

Abstract
A crystal structure of the anaerobic Ni-Fe-S carbon monoxide dehydrogenase (CODH) from Rhodospirillum rubrum has been determined to 2.8-Å resolution. The CODH family, for which the R. rubrum enzyme is the prototype, catalyzes the biological oxidation of CO at an unusual Ni-Fe-S cluster called the C-cluster. The Ni-Fe-S C-cluster contains a mononuclear site and a four-metal cubane. Surprisingly, anomalous dispersion data suggest that the mononuclear site contains Fe and not Ni, and the four-metal cubane has the form [NiFe3S4] and not [Fe4S4]. The mononuclear site and the four-metal cluster are bridged by means of Cys531 and one of the sulfides of the cube. CODH is organized as a dimer with a previously unidentified [Fe4S4] cluster bridging the two subunits. Each monomer is comprised of three domains: a helical domain at the N terminus, an α/β (Rossmann-like) domain in the middle, and an α/β (Rossmann-like) domain at the C terminus. The helical domain contributes ligands to the bridging [Fe4S4] cluster and another [Fe4S4] cluster, the B-cluster, which is involved in electron transfer. The two Rossmann domains contribute ligands to the active site C-cluster. This x-ray structure provides insight into the mechanism of biological CO oxidation and has broader significance for the roles of Ni and Fe in biological systems.
Phototrophic anaerobes such as Rhodospirillum rubrum have the ability to use CO as a sole carbon and energy source (1). This ability derives from the oxidation of CO to CO2 catalyzed by carbon monoxide dehydrogenases (CODHs). Some CODH enzymes also participate in the synthesis or degradation of acetyl-CoA and are referred to as CODH/acetyl CoA synthases (CODH/ACS). The dual role of these CODH/ACSs is found in CO2 fixation to acetyl-CoA by acetogens via the well studied Wood/Ljungdahl pathway (reviewed in ref. 2). Methanogens also use CODH/ACSs in the conversion of acetyl-CoA to methane. As a consequence of these activities, CODHs are essential for the proper regulation of environmental carbon monoxide. Each year, up to 108 tons of CO are oxidized to CO2 by aerobic and anaerobic bacteria containing CODH enzymes (3). Prehistoric environments rich in CO and CO2 and deficient in oxygen (4) prompt speculation that early life forms relied on CO/CO2 as prime sources of both carbon and energy. In an effort to understand the detailed biochemical workings of early and current life on CO, we present the three-dimensional structure of an anaerobic Ni-Fe-S CODH enzyme.
CODHs catalyze the oxidation of carbon monoxide in the two-electron process.
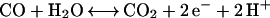 |
1 |
The mechanism of CO oxidation is thought to involve binding and deprotonation of an H2O molecule to form hydroxide at a unique Ni-Fe-S center called the C-cluster (5). CO is believed to bind to a site on the C-cluster adjacent to the hydroxide. Thus, a metal-bound hydroxide may attack the CO carbon. Then the resulting metal–COOH intermediate is deprotonated and CO2 is lost to yield a two-electron-reduced C-cluster (reviewed in ref. 2). In R. rubrum, electrons are passed from the C-cluster by means of an [Fe4S4] center (the B-cluster) to a membrane-associated iron-sulfur protein designated CooF (6). CooF, which is tightly associated with CODH, transfers electrons to a hydrogenase, thereby coupling the oxidation of CO with hydrogen production. This coupling, which is exergonic by 20.1 kJ/mol, is thought to enable R. rubrum to extract energy from CO oxidation.
The Ni-Fe-S cluster responsible for CO oxidation to CO2 has been studied extensively by spectroscopic methods (reviewed in refs. 2 and 7–13). Issues addressed include whether the Ni is part of the cubane (site Ma, Scheme S1) or in a mononuclear site (site Mb, Scheme S1). Extended x-ray absorption fine structure (EXAFS) spectroscopy comparing the R. rubrum CODH (EC 1.2.99.2) with model [NiFe3S4] cubanes (10) and Mössbauer spectroscopy (7) favor a model for the C-cluster that contains one Ni atom in the Mb site bridged to an Fe with usual coordination in the Ma site (Ni-X-[Fe4S4]). This unusual Fe is typically denoted as FCII. It is therefore surprising that the results of anomalous dispersion experiments presented here suggest that Ni is located in the cube (Ma site) and that Fe is located in the Mb site. The presence of an Fe atom in a site coordinated by a histidine residue is consistent with electron nuclear double resonance (ENDOR) spectroscopy on Clostridium thermoaceticum CODH/ACS enzyme (12) and electron paramagnetic resonance (EPR) spectroscopy on the R. rubrum CODH (13). The R. rubrum CODH structure reveals that the only histidine ligand coordinates the Mb site (Scheme I), suggesting that the Mb site contains Fe. It has also been suggested that the Mb site is a [NiFe] binuclear cluster rather than a mononuclear site (11, 13). This possibility is ruled out by the current x-ray analysis.
Scheme 1.

Here, we present the x-ray structure of CODH from R. rubrum, the prototype for the monofunctional CODH enzymes. We compare the x-ray data with the spectroscopic studies of R. rubrum CODH and C. thermoaceticum CODH/ACS. The x-ray structure provides information about the protein topology, the enzyme oligomeric state, and the nature and number of metallocenters in this well characterized member of the CODH family.
Methods
Crystallization and Data Collection.
CODH was purified from R. rubrum in a modification (14) of the original protocol (15). The capillary crystallizations were carried out under argon in a Coy anaerobic chamber by using a modified version of the microcapillary batch diffusion method (16) and precipitating agents (10% polyethylene glycol 8,000/0.1 M Tris buffer, pH 7.5/0.4 M calcium chloride/5% 2-methyl-2,4-pentanediol′. The protein solution, previously incubated in carbon monoxide, was ≈20 mg/ml in concentration before the 50/50 mixing with the precipitating solution. CODH crystallizes in monoclinic space group P21 with cell parameters a = 92.7, b = 200.1, c = 116.8 Å, β = 111.5°, and 6 molecules in the asymmetric unit. For data collection, the crystals were equilibrated in a holding solution containing a final concentration of 35% glycerol and were cooled by quick immersion in liquid N2. Crystals were screened at Stanford Synchrotron Radiation Laboratory (SSRL) beamlines 7–1 and 9–1. Multiwavelength anomalous dispersion (MAD) data were collected at the Advanced Light Source (ALS) beamline 5.02 equipped with an Area Detector Systems Corporation (Poway, CA) charge-coupled device (CCD) x-ray detector. The native data were collected at beamline ×25 at the National Synchrotron Light Source (NSLS) equipped with a Brandeis B4 CCD detector. Data were indexed and processed by using denzo and scalepack (17).
Structure Determination.
The structure was solved by the multiwavelength anomalous dispersion (MAD) phasing method, using data collected primarily near the Fe edge (Table 1). Positions of the iron clusters were identified by using solve (18) and were refined in sharp (19). Because of the resolution limits of the MAD data, experimental phases were calculated to 5.0-Å resolution. However, 6-fold noncrystallographic averaging in dm (20) resulted in interpretable electron density maps. Phase combination in DM with a partial polyalanine model then was used to gradually extend the phases to 3.3-Å resolution. Iterative rounds of model building, refinement, phase combination and extension, and averaging resulted in the 2.8-Å resolution model described in Table 2. This model has 1 Ni atom, 10 Fe atoms, 10 sulfide atoms, and 610 of the 639 residues per monomer. N-terminal residues 1–27 and the C-terminal residues G638 and L639 have poor or no electron density and are not included in the model. Regions present in the model that have high B-factors (70–90 Å2) and/or poor quality electron density include 61–66, 249–250, 393–405, and 457–459. The present model exhibits good geometry with 99.7% of the residues residing in allowed regions of the Ramachrandan plot as calculated by procheck (21). Refinement was carried out in cns (22) by using a bulk solvent correction, two-sigma data cutoff, noncrystallographic symmetry (NCS) restraints, the maximum likelihood target function, and Cartesian molecular dynamics. Of the reflections, 3,716 were used to calculate the Rfree. To determine the position of the nickels and irons in the R. rubrum CODH enzyme, a two-wavelength experiment was carried out on a single crystal at the Advanced Photon Source Consortium for Advanced Radiation Sources (BioCARS, Sector 14; Table 1).
Table 1.
Data refinement and statistics
| Wavelength, Å | Data statistics
|
Rsym | |||
|---|---|---|---|---|---|
| Resolution range, Å | Unique reflections | Total reflections | Completeness, % | ||
| Fe peak 1.7394 | 20.0–3.8 | 71,546* | 132,694 | 92.3 | 0.092 |
| Fe edge 1.7419 | 30.0–5.0 | 30,163* | 59,115 | 88.3 | 0.137 |
| Fe remote 1.4846 | 30.0–4.0 | 62,923* | 140,017 | 95.5 | 0.086 |
| Fe remote 1.6469 | 30.0–5.0 | 29,308* | 61,041 | 83.1 | 0.088 |
| Fe remote 1.8456 | 30.0–4.0 | 64,533* | 140,656 | 96.6 | 0.096 |
| Ni edge 1.4866 | 30.0–3.5 | 90,508* | 147,542 | 91.6 | 0.068 |
| Ni remote 1.3051 | 30.0–3.5 | 47,270 | 140,261 | 94.2 | 0.098 |
| Native 1.3000 | 100.0–2.8 | 90,759 | 223,988 | 94.0 (80.1) | 0.086 (0.35) |
Rsym = [ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|]/ΣhklΣiIi(hkl) for n independent reflections and i observations of a given reflection. 〈I(hkl)〉 is the average intensity of the i observation. Values for data in the highest resolution bin are listed in parentheses.
For these data sets, Friedel pairs were not merged during data processing.
Table 2.
Refinement statistics
| Nonhydrogen atoms in asymmetric unit | 26,904 |
| Resolution range, Å | 100–2.8 |
| No. of reflections >2 σ | 74,708 |
| Rcryst, % | 0.250 |
| Rfree, % | 0.281 |
| rms deviations of protein from | |
| Ideal geometry | |
| Bonds, Å | 0.008 |
| Angles, ° | 1.33 |
| Dihedral angles, ° | 22.1 |
| Average B (Å2) for molecules A and B and molecules C–F | |
| Protein | 62.2 (A and B), 40.3 (C–F) |
| B- and D-clusters | 46.6 (A and B), 27.1 (C–F) |
| C-cluster | 68.6 (A and B), 43.4 (C–F) |
Results and Discussion
Overall Structure.
The six molecules in the asymmetric unit are present as a trimer of dimers, and the dimer is likely to be physiological (see below). Each dimer has 5 metal clusters, two Ni-Fe-S C-clusters, two [Fe4S4] B-clusters, and one [Fe4S4] cluster, bridging the two molecules of the dimer. This bridging cluster will be referred to as the D-cluster [not to be confused with the D-site in C. thermoaceticum CODH/ACS (23)]. Each protein monomer is comprised of three domains: a helical domain at the N terminus, an α/β (Rossmann-like) domain in the middle, and an α/β (Rossmann-like) domain at the C terminus (Fig. 1). The helical domain contributes ligands to the D- and B-clusters. The B- and D-clusters are typical [Fe4S4] clusters with cysteine ligation; Cys41 and Cys49 from each monomer coordinate the bridging cluster, and Cys50, Cys53, Cys58, and Cys72 coordinate the B-cluster. The two Rossmann domains contribute ligands to the C-cluster with loops following the first three β-strands of each domain (Fig. 1). Not surprisingly, the anaerobic CODH from R. rubrum does not resemble the aerobic CODH enzyme (24), given that aerobic CODHs use a molybdopterin cofactor and the protein sequences share no significant sequence homology. However, an intriguing structural similarity exists with the hybrid cluster protein (HCP) from Desulfovibrio vulgaris of unknown function (25). R. rubrum CODH and HCP share the same basic topology: a similar helical domain (rmsd 1.3 for core 71 α-carbons) followed by two Rossmann-like domains (rmsd 0.8–1.6 for core 30–70 α-carbons). In both cases, the Rossmann domains coordinate metalloclusters with 9 of 12 cluster ligands conserved, suggesting some type of evolutionary relationship between these proteins.
Figure 1.

(A) Ribbon drawing of R. rubrum CODH monomer. The N-terminal helical domain is in red and the α/β Rossmann domains are in green and blue. The C-, B-, and D-clusters are in space-filling representations with Fe atoms in white, S in yellow, and the Mb site in orange (see Scheme S1). It should be noted that although the entire bridging D-cluster is shown, only half of the cluster is coordinated by each monomer. All figures were made by using RIBBONS (34). (B) Topology diagram of CODH with same domain coloring as shown in A. The positions of the metalloclusters are shown as yellow circles.
R. rubrum CODH Is a Dimer.
Although gel filtration experiments suggest that R. rubrum CODH is a monomer (15), we now propose R. rubrum CODH to be dimeric based on several lines of structural evidence. First, the existence of a bridging [Fe4S4] cluster covalently coordinated to each subunit supports the assignment of R. rubrum CODH as a dimer. Second, electrons generated at the C-cluster would have to travel 30 Å to the [Fe4S4] B-cluster in the monomer (Fig. 2A). In the dimer, the B-cluster of molecule 2 is only 13 Å center to center (≈8 Å edge to edge) from the C-cluster of molecule 1. Whereas 30 Å is outside the expected distance between redox partners, 13 Å is a common distance (26). Additionally, whereas the C- and B-clusters are buried, the D-cluster could serve as a conduit for electrons from the C-cluster active site to the outside of the molecule (Fig. 2B). Third, the surface area buried at the dimer interface is almost exclusively hydrophobic and extensive, 7,800 Å2, suggesting a tight association of the molecules in the dimer. This area is equivalent to ≈20% of the surface area of each molecule. Finally, the few charged residues that are buried by the dimer interface have charge compensation as expected for a physiological dimer. Of the eight charged residues buried, four are near [Fe4S4] clusters and four are involved in salt bridges. It is interesting to note that the β subunits of C. thermoaceticum CODH/ACS, which are 67% similar (46% identical) to CODH from R. rubrum (27), are present as a dimer in an α2β2 tetramer (28). Therefore, the R. rubrum CODH structure will be a good model for β2 subunits of CODH/ACS from acetogens.
Figure 2.

(A) Ribbon drawing of the top view of the R. rubrum CODH dimer with molecule 1 in blue and molecule 2 in green. The C-, B-, and D-clusters are in space-filling representations with Fe atoms in white, S in yellow, the Mb site in orange, and the Ma site in blue. Note the B-cluster of molecule 2 (B′) is adjacent to the C-cluster of molecule 1 (C), and that the B-cluster of molecule 1 (B) is adjacent to the C-cluster of molecule 2 (C′). Distances between the centers of the clusters are indicated. (B) Ribbon drawing of the side view of the R. rubrum CODH dimer. The D-cluster provides a path for electrons from the buried B- and C-clusters to the surface. The D-cluster may interact with CooF, the iron-sulfur membrane-anchor protein.
Location of Ni and Fe in the C-Cluster.
Electron density of the C-cluster is consistent with a single metal atom and a metal cluster cubane. To identify the positions of Ni and Fe in the C-cluster, a two-wavelength experiment was carried out. Surprisingly, the results suggest that Ni is in the cube (site Ma) and Fe is in the mononuclear site (Mb) (Fig. 3). Difference peaks in a (FNi-remote − FNi-edge) dispersive Fourier map were used to locate the Ni atom. The scattering of Ni is much greater at the remote wavelength (1.300 Å) than at the Ni-edge (1.487 Å), whereas the scattering of Fe is similar at these wavelengths. Therefore, the large difference peaks in the dispersive Fourier map are caused by Ni and not Fe. As shown in Fig. 3, the only large peak in the dispersive Fourier map is located at the corner of the cube (the Ma site). In contrast, the anomalous difference peaks calculated at the Ni-edge should represent the positions of Fe atoms in the structure, because Ni scattering is minimal at this wavelength. The Ni-edge anomalous Fourier maps indicate the presence of Fe in the cube as well as in the mononuclear (Mb) site (Fig. 3). Fig. 3 shows the results of the two-wavelength experiment in one molecule, and the other five molecules in the asymmetric unit have also been analyzed to rule out the possibility that any of the difference density peaks are caused by noise in the data. Each molecule has only one large (4–6 sigma) dispersive peak, and in each case this peak is located at the corner of the cubane. These results strongly suggest that Ni is in the cube, and only in the cube. There is no evidence for a second noncubane Ni site. Also, there is no indication of any dispersive difference density in the B- or D- [Fe4S4] clusters, confirming that the dispersive peaks are caused by Ni and not Fe. In contrast, the Ni-edge anomalous Fourier map shows greater than 4 sigma peaks at each B- and D-cluster, indicating that this Fourier calculation does serve to locate Fe atom positions. The C-clusters of all six molecules have anomalous density peaks, indicating the presence of Fe within the cube and at the mononuclear (Mb) site. Thus, the agreement of the six independent C-cluster electron density maps provides a convincing argument that Fe is in the mononuclear site and Ni is in the cubane.
Figure 3.

Identification of the positions of Ni and Fe in the C-cluster by using data from a two-wavelength Ni-edge experiment. The (FNi-remote − FNi-edge) dispersive Fourier map (magenta) indicates the position of Ni. This map is calculated at 3.5-Å resolution and contoured at 6 sigma. A 3.5-Å resolution anomalous difference Fourier map (cyan) calculated at the Ni-edge wavelength, and contoured at 4 sigma, indicates the position of Fe. See text for discussion. For both Fourier maps, atoms of the clusters are omitted in the phase calculation. Color scheme: C atoms, green; N, blue; Fe of cube, white; S, yellow; Ma site, cyan; Mb site, orange.
The Ni anomalous dispersive data are consistent with some, but not all, of the spectroscopic results on R. rubrum CODHs. It is important to note the R. rubrum CODH protein is isolated from the native organism and kept under anaerobic conditions. Therefore, the apparent presence of the Ni in the cube is not the result of an artifact of recombinant techniques. A series of crystals has been tested for CODH activity by using methylviologen as an indicator, and the crystals are active, indicating that the presence of Ni in the cube is not the result of oxygen or other enzyme damage. Thus, we are left with the task of trying to reconcile the x-ray data with the information from Mössbauer and EXAFS studies. Mössbauer experiments on Ni-deficient R. rubrum CODHs indicate all Fe atoms are located in [Fe4S4] clusters (7). However, Fig. 3 does suggest that in the absence of Ni, the C-cluster could be dramatically altered. Without a structure of Ni-deficient enzyme it is difficult to correlate the Mössbauer and x-ray results. After Ni binding, Mössbauer spectroscopy shows a change in coordination of one Fe (FCII; ref. 7). Because Ni and FCII share ligands as described below, this observation is easy to understand in terms of the current x-ray data. It is difficult to reconcile the published EXAFS results on R. rubrum (10) with the current model if the Ni is thought to occupy a site in a normal cubane, because the EXAFS experiments do not detect the presence of a short (2.7-Å) Ni–Fe distance. If, however, the cubane is distorted by virtue of the link to FCII, it is possible that the two results would be consistent. Unfortunately, the 2.8-Å resolution limit of the x-ray data does not permit a comparison of the distances and geometry of the C-cluster cubane to model complexes at this time. Another issue to consider is that x-ray and spectroscopic data analysis is complicated by protein sample heterogeneity (7). One source of this heterogeneity could be the inherent flexibility of the C-cluster. The C-cluster is coordinated to two different protein domains, and the orientation of these domains is not fixed with respect to each other. A slight movement of one domain with respect to the other could compress or expand the C-cluster, altering side-chain metal interactions. Inherent flexibility of the C-cluster environment may be important for catalysis, as is the case in other metalloenzyme systems; examples of which include the carboxylate shift in methane monoxygenase (29). The B-factors for the C-cluster are higher than for the bridging and B-clusters (Table 2), indicating that the active site cluster is less rigid. Although potentially important in catalysis, ligand rearrangements and flexibility complicate x-ray and spectroscopic analysis and could explain some of the apparent inconsistencies.
The Mononuclear Fe Site.
Fe (FCII) in the mononuclear site (Mb) is coordinated by protein residues His265 and Cys300 (Fig. 4A). Cys531 is also in position to coordinate the Ni and FCII, although the electron density is broad (Fig. 4B) and it is difficult to assign an exact distance between the Fe and S of Cys531. The broad electron density observed may be caused by an oxidation state dependence of the Cys531 side-chain orientation, as the oxidation state of the crystal is unknown and our electron density may represent a mixture of states. It is also possible that side-chain movement, such as a “thiolate shift,” may play a role in catalysis. Substitution of alanine for Cys531 slows the reaction rate 1,000-fold even with full Fe and Ni incorporation (13), suggesting a catalytic or critical structural role for Cys531. Interestingly, the fourth ligand to FCII seems to be a sulfide of the [NiFe3S4] cube. Thus, the current model for FCII has distorted tetrahedral geometry with 1 histidine, 2 cysteines, and 1 sulfide ligand. It is likely that in some oxidation states, an H2O molecule is also a ligand to Fe (12), although it is not visible in this structure.
Figure 4.

(A) Stereoview of the C-cluster. The identity of the ligand to Ni (Ma site) is unknown (position marked with an *). Same color scheme as Fig. 3. (B) A 2.8-Å resolution (2Fobs − Fcalc) electron density map (pink) for the C-cluster. The electron density is spread out near Cys531, and the exact position of the side chain is difficult to determine. Positive difference (Fobs − Fcalc) electron density maps (blue) suggest the presence of an additional ligand to Ni. (C) Model of metal–COOH intermediate. If COOH is placed in the general area of the open coordination sites on Ni and FCII, it can interact with His95 and Lys568. His95, which is part of a hydrogen-bonding network with Asp223 and Trp575 and is ≈2.6 Å away from the modeled intermediate, could act as the catalytic base. Lys568, which is within hydrogen-bonding distance (≈2.5 Å), could stabilize the intermediate. (D) Putative tunnel (shown as a cylinder), identified by visual inspection of electron density maps, leading from the active site to solvent in R. rubrum CODH. His95 and Lys568 (red ball-and-stick) sit at the top of the tunnel. Histidine residues 98, 101, and 108 (red ball-and-stick), conserved among CODH sequences, line the putative tunnel.
The Ni Site.
Ni seems to be coordinated by sulfides of the cube, Cys531, and by a small molecule ligand. In contrast, EXAFS data are best fit by a combination of 2 S and 2–3 N/O atoms (10). As discussed above, the discrepancies between x-ray and EXAFS data could be the results of several factors and await further analysis. Evidence for a small molecule ligand comes from difference electron density calculations (Fig. 4B). The positive difference density is consistent with a ligand to Ni and not the Ni itself, because movement of Ni into the positive difference density is incompatible with the anomalous scattering data. The positive difference density near Ni appears when no ligand is refined in this position. Refinement of H2O also results in positive difference density. Refinement of CO leaves no difference density, indicating that it is approximately the correct size of the unknown ligand. Although CO is a reasonable candidate for this unknown ligand, the x-ray structure suggests that this ligand site does not represent a catalytically relevant position for substrate binding. As opposed to another open coordination site on Ni, a ligand bound in this position is buried in a hydrophobic environment away from proposed catalytic residues Lys568 and His95 (see below). Thus, either substrate binds to this position and rearranges before catalysis or this site represents a noncatalytic ligand site. There is evidence for a noncatalytic CO ligand (COL) site in R. rubrum CODH (14): denaturation of R. rubrum CODH releases a CO molecule, and incubation of the holoenzyme with CO increases enzyme activity (14). Based on the observation that radiolabeled CO binds Ni-deficient enzyme as well as holoenzyme (14), it was originally suggested that the COL is bound to Fe. However, if COL binds to a different site in the presence and absence of Ni, then there would be nothing to rule out an Ni–COL interaction. In support of an Ni–CoL interaction are two pieces of evidence. First, COL binding to Ni would explain why CO incubation of holoenzyme results in significant increase in enzyme activity, whereas CO incubation before Ni addition does not (14). Second, COL protects Ni from Ni-specific chelating agent dimethylglyoxime (14). The structure indicates that filling the ligand-binding site would decrease access to and the conformational flexibility of the C-cluster atoms, protecting Ni. It is important to note that the unknown ligand is unlikely to be an inhibitor such as cyanide, because crystals show catalytic activity as evidenced by the reduction of methylviologen in the presence of CO. Thus, difference in electron density does suggest an additional ligand to Ni; however, the structural resolution does not permit exact identification of this ligand at this time.
Mechanism.
Several mechanistic schemes can and have been proposed for CODH, including involvement of a metal-bound hydroxide (reviewed in refs. 2 and 12) and a metal-bound hydride (11, 13, 30, 31). Although a single x-ray structure cannot definitively assign an enzyme mechanism, it is valuable to consider various schemes in terms of the three-dimensional structure of the active site. One common mechanistic proposal involves the binding and deprotonation of an H2O molecule (5) to form metal-bound hydroxide at either FCII (12), Ni (2), or in a bridging position between metals. CO is believed to bind to a metal site adjacent to the hydroxide. Thus, a metal-bound hydroxide may attack the CO carbon. Then the resulting metal–COOH intermediate is deprotonated, and CO2 is lost to yield a two-electron-reduced enzyme (reviewed in ref. 2). First, in terms of the structure, this mechanism requires open coordination sites on Ni and FCII, located such that metal-bound hydroxide is close enough to react with metal-bound CO. The x-ray structure is consistent with this requirement (see Fig. 4A). Second, an amino acid residue that can act as a catalytic base must be present near the open metal coordination sites to deprotonate the metal–COOH intermediate. His95 is ideally situated to act as a catalytic base. In Fig. 4C, a COOH is modeled such that it could be bound to either the Ni or FCII. In this model, His95 is positioned at an appropriate distance to deprotonate the metal–COOH. Third, the enzyme would be expected to stabilize the anionic metal–COO− intermediate by hydrogen bonding. Lys568 is ideally positioned to carry out this function (Fig. 4C). Fourth, for the next catalytic turnover cycle, [His95−H]+ must deprotonate to generate product H+. Although we cannot assign a path for proton transfer, it is interesting to note that His95 and Lys568 sit on top of a putative cationic tunnel connecting the active site to the solvent (Fig. 4D). Interestingly, histidine residues that are highly conserved among CODHs line this tunnel. Finally, redox active sites must be present on the enzyme to accept the two electrons produced by CO oxidation. Identifying the electron acceptors has been a point of much discussion (2, 5, 7, 8, 11–13, 32). The discovery of a D-cluster provides insight into the issue of electron translocation within CODH. With two locations to deposit electrons (B- and D-clusters) in addition to the C-cluster, the issue of electron distribution now seems less problematic.
Although the cofactors are completely different, the proposed mechanism of the aerobic CODHs is also believed to involve deprotonation of the metal-bound H2O molecule and a subsequent hydroxide attack on CO. In this case, it is believed that CO binds to an S-selanylcysteine residue, and the H2O molecule is bound and activated by Mo (24).
This structure of R. rubrum CODH provides a three-dimensional view of an Ni-Fe-S cluster capable of catalyzing the biological oxidation of CO. The roles of Ni and Fe in the CO oxidation reaction have intrigued chemists for decades. The possibility that an [Fe4S4] moiety was directly involved in substrate binding and catalysis captured attention. The results presented here suggest that the architecture of the cube is Fe-[NiFe3S4], indicating that a classic [Fe4S4] moiety is not involved directly in catalyzing CO oxidation. Although nonnative [NiFe3S4] clusters have been prepared in proteins such as Pyrococcus furiosus ferredoxin (33), such a cluster in CODH would represent the first example of a naturally occurring Ni-Fe-S cubane. This assembly of Ni, Fe, and S may be a relic of a time when CO and CO2 were predominant in the environment, and organisms needed to find ways to derive energy from these carbon sources to survive.
Acknowledgments
We thank Thomas Earnest (Advanced Light Source), Gerry McDermott (Advanced Light Source), Peter Kuhn (SSRL), Tim McPhillips (SSRL), Tina Iverson, and Patricia Takahara for help with data collection; Nathan Spangler for help with protein purification; and Tzanko Doukov for help with crystal activity assays. The data collection facilities at SSRL, the National Synchrotron Light Source, Advanced Light Source, and Advanced Photon Source are funded by the U.S. Department of Energy. In addition, the use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under Grant RR07707. SSRL is also funded by the National Institutes of Health Biomedical Research Technology Program, Division of Research Resources. The crystallization and initial structure determination were conducted at the California Institute of Technology in the laboratory of Professor Douglas C. Rees and supported by National Institutes of Health Grants F32 GM19044 (to C.L.D.) and GM45162 (to D. C. Rees), and by the Department of Energy Sciences Grant DE FGO2-87ER13691 (to P.W.L.). Support for the final stages of the structure determination was provided by the John Templeton Foundation Grant COS182 (to C.L.D.).
Abbreviations
- CODH
carbon monoxide dehydrogenase
- CODH/ACS
CODH/acetyl CoA synthase
- EXAFS
extended x-ray absorption fine structure
- FCII
the unique Fe site on the CODH enzyme
- COL
CO ligand
- SSRL
Stanford Synchrotron Radiation Laboratory
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1JQK).
References
- 1.Uffen R L. Proc Natl Acad Sci USA. 1976;73:3298–3302. doi: 10.1073/pnas.73.9.3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ragsdale S W, Kumar M. Chem Rev. 1996;96:2515–2539. doi: 10.1021/cr950058+. [DOI] [PubMed] [Google Scholar]
- 3.Bartholomew G W, Alexander M. Appl Environ Microbiol. 1979;37:932–937. doi: 10.1128/aem.37.5.932-937.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kasting J F. Science. 1993;259:920–926. doi: 10.1126/science.11536547. [DOI] [PubMed] [Google Scholar]
- 5.Seravalli J, Kumar M, Lu W-P, Ragsdale S W. Biochemistry. 1995;34:7879–7888. doi: 10.1021/bi00024a012. [DOI] [PubMed] [Google Scholar]
- 6.Ensign S A, Ludden P W. J Biol Chem. 1991;266:18395–18403. [PubMed] [Google Scholar]
- 7.Hu Z, Spangler N J, Anderson M E, Xia J, Ludden P W, Lindahl P A, Münck E. J Am Chem Soc. 1996;118:830–845. [Google Scholar]
- 8.Anderson M E, Lindahl P A. Biochemistry. 1996;35:8371–8380. doi: 10.1021/bi952902w. [DOI] [PubMed] [Google Scholar]
- 9.Ralston C Y, Wang H X, Ragsdale S W, Kumar M, Spangler N J, Ludden P W, Gu W, Jones R M, Patil D S, Cramer S P. J Am Chem Soc. 2000;122:10553–10560. [Google Scholar]
- 10.Tan G O, Ensign S A, Ciurli S, Scott M J, Hedman B, Holm R H, Ludden P W, Korszun Z R, Stephens P J, Hodgson K O. Proc Natl Acad Sci USA. 1992;89:4427–4431. doi: 10.1073/pnas.89.10.4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heo J, Staples C R, Telser J, Ludden P W. J Am Chem Soc. 1999;121:11045–11045. [Google Scholar]
- 12.DeRose V J, Telser J, Anderson M E, Lindahl P A, Hoffman B M. J Am Chem Soc. 1998;120:8767–8776. [Google Scholar]
- 13.Staples C R, Heo J, Spangler N J, Kerby R L, Roberts G P, Ludden P W. J Am Chem Soc. 1999;121:11034–11044. [Google Scholar]
- 14.Heo J, Staples C R, Halbleib C M, Ludden P W. Biochemistry. 2000;39:7956–7963. doi: 10.1021/bi992958g. [DOI] [PubMed] [Google Scholar]
- 15.Bonam D, Ludden P W. J Biol Chem. 1987;262:2980–2987. [PubMed] [Google Scholar]
- 16.Georgiadis M M, Komiya H, Chakrabarti P, Woo D, Kornuc J J, Rees D C. Science. 1992;257:1653–1659. doi: 10.1126/science.1529353. [DOI] [PubMed] [Google Scholar]
- 17.Otwiniwski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 18.Terwilliger T C. Acta Crystallogr D. 1994;50:17–23. doi: 10.1107/S0907444993008236. [DOI] [PubMed] [Google Scholar]
- 19.De la Fortelle E, Bricogne G. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 20.Cowtan K. Protein Crystallogr. 1994;31:34–38. [Google Scholar]
- 21.Laskowski R A, McArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 22.Brunger A T, Adams P D, Clore G M, De Lano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Barondeau P, Lindahl P A. J Am Chem Soc. 1997;199:3959–3970. [Google Scholar]
- 24.Dobbek H, Gremer L, Meyer O, Huber R. Proc Natl Acad Sci USA. 1999;96:8884–8889. doi: 10.1073/pnas.96.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arendsen A F, Hadden J, Card G, McAlpine A S, Bailey S, Zaitser V, Duke E H M, Lindley P F, Kro Kel M, Trautwein A X, et al. J Biol Inorg Chem. 1998;3:81–95. [Google Scholar]
- 26.Page C C, Moser C C, Chen X, Dutton P L. Nature (London) 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 27.Kerby R L, Hong S S, Ensign S A, Coppoc L J, Ludden P W, Roberts G P. J Bacteriol. 1992;174:5284–5294. doi: 10.1128/jb.174.16.5284-5294.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia J, Sinclair J F, Baldwin T O, Lindahl P A. Biochemistry. 1996;35:1965–1971. doi: 10.1021/bi9511853. [DOI] [PubMed] [Google Scholar]
- 29.Rosenzweig A C, Nordlund P, Takahara P M, Frederick C A, Lippard S J. Chem Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- 30.Heo J, Halbleib C M, Ludden P W. Proc Natl Acad Sci USA. 2001;98:7690–7693. doi: 10.1073/pnas.141230698. . (First Published June 19, 2001; 10.1073/pnas.141230698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heo J, Staples C R, Ludden P W. Biochemistry. 2000;40:7604–7611. doi: 10.1021/bi002554k. [DOI] [PubMed] [Google Scholar]
- 32.Anderson M E, Lindahl P A. Biochemistry. 1994;33:8702–8711. doi: 10.1021/bi00195a011. [DOI] [PubMed] [Google Scholar]
- 33.Conover R C, Park J-B, Adams M W, Johnson M K. J Am Chem Soc. 1990;112:4562–4564. [Google Scholar]
- 34.Carson M. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]