [PubMed] [Google Scholar]
- 6.Nauseef W M. Hematol Oncol. 1988;2:135–138. [PubMed] [Google Scholar]
- 7.Grisham M B. Lancet. 1994;344:859–861. doi: 10.1016/s0140-6736(94)92831-2. [DOI] [PubMed] [Google Scholar]
- 8.Winterbourn C C, Vissers M C M, Kettle A J. Curr Opin Hematol. 2000;7:53–58. doi: 10.1097/00062752-200001000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Thomas E L, Bozeman P M, Jefferson M M, King C C. J Biol Chem. 1995;270:2906–2913. doi: 10.1074/jbc.270.7.2906. [DOI] [PubMed] [Google Scholar]
- 10.Klebanoff S J. J Bacteriol. 1968;95:2131–2138. doi: 10.1128/jb.95.6.2131-2138.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss S J, Test S T, Eckmann C M, Ross D, Regiani S. Science. 1986;234:200–203. doi: 10.1126/science.3018933. [DOI] [PubMed] [Google Scholar]
- 12.Henderson J P, Byun J, Williams M V, Mueller D M, McCormick M L, Heinecke J W. J Biol Chem. 2001;276:2867–2875. doi: 10.1074/jbc.M005379200. [DOI] [PubMed] [Google Scholar]
- 13.Aratani Y, Koyama H, Nyui S, Suzuki K, Kura F, Maeda N. Infect Immun. 1999;67:1828–1836. doi: 10.1128/iai.67.4.1828-1836.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brennan M-L, Anderson M M, Shih D M, Qu X D, Mehta A C, Lim L L, Shi W, Jacob J S, Crowley J R, Hazen S L, et al. J Clin Invest. 2001;107:419–430. doi: 10.1172/JCI8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deitch E. Shock. 1998;9:1–11. doi: 10.1097/00024382-199801000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Hotchkiss R S, Swanson P E, Cobb J P, Jacobson A, Buchman T G, Karl I E. Crit Care Med. 1997;25:1298–1307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 17.Freeman B D, Reaume A G, Swanson P G, Estein C J, Carlson E J, Buchman T G, Karl I E, Hotchkiss R S. Crit Care Med. 2000;28:1701–1708. doi: 10.1097/00003246-200006000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Cobb J P, Hotchkiss R S, Swanson P E, Chang K, Qui Y, Laubach V E, Karl I E, Buchman T G. Surgery. 1999;126:438–442. [PubMed] [Google Scholar]
- 19.Hope H R, Remsen E E, Lewis C J, Heuvelman D M, Walker M C, Jennings M, Connolly D T. Protein Expression Purif. 2000;18:269–276. doi: 10.1006/prep.1999.1197. [DOI] [PubMed] [Google Scholar]
- 20.van Dalen C J, Whitehouse M W, Winterbourn C C, Kettle A J. Biochem J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas E L, Grisham M B, Jefferson M M. Methods Enzymol. 1986;132:569–585. doi: 10.1016/s0076-6879(86)32042-1. [DOI] [PubMed] [Google Scholar]
- 22.Hazen S L, Hsu F F, Heinecke J W. J Biol Chem. 1996;271:1861–1867. doi: 10.1074/jbc.271.4.1861. [DOI] [PubMed] [Google Scholar]
- 23.Pollock J D, Williams D A, Gifford M A C, Li L L, Du X, Fisherman J, Orkin S H, Doerscshuk C M, Dinauer M C. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 24.Heinecke J W, Hsu F F, Crowley J R, Hazen S L, Leeuwenburgh C, Mueller D M, Rasmussen J E, Turk J. Methods Enzymol. 1999;300:124–144. doi: 10.1016/s0076-6879(99)00121-4. [DOI] [PubMed] [Google Scholar]
- 25.Frost M, Halliwell B, Moore K. Biochem J. 2000;345:453–458. [PMC free article] [PubMed] [Google Scholar]
- 26.Byun J, Henderson J P, Mueller D M, Heinecke J W. Biochemistry. 1999;38:2590–2600. doi: 10.1021/bi9822980. [DOI] [PubMed] [Google Scholar]
- 27.Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley T J, Abraham S N, Shapiro S D. Nat Med. 1998;4:615–618. doi: 10.1038/nm0598-615. [DOI] [PubMed] [Google Scholar]
- 28.Hazen S L, Hsu F F, Mueller D M, Crowley J R, Heinecke J W. J Clin Invest. 1996;98:1283–1289. doi: 10.1172/JCI118914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakamaki K, Kanda N, Ueda T, Aikawa E, Nagata S. Cytogenet Cell Genet. 2000;88:246–248. doi: 10.1159/000015529. [DOI] [PubMed] [Google Scholar]
- 30.Klebanoff S. J Clin Invest. 1967;46:1078. [Google Scholar]
- 31.Mayeno A N, Curran A J, Roberts R L, Foote C S. J Biol Chem. 1989;264:5660–5668. [PubMed] [Google Scholar]
- 32.Wu W, Samoszuk M K, Comhair S A A, Thomassen M J, Farver C F, Dweik R A, Kavuru M S, Erzurum S C, Hazen S L. J Clin Invest. 2000;105:1455–1463. doi: 10.1172/JCI9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arlandson M, Decker T, Roongta V A, Bonilla L, Mayo K H, MacPherson J C, Hazen S L, Slungaard A. J Biol Chem. 2001;276:215–224. doi: 10.1074/jbc.M004881200. [DOI] [PubMed] [Google Scholar]
- 34.Wu W, Chen Y, d'Avignon A, Hazen S L. Biochemistry. 1999;38:3538–3548. doi: 10.1021/bi982401l. [DOI] [PubMed] [Google Scholar]